 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted solvothermal synthesis of zirconium oxide based metal–organic frameworks†
Weibin
Liang
and
Deanna M.
D'Alessandro
*
School of Chemistry, Building F1, University of Sydney, NSW 2006, Australia. E-mail: deanna@chem.usyd.edu.au; Tel: +61 (2)93513777
First published on 15th March 2013
Abstract
Zirconium oxide based Metal–Organic Frameworks were synthesised using a rapid and efficient microwave-assisted solvothermal method that produced purer phases and higher quality crystalline products in significantly (>95%) less time than the conventional heating method. A new amino-functionalised analogue has been synthesised exclusively using this microwave-assisted methodology.
Metal–Organic Frameworks (MOFs) are a rapidly expanding class of microporous materials which exhibit enormous structural and chemical diversity.1–3 By taking advantage of the vast array of inorganic and organic building blocks, a plethora of functional materials with promising applications have been obtained.2,4,5 One issue that has impeded the real-world application of many frameworks is their low stability, particularly under ambient conditions;6 for example, the archetypal framework [Zn4O(bdc)3], MOF-5, where bdc = 1,4-benzenedicarboxylate, is prone to hydrolysis in air at ambient temperature.7–10
Recently, Serre and coworkers reported a series of porous zirconium MOFs with the general formula [ZrO(O2C–X–CO2)] (X = C6H4, C10H6, C12H8 and C12N2H6Cl2 for MIL-140A, MIL-140B, MIL-140C and MIL-140D, respectively), which comprise one-dimensional zirconium oxide chains connected by linear dicarboxylic acid ligands.11 This class of materials combines exceptional hydrophobicity with high chemical and mechanical stability which paves the way for their industrial application in areas such as catalysis, gas adsorption and gas separation processes.
Typically, MOFs are synthesised by mass diffusion, conventional electric heating (CE) and microwave heating (MW),12 while electrochemical, mechanochemical and sonochemical methods have been less commonly employed.12,13 Compared with other methods, the MW synthesis protocol offers distinct advantages in terms of the reduction in the synthesis time and the increase in energy efficiency that favours industrial production processes.14–19 The present work demonstrates the efficient synthesis of the isoreticular frameworks MIL-140A-MW and MIL-140B-MW using microwave irradiation, in addition to the synthesis of a new framework, MIL-140A-NH2-MW ([ZrO(abdc)], where abdc = 2-amino-1,4-benzenedicarboxylate).
The reaction parameters employed for the MW synthesis of the MIL-140 frameworks were analogous to those reported previously in the literature for the CE heating method,11 with only the synthesis time being varied. The reaction medium consisting of 0.25 mmol ZrCl4, 0.5 mmol of a given ligand (1,4-benzenedicarboxylic acid (H2bdc), 2,6-naphthalenedicarboxylic acid (H2ndc) or 2-amino-1,4-benzenedicarboxylic acid (H2abdc)) and 2.5 mL DMF was heated at a temperature of 220 °C to produce MIL-140A-CE and MIL-140B-CE after 16 and 6 h, respectively, under CE heating, compared with MIL-140A-MW and MIL-140B-MW produced after 15 minutes under MW heating (ramp time = 2 min, hold time = 15 min) (ESI†). Initial attempts to produce the amino-functionalised framework incorporating the abdc ligand under CE heating using the same conditions (temperature, concentration, stoichiometry) and only varying the acetic acid/ZrCl4 ratio produced the UiO-66-NH2 framework exclusively (ESI†),20 rather than the desired MIL-140A-NH2 phase; the possibility of generating the latter phase using alternate conditions under CE heating cannot, however, be excluded.
Previous reports concerning the MW synthesis of MOFs have described order of magnitude increases in the reaction rates compared with CE heating, principally due to localised superheating resulting from (i) dipolar polarisation (the principal heat-generation pathway for the solvents which results in rapid molecular rotation induced by the oscillation of the electric field), (ii) enhanced refluxing within the sealed vessel, (iii) the presence of electrically conducting materials, and (iv) the generation of gas plasma from absorbed gases.15,21 In the present case, the superheating effect explains why it is possible to perform the MW synthesis using shorter reaction times compared with the conventional solvothermal synthesis method.
The crystallinity of the frameworks prepared using the two synthesis protocols was examined using powder X-ray diffraction (PXRD) analysis (Fig. 1). The frameworks synthesised via MW heating exhibited higher crystallinities than their counterparts obtained from CE heating, as evidenced by the reduced linewidths and more intense diffraction peaks in the former case. The positions and relative intensities of the peaks in the experimental PXRD patterns of MIL-140A-MW and MIL-140B-MW along with the cell parameters calculated from Le Bail refinement were in very good agreement with those obtained for MIL-140A-CE and MIL-140B-CE, respectively (ESI†). The PXRD patterns and cell parameters for the two frameworks synthesised by CE heating were also in good agreement with those reported in the literature,11 however, compared with the simulated pattern, the PXRD for MIL-140A-CE exhibited a mixture of two phases: additional peaks at 2θ = 12.1°, 17.1°, and 25.9° corresponded to the UiO-66 impurity (ESI†).22
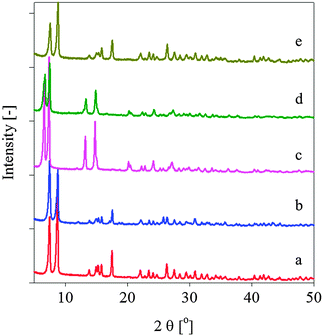 | ||
| Fig. 1 PXRD patterns of (a) MIL-140A-MW, (b) MIL-140A-CE, (c) MIL-140B-MW, (d) MIL-140B-CE and (e) MIL-140A-NH2-MW. | ||
The rapid heating rate employed in the MW synthesis method (25 °C to 220 °C within 2 min) was found to be critical to the improvement in the product purity compared with CE heating. Recently, Serre and coworkers hypothesised that the MIL-140 phase may be the recrystallisation product of the UiO phase.11 While the latter is initially formed at low temperatures (150 °C for UiO-66),22 a further increase in the temperature leads to dissolution of this phase and recrystallisation of MIL-140 at 220 °C. In the present case, even after 16 h under CE heating, the UiO-66 phase persisted in MIL-140A-CE, and a further increase in the reaction time to 18 h did not improve the phase purity of the product (ESI†). Furthermore, under MW heating, an increase in the ramp time from 2 min to 1 h (target temperature: 220 °C) led to the appearance of the UiO-66 phase impurity in the PXRD patterns of MIL-140A-MW (ESI†). Thus, the use of a rapid ramp rate in the MW heating protocol is key to the improved phase purity and crystallinity of the MIL-140 frameworks synthesised herein.
The importance of the microwave methodology was further demonstrated by the synthesis of a new isoreticular framework incorporating abdc, MIL-140A-NH2-MW, which could not be attained in initial trials using the CE heating method (varying the acetic acid/ZrCl4 ratio). Regardless of the reaction time (12 and 24 h) and the concentration of the acetic acid modulator, only UiO-66-NH2 could be obtained using the CE heating method (ESI†).20 The close similarity of the PXRD patterns of MIL-140A-MW and MIL-140A-NH2-MW confirmed the isostructural nature of the frameworks (Fig. 1a and e).
Fig. 2 shows the SEM images of the frameworks synthesised under MW irradiation at 220 °C for 15 min and using CE heating at 220 °C for more than 6 h (16 h for MIL-140A-CE and 6 h for MIL-140B-CE). The materials synthesised by MW heating appeared as plate-like crystallites (Fig. 2a, c and e), in comparison with the relatively more disordered nature of the materials obtained by CE heating (Fig. 2b and d). In particular, the irregular clusters observed in the latter cases may be assigned to the UiO impurities which were detected in the PXRD patterns (ESI†). A comparison of the SEM images shows that the materials synthesised under MW irradiation (Fig. 2a and c) exhibit a higher degree of crystallinity than those synthesised via the CE heating method (Fig. 1b and d).
 | ||
| Fig. 2 SEM images of (a) MIL-140A-MW, (b) MIL-140A-CE, (c) MIL-140B-MW, (d) MIL-140B-CE and (e) MIL-140A-NH2-MW (scale bar = 3 μm). | ||
All frameworks, including the newly synthesised MIL-140A-NH2-MW, exhibited pronounced thermal stabilities up to ca. 450 °C. In general, thermogravimetric analysis (TGA) of the CE and MW synthesised samples revealed two distinct weight loss steps corresponding to the loss of methanol employed in the washing procedure, and to the decomposition of the organic linkers and the formation of zirconium oxide, respectively (ESI†).23,24 The theoretical and experimental weight loss profiles for MIL-140A-MW, MIL-140A-CE, MIL-140B-MW and MIL-140A-NH2-MW (calculated from their chemical formulae, ESI†) were in good agreement; however, the experimentally determined weight loss for MIL-140B-CE (57.9%) was less than the theoretical value (61.6%).
The experimental and theoretical Brunauer–Emmett–Teller (BET) surface areas for all synthesised samples are summarised in Table 1 (the N2 sorption isotherms are shown in the ESI†). The experimental BET surface areas were determined as 337(8) and 438(8) m2 g−1, respectively, for MIL-140A-MW and MIL-140B-MW, which were in good agreement with the theoretical values (360 and 429 m2 g−1 for MIL-140A and MIL-140B, respectively).11 For MIL-140A-CE and MIL-140B-CE synthesised by CE heating, the BET surface areas were calculated to be 585(16) and 398(9) m2 g−1, respectively. The discrepancy between the experimental and theoretical surface areas for MIL-140A-CE (62.5% higher) is attributed to the presence of the relatively more porous UiO-66 impurity within the product (the BET surface area for UiO-66 was reported to be 969 m2 g−1).25 The surface area for MIL-140A-NH2-MW was 234(6) m2 g−1, which was 30% lower than that for MIL-140A-MW, consistent with the reduced pore space and the increased overall weight of the functionalised framework which results from the introduction of the amino group.
| Phasea | Theoretical SBET [m2 g−1]11 | Reported SBET [m2 g−1]11 | Experimental SBET [m2 g−1] |
|---|---|---|---|
| a MW: microwave heating, temperature ramp to 220 °C within 2 min; CE: conventional electric heating; the reaction vessel was placed in a pre-heated oven at 220 °C. | |||
| MIL-140A-MW | 360 | — | 337(8) |
| MIL-140A-CE | 360 | 415(10) | 585(16) |
| MIL-140B-MW | 429 | — | 438(8) |
| MIL-140B-CE | 429 | 460(10) | 398(9) |
| MIL-140A-NH2-MW | — | — | 234(6) |
In summary, the application of a microwave-assisted solvothermal method to frameworks in the MIL-140 series produces purer phases and higher quality crystalline products in significantly (>95%) less time than the conventional heating method. A new amino-functionalised analogue, which was unattainable using the CE heating method within the range of synthesis conditions explored in this work, was exclusively synthesised using this microwave-assisted methodology. The extension of the MW method to a wider range of MIL-140 analogues with other functional groups will pave the way towards new materials for gas separation and catalysis, amongst other applications. The development of rapid and efficient synthesis methods for MOFs which favour industrial production represents a critical aspect in their eventual deployment at the industrial scale.
Notes and references
- M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef CAS.
- A. Bétard and R. A. Fischer, Chem. Rev., 2012, 112, 1055–1083 CrossRef.
- F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- A. Corma, H. García, F. X. Llabrés and I. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS.
- S. J. Yang, J. Y. Choi, H. K. Chae, J. H. Cho, K. S. Nahm and C. R. Park, Chem. Mater., 2009, 21, 1893–1897 CrossRef CAS.
- M. Sabo, A. Henschel, H. Frode, E. Klemm and S. Kaskel, J. Mater. Chem., 2007, 17, 3827–3832 RSC.
- J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2006, 128, 10678–10679 CrossRef CAS.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Ferey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9188 CrossRef CAS.
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2012, 113, 734–777 CrossRef.
- E. Haque, N. A. Khan, C. M. Kim and S. H. Jhung, Cryst. Growth Des., 2011, 11, 4413–4421 CAS.
- J. Klinowski, F. A. Almeida Paz, P. Silva and J. Rocha, Dalton Trans., 2011, 40, 321–330 RSC.
- M. Kim, S. J. Garibay and S. M. Cohen, Inorg. Chem., 2011, 50, 729–731 CrossRef CAS.
- Z. Xiang, D. Cao, X. Shao, W. Wang, J. Zhang and W. Wu, Chem. Eng. Sci., 2010, 65, 3140–3146 CrossRef CAS.
- X.-F. Wang, Y.-B. Zhang, H. Huang, J.-P. Zhang and X.-M. Chen, Cryst. Growth Des., 2008, 8, 4559–4563 CAS.
- O. Karagiaridi, M. B. Lalonde, W. Bury, A. A. Sarjeant, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 18790–18796 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- Y. Yoo and H. K. Jeong, Chem. Commun., 2008, 2441–2443 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS.
- A. Schaate, S. Dühnen, G. Platz, S. Lilienthal, A. M. Schneider and P. Behrens, Eur. J. Inorg. Chem., 2012, 790–796 CrossRef CAS.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, PXRD patterns, Le Bail refinements, TGA curves and sorption data. See DOI: 10.1039/c3cc40368h |
| This journal is © The Royal Society of Chemistry 2013 |