Molecular iodine-catalyzed multicomponent reactions: an efficient catalyst for organic synthesis
Yi-Ming Ren*a, Chun Cai*b and Ren-Chun Yanga
aDepartment of Biochemical Engineering, Anhui Polytechnic University, Wuhu, 241000, P. R. China. E-mail: yimingren@ahpu.edu.cn; Fax: +86-553-2871255; Tel: +86-553-2871255
bChemical Engineering College, Nanjing University of Science & Technology, Nanjing, 210094, P. R. China. E-mail: c.cai@mail.njust.edu.cn; Fax: +86-25-84315030; Tel: +86-25-84315514
First published on 6th February 2013
Abstract
The multicomponent reactions (MCRs) consist of two or more synthetic steps which are carried out without isolation of any intermediate thus reducing time, saving money, energy and raw materials. The development of MCRs in the presence of molecular iodine is an efficient approach that meets with the requirements of sustainable chemistry. The aim of this review is to highlight the synergistic effect of the combined use of MCRs and molecular iodine for the development of new eco-compatible methodologies for organic chemistry.
 Yi-Ming Ren | Yi-Ming Ren received his Ph.D. degree from the Nanjing University of Science & Technology in 2009 under the supervision of the Professor Chun Cai. He is currently an Associate Professor in the Department of Biochemical Engineering at Anhui Polytechnic University, Wuhu, Anhui, China. His research interests center on Green Chemistry with an emphasis on iodine catalysis and multicomponent reactions. |
 Chun Cai | Chun Cai is a Professor and the Director for Department of Pharmaceutical and Fine Chemicals at Nanjing University of Science & Technology, Nanjing, China. He received his Undergraduate Diploma in Organic Synthesis (1988) and his Ph.D. in Energetic Materials (1993), both at East China Institute of Technology. He was a visiting scientific co-worker at the Australian National University (2002–2003). His research interests center on Green Chemistry with an emphasis on fluorous biphasic catalysis, fluorine chemistry, metal catalysis, organometallic chemistry and iodine catalysis. He has published over 150 scientific papers. |
 Ren-Chun Yang | Ren-Chun Yang was born in Anhui, China, in 1979. He received a Ph.D. degree in material science from University of Science and Technology Beijing (with Professor Xiao-Gang Li) in 2010 and then worked as an instructor at Anhui Polytechnic University in Wuhu. His research interests include catalysis, environmental protection, and porous materials. |
1. Introduction
The length of a synthesis is dependent upon the average molecular complexity produced per operation, which depends in turn on the number of chemical bonds being created. Therefore, devising reactions that achieve multi-bond formation in one operation is becoming one of the major challenges in searching for step-economic syntheses. Multicomponent reactions (MCRs) processes, in which three or more reactants are combined in a single chemical step to produce products that incorporate substantial portions of all the components, naturally comply with many of these stringent requirements for ideal organic syntheses.1In fact, there have been various classification systems for MCRs. For example, three components can be divided into three different categories depending on the nature of the starting materials (Fig. 1). ABC designates a MCRs involving three different reagents; ABB (or ABB′),2 a reaction that involves one molecule of reagent A and two molecules of reagent B; and finally, AAA, a reaction that involves three molecules of the same reagent.
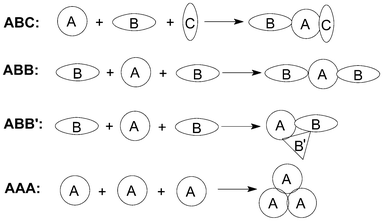 | ||
| Fig. 1 The classification of MCRs. | ||
In recent years, molecular iodine has been used for various organic transformations including Lewis acid catalyst.3–5 Owing to numerous advantages associated with this eco-friendly element, molecular iodine has been explored as a powerful catalyst for MCRs. To the best of our knowledge, reviews on the MCRs use of molecular iodine have not been reported recently. This article will be divided in two sections. The first one concerns the use of molecular iodine as catalyst for ABC(D..) MCRs, the second one presents molecular iodine catalyzed ABB and ABB′ MCRs.
2. Molecular iodine catalyzed ABC(D..) multicomponent reactions
2.1 Synthesis of highly substituted imidazoles from functionalized carbonyl compounds
Highly substituted imidazoles are very useful intermediates for the development of molecules of pharmaceutical or biological interest. The synthesis of highly substituted imidazoles via MCRs involving the cyclocondensation of a 1,2-diketone (or a-hydroxy ketones), an aldehyde and ammonia or ammonium acetate is a useful process. The catalytic activity of molecular iodine is remarkable and the use of low cost, commercially available iodine as catalyst for the synthesis of highly substituted imidazoles in excellent yields is also significant under the aspect of environmentally benign processes.Kidwai et al.6,7 reported the one-pot synthesis of 2,4,5-triarylimidazoles 1 using benzil (or benzoin), aldehydes and ammonium acetate using molecular iodine as catalyst under 75 °C (Scheme 1). The condensation of benzil, benzaldehyde and ammonium acetate in the presence of 5 mol% of catalyst give 99% yield of 2,4,5-triphenylimidazole after 15 min using ethanol as solvent. However, a longer reaction time (2 h) and more amount catalyst (10 mol%) would be necessary using benzoin as reagent, because the benzoin undergoes aerial oxidation to benzil under these conditions.
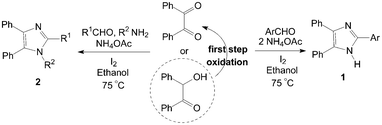 | ||
| Scheme 1 Iodine catalyzed synthesis of highly substituted imidazoles. | ||
The authors7,8 had also reported the one-pot synthesis of 1,2,4,5-tetraarylimidazoles 2 using benzil (or benzoin), aldehyde, amine and ammonium acetate under the same reaction conditions (Scheme 1). This method is effective for the preparation of 1,2,4,5-tetraarylimidazoles 2 from both electron efficient as well as electron deficient aromatic aldehydes. The aryl groups substituted with different groups and also the same groups located at different positions of the aromatic ring did not show any effect on the formation of 1,2,4,5-tetraarylimidazoles 2. Similarly, a longer reaction time (2 h) and more amount catalyst (10 mol%) would be necessary using benzoin as reactant. A series of some new tetrazolo[1,5-a]quinoline based tetrasubstituted imidazole derivatives had also been synthesized by a reaction of tetrazolo[1,5-a]quinoline-4-carbaldehyde, benzil, aromatic amine and ammonium acetate in the presence of iodine.9 And one-pot synthesis of 1H-phenanthro[9,10]imidazol-2-yl from phenantherenquinone, aldehydes and ammonium acetate using molecular iodine as catalyst in ethanol was also described.10
With the increasing public concern over environmental degradation, the use of environmentally benign solvents like water and solvent-free reactions represent very powerful green chemical technology procedures from both the economical and synthetic point of view. We11 reported the synthesis of highly substituted imidazoles 1 and 2 by the reaction of 1,2-diketone, aldehydes and ammonium acetate or via one-pot four-component condensation of 1,2-diketone, aldehydes, amines and ammonium acetate in the presence of a catalytic amount of molecular iodine under solvent-free conditions. And Jayabharathi et al.12 had also reported synthesis of 2,4,5-triarylimidazoles 1 under solvent-free conditions. The advantages of the “solvent-free” method are the elimination of the metals, organic solvents and toxic reagents, operational simplicity and high yields of products.
We13 had also reported the synthesis of 1,2,4,5-tetraarylimidazoles 2 by three-component condensation of benzil, benzonitrile derivatives and primary amines in the presence of a catalytic amount of molecular iodine (10 mol%) under solvent-free conditions (Scheme 2). It was found that primary aliphatic amines reacted efficiently with benzil and benzonitrile derivatives promoted by molecular iodine within 1 h. However, the reaction failed to proceed with aniline derivatives under the same reaction conditions, this may be due to the conjugated Schiff base formation by aniline and carbonyl being relatively stable and difficult to further react with the benzonitrile derivatives.
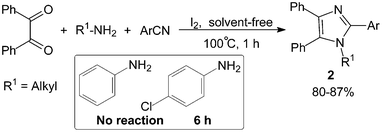 | ||
| Scheme 2 Iodine catalyzed solvent-free synthesis of 1,2,4,5-tetraarylimidazoles. | ||
2.2 Biginelli reaction and Biginelli-like reaction
Dihydropyrimidinones and their sulfur analogues have attracted much attention in previous years due to the large range of biological activities such as calcium channel blockers, antiviral, antitumor, and anti-inflammatory drugs. via the Biginelli reaction, the synthesis of 3,4-dihydropyrimidin-2(1H)-ones and thione have received renewed interest, and several improved procedures have recently been reported. Bhosale et al.14 reported the synthesis of 3,4-dihydropyrimidin-2(1H)-ones by iodine-catalyzed cyclocondensation of aldehyde, ethyl acetoacetate and urea. The reaction was carried out in toluene at reflux temperature for 3–5 h with high yields. This one-pot protocol has a simple work-up with excellent yields for substituted aromatic aldehydes. For example, the reaction proceeded very cleanly to afford 3 containing both guanosine and dihydropyrimidine moieties in 90% yield (Scheme 3). However, the cyclocondensation with aliphatic aldehydes such as n-butanal and n-hexanal were sluggish under the present reaction conditions and afforded the corresponding dihydropyrimidin-2(1H)-ones in 76% and 69% yields, respectively.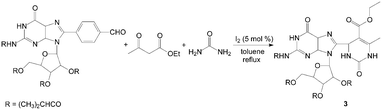 | ||
| Scheme 3 Iodine catalyzed synthesis of 3,4-dihydropyrimidin-2(1H)-ones. | ||
The three-components, aldehyde, 1,3-dicarbonyl compound and urea or thiourea were refluxed under N2 in the presence of iodine as catalyst using CH3CN as a solvent (Scheme 4, Method A).15 Most importantly aromatic aldehydes carrying either electron-donating or electron-withdrawing substituents reacted very well to give the desired products 4 in excellent yields. Even aliphatic aldehydes, which normally show poor yields in the Biginelli reaction afforded the products with high yields.16 Thiourea also reacted in a similar manner like urea.
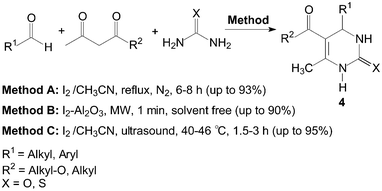 | ||
| Scheme 4 Iodine catalyzed synthesis of 3,4-dihydropyrimidin-2(1H)-ones and thione. | ||
Meanwhile, some instruments were employed for the Biginelli reaction using molecular iodine as catalyst (Scheme 4, Method B and C). Saxena et al.17 reported a quick and efficient one-pot method for the three component condensation of an aldehyde, urea/thiourea and a 1,3-dicarbonyl compound to synthesize 3,4-dihydropyrimidin-2(1H)-ones and thione 4 using iodine–alumina as the catalyst under microwave irradiation. The reactions were completed thoroughly and irradiated in a Prolabo fixed focus microwave reactor at a temperature of 90 °C for 1 min. Wang et al.18 reported the Biginelli reaction catalyzed by iodine in CH3CN under ultrasound irradiation. When the frequency was 25 kHz, the condensation of 4-chlorobenzaldehyde, ethyl acetoacetate and urea gave the desired product in 95% yield within 2 h using iodine (40 mol%) as catalyst.
However, the scope of substrates for the Biginelli reaction is limited to aromatic aldehydes, acetoacetate (or acetylacetone) and urea or thiourea. Zalavadiya et al.19 described the synthesis of some new 3,4-dihydropyrimidin-2(1H)-ones 5 from N-(3-chloro-4-fluorophenyl)urea (Scheme 5). Furthermore, the first Biginelli-like reaction, reported by Wang et al.20 was conducted in CH3CN by condensation of aldehydes, ketones, and urea, using FeCl3·6H2O and TMSCl as catalysts, which remarkably broadened the Biginelli reaction. We21 reported the synthesis of 5-unsubstituted-3,4-dihydropyrimidin-2(1H)-ones 6via the Biginelli-like reaction in the presence of molecular iodine under solvent-free conditions (Scheme 5). The results showed that both various aromatic aldehydes and several aromatic ketones were converted to the corresponding products 6 in good yields and in a short time. However, the dehydrogenated products 7 were obtained when aromatic aldehydes with electron-donating groups, such as methoxy, methyl, were used (Scheme 6).21,22
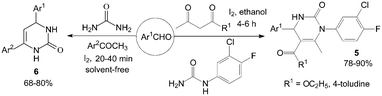 | ||
| Scheme 5 Iodine catalyzed synthesis of new 3,4-dihydropyrimidin-2(1H)-ones and 5-unsubstituted-3,4-dihydropyrimidin-2(1H)-ones. | ||
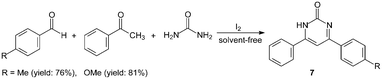 | ||
| Scheme 6 Iodine catalyzed Biginelli-like reaction. | ||
Cyclic 1,3-dicarbonyl compounds were also employed with urea and aromatic aldehydes to produce spiropyrimidines 8 and dihydropyrimidinones 9 under microwave irradiation in a solvent-free condition (Scheme 7).23 Notably, The Biginelli-like reactions were carried out in presence of 1 mol% iodine under microwave irradiation within 4.5 min.
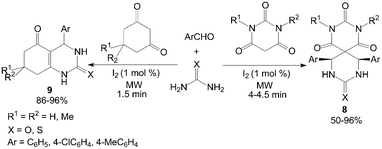 | ||
| Scheme 7 Iodine catalyzed Biginelli-like reaction. | ||
2.3 Synthesis of 2,4,6-triarylpyridines
One-pot, three-components synthesis of 2,4,6-triarylpyridines 10 were performed under solvent-free conditions using molecular iodine as the catalyst (Scheme 8).21 The reaction medium had an influence on the reaction; the results showed that solvent-free conditions provided better yield than if solvents were used (Table 1). And the results showed that both electron-deficient and electron-rich aromatic aldehydes were converted to the corresponding 2,4,6-triarylpyridines 10 in moderate yields (48–61%).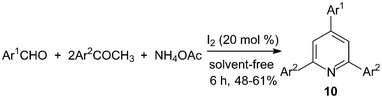 | ||
| Scheme 8 Iodine catalyzed synthesis of the 2,4,6-triarylpyridines. | ||
| Entry | Solvent | T (°C) | Yield (%) |
|---|---|---|---|
| 1 | — | 120 | 56 |
| 2 | EtOH | 78 | 22 |
| 3 | CH3CN | 82 | 17 |
| 4 | DMF | 140 | 9 |
| 5 | CH2Cl2 | 40 | 10 |
| 6 | Et3N | 90 | Tace |
| 7 | PhMe | 110 | 11 |
2.4 Synthesis of Hantzsch 1,4-dihydropyridine compounds
In recent years, an increasing interest has been focused on the synthesis of Hantzsch 1,4-dihydropyridine compounds (1,4-DHPs) owing to their significant biological activity. Ko et al.24 reported a novel synthesis of 1,4-DHPs 11 and 12 promoted by the catalytic amount of iodine under ambient conditions with excellent yields (Scheme 9). Both aliphatic and aromatic aldehydes react equally good to give the products with excellent yields. The aryl group substituted with different groups and same groups located at different positions of the aromatic ring has not shown much effect on the formation of the final product.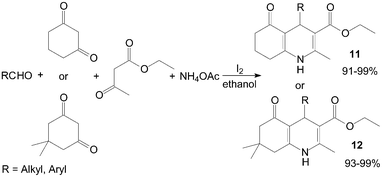 | ||
| Scheme 9 Iodine catalyzed synthesis of the 1,4-DHPs. | ||
The first example of the synthesis of different kinds of methyl-1-hydroxyethyl-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylates 13 and bis(methy-1-hydroxyethyl-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylates) 14 under mild and solvent-free conditions was reported by Zolfigol et al. (Scheme 10).25 Although reactions occurred both in the presence and in the absence of molecular iodine but iodine catalyzed the described reaction efficiently and also improved the reactions yields (Table 2). Meanwhile, the reaction times and yields of reactions were not seriously changed but purification of crude products of the method A was more easier than methods B and C. Similarly, aniline also was employed for synthesis of N-aryl-1,4-dihydropyridine 15 and 16 with good yields (Scheme 11).26,27
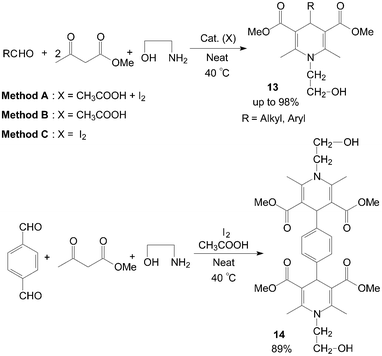 | ||
| Scheme 10 Synthesis of the N-hydroxyethyl 1,4-dihydropyridine compounds. | ||
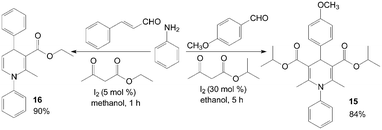 | ||
| Scheme 11 Synthesis of N-substituted 1,4-DHPs using aniline as reagent. | ||
| R | Time (h) | Yield (%) | ||||
|---|---|---|---|---|---|---|
| Method A | Method B | Method C | Method A | Method B | Method C | |
| 4-Cl-C6H4 | 0.75 | 2 | 0.75 | 85 | 60 | 84 |
| 2-Furyl | 3 | 4.25 | 3 | 89 | 73 | 89 |
2.5 Strecker-type reaction
α-Aminonitriles are usually synthesized by the nucleophilic addition of a cyanide anion to imines. The Strecker-type reaction provides one of the most efficient methods for the synthesis of α-aminonitriles 17 and 18 (Scheme 12). The treatment of benzaldehyde and benzyl amine with TMSCN in the presence of a catalytic amount of iodine afforded 2-(N-benzylamino)-1-phenylacetonitrile in 94% yield using acetonitrile as solvent at room temperature.28 Wang et al.29 reported a mild and convenient method for Strecker-type reactions of aldehydes, amines and tributyltin cyanide (Bu3SnCN) under the same reaction conditions. A longer reaction time (1–8 h) would be necessary using TMSCN as reagent.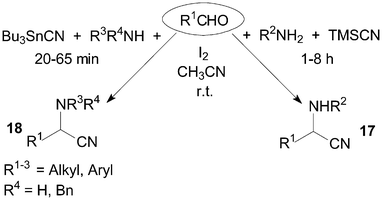 | ||
| Scheme 12 Iodine catalyzed Strecker-type reaction. | ||
2.6 Synthesis of homoallyl ethers and protected homoallylic amines
Aldehydes and aldimines react with allyltrimethylsilane in the presence of a catalytic amount of iodine to give the corresponding protected homoallyl amines 19 efficiently (Scheme 13).30 In general, excellent yields of homoallyl amines 19 were obtained with 10 mol% of iodine at room temperature in acetonitrile. Both aromatic and aliphatic aldehydes undergo homoallylation with 66–82% yield irrespective of the nature of the substrate. Interesting, in this research, no traces of the corresponding homoallylic alcohol 20 resulting from direct addition of allyltrimethylsilane to the aldehyde were observed, even if the article by Yadav et al.31 had shown that iodine could efficiently catalyse the allylation of aldehydes with allyltrimethylsilane in acetonitrile at 0 °C to afford the corresponding homoallyl alcohols 20 in high yields in about 1 min (Scheme 13).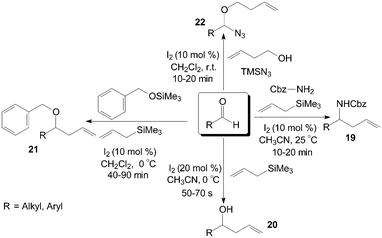 | ||
| Scheme 13 Synthesis of the homoallyl ethers and protected homoallylic amines. | ||
Various homoallyl benzyl ethers 21 were synthesized in moderate to high yields by three-component condensation of aldehydes, benzyloxytrimethylsilane, and allyltrimethylsilane in presence of iodine (10 mol%) in dichloromethane at 0 °C (Scheme 13).32 The reaction (based on benzaldehyde) was complete in 45 min with 86% yield of the corresponding homoallyl benzyl ether at 0 °C. However, the best result with a yield of 67% was obtained after 210 min of reaction at 27 °C. Yadav et al.33 also reported a direct approach for the one-pot preparation of a range of α-alkoxy azides 22 and homoallyl ethers 21 including benzyl, allyl, and propargyl ethers. In all cases, the reactions proceeded rapidly at room temperature under mild conditions (Scheme 13).
2.7 Mannich reaction
Iodine has been found to be very effective catalyst for a Mannich reaction34 between an aromatic aldehyde, an aromatic ketone and benzyl carbamate, even though this is a less reactive amine, to produce Cbz-protected β-aryl β-amino carbonyl compounds 23 in high yields (Scheme 14).35 According to a systematic study, moderate to high yields of β-amino carbonyl compounds 23 were obtained with 10 mol% of iodine at room temperature in acetonitrile for 24 h. Substrates bearing various functional groups such as CH3, OMe, Cl and Br all reacted successfully to produce the corresponding β-amino carbonyl compounds 23. To our surprise, ortho and para substituted aldehydes gave good results, meta-substituted aldehydes such as 3-methoxybenzaldehyde failed to yield any product.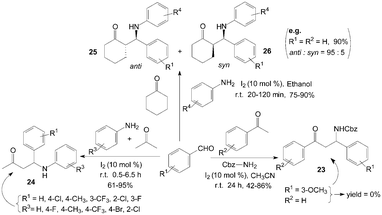 | ||
| Scheme 14 Iodine catalyzed Mannich reaction. | ||
Owing to some special biological activity of fluorinated compounds, Xia et al.36 described the direct three-component transformation into β-aminobutanones 24 containing fluorine atoms via iodine catalyzed coupling of aldehydes and amines in acetone (Scheme 14). There was remarkable electronic effect from the substituents on aniline moiety. For weak electron-withdrawing and electron-donating groups such as chloro- and methoxy, the three-component coupling could be executed smoothly in good yields within 1 h. And aliphatic amines were inert and unable to undergo such direct Mannich reactions. When butylamine was used with 4-fluorobenzaldehyde in acetone under 5 mol% iodine catalysis, none of expected product was obtained except the recycled substrates.
Recently, a variety of β-amino ketones 25 and 26 were readily prepared in high yields and with good to excellent anti selectivity under extremely mild conditions by means of three component coupling of aromatic aldehydes, aryl amines and ketones using 10 mol% of molecular iodine in ethanol (Scheme 14).37
β-Amino ketones 27–30 were synthesized via a three-component reaction of benzaldehyde, aniline and silyl enol ether in the presence of catalytic amount of iodine at room temperature (Scheme 15).38 All reactions were completed within three hours at room temperature, and corresponding products were obtained in good to excellent yields from aromatic aldehydes and aniline. However, no reaction was observed with aliphatic amines or aldehydes, the authors attribute this to the slow formation and unstable nature of the imine formed from either the aliphatic amine examined or the aliphatic aldehyde.
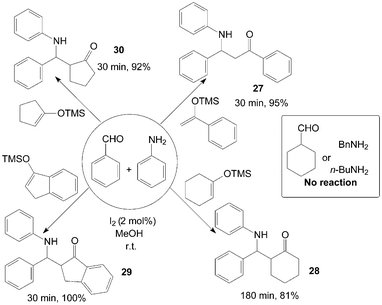 | ||
| Scheme 15 Iodine catalyzed synthesis of the β-amino ketone. | ||
Kataki et al.39 described the Mannich-type reaction of aldehydes, benzyl carbamate and silyl ketene acetal in a three component condensation to afford the corresponding Cbz protected β-amino esters 31 in moderate to high yields (Scheme 16). And Villano et al.40 reported the one-pot, two steps synthesis of multifunctionalized β-ketoesters 32 using Chan's diene as substrate.
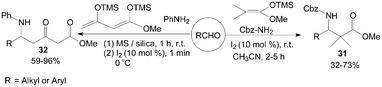 | ||
| Scheme 16 Iodine catalyzed Mannich-type reaction. | ||
A variety of functionalized flavanone 33 and tetrahydropyrimidine derivatives 34 were achieved under remarkably mild conditions by Mannich type reaction (Scheme 17).41 The reaction for synthesis of flavanone 33 did not work when no amine was used. After systematic screening, the reaction trend suggests that the formation of imine as well as the elimination of amine is the key steps involved in the formation of flavanone derivatives. During the screening of the reaction conditions with various amines for synthesis of flavanone 33, the authors obtained an interesting five component condensed products 34, which comprised two aldehyde units, two ammonia molecules, and one 2-hydroxyacetophenone moiety. In the paper, plausible reaction mechanisms for the formation of flavanone 33 and tetrahydropyrimidine derivatives 34 were proposed as well.
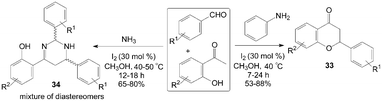 | ||
| Scheme 17 Synthesis of flavanone and tetrahydropyrimidine by Mannich type reaction. | ||
One-pot synthesis of primary 1-aminophosphonates 35 from coupling reaction of aldehydes/ketones, HMDS (1,1,1,3,3,3-hexamethyldisilazane) and diethyl phosphite under solvent-free conditions was developed by Sobhani et al. (Scheme 18).42
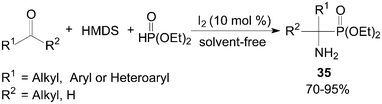 | ||
| Scheme 18 Synthesis of the primary 1-aminophosphonate compounds. | ||
Recently, Jia et al.43,44 had reported that iodine could effectively catalyze the double Mannich reaction. A series of 4-piperidones 36–38 were prepared by treatment of aromatic aldehydes, amines with ketones (Scheme 19). In the “double” Mannich reaction, four new bonds could be formed via only one transformation with high bond forming efficiency.
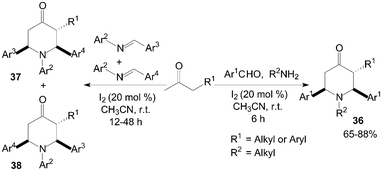 | ||
| Scheme 19 Iodine catalyzed double Mannich reaction. | ||
2.8 Synthesis of quinoline and quinolinone derivatives from aldehydes and amines
Many quinoline and quinolinone derivatives from aldehydes and amines were prepared under mild conditions using molecular iodine as catalyst (Scheme 20). The diastereoselective synthesis of trans-endo-decahydroquinolin-4-one 39via a three-component reaction of aldehydes, anilines and 1-acetylcyclohexene in the presence of iodine at room temperature was described by Lin et al.45 The experiment results showed that most of the aldehydes reacted readily to produce the products 39 in excellent yields and high diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Cyclohexenone was also employed with benzaldehyde and aniline under the same conditions. 3-Exo-phenyl-2-phenyl-2-azabicyclo[2.2.2]octan-5-one 40 and 3-endo-phenyl-2-phenyl-2-azabicyclo[2.2.2]octan-5-one isomers 41 were obtained in high yields (Scheme 20). Furthermore, a two-step, one-pot synthesis of 2-arylquinolines 42 from an aniline, 4-bomobenzaldehyde and decanal was examined.46
:1). Cyclohexenone was also employed with benzaldehyde and aniline under the same conditions. 3-Exo-phenyl-2-phenyl-2-azabicyclo[2.2.2]octan-5-one 40 and 3-endo-phenyl-2-phenyl-2-azabicyclo[2.2.2]octan-5-one isomers 41 were obtained in high yields (Scheme 20). Furthermore, a two-step, one-pot synthesis of 2-arylquinolines 42 from an aniline, 4-bomobenzaldehyde and decanal was examined.46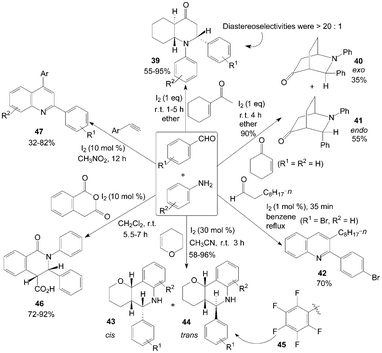 | ||
| Scheme 20 Synthesis of quinoline and quinolinone derivatives from aromatic aldehydes and aniline derivatives. | ||
The first example of the preparation for pyrano[3,2-c]quinolines 43 and 44via an iodine-catalyzed three-component reaction of aldehydes, anilines, and 2,3-dihydropyran in a one-pot operation was described by Xia et al.47 and Rai et al.48 (Scheme 20). The most products were obtained as cis/trans isomers without any other isomer detected except for using 3-chloroaniline or 3-trifluoromethylaniline as reagent, and the trans isomer 44 was always the major product. Similarly, Jin et al.49 had also reported synthesis of 2-perfluorophenyl tetrahydroquinolines 45 from pentafluorophenylaldehyde, anilines, and dihydropyran in CF3CH2OH by stirring for 14 h at room temperature using molecular iodine as catalyst (15 mol%). Similarly, the dihydrofuran was also employed for synthesis of furo[3,2-c]quinolines using molecular iodine as catalyst.50
Yadav et al.51 reported the direct synthesis of highly substituted cis-oxoisoquinolinecarboxylic acids 46 from homophthalic anhydride, aromatic aldehydes and anilines in the presence of 10 mol% molecular iodine under neutral conditions (Scheme 20). It is very interesting that the only products were obtained as a cis-diastereomer.
Recently, a one-pot synthesis of quinolines 47via iodine-catalyzed and air-mediated tandem condensation/imino-Diels–Alder/isomerization/oxidation of simple readily available amines, aldehydes, and alkynes was investigated (Scheme 20).52 Based on the experimental results, the authors proposed a possible mechanism for the formation of quinolines 47. In the possible mechanism, via imino-Diels–Alder reaction, intermediate is oxidized finally by O2 in air to give the quinolines 47.
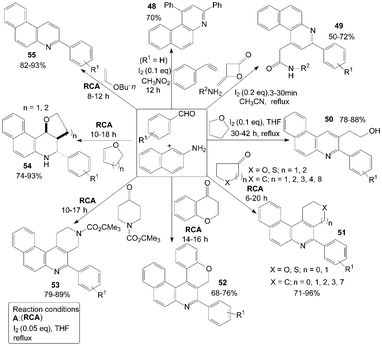
Furthermore, naphthalene-2-amine was employed for the synthesis of a variety of functionalized quinolines (Scheme 21). According to the route of synthesis of 47, benzo[f]quinoline 48 was produced in 70% yield by three-component reaction of naphthalenamine, phenylacetylene and benzaldehyde in a one-pot operation.52
 | ||
| Scheme 21 Synthesis of quinolines from aromatic aldehydes and naphthalenamine. | ||
The domino synthesis of benzo[f]quinolinyl 49 from diketene, amines, aromatic aldehydes and naphthalene-2-amine was developed by Zeng et al. (Scheme 21).53 Meanwhile, the transformation proceeded successfully and benzo[h]quinolinyl acetamides were obtained when 4-chlorobenzylaldehye or 4-methoxybenzaldehyde were employed with naphthalene-1-amine under the same conditions. However, because of the steric hindrance of ortho-substitution, the reaction was failed when 2-chlorobenzaldehyde was employed.
Initially, the unexpected products 50 were confirmed as “byproducts” in the reaction of 4-bromobenzaldehyde with naphthalen-2-amine in THF at room temperature to gain Schiff base.54 In a systematic study, an efficient synthesis of 2-(3-arylbenzo[f]quinolin-2-yl)ethanol derivatives 50 by an unusual THF-involved reaction of aromatic aldehyde and naphthalen-2-amine promoted by iodine was demonstrated with good yields (Scheme 21). But the authors failed to get the expected products when naphthalen-1-amine or p-toluidine was used. Owing to the activity of naphthalen-1-amine or p-toluidine is less than that of naphthalene-2-amine.
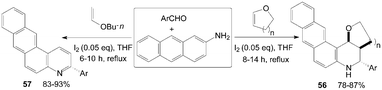
A convenient, effective, and eco-friendly process had been developed by Wang et al.55–58 leading to a library of quinolines derivatives, such as pyranoquinoline, thiopyranoquinoline, thienoquinoline, benzo(naphtho)quinoline and benzo[f]quinoline derivatives (51–57) (Scheme 21 and 22). The processes, which had similarly reaction conditions, provided a powerful tool toward the one-pot synthesis of diverse and complex compounds as well as small and drug-like heterocycles. Owing to their convergence and productivity, the novel synthetic method could attract considerable attention from the point of view of “combinatorial chemistry”.
 | ||
| Scheme 22 Synthesis of quinolines from aromatic aldehydes and anthracen-2-amine. | ||
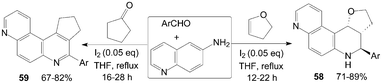
The quinolin-6-amine was also employed for synthesis of phenanthrolines 58 and 59 using molecular iodine as catalyst (Scheme 23).59,60
 | ||
| Scheme 23 Synthesis of phenanthrolines from aromatic aldehydes and quinolin-6-amine. | ||
Wang et al. also reported the synthesis of benzo[f]pyrimido[4,5-b]quinoline derivatives 60via one-pot three-component condensation of benzaldehydes, naphthalen-2-amine, and barbituric acid at room temperature in aqueous media (Scheme 24).61
![Synthesis of benzo[f]pyrimido[4,5-b]quinoline derivatives in aqueous media.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s24.gif) | ||
| Scheme 24 Synthesis of benzo[f]pyrimido[4,5-b]quinoline derivatives in aqueous media. | ||
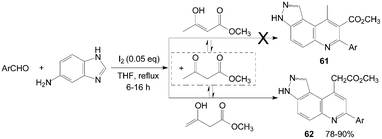
In an initial assumption, 3H-pyrazolo[4,3-f]quinoline-8-carboxylate derivatives 61 might be obtained in high yields by reaction of aromatic aldehyde, 1H-indazol-5-amine, and methyl 3-oxobutanoate as reactants (Scheme 25).62 However, the reaction gave 3H-pyrazolo[4,3-f]quinolin-9-yl acetate derivatives 62 in high yields and with high regio-selectivity rather than the products 61.
 | ||
| Scheme 25 Synthesis of quinolines from aromatic aldehydes and anthracen-2-amine. | ||
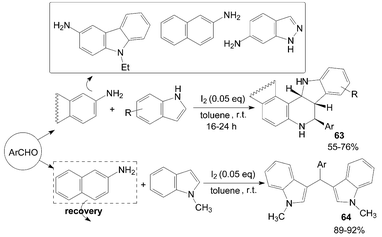
Wang et al.63 reported an iodine-catalyzed Povarov reaction of synthesis of exo-indolo[3,2-c]quinoline 63 using indole as dienophile carried out in toluene at room temperature (Scheme 26). N-methylindole was also chosen as a dienophile to react with aldehyde and naphthalen-2-amine under the same conditions. It is interesting that the above reaction gave 3-[(aryl)(1-methyl-1H-indol-3-yl)methyl]-1-methyl-1H-indole derivatives 64 in high yields.
 | ||
| Scheme 26 Iodine catalyzed Povarov reaction. | ||
A series of benzo[h]pyrazolo[3,4-b]quinolines 65 were also prepared from 3-methyl-1-phenyl-1H-pyrazol-5-amine, 2-hydroxynaphthalene-1,4-dione and aromatic aldehydes in the presence of molecular iodine in refluxing H2O (Scheme 27).64
![Synthesis of benzo[h]pyrazolo[3,4-b]quinolines from aromatic aldehydes.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s27.gif) | ||
| Scheme 27 Synthesis of benzo[h]pyrazolo[3,4-b]quinolines from aromatic aldehydes. | ||
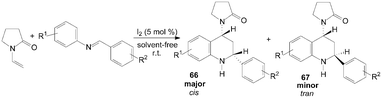
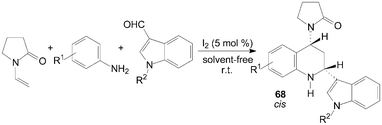
A two component aza-Diels–Alder cyclization of N-vinyl-2-pyrrolidinone with N-arylimine gave tetrahydroquinoline derivatives 66 and 67 in good yields and high stereo-selectivity under solvent-free conditions was described by Shen et al. (Scheme 28).65 Interestingly, the three components aza-Diels–Alder reaction of N-vinyl-2-pyrrolidinone, anilines and indole-3-carbaldehydes under the same condition afford only cis-product 68 in good yields (Scheme 29).65
 | ||
| Scheme 28 Iodine catalyzed two component aza-Diels–Alder reaction. | ||
 | ||
| Scheme 29 Iodine catalyzed three components aza-Diels–Alder reaction. | ||
2.9 Synthesis of quinolines, piperidines and β-amino carbonyl compounds from aldehydes, β-ketoesters and amines
A mild, efficient and highly selective method for the synthesis of benzo[f]quinoline derivatives 69via three-component reactions of aromatic aldehydes, naphthalen-2-amine and β-keto esters using iodine as catalyst in refluxing tetrahydrofuran (THF) was described (Scheme 30).66 Interestingly, in methanol, functionalized piperidines 70 and 72 were obtained via five-component reactions of 1,3-dicarbonyl compounds, amines and aromatic aldehydes at room temperature (Scheme 30).67 However, in case of diethylmalonate, the corresponding β-amino carbonyl compounds 71 was yielded under above reaction conditions (Scheme 30).67 This can be attributed to the lack of enolizable alkyl group in the β-position.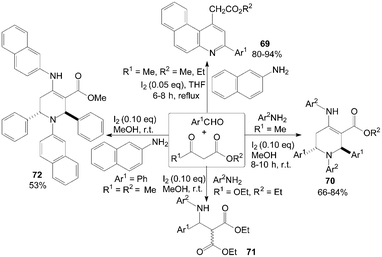 | ||
| Scheme 30 Synthesis of quinolines, piperidines and β-amino carbonyl compounds from aldehydes, β-ketoesters and amines. | ||
2.10 Synthesis of β-acetamido ketones, amidophenols and 4-amido tetrahydropyrans from carbonyl compounds and nitrile
A one-pot coupling of aromatic aldehydes, enolizable ketones or ketoesters, and acetonitrile in the presence of acetyl chloride to form β-acetamido ketones 73 at room temperature was described by Das et al. (Scheme 31).68 In the reaction, When propiophenones (instead of acetophenones68,69) were used in the reaction both anti and syn products were formed.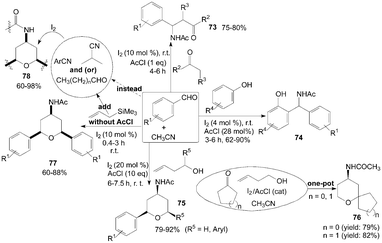 | ||
| Scheme 31 Synthesis of β-acetamido ketones, amidophenols and 4-amido tetrahydropyrans from carbonyl compounds and nitrile. | ||
Amidophenols derivatives 74 from acetonitrile, substituted phenol and aromatic aldehyde in the presence of catalytic amount of iodine and acetyl chloride were prepared under mild conditions (Scheme 31).70 The reaction was amenable to a wide variation in phenol and aldehydes. The reaction proceeded well irrespective of the presence of electron withdrawing or electron donating group on aromatic aldehydes and phenols.
Three-component coupling of carbonyl compounds, homoallylic alcohols and acetonitrile was achieved using iodine and acetyl chloride at ambient temperature via the Prins–Ritter reaction sequence to furnish 4-amido tetrahydropyrans 75 and 76 in high yields with all cis selectivity (Scheme 31).71 The formation of the products 75 and 76 could be explained by hemi-acetal formation followed by Prins-cyclization and subsequent Ritter amidation.
In the above reactions for the synthesis of compounds 73–76, the acetyl group of the products was derived from acetonitrile. The presence of acetyl chloride was vital. In the absence of acetyl chloride, the reaction did not proceed. The role of acetyl chloride for the formation of product is not clear.
Sakurai–Prins–Ritter reaction,72 which involved an aromatic aldehyde, allyltrimethylsilane and acetonitrile (without acetyl chloride), afforded the products of symmetrical 4-amido tetrahydropyrans 77 in satisfactory yields within a short time (Scheme 31). The reactions were also successful with aliphatic aldehydes and other nitriles (such as benzonitrile, 4-methyl benzonitrile, and isobutyronitrile) to produce the corresponding 4-amido tetrahydropyrans 78 in good yields.
2.11 Synthesis of amidoalkyl naphthols and bisnaphthols from carbonyl compounds and 2-naphthol
Similarly, based on the route of synthesis of 74 (Scheme 31), amidoalkyl naphthols 79 were obtained in 45–70% yields by three-component reaction of nitrile, aldehydes and 2-naphthol in the presence of catalytic amount of iodine and acetyl chloride (Scheme 32).70 However, only α-hydrogenated nitriles involved in the reaction and yielded the corresponding amidoalkyl naphthols 79, the reactions with aromatic nitriles did not yield the desired product, while bisnaphthols 80 were formed exclusively. The results showed that aromatic nitrile moiety was not involved in the reaction pathway.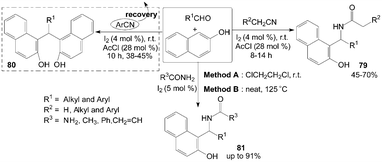 | ||
| Scheme 32 Synthesis of amidoalkyl naphthols and bisnaphthols from carbonyl compounds and 2-naphthol. | ||
Amidoalkyl naphthols 81 were readily prepared using urea or amides in high yields with two methods (Scheme 32).73 The yields of the products were somewhat higher in Method A but the conversion times were much lower in Method B (Table 3). Aromatic aldehydes underwent facile conversions but aliphatic aldehydes afforded the products in low yields (Table 3). On the other hand, the reactions with thiourea were considered,74 but no corresponding products were produced. Also, amines such as ethylamine and aniline were utilized and no aminoalkyl naphthol was obtained.
| Entry | R1 | R3 | Time (h) | Yield (%) | ||
|---|---|---|---|---|---|---|
| Method A | Method B | Method A | Method B | |||
| 1 | Ph | NH2 | 12 | 4.5 | 91 | 87 |
| 2 | C2H5 | NH2 | 26 | 9 | 35 | 20 |
2.12 Synthesis of tetrahydrobenzo[b]pyrans, 2-amino-2-chromenes and pyranopyrazoles from aromatic aldehydes with malononitrile
A convenient and efficient method for synthesis of tetrahydrobenzo[b]pyran 82via one-pot three-component condensation of aromatic aldehydes with malononitrile and 1,3-cyclic dimedones had been developed by using iodine/DMSO system (Scheme 33).75 The I2/K2CO3/KI/H2O system was introduced as an environmentally friendly catalyst system in three component coupling reactions of substituted 2-amino-2-chromenes 83 synthesis.76 The reaction for the synthesis 83 proceeds quickly (12–20 min) under very mild reaction conditions (aqueous medium). Various pyranopyrazoles 84 were also synthesized by an iodine catalyzed four component reaction at room temperature in water.77![Synthesis of tetrahydrobenzo[b]pyrans, 2-amino-2-chromenes and pyranopyrazoles from aromatic aldehydes with malononitrile.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s33.gif) | ||
| Scheme 33 Synthesis of tetrahydrobenzo[b]pyrans, 2-amino-2-chromenes and pyranopyrazoles from aromatic aldehydes with malononitrile. | ||
In the above reactions for the synthesis of compounds 82–83, the reactions occur via initial formation of arylidenemalononitrile 85 in quantitative yield by the Knoevenagel condensation78,79 of malononitrile to the aldehyde by loss of water molecules (Scheme 33). On the other hand, iodine catalyzed formation of pyrazolone 86 is expected to occur by the reaction between hydrazine hydrate and ethyl acetoacetate.77
2.13 Synthesis of quinazolinones and quinazolin-4-amines from isatoic anhydride or ortho esters
Isatoic anhydride reacted with ammonium acetate or primary amines and aldehydes in the presence of iodine and acetic acid to produce the corresponding quinazolinones 87 in moderate to good yields (Scheme 34).80 After systematic screening, a mixture of water and acetonitrile (1:1) was utilized as the solvent, and no quinazolinones 87 were formed in the absence of acetic acid when using amine as reactant. However, using ammonium acetate instead of amine in the reaction (without acetic acid) produced 2-substituted quinazolinones 88 successfully in higher yields and shorter times compared to disubstituted analogues.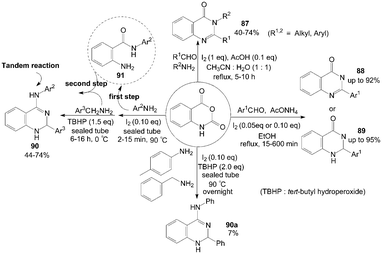 | ||
| Scheme 34 Synthesis of quinazolinones and quinazolin-4-amines from isatoic anhydride. | ||
Zeng et al.81 reported the same experiment results. The authors described an efficient one-pot three-component procedure for the selective synthesis of mono substituted quinazolin-4(3H)-ones 88 and 2,3-dihydroquinazolin-4(1H)-ones 89 using ammonium acetate instead of amine without acetic acid (Scheme 34). Initially, the unexpected product 88 was obtained using 20 mol% of molecular iodine as catalyst. Interestingly, the pure products 89 could be obtained with high yields when 5 mol% of molecular iodine as catalyst. The above result indicated that the catalytic iodine played the roles of both catalyst and oxidant. Similarly, under solvent-free conditions, one-pot synthesis of 2,3-dihydroquinazolin-4(1H)-ones 89 from coupling reaction of isatoic anhydride, aromatic aldehyde and ammonium acetate was developed by Rostamizadeh et al.82
Initially, Zeng et al.83 tried to combine three reactants isatoic anhydride, p-toluidine and benzylamine for synthesis of 2-phenyl-N-(p-tolyl)-1,2-dihydroquinazolin-4-amine 90a (Scheme 34). However, the one-pot three-component reaction gave a poor yield (only 7%) of product 90a. After a series of attempts, the authors found an efficient two step route for the synthesis of products 90 (but one pot, avoiding the isolation of intermediate 91) with moderate yields.
Furthermore, 3,4-dihydroquinazolin-4-ones 92 were also synthesized in high yields by three-component condensation of anthranilic acids, ortho esters and amines under solvent-free conditions (Scheme 35).84,85
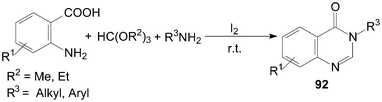 | ||
| Scheme 35 Synthesis of quinazolinones from ortho esters. | ||
2.14 Synthesis of tetrahydropyrimidines, pyrrolidines, tetrasubstituted pyrroles and dihydro-2-oxypyrroles from dialkylacetylene dicarboxylate
A short and simple synthesis of tetrahydropyrimidine 93 and pyrrolidine 94 derivatives were accomplished in good to excellent yields at room temperature by the reaction of dialkylacetylene dicarboxylate (DAAD) A, amines B, and formaldehyde C in the presence of molecular iodine at room temperature (Scheme 36).86,87 Interestingly, when the molar ratios of A, B and C were 1:2:4 and 1:1:4, tetrahydropyrimidine 93 and pyrrolidines 94 respectively, were formed.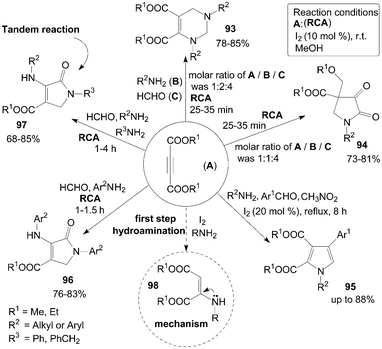 | ||
| Scheme 36 Synthesis of various compounds from DAAD. | ||
A series of 1,2,3,4-tetrasubstituted pyrroles 95 were subsequently prepared from aromatic aldehydes, amines, DAAD and nitromethane in the presence of molecular iodine as a catalyst (Scheme 36).88 However, only a trace amount of product was detected when aliphatic aldehyde or nitroethane was employed.
On the other hand, polysubstituted pyrroles 99 would also be prepared using 1,3-dicarbonyl compound instead of DAAD in the reaction (Scheme 37).89 The role of iodine in the present reaction was possibly the Michael addition reaction of intermediate β-enamino carbonyl compounds 100 with intermediate nitroalkenes 101 which was accelerated form intermediate 102 in presence of iodine.
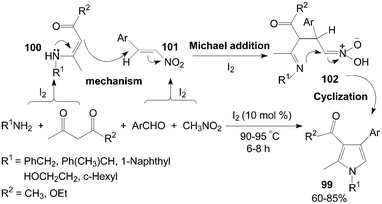 | ||
| Scheme 37 Synthesis of polysubstituted pyrroles from 1,3-dicarbonyl compounds. | ||
A one-pot four-component coupling of DAAD, amines, and formaldehyde in the presence of molecular iodine to form multi-functionalized dihydro-2-oxypyrrole derivatives 96 and 97 at room temperature was described by Khan et al. (Scheme 36).90 Initially, the reaction of DAAD, aniline derivatives, and formaldehyde for synthesis of products 96 were carried out in the presence of iodine in methanol with high yields. Next, the authors explored the methods using two different amines for the synthesis of different substituted dihydro-2-oxypyrroles 97 in high yields via the tandem reaction.
In the above reactions for the synthesis of compounds 93–97, the hydroamination of DAAD with amines could rapidly form the active intermediate 98 (Scheme 36), which would then undergo nucleophilic addition reaction (or Michael reaction, or Mannich type reaction) to afford the desired products.
According to the above plausible mechanism, the desirable different substituted dihydro-2-oxypyrrole derivatives 97 could be obtained by changing the sequence of addition of amine with DAAD due to the formation of selective hydroamination products 98 (Scheme 38). For example, the mixture of dimethylacetylene dicarboxylate (DMAD) and benzylamine was stirred for 10 min to afford intermediate 98a, and then aniline, formaldehyde, and iodine were added sequentially into the above reaction mixture. The product 97a was isolated in 83% yield. Comparatively, the intermediate 98b would be obtained firstly when DMAD and aniline was stirred for 10 min, and the product 97b was isolated in 68% yield under the same conditions.
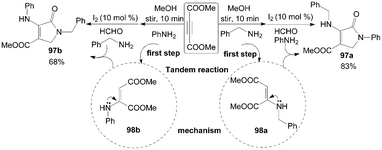 | ||
| Scheme 38 Synthesis of different substituted dihydro-2-oxypyrroles from DMAD. | ||
2.15 Synthesis of pyrimidine derivatives from 5-aminotetrazole
5-Aminotetrazole (5-AT) reacted with aromatic aldehydes and aromatic ketones in the presence of 10 mol% iodine in refluxing i-PrOH to produce the corresponding 5,7-diaryl-4,7-dihydrotetrazolo[1,5-a]pyrimidines 103 in moderate yields (Scheme 39).91 Most of the substrates selected, including para-substituted benzaldehydes and meta-substituted acetophenone, participated in the reaction smoothly, and the desired products 103 were obtained in moderate yields. However, the ortho-substituted benzaldehydes failed to provide the expected products.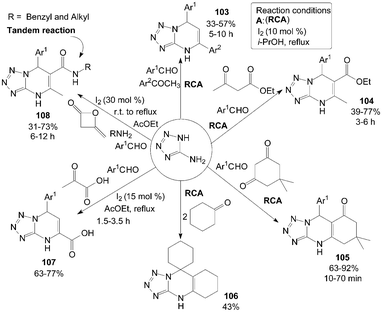 | ||
| Scheme 39 Synthesis of pyrimidine derivatives from 5-aminotetrazole. | ||
Under the above reaction conditions, using ethyl acetoacetate or dimedone instead of aromatic ketones in the reaction produced the desired products 104 and 105 successfully in moderate to high yields (Scheme 39).91 Notably, the product 106 was obtained in 43% yield via treating 5-AT and cyclohexanone (2 equiv.) under the previous modified conditions (Scheme 39).91
Zeng et al.91,92 also reported the direct synthesis of 5-aryl-5,8-dihydrotetrazolo[1,5-a]pyrimidine-7-carboxylic acids 107 from pyruvic acid, 5-AT and aromatic aldehydes in the presence of 15 mol% iodine in refluxing AcOEt (Scheme 39).91 Both electron-deficient and electron-rich aromatic aldehydes were smoothly converted to the corresponding products 107 in moderate to good yields. However, sterically hindered aromatic aldehydes bearing groups such as 2-NO2 and 2-Cl proceeded at a relatively slow rate and gave lesser yields than para-substituted aromatic aldehydes.
Four-component tandem procedure to prepare a series of dihydrotetrazolopyrimidinyl carbamides 108 starting from diketene, amines, 5-AT, and aromatic aldehydes was developed by Zeng et al. (Scheme 39).93 In the procedure of tandem reaction, a solution of amine and diketene was stirred in AcOEt at room temperature for 2 h. Then, the aldehyde, 5-AT and iodine were added, and the temperature was increased to 78 °C. However, the expected product was not detected in the final mixture in the case of anilines or aliphatic aldehydes.
2.16 Synthesis of spiro[indene-2,3′-piperidine] derivatives
Dai et al.94 described a facile, multi-component reaction involving condensation of ethyl trifluoroacetoacetate, 1,3-indanedione, ammonium acetate, and aromatic aldehydes in the presence of iodine at room temperature to afford a series of novel trifluoromethyl-containing spiro[indene-2,3′-piperidine] scaffolds 109 in moderate yields, along with the minor products 110, respectively (Scheme 40). Interestingly, the corresponding dehydrated products 111 as a major product in 69% yield, along with a minor dehydrated products 112 in 9% yield were produced when using non-fluorinated substrate ethyl acetoacetate instead of ethyl trifluoroacetoacetate in the reaction under the same reaction conditions.![Synthesis of spiro[indene-2,3′-piperidine] derivatives.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s40.gif) | ||
| Scheme 40 Synthesis of spiro[indene-2,3′-piperidine] derivatives. | ||
2.17 Synthesis of 7-arylbenzopyrano[1,3]diazepines and 3,3′-phenylmethylenebis-(4-hydroxycoumarin) from 4-hydroxycoumarin
A novel and efficient method for 7-arylbenzopyrano[1,3]diazepines scaffold 113 using molecular iodine as a catalyst in non-protic solvents was reported by Kidwai et al. (Scheme 41).95 The reactions in non-protic solvents like dichloromethane, acetonitrile and tetrahydrofuran leads to the formation of the products 113 with excellent yields. However, the surprising results were obtained when the same reaction was tried with protic polar solvents. In protic polar solvents like methanol, ethanol the condensation of 4-hydroxycoumarin with aromatic aldehydes facilitates bis adduct 3,3′-arylmethanebis-(4-hydroxycoumarin) 114. Thus, it is concluded that formation of the products 113 are favoured only in the non-protic solvents.![Synthesis of 7-arylbenzopyrano[1,3]diazepines and 3,3′-phenylmethylenebis-(4-hydroxycoumarin) from 4-hydroxycoumarin.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s41.gif) | ||
| Scheme 41 Synthesis of 7-arylbenzopyrano[1,3]diazepines and 3,3′-phenylmethylenebis-(4-hydroxycoumarin) from 4-hydroxycoumarin. | ||
2.18 Synthesis of pyrrolidin-2-one derivatives and (phenylmethylene)bis((4-methoxyphenyl)sulfane)
A simple and efficient three components domino reaction of γ-butyrolactam, aromatic aldehydes and substituted thiophenols catalyzed by iodine resulted in the formation of 1-((phenylthio)(phenyl)methyl)pyrrolidin-2-one derivatives 115 (Scheme 42).96 The reaction was allowed to proceed without adding thiophenol giving 1,1′-((2-bromophenyl)methylene)bis(pyrrolidin-2-one) product 116 (Scheme 42).97 Similarly, The reaction was also allowed to proceed without adding γ-butyrolactam it gives (phenylmethylene)bis((4-methoxyphenyl)sulfane) product 117 (Scheme 42).98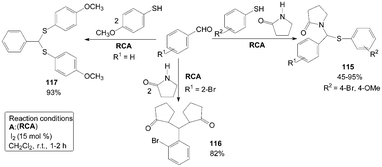 | ||
| Scheme 42 Synthesis of pyrrolidin-2-one derivatives and (phenylmethylene)bis((4-methoxyphenyl)sulfane). | ||
The authors think two mechanisms are possible for the synthesis of product 115 (Scheme 43). One possible mechanism is the formation of thiohemiacetal intermediate 118 in the first step and the second step is the attack of amide on intermediate 118 to yield the desired product 115. Another possible mechanism is the generation of an N-acyliminium ion intermediate 119 in the first step followed by the attack of nucleophile (thiophenol) on the intermediate 119 leading to the formation of the desired product 115. The reaction between γ-butyrolactam and benzaldehyde was stirred for 30 min firstly, and then thiophenol was added to the reaction mixture to get the product 115. However, added γ-butyrolactam to the reaction mixture (thiopheno and benzaldehyde) after 30 min but did not get the product 115. So, in the one-pot three-components reaction, the possible mechanism is iodine attacks the carbonyl oxygen of aldehyde and gives rise to the reaction, thereby carbonyl carbon gets bonded with nitrogen of γ-butyrolactam to form intermediate 119. Nucleophilic attack of thiophenol on the intermediate 119 yields the desired product 115.
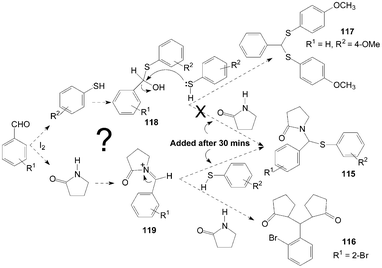 | ||
| Scheme 43 A possible mechanism for the formation of pyrrolidin-2-one derivatives. | ||
2.19 (aza-)Friedel–Crafts reaction
Jaratjaroonphong et al.99 reported a highly efficient one-pot, three-component aza-Friedel–Crafts reaction of electron-rich arenes or heteroarenes, aldehydes, and benzyl or tert-butyl carbamates in toluene under “open-flask” and mild conditions (Scheme 44). After systematic screening, it was observed that when non-polar and weakly polar solvents such as toluene, CH2Cl2, or THF were used, the reaction gave the corresponding N-protected diarylmethylamines 120 as the major product and only a trace amount of double-addition products diarylalkane derivatives 121. Notably, the products 121 were obtained in moderate to high yields without benzyl or tert-butyl carbamates (Scheme 44).100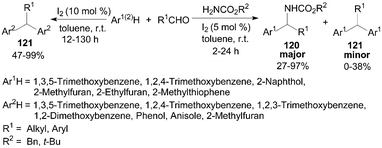 | ||
| Scheme 44 Iodine catalyzed (aza-)Friedel–Crafts reaction. | ||
2.20 Synthesis of 1,3-oxazinan-2-ones, xanthenes and anthracene-9,11-diones
Molecular iodine catalyzed highly efficient one-pot three component coupling of β-naphthol, aromatic aldehydes and urea to produce 1-aryl-1,2-dihydro-naphtho[1,2-e][1,3]oxazin-3-one derivatives 122 under solvent free conditions was described by Sharma101 and Nizam102et al. (Scheme 45). Interestingly, the product formation 122 is possible only at very high temperature (140–150 °C) and at lower temperature (90–100 °C) formation of 14-aryl-14-H-dibenzo[a,j]xanthenes 123 was observed. Previously, the articles103,104 had shown that iodine could efficiently catalyse the reaction of 2-naphthol with aryl or alkyl aldehydes under neat conditions in acetonitrile at 90–100 °C to afford the corresponding products 123 in high yields (up to 95%) within 5 h.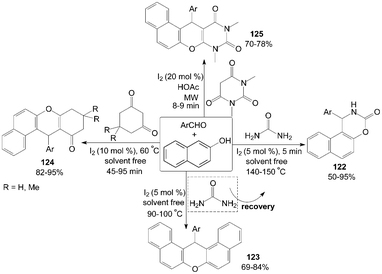 | ||
| Scheme 45 Synthesis of 1,3-oxazinan-2-ones, xanthenes and anthracene-9,11-diones. | ||
Cyclic 1,3-dicarbonyl compounds were also employed with β-naphthol and aromatic aldehydes to produce tetrahydrobenzo[a]xanthene-11-one derivatives 124 and diazabenzo[a]anthracene-9,11-dione derivatives 125 (Scheme 45).105–107
A variety of new tetrahydrobenzo[c]xanthene-1,11-dione 126 derivatives were also prepared by condensation of 4-hydroxylcoumarin, aromatic aldehydes, and 5,5-dimethylcyclohexane-1,3-dione under microwave irradiation in good yields (Scheme 46).108
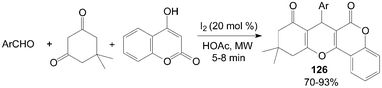 | ||
| Scheme 46 Iodine catalyzed synthesis of xanthenes. | ||
2.21 One-pot tandem reactions
A tandem reaction is a reaction in which several bonds are formed in sequence without isolating intermediates, changing reaction conditions, or adding reagents. So, the tandem reactions are commonly referred to under the nebulous phrase “multistep one-pot reactions.” Some instances of the tandem reaction have been presented in the above Schemes (Schemes 16, 34, 36, 38 and 39).A variety of aldehydes reacted with iodine in ammonia water at room temperature to give the intermediate nitrile 127, which were trapped by addition of hydrogen peroxide, dicyandiamide, or sodium azide to produce their corresponding amides 128, 1,3,5-triazines 129, and tetrazoles 130 in moderate to high yields (Scheme 47).109,110
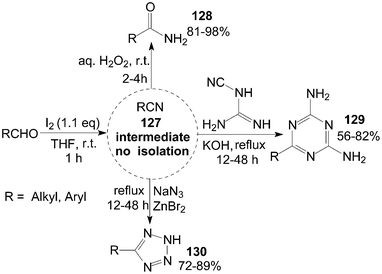 | ||
| Scheme 47 Iodine catalyzed tandem reaction. | ||
A highly efficient synthesis of hydantoins 132 had been developed from 1,3-dicarbonyl compounds, ureas, and methyl ketones or terminal aryl alkenes (Scheme 48).111 This protocol involves a sustainable integration of two coupled domino processes: iodine-promoted synthesis of unsymmetrical intermediates 1,4-enediones 131 (domino process I)112,113 and the sequential transformation into hydantoins 132 (domino process II). In the reaction, the presence of iodine is important for accelerating equilibrium toward the hydantoins 132; otherwise, a mixture of products (without hydantoins 132) would be obtained in the absence of iodine.
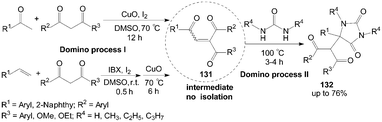 | ||
| Scheme 48 Iodine catalyzed synthesis of hydantoins via tandem reaction. | ||
Kumar et al.114 described an interesting iodine promoted sp3 C–H bond activation of an alkyl azaarene to form alkyl azaarene pyridinium (AAP) zwitterions 135 in a new multicomponent tandem reaction. 1-(Quinolin-2-ylmethyl)pyridinium iodide 133 is formed firstly by the reaction of methyl azaarenes and pyridine, which attacks an arylidene dione 134 (obtained from aldehydes and meldrum acid via Knoevenagel condensation in the presence of triethylamine) to form AAP zwitterions (Scheme 49).
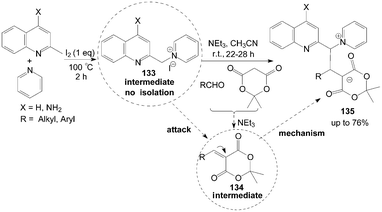 | ||
| Scheme 49 Synthesis of alkyl azaarene pyridinium zwitterions via tandem reaction. | ||
The intermediate products of hydrazones 136 were obtained firstly by the reaction mixture of 2-aminobenzohydrazide and acetophenone (or 3-nitrobenzaldehyde) stirring at room temperature (or 0 °C) for 2–3 h in ionic liquids (IL), then the aromatic aldehyde and iodine were added to the mixture for 2–8 h at 50 °C. The above tandem reaction provided structural diversification of 3-arylideneaminoquinazolin-4(1H)-one derivatives 137 in high yields (Scheme 50).115
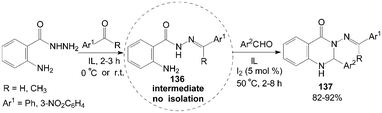 | ||
| Scheme 50 Synthesis of 3-arylideneaminoquinazolin-4(1H)-ones in ionic liquids. | ||
3. Molecular iodine catalyzed ABB and ABB′ multicomponent reactions
3.1 Synthesis of 1,5-benzodiazepine from o-phenylenediamine and ketones
2.3-Dihydro-lH-1.5-benzodiazepines 138 have been synthesized from o-phenylenediamine and ketones under solvent-free conditions (Scheme 51).116 All of the ketones, including cyclic ketones and acyclic ketones reacted smoothly with o-phenylenediamine to furnish products 138 in the presence of a catalytic amount of iodine within 10 min. In the reactions, the amino group of o-phenylenediamine attacks the carbonyl group of the ketone, which is activated by iodine, giving the intermediate diimine 139. A 1,3-shift of the hydrogen attached to the methyl group then occurs to form an isomeric enamine 140, which cyclizes to afford products 138.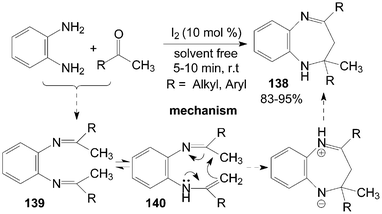 | ||
| Scheme 51 Synthesis of 1,5-benzodiazepine from o-phenylenediamine and ketones. | ||
3.2 Synthesis of 4,9-dihydro-2H-benzo[f]isoindoles
In the presence of iodine, 4,9-dihydro-2H-benzo[f]isoindole derivatives 141 could be efficiently constructed from propargyl alcohols and phosphoramides in the presence of iodine in a single step via ABB′ MCRs (Scheme 52).117![Iodine catalyzed synthesis of 4,9-dihydro-2H-benzo[f]isoindoles.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s52.gif) | ||
| Scheme 52 Iodine catalyzed synthesis of 4,9-dihydro-2H-benzo[f]isoindoles. | ||
3.3 Synthesis of 1,2,3-triaroylindolizines
A very convenient and efficient method for the synthesis of 1,2,3-triaroylindolizines 142 directly from methyl ketones and pyridines was developed by Yang et al. (Scheme 53).118 All methyl ketones, regardless of their electronic or steric properties, proceeded efficiently to afford their corresponding products 142 in moderate to excellent yields.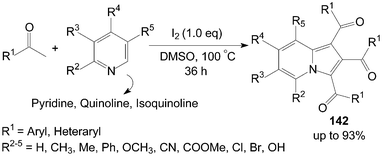 | ||
| Scheme 53 Synthesis of 1,2,3-triaroylindolizines. | ||
The regioselectivity of 3-substituted pyridines was addressed and the ratio of the corresponding indolizine isomers is highly dependent on the substituent nature (Scheme 54).118 Although the electron-withdrawing group (CO2CH3) led to product 142a in preference over 142 (Table 4, entry 1), the regioselectivity was reversed for 3-methylpyridine, 3-chloropyridine, and 3-bromopyridine, which yielded indolizines 142 as the major isomers (Table 4, entries 2–4). Interestingly, the active hydroxyl group was not only compatible under the same reaction conditions but also exclusively led to the regioisomer 142 (Table 4, entry 5).
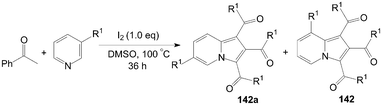 | ||
| Scheme 54 Synthesis of 1,2,3-triaroylindolizines. | ||
| Entry | R1 | 142a:142 | Yield (%) |
|---|---|---|---|
| 1 | CO2CH3 | 1.2:1 | 77 |
| 2 | CH3 | 1:3.3 | 72 |
| 3 | Cl | 1:4.8 | 58 |
| 4 | Br | 1:3.9 | 54 |
| 5 | OH | 0:1.0 | 43 |
3.4 Synthesis of xanthene, spiro dihydrofuran, cyclopropane and xanthenedione derivatives from 1,3-dicarbonyl compounds
Molecular iodine facilitated the reaction of 5,5-dimethyl-1,3-cyclohexanedione with aromatic aldehydes in iso-propanol affording a variety of 1,8-dioxo-octahydroxanthenes 143 in high yields (Scheme 55).119 However, spiro dihydrofurans 144 were obtained under the different reaction condition (Scheme 55).120 Surprisingly, a new type of compound, cyclopropane derivative 145 were obtained rather than the expected products 144via the reaction of aromatic aldehydes with 1,3-indandione in the presence of molecular iodine and dimethylaminopyridine (DMAP) under mechanical milling conditions (Scheme 55).120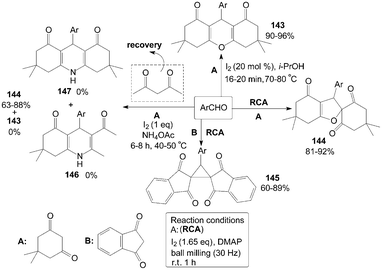 | ||
| Scheme 55 Synthesis of xanthene, spiro dihydrofuran and cyclopropane derivatives. | ||
Initially, Sahu et al.121 tried to combine four reactants dimedone, 4-anisaldehyde, acetylacetone and ammonium acetate in ethanol for synthesis of Hantzsch 1,4-dihydropyridine compound 146. Interestingly, no desired product 146 could be isolated, and the products 143 and 147 were also not obtained under the same reaction conditions. The product 144 was the sole product isolated from the reaction mixture and acetylacetone was not incorporated in the reaction (Scheme 55). However, instead of the product 144, the product 143 was isolated in good yield when the authors condensed dimedone and 4-chlorobenzaldehyde in refluxing methanol in the presence of iodine alone (without ammonium acetate). So, ammonium acetate is a key “promoter” for synthesis of the products 144 in the above reaction.
Similarly, Rong et al.122 want to synthesize 1,4-dihydropyridine derivatives 148 by the reaction of imines with dimedone with the catalyst iodine and zinc powder in methanol (Scheme 56). However, they could not get the anticipated products 148; surprisingly, xanthenediones derivatives 149 were obtained with excellent yields. And when the reaction was carried out without a catalytic amount of molecular iodine, ring-opening derivatives of xanthenediones 150 were obtained in high yields.
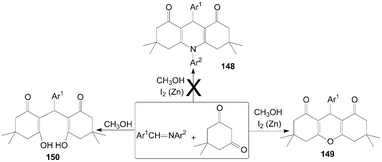 | ||
| Scheme 56 Synthesis of xanthenediones from dimedone. | ||
3.5 Synthesis of quinazolin-4-(1H)-one and quinoline derivatives
Similarly, based on the route of synthesis of 137 (Scheme 50), a combinatorial synthesis of quinazolin-4-(1H)-one derivatives 151 were accomplished by a reaction of 2-aminobenzohydrazides with two equivalents of aldehydes or ketones in ionic liquids (IL) catalyzed by iodine with good to excellent yields (Scheme 57).115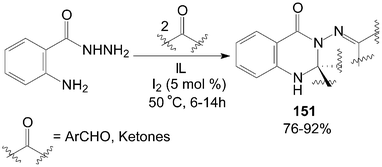 | ||
| Scheme 57 Synthesis of quinazolin-4-(1H)-one derivatives from carbonyl compounds. | ||
One-pot reactions of naphthalen-2-amine and alkyl aldehydes in the presence of iodine in refluxing THF to produce the corresponding 2,3-dialkylbenzo[f]quinolines 152 in moderate to good yields (Scheme 58).123
![Synthesis of 2,3-dialkylbenzo[f]quinolines from naphthalen-2-amine and alkyl aldehydes.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s58.gif) | ||
| Scheme 58 Synthesis of 2,3-dialkylbenzo[f]quinolines from naphthalen-2-amine and alkyl aldehydes. | ||
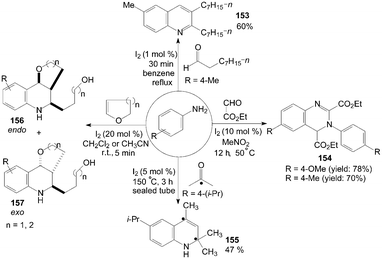
Anilines were also treated with aldehydes in the presence of iodine to give the corresponding 3,4-dialkyl-substituted quinolines 153 and quinazolines 154 in good yields (Scheme 59).46,52
 | ||
| Scheme 59 Synthesis of quinolines and quinazolines from anilines. | ||
The mechanism of the formation of substituted quinolines from anilines and ketones has been studied by the use of 13C-labeled ketones in the experiments. The reaction of 4-isopropylaniline with 5 equiv of acetone containing 20 mol% of 13C(2) acetone (100% labeled) at 150 °C for 3 h in a sealed tube gave the quinoline 155 in 47% yield. The result showed clearly that the incorporation of two acetone molecules to form the quinoline 155 (Scheme 59).124
1,2,3,4-Tetrahydroquinoline derivatives 156 and 157 were obtained when cyclic enol ethers were reacted with anilines (Scheme 59).125 After systematic screening, electron-rich anilines were more reactive than electron-deficient anilines. The strong electron-withdrawing group substituted 4-nitroaniline only gave a trace of product. In all cases, the products were obtained as a mixture of endo/exo-isomers.
3.6 Cross-aldol condensation
Cross-aldol condensation of aromatic aldehydes with cyclic ketones is an important synthetic reaction for the preparation of α,α′-bis(substituted-benzylidene) cycloalkanones 158, 3,5-bis-(arylmethylidene)-tetrahydropyran-4-ones 159 and α,α′--bis(substituted benzylidene)-1-carbethoxy-4-piperidones 160. The above reactions were accomplished at room temperature using iodine as catalyst with good to excellent yields (Scheme 60).126–128 Notably, in all cases of the synthesis of products 159, the reactions proceed rapidly and go to completion within 30–60 min at room temperature.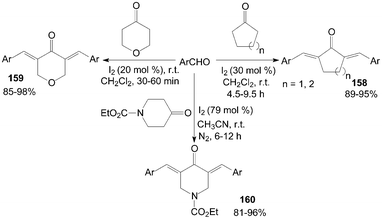 | ||
| Scheme 60 Iodine catalyzed cross-aldol condensation. | ||
3.7 Synthesis of dipyrromethanes, 2,5-dialkylated pyrroles, porphyrins and cyclononatripyrrole from pyrroles
Dipyrromethanes 161 were readily prepared by the reaction of pyrrole and aromatic aldehydes in moderate to high yields with two methods (Scheme 61).129,130 In the above two methods, all the reactions were accomplished within several minutes. However, the yields of the products were somewhat higher in Method B.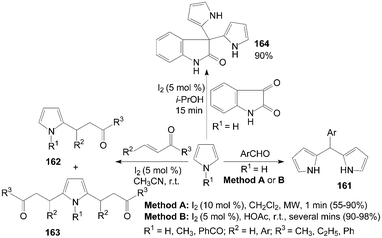 | ||
| Scheme 61 Synthesis of dipyrromethanes and 2,5-dialkylated pyrroles from pyrroles. | ||
2-Alkyl pyrroles 162 and 2,5-dialkyl pyrroles 163 were obtained when the conjugate addition of pyrroles with methyl vinyl ketone using an equimolar ratio of reagents (Scheme 61).131 Only products 163 were obtained by increasing the molar ratio of alkenes to pyrroles (3:1) in high yields within a short reaction time at room temperature. However, N-benzoyl pyrrole (R1 = PhCO) afforded only monoalkylated products 162 in somewhat low yields. This can be attributed to the lower electron density on the ring carbon due to the electron withdrawing benzoyl group on the ring nitrogen.
Furthermore, pyrrole also reacted efficiently with isatin in presence of iodine in i-PrOH to afford 3,3-dipyrrolyl-2-oxindole 164 (Scheme 61).132
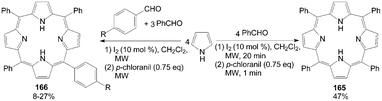
The development of pyrrole chemistry has largely been associated with the synthesis of natural products. One of these natural products is porphyrins. The application is the well-known use of porphyrins as photosensitizers in photodynamic therapy.133 However, the synthesis of unsymmetrical porphyrins presents a real challenge especially when practicable yields are needed. The well-known Little's mixed aldehyde method134 leads to mono functionalized meso-substituted porphyrins in low yields (4-7%). Boëns et al.133 reported an iodine-catalyzed, one-pot synthesis of functionalized porphyrins 165 and 166 (Scheme 62), a method that, contrary to the previous protocol, didn't require prior reactant or solvent distillation, and had higher yields (up to 47%) within a shorter reaction time.
 | ||
| Scheme 62 Synthesis of porphyrins from pyrroles. | ||
Initially, Stępień et al.135 tried to synthesize dicarbinol 167 from ethyl 3,3-dimethyl-5,7-dihydro[1,3]dioxepino[5,6-c]pyrrole-6-carboxylate using iodine as catalyst. Interestingly, the unexpected major product formed was the cyclononatripyrrole 168, accompanied by smaller amounts of the dipyrromethane derivative 169 and the monopyrrole 170 (Scheme 63).
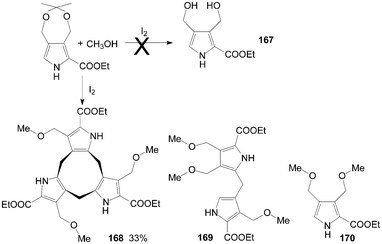 | ||
| Scheme 63 Synthesis of cyclononatripyrrole from pyrroles. | ||
3.8 Synthesis of di(indolyl)indolin-2-ones, bis(indolyl)methanes, indolo[2,3-b]carbazole and trisindolylalkanes from indoles
One-pot synthesis of di(indolyl)indolin-2-ones 171 was performed by the reaction of isatin with 1- or 2-substituted indoles in good to excellent yields with two methods (Scheme 64).132,136 The yields of the products were somewhat higher in Method A. Meanwhile, the reaction times and amount of iodine were much lower in Method A. Interestingly, 3-substituted indoles underwent smooth coupling with isatin to give the corresponding bisindolyl oxindoles 172 in 85% yields (Scheme 64).132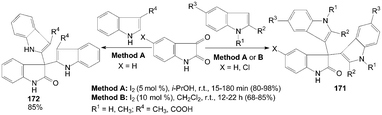 | ||
| Scheme 64 Synthesis of di(indolyl)indolin-2-ones from isatin and indoles. | ||
In aqueous medium, molecular iodine had been used an efficient catalyst for one-pot synthesis of 3,3′-arylmethylenebis-(4-hydroxycoumarin) 173 and 2,2′-arylmethylenebis(3-hydroxyl-5,5-dimethyl-2-cyclohexen-1-one) 174 in excellent yields (Scheme 65).137
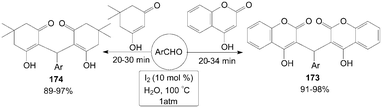 | ||
| Scheme 65 Iodine catalysed aqueous mediated synthesis of Michael adduct. | ||
A highly efficient synthesis of bis(indolyl)methanes 175 has been developed from carbonyl compounds and indole in good to excellent yields with three methods (Scheme 66).138–140 In the above three methods, most of products 175 were prepared within 10 min. Notably, the synthesis of products 175 by Method B139 is in a very short time (<1 min). However, in Method C,140 steric congestion around carbonyl carbon impedes the reaction rate substantially as reflected in the longer reaction time required (3–4.5 h) for sterically encumbered aromatic ketones.
![Synthesis of bis(indolyl)methanes and indolo[2,3-b]carbazole from indole.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s66.gif) | ||
| Scheme 66 Synthesis of bis(indolyl)methanes and indolo[2,3-b]carbazole from indole. | ||
The products 175 could be directly synthesized from benzyl alcohol and indole in up to 86% yield using molecular oxygen, visible light, and catalytic iodine (Scheme 66).141 In the reaction, the catalytic iodine played the roles of both catalyst and oxidant. A new type of Schiff base N-tert-butanesulfinyl aldimine was also employed with indole to produce products 175 in good to excellent yields (Scheme 66).142
Interestingly, Deb et al.143 reported the reaction using equimolar amounts of indole and an aldehyde under thermal conditions without giving time for isomerization of the products 175 formed during the reaction and allowing a second aldehyde molecule to become involved in the reaction to afford indolo[2,3-b]carbazoles 176 (Scheme 66). In a systematic study, it was observed that substituents in the aromatic ring of the aldehyde have a tremendous effect on the reaction process. Electron-withdrawing groups in the aromatic ring of the aldehyde retarded the reaction while electron-donating groups accelerated the reaction. And in the case of aliphatic aldehydes, the yields were low (20–25%).
Interestingly, the 3,3′-bis(indolyl)methanes 175 isomerized to 2,3′-bis-(indolyl)methanes 177 when the condensations of indole and aldehyde were carried out for a longer time (14 h). Then acid-catalyzed intramolecular cyclizations were accomplished directly by treating the crude product 177 with different ortho esters in the presence of sulfuric acid or methanesulfonic acid as a catalyst to afford the corresponding indolo[3,2-b]carbazoles 178 (Scheme 67).144
![Synthesis of indolo[3,2-b]carbazoles from indole.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s67.gif) | ||
| Scheme 67 Synthesis of indolo[3,2-b]carbazoles from indole. | ||
Bis(indolyl)carbonyl compounds 179 were synthesized from 2-unsubstituted indoles such as indole, 5-bromoindole, and 5-methoxyindole, with 1,3-dicarbonyl compounds (Scheme 68).145
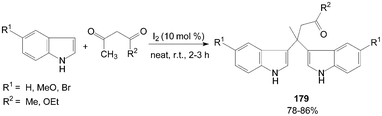 | ||
| Scheme 68 Synthesis of bis(indolyl)carbonyl compounds from indole. | ||
Furthermore, 4-[(indol-3-yl)-arylmethyl]-1-phenyl-3-methyl-5-pyrazolones 180 could be smoothly and effectively obtained in good yields by the three-component reactions of aldehydes, indole, and 1-phenyl-3-methyl-5-pyrazolones under solid-state conditions at room temperature (Scheme 69).146 However, in the absence of iodine as a catalyst, the above three-component mixture could provide only the product 4-chlorophenyl-bis(1-phenyl-3-methyl-5-pyrazolon-4-yl)methane 181 at room temperature. And it was well known142 that products 175 were obtained in low yields in the absence of Lewis acids. So, the authors gave the likely mechanism for the three-component reaction. Firstly, the intermediate 182 was offered in preference to the intermediate 183, because the latter was too unstable to undergo the further transformation. Then, the subsequent Michael addition of indole with the intermediate 182 was successfully carried out to afford the target compounds 180 in excellent yields.
![Synthesis of 4-[(indol-3-yl)-arylmethyl]-1-phenyl-3-methyl-5-pyrazolones from indole.](/image/article/2013/RA/c3ra23461d/c3ra23461d-s69.gif) | ||
| Scheme 69 Synthesis of 4-[(indol-3-yl)-arylmethyl]-1-phenyl-3-methyl-5-pyrazolones from indole. | ||
Trisindolylalkanes 184–187 had also been synthesized from indole or 1-methylindole with 2-cyclohexen-1-one, or α,β-unsaturated aldehyde, or orthoformate at room temperature (Scheme 70).147–150
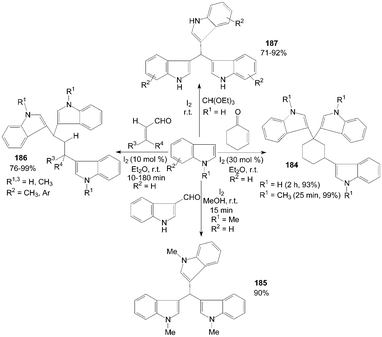 | ||
| Scheme 70 Synthesis of trisindolylalkanes from indoles. | ||
4. Conclusions
A large number of MCRs in the presence of molecular iodine have been presented, most of which show quite good conversion and selectivity for preparing products of industrial and pharmacological interest. Molecular iodine has broad transformation ability of functional groups and can be used widely in MCRs.MCRs generally benefit from other aspects such as atom-economy, the use of readily available starting materials, resource effectiveness and bond-forming efficiency, which render these reactions useful environmentally friendly alternatives, in keeping with the greener direction in which organic chemistry is proceeding. Future research in the area of MCRs will include the discovery and design of novel MCRs, experimental improvements, and more and more applications in drug discovery, materials science, bioconjugates, and agrochemical compounds. It will not be surprising if more and more applications of molecular iodine in MCRs appear in near future.
The advantages of iodine are operational simplicity, low cost, and less toxicity. But the reuse of iodine is difficult because it has a good solubility in most organic solvents and easy sublimation at high temperature. Solid supported iodine with unreduced activity could considerably contribute to green chemistry. There have been a few reports of solid-supported iodine as a catalyst thus far. Such as the iodine supported on polyvinylpyrrolidone (I2/PVP),151 aminopropyl silica gel (I2/APSG),152 neutral alumina surface (I2/Al2O3)17,153 or natural phosphate (I2/NP)154etc. However, the above supports were the preferred choice as supports to keep the reaction medium under mild and neutral conditions, and the reuse of iodine was also difficult. So simple, efficient supports for reusable iodine are still desirable.155,156
Acknowledgements
This project is sponsored by the National Natural Science Foundation of China (No.51202003)References
- J. Zhu and H. Bienaymé, Multicomponent reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
- D. Tejedor and F. García-Tellado, Chem. Soc. Rev., 2007, 36, 484–491 RSC.
- H. Togo and S. Iida, Synlett, 2006, 2159–2175 CrossRef CAS.
- M. Jereb, D. Vražič and M. Zupan, Tetrahedron, 2011, 67, 1355–1387 CrossRef CAS.
- P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Chem.–Eur. J., 2012, 18, 5460–5489 CrossRef CAS.
- M. Kidwai, P. Mothsra, V. Bansal and R. Goyal, Monatsh. Chem., 2006, 137, 1189–1194 CrossRef CAS.
- M. Kidwai, P. Mothsra, V. Bansal, R. K. Somvanshi, A. S. Ethayathulla, S. Dey and T. P. Singh, J. Mol. Catal. A: Chem., 2007, 265, 177–182 CrossRef CAS.
- M. Kidwai and P. Mothsra, Tetrahedron Lett., 2006, 47, 5029–5031 CrossRef CAS.
- D. C. Mungra, H. G. Kathrotiya, N. K. Ladani, M. P. Patel and R. G. Patel, Chin. Chem. Lett., 2012, 23, 1367–1370 CrossRef CAS.
- H. Behmadi, M. Roshani and S. M. Saadati, Chin. Chem. Lett., 2009, 20, 5–8 CrossRef CAS.
- Y.-M. Ren and C. Cai, Adv. Mater. Res., 2012, 396–398, 1871–1874 CAS.
- J. Jayabharathi, V. Thanikachalam and K. Jayamoorthy, Spectrochim. Acta, Part A, 2012, 89, 168–176 CrossRef CAS.
- Y.-M. Ren and C. Cai, J. Chem. Res., 2010, 34, 133–134 CrossRef CAS.
- R. S. Bhosale, S. V. Bhosale, S. V. Bhosale, T. Wang and P. K. Zubaidha, Tetrahedron Lett., 2004, 45, 9111–9113 CrossRef CAS.
- K. V. N. S. Srinivas and B. Das, Synthesis, 2004, 2091–2093 CAS.
- J. J. V. Eynde, N. Audiart, V. Canonne, S. Michel, Y. V. Haverbeke and C. O. Kappe, Heterocycles, 1997, 45, 1967–1978 CrossRef.
- I. Saxena, D. C. Borah and J. C. Sarma, Tetrahedron Lett., 2005, 46, 1159–1160 CrossRef CAS.
- J.-S. Wang, J.-T. Li and Z.-P. Lin, Lett. Org. Chem., 2006, 3, 523–525 CrossRef CAS.
- P. Zalavadiya, S. Tala, J. Akbari and H. Joshi, Arch. Pharm., 2009, 342, 469–475 CrossRef CAS.
- Z.-T. Wang, L.-W. Xu, C.-G. Xia and H.-Q. Wang, Tetrahedron Lett., 2004, 45, 7951–7953 CrossRef CAS.
- Y.-M. Ren and C. Cai, Monatsh. Chem., 2009, 140, 49–52 CrossRef CAS.
- M. B. Madhusudana Reddy and M. A. Pasha, Synth. Commun., 2011, 41, 1875–1880 CrossRef CAS.
- D. Prajapati, D. Bhuyan, M. Gohain and W. Hu, Mol. Diversity, 2011, 15, 257–261 CrossRef CAS.
- S. Ko, M. N. V. Sastry, C. Lin and C.-F. Yao, Tetrahedron Lett., 2005, 46, 5771–5774 CrossRef CAS.
- M. A. Zolfigol, P. Salehi, A. Khorramabadi-Zad and M. Shayegh, J. Mol. Catal. A: Chem., 2007, 261, 88–92 CrossRef CAS.
- J. D. Akbari, S. D. Tala, M. F. Dhaduk and H. S. Joshi, Arkivoc, 2008, 126–135 CAS.
- A. Kumar, R. A. Maurya, S. Sharma, M. Kumar and G. Bhatia, Eur. J. Med. Chem., 2010, 45, 501–509 CrossRef CAS.
- L. Royer, S. K. De and R. A. Gibbs, Tetrahedron Lett., 2005, 46, 4595–4597 CrossRef CAS.
- H.-S. Wang, L.-F. Zhao and Z.-M. Du, Chin. J. Chem., 2006, 24, 135–137 CrossRef CAS.
- P. Phukan, J. Org. Chem., 2004, 69, 4005–4006 CrossRef CAS.
- J. S. Yadav, P. K. Chand and S. Anjaneyulu, Tetrahedron Lett., 2002, 43, 3783–3784 CrossRef CAS.
- D. Kataki and P. Phukan, Tetrahedron Lett., 2009, 50, 1958–1960 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, G. M. Reddy and R. Narender, Synthesis, 2009, 963–968 CrossRef CAS.
- G. Li, R. Long and J. Yang, Kinet. Catal., 2011, 52, 397–400 CrossRef CAS.
- P. Phukan, D. Kataki and P. Chakraborty, Tetrahedron Lett., 2006, 47, 5523–5525 CrossRef CAS.
- M. Xia and Y.-D. Lu, Synth. Commun., 2007, 37, 725–735 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, K. S. Shankar, K. Premalatha and T. Swamy, Lett. Org. Chem., 2008, 5, 353–359 CrossRef CAS.
- B. S. Lee, S. Mahajan and K. D. Janda, Synlett, 2005, 1325–1327 CAS.
- D. Kataki and P. Phukan, Indian J. Chem. B, 2006, 45, 1759–1761 Search PubMed.
- R. Villano, M. R. Acocella and A. Scettri, Tetrahedron, 2011, 67, 2768–2772 CrossRef CAS.
- V. Kavala, C. Lin, C.-W. Kuo, H. Fang and C.-F. Yao, Tetrahedron, 2012, 68, 1321–1329 CrossRef CAS.
- S. Sobhani and A. Vafaee, J. Iran. Chem. Soc., 2010, 7, 227–236 CrossRef CAS.
- X. Jia, X. Chen, C. Huo, F. Peng, C. Qing and X. Wang, Chin. J. Chem., 2012, 30, 1504–1510 CrossRef CAS.
- X. D. Xiao, X. N. Chen, C. D. Huo, F. F. Peng, C. Qing and X. C. Wang, Chin. Chem. Lett., 2012, 23, 309–312 CrossRef.
- C. Lin, H. Fang, Z. Tu, J.-T. Liu and C.-F. Yao, J. Org. Chem., 2006, 71, 6588–6591 CrossRef CAS.
- X.-F. Lin, S.-L. Cui and Y.-G. Wang, Tetrahedron Lett., 2006, 47, 3127–3130 CrossRef CAS.
- M. Xia and Y.-D. Lu, Synlett, 2005, 2357–2361 CrossRef CAS.
- N. P. Rai, S. Shashikantha and P. N. Arunachalam, Synth. Commun., 2009, 39, 2125–2136 CrossRef CAS.
- G. Jin, J. Zhao, J. Han, S. Zhu and J. Zhang, Tetrahedron, 2010, 66, 913–917 CrossRef CAS.
- Y.-C. Li, J.-M. Zhang, L.-T. Dong and M. Yan, Chin. J. Chem., 2006, 24, 929–932 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, A. R. Reddy and A. V. Narsaiah, Synthesis, 2007, 3191–3194 CrossRef CAS.
- X. Li, Z. Mao, Y. Wang, W. Chen and X. Lin, Tetrahedron, 2011, 67, 3858–3862 CrossRef CAS.
- L.-Y. Zeng and C. Cai, Org. Biomol. Chem., 2010, 8, 4803–4805 CAS.
- X.-S. Wang, J. Zhou, K. Yang and Y.-L. Li, Tetrahedron Lett., 2011, 52, 612–614 CrossRef CAS.
- X.-S. Wang, Q. Li, J.-R. Wu and Y.-L. Li, Synth. Commun., 2009, 39, 702–715 CrossRef CAS.
- X.-S. Wang, Q. Li, J.-R. Wu and S.-J. Tu, J. Comb. Chem., 2009, 11, 433–437 CrossRef CAS.
- X.-S. Wang, J. Zhou, M.-Y. Yin, K. Yang and S.-J. Tu, J. Comb. Chem., 2010, 12, 266–269 CrossRef CAS.
- X.-S. Wang, Q. Li, C.-S. Yao and S.-J. Tu, Eur. J. Org. Chem., 2008, 3513–3518 CrossRef CAS.
- X.-S. Wang, M.-Y. Yin, S.-L. Wang, W. Wang and Y.-L. Li, J. Heterocycl. Chem., 2012, 49, 585–588 CrossRef CAS.
- M.-Y. Yin, M.-M. Zhang, W. Wang, Y.-L. Li and X.-S. Wang, Arkivoc, 2011, 51–59 CAS.
- X.-S. Wang, Q. Li, J.-R. Wu and M.-M. Zhang, Synth. Commun., 2009, 39, 3069–3080 CrossRef CAS.
- M.-M. Zhang, W. Wang, T.-J. Li, C.-S. Yao and X.-S. Wang, Res. Chem. Intermed., 2012 DOI:10.1007/s11164-012-0712-9 In press.
- X.-S. Wang, M.-Y. Yin, W. Wang and S.-J. Tu, Eur. J. Org. Chem., 2012, 4811–4818 CrossRef CAS.
- L. Wu, L. Yang, F. Yan, C. Yang and L. Fang, Bull. Korean Chem. Soc., 2010, 31, 1051–1054 CrossRef CAS.
- S.-S. Shen and S.-J. Ji, Chin. J. Chem., 2008, 26, 935–940 CrossRef CAS.
- X.-S. Wang, Q. Li, J.-R. Wu, Y.-L. Li, C.-S. Yao and S.-J. Tu, Synthesis, 2008, 1902–1910 CrossRef CAS.
- A. T. Khan, M. M. Khan and K. K. R. Bannuru, Tetrahedron, 2010, 66, 7762–7772 CrossRef CAS.
- B. Das, K. R. Reddy, R. Ramu, P. Thirupathi and B. Ravikanth, Synlett, 2006, 1756–1758 CrossRef CAS.
- M. A. Pasha, V. P. Jayashankara and N. Ramachandraswamy, Synth. Commun., 2007, 37, 1551–1556 CrossRef CAS.
- N. P. Selvam and P. T. Perumal, Tetrahedron, 2008, 64, 2972–2978 CrossRef CAS.
- P. Srinivasana, P. T. Perumal and S. Raja, Indian J. Chem. B, 2011, 50, 1083–1091 Search PubMed.
- G. Sabitha, M. Bhikshapathi, S. Nayak and J. S. Yadav, Synth. Commun., 2011, 41, 8–15 CrossRef CAS.
- B. Das, K. Laxminarayana, B. Ravikanth and B. R. Rao, J. Mol. Catal. A: Chem., 2007, 261, 180–183 CrossRef CAS.
- R. R. Nagawade and D. B. Shinde, Mendeleev Commun., 2007, 17, 299–300 CrossRef CAS.
- R. S. Bhosale, C. V. Magar, K. S. Solanke, S. B. Mane, S. S. Choudhary and R. P. Pawar, Synth. Commun., 2007, 37, 4353–4357 CrossRef CAS.
- Y.-M. Ren and C. Cai, Catal. Commun., 2008, 9, 1017–1020 CrossRef CAS.
- M. B. M. Reddy and M. A. Pasha, Indian J. Chem. B, 2012, 51, 537–541 Search PubMed.
- Y.-M. Ren and C. Cai, Synth. Commun., 2007, 37, 2221–2225 Search PubMed.
- Y.-M. Ren and C. Cai, Catal. Lett., 2007, 118, 134–138 CrossRef CAS.
- M. Dabiri, P. Salehi, M. Bahramnejad and M. Alizadeh, Monatsh. Chem., 2010, 141, 877–881 CrossRef CAS.
- L.-Y. Zeng and C. Cai, J. Heterocycl. Chem., 2010, 47, 1035–1039 CrossRef CAS.
- S. Rostamizadeh, A. M. Amani, R. Aryan, H. R. Ghaieni and N. Shadjou, Synth. Commun., 2008, 38, 3567–3576 CrossRef CAS.
- L.-Y. Zeng, W.-B. Yi and C. Cai, Eur. J. Org. Chem., 2012, 559–566 CrossRef CAS.
- H.-S. Wang and J.-E. Zeng, Chin. J. Chem., 2008, 26, 175–178 CrossRef CAS.
- M. Wang, Z.-G. Song and T.-T. Zhang, Org. Prep. Proced. Int., 2010, 42, 169–173 CrossRef CAS.
- B. Das, B. S. Kanth, D. B. Shinde and V. T. Kamble, Helv. Chim. Acta, 2011, 94, 2087–2091 CrossRef CAS.
- L. Nagarapu, H. R. Vulupala, Ch. Venkatanarsimhaji, R. Bantu and A. R. Reddy, Synth. Commun., 2012, 42, 2131–2138 CrossRef CAS.
- B. Das, N. Bhunia and M. Lingaiah, Synthesis, 2011, 3471–3474 CrossRef CAS.
- G. R. Reddy, T. R. Reddy, S. C. Joseph, K. S. Reddy and M. Pal, RSC Adv., 2012, 2, 3387–3395 RSC.
- A. T. Khan, A. Ghosh and Md. M. Khan, Tetrahedron Lett., 2012, 53, 2622–2626 CrossRef CAS.
- L.-Y. Zeng and C. Cai, J. Comb. Chem., 2010, 12, 35–40 CrossRef CAS.
- L.-Y. Zeng, Y.-M. Ren and C. Cai, Synth. Commun., 2011, 41, 3635–3643 CrossRef CAS.
- L.-Y. Zeng, F. Ji and C. Cai, J. Heterocycl. Chem., 2012, 49, 237 CrossRef CAS.
- B. Dai, Y. Duan, X. Liu, L. Song, M. Zhang, W. Cao, S. Zhu, H. Deng and M. Shao, J. Fluorine Chem., 2012, 133, 127–133 CrossRef CAS.
- M. Kidwai, V. Bansal and P. Mothsra, J. Mol. Catal. A: Chem., 2007, 266, 43–46 CrossRef CAS.
- G. Ramachandran, N. S. Karthikeyan, P. Giridharan and K. I. Sathiyanarayanan, Org. Biomol. Chem., 2012, 10, 5343–5346 CAS.
- H.-Q. Li, G. Ramachandran, V. Satheesh, K. Sathiyanarayanan and R. S. Rathore, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o768 Search PubMed.
- H.-Q. Li, G. Ramachandran, M. Sathishkumar, K. Sathiyanarayanan and R. S. Rathore, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o782 Search PubMed.
- J. Jaratjaroonphong, S. Krajangsri and V. Reutrakul, Tetrahedron Lett., 2012, 53, 2476–2479 CrossRef CAS.
- J. Jaratjaroonphong, S. Sathalalai, P. Techasauvapak and V. Reutrakul, Tetrahedron Lett., 2009, 50, 6012–6015 CrossRef CAS.
- M. Sharma, S. Manohar and D. S. Rawat, J. Heterocycl. Chem., 2012, 49, 589–595 CrossRef CAS.
- A. Nizam and M. A. Päsha, Synth. Commun., 2010, 40, 2864–2868 CrossRef CAS.
- B. Das, B. Ravikanth, R. Ramu, K. Laxminarayana and B. V. Rao, J. Mol. Catal. A: Chem., 2006, 255, 74–77 CrossRef CAS.
- K. V. Sashidhara, A. Kumar, R. P. Dodda and B. Kumar, Tetrahedron Lett., 2012, 53, 3281–3283 CrossRef CAS.
- R.-Z. Wang, L.-F. Zhang and Z.-S. Cui, Synth. Commun., 2009, 39, 2101–2107 CrossRef CAS.
- X. J. Sun, J. F. Zhou and P. S. Zhao, J. Heterocycl. Chem., 2011, 48, 1347–1350 CrossRef CAS.
- K. P. Kumar, S. Satyanarayana, P. L. Reddy, G. Narasimhulu, N. Ravirala and B. V. S. Reddy, Tetrahedron Lett., 2012, 53, 1738–1741 CrossRef.
- X.-J. Sun, J.-F. Zhou and S.-J. Zhi, Synth. Commun., 2012, 42, 1987–1994 CrossRef CAS.
- J.-J. Shie and J.-M. Fang, J. Org. Chem., 2003, 68, 1158–1160 CrossRef CAS.
- M. B. M. Reddy and M. A. Pasha, Synth. Commun., 2011, 41, 2081–2085 CrossRef.
- M. Gao, Y. Yang, Y.-D. Wu, C. Deng, W.-M. Shu, D.-X. Zhang, L.-P. Cao, N.-F. She and A.-X. Wu, Org. Lett., 2010, 12, 4026–4029 CrossRef CAS.
- M. Gao, Y. Yang, Y.-D. Wu, C. Deng, L.-P. Cao, X.-G. Meng and A.-X. Wu, Org. Lett., 2010, 12, 1856–1859 CrossRef CAS.
- Y. Yang, M. Gao, L.-M. Wu, C. Deng, D.-X.E Zhang, Y. Gao, Y.-P. Zhu and A.-X. Wu, Tetrahedron, 2011, 67, 5142–5149 CrossRef CAS.
- A. Kumar, G. Gupta and S. Srivastava, Org. Lett., 2011, 13, 6366–6369 CrossRef CAS.
- X.-S. Wang, J. Sheng, L. Lu, K. Yang and Y.-L. Li, ACS Comb. Sci., 2011, 13, 196–199 CrossRef CAS.
- W.-Y. Chen and J. Lu, Synlett, 2005, 1337–1339 CrossRef CAS.
- G. Yin, Y. Zhu, L. Zhang, P. Lu and Y. Wang, Org. Lett., 2011, 13, 940–943 CrossRef CAS.
- Y. Yang, M. Gao, D.-X. Zhang, L.-M. Wu, W.-M. Shu and A.-X. Wu, Tetrahedron, 2012, 68, 7338–7344 CrossRef CAS.
- N. Mulakayala, P. V. N. S. Murthy, D. Rambabu, M. Aeluri, R. Adepu, G. R. Krishna, C. M. Reddy, K. R. S. Prasad, M. Chaitanya, C. S. Kumar, M. V. B. Rao and M. Pal, Bioorg. Med. Chem. Lett., 2012, 22, 2186–2191 CrossRef CAS.
- G.-W. Wang and J. Gao, Org. Lett., 2009, 11, 2385–2388 CrossRef CAS.
- D. P. Sahu, S. K. Giri, V. Varshney and S. Kumar, Synth. Commun., 2009, 39, 3406–3419 CrossRef CAS.
- L. Rong, X. Li, H. Wang, D. Shi, S. Tu and Q. Zhuang, Synth. Commun., 2006, 36, 2345–2353 CrossRef CAS.
- X.-S. Wang, Q. Li, M.-M. Zhang, C.-S. Yao and S.-J. Tu, J. Heterocycl. Chem., 2008, 45, 1027–1031 CrossRef CAS.
- S. E. Denmark and S. Venkatraman, J. Org. Chem., 2006, 71, 1668–1676 CrossRef CAS.
- X.-F. Lin, S.-L. Cui and Y.-G. Wang, Tetrahedron Lett., 2006, 47, 4509–4512 CrossRef CAS.
- B. Das, P. Thirupathi, I. Mahender and K. R. Reddy, J. Mol. Catal. A: Chem., 2006, 247, 182–185 CrossRef CAS.
- M. B. M. Reddy, A. Nizam and M. A. Pasha, Synth. Commun., 2010, 40, 3728–3733 CrossRef CAS.
- M. A. Ameen and E. Kh. Ahmed, J. Heterocycl. Chem., 2008, 45, 579–581 CrossRef CAS.
- P.-A. Faugeras, B. Boëns, P.-H. Elchinger, J. Vergnaud, K. Teste and R. Zerrouki, Tetrahedron Lett., 2010, 51, 4630–4632 CrossRef CAS.
- S. S. Ali, Arch. Appl. Sci. Res., 2010, 2, 121–125 CAS.
- B. Das, N. Chowdhury and K. Damodar, Tetrahedron Lett., 2007, 48, 2867–2870 CrossRef CAS.
- P. Paira, A. Hazra, S. Kumar, R. Paira, K. B. Sahu, S. Naskar, P. Saha, S. Mondal, A. Maity, S. Banerjee and N. B. Mondal, Bioorg. Med. Chem. Lett., 2009, 19, 4786–4789 CrossRef CAS.
- B. Boëns, P.-A. Faugeras, J. Vergnaud, R. Lucas, K. Teste and R. Zerrouki, Tetrahedron, 2010, 66, 1994–1996 CrossRef.
- R. G. Little, J. A. Anton, P. A. Loach and J. A. J. Ibers, J. Heterocycl. Chem., 1975, 12, 343–349 CrossRef CAS.
- M. Stępień and J. L. Sessler, Org. Lett., 2007, 9, 4785–4787 CrossRef.
- B. V. S. Reddy, N. Rajeswari, M. Sarangapani, Y. Prashanthi, R. J. Ganji and A. Addlagatta, Bioorg. Med. Chem. Lett., 2012, 22, 2460–2463 CrossRef.
- M. Kidwai, V. Bansal, P. Mothsra, S. Saxena, R. K. Somvanshi, S. Dey and T. P. Singh, J. Mol. Catal. A: Chem., 2007, 268, 76–81 CrossRef CAS.
- S.-J. Ji, S.-Y. Wang, Y. Zhang and T.-P. Loh, Tetrahedron, 2004, 60, 2051–2055 CrossRef CAS.
- B. P. Bandgar and K. A. Shaikh, Tetrahedron Lett., 2003, 44, 1959–1961 CrossRef CAS.
- N. C. Ganguly, P. Mondal and S. K. Barik, Green Chem. Lett. Rev., 2012, 5, 73–81 CrossRef CAS.
- T. Nobuta, A. Fujiya, N. Tada, T. Miura and A. Itoh, Synlett, 2012, 2975–2979 CAS.
- B. Ke, Y. Qin, Q. He, Z. Huang and F. Wang, Tetrahedron Lett., 2005, 46, 1751–1753 CrossRef CAS.
- M. L. Deb, B. Baruah and P. J. Bhuyan, Synthesis, 2008, 286–292 CAS.
- R. Gu, A. Hameurlaine and W. Dehaen, J. Org. Chem., 2007, 72, 7207–7213 CrossRef CAS.
- N. Singh and K. N. Singh, Synlett, 2012, 23, 2116–2120 CrossRef CAS.
- M. Xia and Y. Lu, Synth. Commun., 2006, 36, 2389–2399 CrossRef CAS.
- S. Ko, C. Lin, Z. Tu, Y.-F. Wang, C.-C. Wang and C.-F. Yao, Tetrahedron Lett., 2006, 47, 487–492 CrossRef CAS.
- Z.-H. Zhang and J. Lin, Synth. Commun., 2007, 37, 209–215 CrossRef CAS.
- X.-F. Zeng, S.-J. Ji and X.-M. Su, Chin. J. Chem., 2008, 26, 413–416 CrossRef CAS.
- A. Hazra, P. Paira, K. B. Sahu, S. Banerjee and N. B. Mondal, Catal. Commun., 2008, 9, 1681–1684 CrossRef CAS.
- N. Iranpoor, B. Tamami and K. Niknam, Can. J. Chem., 1997, 75, 1913–1919 CrossRef CAS.
- B. Tamami, N. Iranpoor and H. Mahdavi, Synth. Commun., 2002, 32, 1251–1258 CrossRef CAS.
- N. Deka and J. C. Sarma, Chem. Lett., 2001, 794–795 CrossRef CAS.
- M. Z. Zahouily, A. Mezdar, J. Rakik, A. Elmakssoudi, A. Rayadh and S. Sebti, J. Mol. Catal. A: Chem., 2005, 233, 43–47 CrossRef CAS.
- Y.-M. Ren and C. Cai, Tetrahedron Lett., 2008, 49, 7110–7112 CrossRef CAS.
- Y.-M. Ren and C. Cai, Synth. Commun., 2010, 40, 1670–1676 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |