De novo design of small molecule inhibitors targeting the LEDGF/p75-HIV integrase interaction†
Claudia
Cavalluzzo‡
a,
Arnout
Voet‡
b,
Frauke
Christ
c,
Brajendra Kumar
Singh
ad,
Ajendra
Sharma
a,
Zeger
Debyser
c,
Marc De
Maeyer
b and
Erik Van der
Eycken
*a
aLaboratory for Organic & Microwave-Assisted Chemistry (LOMAC); Katholieke Universiteit Leuven, Celestijnenlaan 200F, B-3001, Leuven, Belgium. E-mail: erik.vandereycken@chem.kuleuven.be; Fax: +3216327990
bLaboratory for Biomolecular modeling; Katholieke Universiteit Leuven, Celestijnenlaan 200G, B-3001, Leuven, Belgium
cMolecular Medicine, Katholieke Universiteit Leuven, Kapucijnenvoer 33, B-3000, Leuven, Flanders, Belgium
dPresent address: Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi, 110 007, India
First published on 2nd December 2011
Abstract
The integration of the viral DNA into the host genome is one of the essential steps in the HIV replication cycle. This multistep process mediated by the viral enzyme integrase (IN) allows identification and development of inhibitors targeting different integrase activities. Lens epithelium-derived growth factor (LEDGF/p75) has recently been identified as a crucial cellular co-factor of integration that acts by tethering IN to the cellular chromatin. Small molecules inhibiting the LEDGF/p75-IN interaction may become new and highly active antiretroviral therapeutic agents. In this paper we report the rational design, synthesis and evaluation of inhibitors that target the LEDGF/p75 protein and compete with IN binding. These molecules are designed to mimic the integrase alpha-3 helix, which interacts with LEDGF/p75, using pharmacophore guided scaffold replacement. The inhibitor 3-(1H-indol-3-ylthio)-N-(2-isopropoxy-6-methoxypyridin-3-yl)benzamide (CAB1) and its derivatives (CAB2–13) inhibit the LEDGF/p75-IN protein–protein interaction with moderate potency. These CAB inhibitors are the first reported example of small molecules targeting the LEDGF/p75 partner of the protein–protein interaction, in contrast to the previously reported compounds which target the integrase partner.
Introduction
Since the discovery of HIV as the causative agent of AIDS more than 25 years ago, virus spreading in pandemic proportions resulted in the death of more than 25 million people.1 More than 20 drugs have been approved for the treatment of HIV infection, significantly improving patients' life expectancy. Most drugs target the viral proteins such as Reverse Transcriptase, Protease or Integrase. Enfuvirtide and Maraviroc on the other side inhibit the entry of the viral particle into the host cell.2 Unfortunately all drugs approved for use in patients have given rise to resistance occurrence. Hence there is a necessity for the development of new antiretroviral agents with high potency against a wider range of viral variants and potentially a higher barrier towards resistance development.3 The viral integrase enzyme (IN), with its multiple functions in the early HIV replication cycle, is an appealing target for the development of new antiretrovirals. Currently only the strand transfer inhibitor Raltegravir has been approved for clinical use, but resistance mutations rapidly occur.4IN is a 32 kDa protein responsible for the integration of the viral cDNA into the host genome.5 It is subdivided into three structural domains: the N-terminal domain (NTD), the catalytic core domain (CCD) and the C-terminal domain (CTD) and forms multimeric complexes.6–9 IN mediates two enzymatic reactions: (i) the cleavage of a dinucleotide from the 3′ ends of linear viral cDNA, and (ii) the transesterification of the processed cDNA into the human chromatin.5
The multistep integration process is an interesting target for the development of various inhibitor classes due to its complexity and the multimeric quaternary structure including the interaction with a human protein, LEDGF/p75.10 This cofactor was identified as a tight binding partner of HIV IN and found to be crucial for the insertion of the viral cDNA into the host genome.11–13 The LEDGF/p75–IN interaction is mediated by the CCD of IN that binds to the IN binding domain (IBD) present in the C-terminal region of LEDGF/p75. Through this interaction LEDGF/p75 functions as a tethering factor linking the integration complex to the chromatin.10,11,14 Of major importance for the validation of LEDGF/p75 as a target for antiretroviral therapy was the demonstration that the overexpression of the IBD of LEDGF/p75 can efficiently compete with endogenous LEDGF/p75 and potently inhibits HIV replication.15,16
Targeting the LEDGF/p75-IN interaction with Small Molecule Protein–Protein Interaction Inhibitors (SMPPIIs) is a new strategy for the development of antiretroviral therapeutics. Up to now, only a few inhibitors of the HIV-1 IN-LEDGF/p75 complex have been reported, all binding to the IN component of the protein–protein interactions.17 The first reported molecule, D77, was identified during a screening approach and is reported to inhibit the LEDGF/p75–IN interaction in vitro.18 Next, CHIBA-3003, an IN strand transfer inhibitor derivative displaying micromolar inhibition of the LEDGF/p75–IN interaction, was reported.19 Both compounds bind to the dimer interface of IN but did not display antiviral activity in cell culture. Recently LEDGINs were reported as the first class of LEDGF/p75–IN inhibitors, with potent antiviral activity in cell culture.20 These compounds were derived from molecules identified by a virtual screening strategy. The LEDGINs target the CCD interfere with its interaction with LEDGF/p75–IBD. They provide proof-of-principle that rational design is a valid approach to develop protein–protein interaction inhibitors with antiviral activity. Crystal structures of the compounds soaked in the CCD dimer confirmed their binding mode and mechanism of action on a structural level. Resistance selection resulted in the integrase Ala-128-Thr mutation, indicating that LEDGINs indeed bind in the integrase pocket as required during the computational drug discovery setup. This occurrence of resistance however opened the search for molecules targeting the LEDGF/p75–IN interaction with a potentially higher genetic barrier to resistance. It is generally believed that binding to the human protein (LEDGF/p75) and not the viral protein (IN) will make resistance selection difficult for the virus.20 Nevertheless, this approach may induce increased toxicity since the human proteins can interact with other cellular binding partners. In fact, LEDGF/p75 interacts with several human proteins such as JPO2,21 PogZ22, the S-phase kinase23 and Menin.24
Here we report the rational design, synthesis and evaluation of small molecules binding to LEDGF/p75 and inhibiting the LEDGF/p75–IN interaction in vitro. These molecules are the first example of SMPPIIs targeting LEDGF/p75 and not IN, thus giving the possibility of developing new antiretrovirals without rising resistance. Molecular modeling allowed us to design a compound (referred to as CAB1), that mimics the alpha-3 helix of IN, a major contributor of the IBD–CCD interaction. The inhibitory activities of the small molecules were determined using a proximity based luminescence method.
Results and discussion
Design of the molecules
The CAB inhibitors were designed to mimic the interaction of the crucial amino acids (Thr-124, Ala-127 and Trp-131) of the integrase alpha-3 helix (Fig. 1) with LEDGF/p75 without resembling a classical alpha helix mimetic.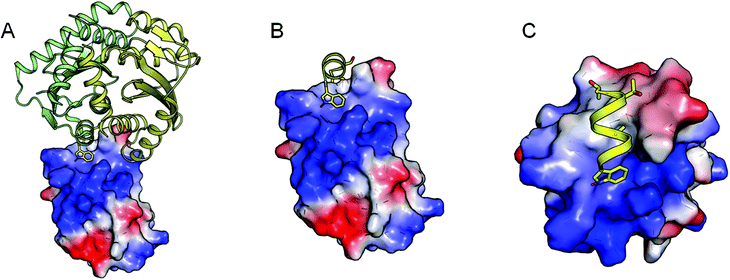 | ||
| Fig. 1 Schematic representation of the IBD structure (electrostatics are mapped on the molecular surface) in complex with the integrase CCD dimer (green-yellow cartoon A). The key interacting amino acid Trp-131 (sticks) of the alpha 3 helix (yellow cartoon) is depicted in panel B and C in different orientations to highlight the interaction of the residue in a subducted surface of the IBD. | ||
This rational approach was preferred over other classical methodologies, which also employ the structural information as available in the crystal structure of the protein complex, such as the discovery through virtual screening of small molecule ligands (employing pharmacophore modeling or molecular docking) or the design and synthesis of an alpha helix mimetic.25 In fact, a virtual screening experiment, similar to the approach previously used to discover IN-binding molecules for this interaction,20 was performed. Molecules that passed the pharmacophore query (based on the protein interaction interface) were subsequently docked to the LEDGF/p75 interface; however none of these docked molecules fulfilled the prerequisite binding mode according to the pharmacophore model, and their predicted affinity was much lower than classically observed in docking simulations. As such this approach was abandoned.
A second attempt focused on the design of an alpha helix mimetic incorporating the key interacting groups (Thr-124, Ala-127 and Trp-131, see Fig. 2A) such that the functionalities grafted on a scaffold overlap with the geometry of the interacting amino acids in a relaxed conformation (see Fig. 2B and C). This option was modelled, but the interaction of a functionalized alpha helix mimetic scaffold in complex with the IBD structure indicated that over 70% of the compounds' solvent accessible surface area was solvent exposed (see Fig. 2D). Furthermore, during docking simulations such compounds bound in a completely different conformation than intended. As the required binding mode is rather unlikely to exist, we opted for another de novo design strategy.
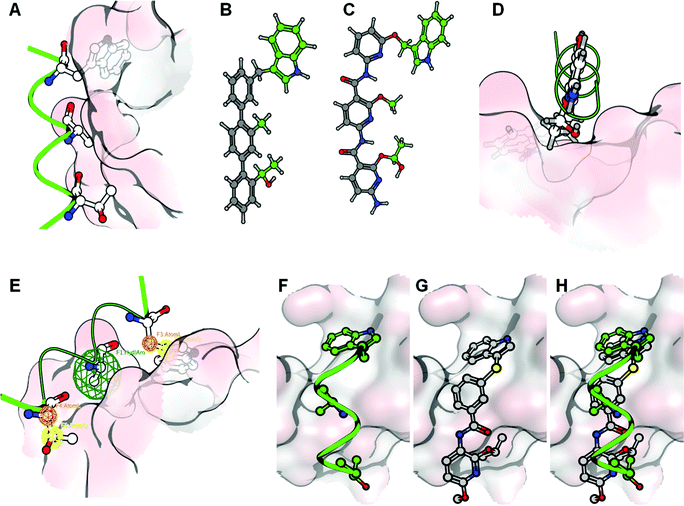 | ||
| Fig. 2 From the alpha-3 helix mimic to the de novo design strategy: the interaction of the alpha-3 helix with the IBD is mainly exerted by 3 amino acids (Thr-124, Ala- 127 and Trp-131) (A). These can be transferred onto helix-mimetic scaffolds such as the molecules depicted in B and C. These molecules however are unlikely to bind to the IBD surface since the major part of the molecule is solvent exposed (D). Therefore a scaffold incorporating the key amino acid interactions using pharmacophore features was used to design an inhibitory molecule (E). In frames F, G and H, the alpha-3 helix, a docked conformation of the designed compound and their superpositions, are represented respectively. In these frames the IBD surface is colored according to the electrostatic interactions. | ||
This strategy combines the knowledge gained from the two previously explored methods. The pharmacophore model, used for pharmacophore guided scaffold replacement, describes the projection of the interacting amino acids (Thr-124 and Trp-131) onto a small molecule scaffold in such a way that the core has maximal contact with the IBD surface, by translating the Ala-127 into a hydrophobic or aromatic feature (see Fig. 2E). Several core options are plausible, and the compound presented here was chosen since it showed the highest electrostatic field similarity with the mimicked alpha helix. Additionally this compound’s docked binding mode was in agreement with the molecules conformation and interactions according to the pharmacophore guided scaffold replacement strategy (Fig. 2F, G and H). Furthermore, in the designed compound Thr-124 was replaced with an isopropyl functionality to increase the hydrophobic interacting surface of the inhibitor as well as the chemical feasibility. The sulfur linker was chosen because its atom’s size was found to be optimal to span the distance between the aromatic scaffold and the indole moiety.
To explore the necessity of the functional groups on the molecules and to explore a putative Structure Activity Relationship (SAR), additional derivatives of the de novo designed CAB-inhibitor were generated.
Synthesis of the molecules
The synthesis of the de novo designed CAB-inhibitors has been accomplished by amide coupling of an appropriately substituted 3-(1H-indol-3-ylthio)benzoic acid with a 2,6-dialkoxypyridin-3-amine. These compounds were generated as indicated in the retrosynthetic Scheme 1.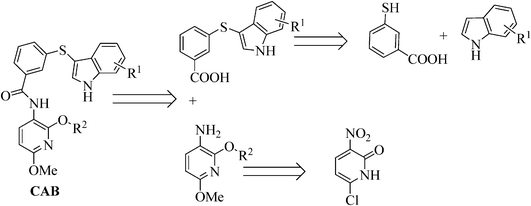 | ||
| Scheme 1 Retrosynthetic analysis of the designed compounds. | ||
The sulfynilation of the variously substituted indoles 1a–d was accomplished with FeCl3 as catalyst in acetonitrile26 under microwave irradiation27,28 at a ceiling temperature of 140 °C and a maximum power of 150 W for 30 min, yielding the desired compounds with a 90–96% yield (Scheme 2). Remarkably, under conventional heating at reflux temperature the reaction requires 6 h to reach completion and the desired compound 3a could only be obtained in a moderate yield of 50% due to the formation of an unidentified by-product.
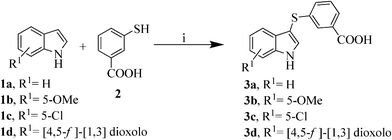 | ||
| Scheme 2 Synthesis of intermediates 3a–d. Reagents and conditions: i) FeCl3 (20% mol), acetonitrile, MW: 140 °C, 150 W, 30 min, 90–96%. | ||
According to our previously optimized protocol,29 pyridone 4 has been subjected to O-alkylation using different alkyl halides in the presence of Ag2CO3 under microwave irradiation to afford compounds 5a,b. Treatment of 5a,b with NaOMe in MeOH at reflux temperature for 1 h resulted in the formation of the intermediates 7a. Compound 7c could directly be formed from 2,6-dichloro-3-nitropyridine (6) using an excess of NaOMe in MeOH for 2 h at reflux temperature. Compounds 7a–c were subsequently reduced to the corresponding 3-aminopyridines 8a–cvia a Pd/C-catalysed hydrogenation at 25 psi pressure at r.t. for 2 h (Scheme 3).
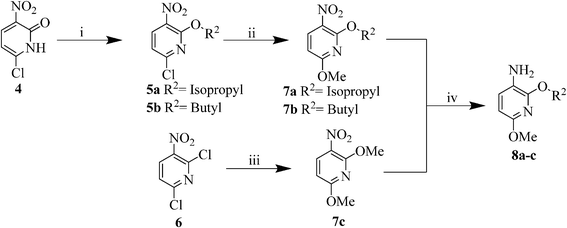 | ||
| Scheme 3 Synthesis of intermediates 8a–c. | ||
The two key building blocks 3 and 8 were then coupled using HOAt, EDC.HCl and N-methyl morpholine under microwave irradiation at a ceiling temperature of 80 °C and a maximum power of 80 W for 20 min to give the desired final compounds CAB1–8 with 55–75% yields (Table 1).
|
|
|||
|---|---|---|---|
| Compound | R1 | R2 | Yieldb (%) |
| a Reactions were performed with benzoic acids 3 (0.25 mmol) in DCM (2 mL) using 3-aminopyridines 8 (1.3 equiv), EDC.HCl (1.3 equiv), HOAt (1.3 equiv) and N-methyl morpholine (1.3 equiv) under microwave irradiation at a ceiling temperature of 80 °C and a maximum power of 80 W for 20 min. EDC.HCl = 1-Ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride; HOAt = 1-hydroxy-7-azabenzotriazole. b isolated yields are given. | |||
| CAB1 | H | Isopropyl | 75 |
| CAB2 | H | Butyl | 63 |
| CAB3 | H | Methyl | 65 |
| CAB4 | 5-OMe | Isopropyl | 70 |
| CAB5 | 5-OMe | Methyl | 50 |
| CAB6 | 5-Cl | Isopropyl | 65 |
| CAB7 | 5-Cl | Methyl | 60 |
| CAB8 | [4,5-f]-[1,3] dioxolo | Methyl | 55 |
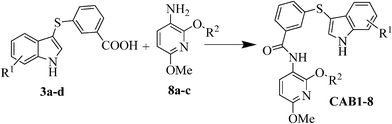
Next we extended the series by using two commercially available anilines following the previous amide coupling procedure. In this way compounds CAB9, 10 were obtained (Fig. 3). The synthesis of the 2-isopropoxy analogue of CAB9 was attempted, however we were unable to isolate the desired compound. To verify the importance of the indole moiety we also synthesized compounds CAB11, 12 in which the indole ring has been omitted. Condensation of the intermediates 8a,c with the commercially available 3-(methylthio)-benzoic acid or benzoic acid respectively, resulted in the desired compounds (Fig. 3).
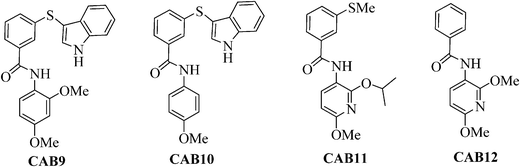 | ||
| Fig. 3 Structures of compounds CAB9-12. Reagents and conditions: i) Ag2CO3 (2 equiv), R2I (1.2 equiv), hexane, MW: 150 °C, 200 W, 10 min, 89–93%; ii) NaOMe (1.05 equiv), MeOH, reflux, 2 h, 80–85%; iii) NaOMe (2.5 equiv), MeOH, reflux, 2 h, 90%; iv) H2, Pd/C, EtOH, 25 psi, r.t., 2 h, 75–82%. | ||
Since the indole binding area lays on a downwardly bent surface with respect to the other part of the inhibitor binding site, a rigidification of the linker of the indole moiety was attempted, while taking the correct 3D organization of the chemical functionalities into account. As such, the thioether linkage in CAB1, has been replaced by a carbonyl, leading to the synthesis of compound CAB13 (Scheme 4). Friedel–Crafts acylation of indole with the acid chloride of the mono-methyl isophtalate (9) was carried out with diethylalluminiumchloride30 resulting in the formation of compound 10. After saponification with NaOH (2M) the benzoic acid derivative 11 was coupled with 8a applying the previously described conditions, resulting in the formation of the desired compound CAB13.
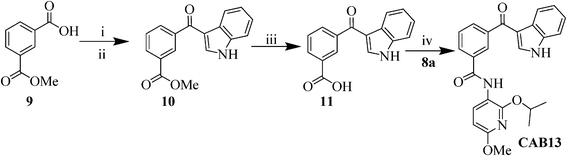 | ||
| Scheme 4 Synthesis of 3-(1H-indole-3-carbonyl)-N-(2-isopropoxy-6-methoxypyridin-3-yl)benzamide (CAB13). Reagents and conditions: i) SOCl2, reflux, 1 h, 80%; ii) indole, Et2AlCl (0.38 mL, 1.8 M, 0.7 mmol), toluene, 0 °C, 2 h, 50%; iii) NaOH (2M), reflux, 4 h, 85%; iv) HOAt (1.3 equiv), EDC.HCl (1.3 equiv), 4-methyl morpholine (1.3 equiv), DCM, MW: 80 °C, 80 W, 20 min, 51%. | ||
Biological evaluation
All compounds CAB1–13 were evaluated for their ability to inhibit the LEDGF/p75–IN interaction in a test tube protein–protein interaction assay.The synthesized CAB-compounds were first tested in the AlphaScreen assay to evaluate inhibition of the interaction between LEDGF/p75 and HIV-1 IN. The originally designed CAB1 showed a 50% inhibition of the interaction (Table 2). To improve the potency and to investigate the SAR we modified both the indole and the pyridine core. We first decided to introduce different substituents at the C-5 position of the indole such as Cl, OMe and the bulkier [4,5-f]-[1,3] dioxolo. The electron-donating methoxy group (Table 2, CAB4) was equally tolerated as well as the [4,5-f]-[1,3] dioxolo (Table 2, CAB8) whereas the introduction of the electronegative chlorine caused a complete loss of activity (Table 2, CAB6). The changes in the substitution of the indole core were combined with the replacement of the isopropoxy- with a methoxy-group on the pyridine core; no relevant biological difference was observed (Table 2, CAB3,5,7). The introduction of the bulkier butoxy group on the pyridine core, to enhance the hydrophobic interactions, did not result in an important change of the activity (Table 2, CAB2). We speculate that the electronic nature of the pyridine ring rather than the hydrophobic interaction appears to be crucial for the biological activity. In fact the replacement of the pyridine system with a benzene ring, combined with the introduction of the less bulky methoxy side chain group, showed a slight activity improvement (Table 2, CAB9). The absence of a substituent at the C-2 position of the benzene caused an activity decrease (Table 2, CAB10).
Next we examined the importance of the indole. Therefore two derivatives were synthesized in which the indole core was omitted or replaced by a methyl group (Table 2, CAB12 and 11, respectively). However, both modifications appeared to be detrimental for the inhibition underlining the relevance of the indole moiety for the biological activity. To give more rigidity to the indole side chain we replaced the thioether linker with a carbonyl moiety. Unfortunately the analogue CAB13 did not inhibit the IN–LEDGF/p75 interaction (Table 2, CAB13).
For the compounds showing 50% inhibition or better in the IN–LEDGF/p75 protein–protein interaction assay, the EC50 values for inhibition of HIV-1 replication in cell culture were measured, as well as their potency to inhibit the interaction of LEDGF/p75 with its cellular interactor JPO2 (Table 3). The most active compound of our series, CAB9 (Table 3), bears an indole linked to the central core of the molecule via a thioether bond; the substituted pyridine ring has been replaced by a 2,4-dimethoxyaniline, thus simplifying the chemistry.
| Compound | IC 50 value (μM) | MTT:MT-4 EC50b (μM) | MTT:MT-4 CC50c (μM) | JPO2 %d inhibition |
|---|---|---|---|---|
| a 50% inhibitory concentration as determined by AlphaScreen. b Effective concentration required to reduce HIV-1 induced cytopathic effect by 50% in MT-4 cells. c Cytotoxic concentration reducing MT-4 cell viability by 50%. d Measured at 100 μM. All values represent the average of triplicates. | ||||
| CAB1 | 64 | >48 | 48 | 42 |
| CAB4 | >100 | >38 | 38 | 32 |
| CAB8 | >100 | >29 | 29 | 36 |
| CAB9 | 30 | >10 | 10 | 37 |
Furthermore, the AlphaScreen based LEDGF/p75–JPO2 inhibition assay indicated that these inhibitors not only inhibit the interaction between LEDGF/p75 and the viral IN, but also its natural interacting protein JPO2. This expected result originates from the fact that targeting the LEDGF/p75 protein with a small molecule on the IBD surface will also disrupt the interaction of its cellular binding partners, since Maertens, et al.13 have indicated that JPO2 binds to LEDGF/p75 on the same region as IN does. As such this proves that the CAB derivatives truly inhibit the LEDGF/p75–IN interaction by binding the IBD and not by binding on the IN protein such as the previously identified LEDGINS.20 In fact the IN protein and the JPO2 are sequentially and structurally not related to each other, excluding that the inhibitors reported here act by binding to IN as well as JPO2. The MTT drug susceptibility assay revealed that all evaluated compounds clearly enter the cell, which is demonstrated by the observed cytotoxicity. This, together with the in vitro influence of our compounds on the natural binding partner of LEDGF/p75, indicates that the compounds truly inhibit the cellular cofactor LEDGF/p75 and not the HIV-IN, as previously published inhibitors do.
Although not very high, the cytotoxicity expressed by the CAB-compounds is sufficient to reveal that this series of compounds is inhibiting the cellular role of LEDGF/p75.
For instance it is known that like HIV-1 IN, JPO2 also interacts with the IBD of LEDGF/p75.22 The amino acids in the IBD that are critical for IN interaction were proven not crucial for the interaction with JPO2, underlining the differential structural constraints for both interactions. Consequently, more potent compounds could be designed taking into consideration these differences between the specific LEDGF/p75 binding domains targeting only the regions involved into the binding with IN.
Conclusion
The design and synthesis of novel SMPPIIs inhibiting the LEDGF/p75–IN interaction responsible for the integration of the viral genome into the host DNA is presented. The CAB-compounds were rationally designed in agreement with the pharmacophore scaffold replacement model (Fig. 2D). In this model the amino acid residues of IN involved in the interaction with LEDGF/p75, as demonstrated by the crystal structure of the complex, were projected onto a small scaffold to optimize the contacting surface of the IBD of LEDGF/p75. Out of thirteen synthesized analogues, compound CAB9 showed the strongest inhibition of the targeted protein–protein interaction. Experimental evidence is provided that these compounds are in fact LEDGF/p75 binding molecules disrupting the LEDGF/p75 HIV-1 integrase interaction. In this study we wanted to indicate the possibility to rationally design inhibitors that selectively target the LEDGF/p75 partner of the protein–protein interaction, rather than the previously reported structures where the integrase partner was targeted. Currently the potency of the described compounds is not sufficient to inhibit HIV-replication in cell culture, but further efforts, in order to increase the specificity of such compounds towards the critical region of IBD binding to IN and not to other cellular co-factors, are undertaken. We hope, in this way, to improve the activity of this novel and promising series of SMPPII.Experimental section
Molecular modelling
Analysis of the protein–protein interaction between the CCD of HIV-1 Integrase with the IBD of LEDGF/p75 was done using the Brugel software package.31 The crystal structures of the interacting domains of interest (CCD and IBD) were retrieved from the PDB repository (PDB entry 2B4J).32 The non-bonded interactions between the two partners were determined per amino acid using a CHARMM based force field.33All molecular modeling experiments involving small molecules were performed using the Molecular Operating Environment (MOE) (The Chemical Computing Group, Montreal, Canada) and the latest available version of the GOLD docking algorithm.
All alpha helix mimetic molecules (Fig. 2, panel B and C) were modeled by sketching and their optimal conformation was analyzed by conformational analysis. The docking of the ligand compounds was performed using the pharmacophore constrained docking method as implemented in MOE. Subsequently the Trp-131 residue was described with 3 pharmacophore features and the Thr-124 and the Ala-127 each with one hydrophobic feature.
The de novo design of a non alpha helix-mimetic molecule was done using the pharmacophore based scaffold replacement method implemented in MOE. The interacting amino acids Thr-124 and Trp-131 were retained as molecular functionality with their linking atom bonds as pharmacophore linking features, while amino acid Ala-127 was described as a “hydrophobic OR aromatic” pharmacophore feature. This pharmacophore query was used to find a scaffold replacement, from the provided scaffold database [MOE2009.10 distribution, containing approximately 750,000 scaffolds] that links the two chemical groups. Possible chemical solutions were analyzed for synthetic feasibility as well as retaining the desired conformation during conformational analysis. To fulfil the pharmacophore, the class of CAB-compounds was designed in such a way that the indole moiety is linked with the alkoxy group via a thio-ether bond.
Chemistry
General remarks
1H and 13C NMR spectra were recorded on a Bruker Avance 300 (300 MHz, 1H; 75 MHz 13C) and 400 (400 MHz, 1H; 100 MHz 13C) instruments. The chemical shifts are reported in parts per million relative to tetramethylsilane using the residual solvent signal as internal reference. Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), qt (quintet), m (multiplet) and b (broad). Coupling constants, J, are reported in Hertz. Mass spectra were recorded by using a Kratos MS50TC and a Kratos Mach III data system. The ion source temperature was 150–250 °C as required. High resolution EI-mass spectra were performed with a resolution of 10000. The low resolution spectra were obtained with a HP5989A MS instrument. For thin layer chromatography, analytical TLC plates (Alugram SIL G/UV254 and 70–230 mesh silica gel (E.M.Merck)) were used. Visualization was accomplished with UV (254).Microwave irradiation experiments
All microwave irradiation experiments were carried out in a dedicated CEM-Discover monomode microwave apparatus (CEM Corporation P.O. Box 200 Matthews, NC 28106), operating at a frequency of 2.45 GHz with maximum irradiation power of 300 W. Reaction mixtures were efficiently stirred with a magnetic stirrer. The reactions were carried out in a sealed 10 mL glass vial. The temperature of the reactions was measured by infrared.General procedure for the thioarylation of the indoles
A mixture of the indole 1 (0.2 mmol), mercaptobenzoic acid (3) (0.2 mmol) and anhydrous FeCl3 (7 mg, 20 mol%) in acetonitrile (2 mL) was irradiated at a ceiling temperature of 140 °C and a maximum power of 150 W for 30 min. The progress of the reaction was monitored by TLC and CI-MS. After complete conversion the reaction mixture was cooled by compressed air and brought to r.t.. The FeCl3 was filtered off on Celite, the acetonitrile was evaporated under reduced pressure and the resulted crude product was purified by column chromatography over silica gel (heptane–EtOAc 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 containing acetic acid (0.1%)) yielding the desired products 3a–3d in 90–96% yield.
:2 containing acetic acid (0.1%)) yielding the desired products 3a–3d in 90–96% yield.
Compounds 5a,b were prepared and characterized in accordance with the published procedure.20
General procedure for the synthesis of compounds 7a,b
A mixture of the 6-chloro-2-alkoxy-3-nitropyridine (5 (0.5 mmol) and anhydrous NaOMe (28 mg, 0.525 mmol) was refluxed for 2 h in dry MeOH (6 mL). The progress of the reaction was monitored by TLC and CI-MS. After complete conversion the mixture was cooled to r.t. and the MeOH was evaporated under reduced pressure. The organic residue was washed with H2O, extracted with EtOAc and the extract was dried on Na2SO4. The crude compound was purified by column chromatography over silica gel (heptane–EtOAc 8:2) giving the desired products 7a,b in 72–80% yield.
Procedure for the synthesis of compound 7c
Compound 7c was obtained using a slightly modified procedure which consisted of directly refluxing the commercially available 2,6-dichloro-3-nitropyridine (6) (96 mg, 5 mmol) with anhydrous NaOMe (80 mg, 12.5 mmol) for 2 h in dry MeOH (6 mL). The progress of the reaction was monitored by TLC and CI-MS. After complete conversion the reaction mixture was cooled to r.t., the MeOH was evaporated under reduced pressure and the organic residue was washed with H2O and extracted with EtOAc. The extract was dried on Na2SO4 and the crude product was purified by column chromatography over silica gel (heptane–EtOAc 8:2) giving the desired product 7c in 93% yield.
General method for the synthesis of compounds 8a–c
A mixture of compound 7 (0.2 mmol) and 5% Pd/C (5 mol%) in MeOH (5 mL) was agitated at r.t. under 25 psi hydrogen for 2 h. The catalyst was removed by filtration on Celite and the filtrate was evaporated to dryness to give the pure products (75–82%).General procedure for the synthesis of compound 10
To perform the Friedel–Crafts acylation, the commercially available mono-methyl isophtalate (9) was converted to the corresponding acid chloride in accordance with the published procedure.30 To a DCM (2 mL) solution of indole (1a) (54 mg, 0.46 mmol) was added diethylalluminum chloride (0.38 mL, 0.7 mmol, 1.8 M in toluene) at 0 °C. The mixture was stirred at 0 °C for 30 min and then the acid chloride derivative of compound 9 (81 mg, 0.69 mmol) in DCM (2 mL) was added drop wise. The resulting solution was stirred at 0 °C for 2 h and then brought to r.t.. A pH 7 phosphate buffer was added to quench the reaction and the mixture was extracted with EtOAc. The combined organic layers were concentrated under vacuum and the residue was purified by column chromatography over silica gel (heptane–EtOAc 9:1) yielding the desired compound in 63%.
General procedure for the synthesis of compound 11
A suspension of the ester 10 (50 mg, 0.178 mmol) in NaOH (5 mL, 2M) was refluxed until no more starting material was observed (TLC and CI-MS monitoring). The mixture was neutralized with HCl (1 M) and extracted with EtOAc. The organic layers were combined and concentrated under vacuum. The crude product 11 (40 mg) was directly used without further purification.:2) giving the desired products CAB1–13 in 50–80% yields.
LEDGF/p75-HIV-1 Integrase interaction screening (AlphaScreen technology)
AlphaScreen (Amplified Luminiscent Proximity Homogeneous Assay) is a bead-based technology used to study biomolecular interactions in a microtiter plate format. Binding of integrase to LEDGF/p75 leads to an energy transfer from the donor to the acceptor bead. This transfer induces the emission of luminescence. Inhibition of the interaction decreases the emission in a concentration dependent manner.The AlphaScreen assay was performed according to the manufacturer's protocol (Perkin Elmer, Benelux). Reactions were performed in 25 μL final volume in 384-well Optiwell™ microtiter plates (Perkin Elmer). The reaction buffer contained 25 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.01% (v/v) Tween-20 and 0.1% (w/v) bovine serum albumin. His6-tagged integrase (300 nM final concentration) was incubated with the compounds for 30 min at 4 °C. The compounds were added in varying concentrations ranging from 0.1 up to 100 μM. Afterwards 100 nM flag-LEDGF/p75 was added and incubation was prolonged for an additional hour at 4 °C. Subsequently 5 μL of Ni-chelate-coated acceptor beads and 5 μL anti-flag donor beads were added to a final concentration of 20 μg mL−1 of both beads. Proteins and beads were incubated for 1 h at 30 °C to allow association. Exposure of the reaction to direct light was omitted as much as possible and the emission of light from the acceptor beads was measured in the EnVision plate reader (Perkin Elmer, Benelux) and analyzed using the EnVision manager software. In order to analyse the inhibition of the JPO2-LEDGF/p75 interaction a modified protocol was used. Streptavidin-coated donor beads were incubated with biotinylated anti-MBP antibody (Vector laboratories, Burlingame, CA) for 1 h at room temperature. MBP-JPO2 was bound through the biotinylated anti-MBP antibody to the donor beads. Subsequently the assay was performed as described above.
Drug susceptibility assay
The inhibitory effect of the compounds on the HIV-induced CPE in MT-4 cell culture was determined by the MTT-assay.34 This assay is based on the reduction of the yellow colored 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) by mitochondrial dehydrogenase of metabolically active cells to a blue formazan derivative, which can be measured spectrophotometrically. The 50% cell culture infective dose of the HIV strains was determined by titration of the virus stock using MT-4 cells. For the drug susceptibility assays, MT-4 cells were infected with 100 to 300 50% cell culture infective doses of the HIV strain in the presence of five-fold serial dilutions of the antiviral drugs. The concentration of the compound achieving 50% protection against the CPE of HIV, which is defined as the 50% effective concentration (IC50 value), was determined. The concentration of the compound that was toxic for 50% of the MT-4 cells, which is defined as the 50% cytotoxic concentration (CC50), was determined as well.Virus strains
The origin of the HIV-1 strain IIIB has been described previously.35Expression and purification of recombinant proteins
His6-tagged HIV-1 integrase, 3×flag-tagged LEDGF/p75 and MBP-JPO2 were purified for AlphaScreen applications as described previously.21,36Acknowledgements
Financial Support was provided by the Research Fund of the Katholieke Universiteit Leuven (IDO 06/006) and the Fund for Scientific Research–Flanders (Belgium) (F.W.O). A. V. wishes to acknowledge the Research Fund of Katholieke Universiteit Leuven for the postdoctoral fellowship. The authors wish to thank Mr. B. Demarsin for the HRMS and Mr. S. G. Modha for his fruitful discussions and help. We would also like acknowledge the technical help of N. J. Vander Veken (AlphaScreen) and B. Van Remoortel (anti-HIV testing). Dr. B. K. Singh wishes to acknowledge the Department of Science and Technology (DST, India) for funding supportReferences
- WHO, WHO AIDS Update 2008. 2008 Search PubMed.
- J. C. Tilton and R. W. Doms, Antiviral Res., 2010, 85, 91–100 CrossRef CAS.
- C. S. Adamson and E. O. Freed, Drug Discovery Today, 2008, 13, 424–432 CrossRef CAS.
- I. Malet, O. Delelis, M. A. Valantin, B. Montes, C. Soulie, M. Wirden, L. Tchertanov, G. Peytavin, J. Reynes, J. F. Mouscadet, C. Katlama, V. Calvez and A. G. Marcelin, Antimicrob. Agents Chemother., 2008, 52, 1351–1358 CrossRef CAS.
- P. O. Brown, Curr. Top. Microbiol. Immunol., 1990, 157, 19–48 CrossRef CAS.
- A. Engelman and R. Craigie, J. Virol., 1992, 66, 6361–6369 CAS.
- F. D. Bushman, A. Engelman, I. Palmer, P. Wingfield and R. Craigie, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3428–3432 CrossRef CAS.
- R. A. Lutzke, C. Vink and R. H. Plasterk, Nucleic Acids Res., 1994, 22, 4125–4131 CrossRef CAS.
- A. P. Eijkelenboom, R. A. Lutzke, R. Boelens, R. H. Plasterk, R. Kaptein and K. Hard, Nat. Struct. Biol., 1995, 2, 807–810 CrossRef CAS.
- K. Busschots, A. Voet, M. De Maeyer, J. C. Rain, S. Emiliani, R. Benarous, L. Desender, Z. Debyser and F. Christ, J. Mol. Biol., 2007, 365, 1480–1492 CrossRef CAS.
- P. Cherepanov, G. Maertens, P. Proost, B. Devreese, J. Van Beeumen, Y. Engelborghs, E. De Clercq and Z. Debyser, J. Biol. Chem., 2003, 278, 372–381 CrossRef CAS.
- M. Llano, D. T. Saenz, A. Meehan, P. Wongthida, M. Peretz, W. H. Walker, W. Teo and E. M. Poeschla, Science, 2006, 314, 461–464 CrossRef CAS.
- G. Maertens, P. Cherepanov, Z. Debyser, Y. Engelborghs and A. Engelman, J. Biol. Chem., 2004, 279, 33421–33429 CrossRef CAS.
- P. Cherepanov, E. Devroe, P. A. Silver and A. Engelman, J. Biol. Chem., 2004, 279, 48883–48892 CrossRef CAS.
- A. Hombrouck, J. De Rijck, J. Hendrix, L. Vandekerckhove, A. Voet, M. De Maeyer, M. Witvrouw, Y. Engelborghs, F. Christ, R. Gijsbers and Z. Debyser, PLoS Pathog., 2007, 3, 418–430 CAS.
- J. De Rijck, L. Vandekerckhove, R. Gijsbers, A. Hombrouck, J. Hendrix, J. Vercammen, Y. Engelborghs, F. Christ and Z. Debyser, J. Virol., 2006, 80, 11498–11509 CrossRef CAS.
- L. Q. Al-Mawsawi and N. Neamati, ChemMedChem, 2011, 6(2), 228–241 CrossRef CAS.
- L. Du, Y. Zhao, J. Chen, L. Yang, Y. Zheng, Y. Tang, X. Shen and H. Jiang, Biochem. Biophys. Res. Commun., 2008, 375, 139–144 CrossRef CAS.
- L. De Luca, M. L. Barreca, S. Ferro, F. Christ, N. Iraci, R. Gitto, A. M. Monforte, Z. Debyser and A. Chimirri, ChemMedChem, 2009, 4, 1311–1316 CrossRef CAS.
- F. Christ, A. Voet, A. Marchand, S. Nicolet, B. A. Desimmie, D. Marchand, D. Bardiot, N. J. Van der Veken, B. Van Remoortel, S. V. Strelkov, M. De Maeyer, P. Chaltin and Z. Debyser, Nat. Chem. Biol., 2010, 6, 442–448 CrossRef CAS.
- K. Bartholomeeusen, J. De Rijck, K. Busschots, L. Desender, R. Gijsbers, S. Emiliani, R. Benarous, Z. Debyser and F. Christ, J. Mol. Biol., 2007, 372, 407–421 CrossRef CAS.
- K. Bartholomeeusen, F. Christ, J. Hendrix, J. C. Rain, S. Emiliani, R. Benarous, Z. Debyser, R. Gijsbers and J. De Rijck, J. Biol. Chem., 2009, 284, 11467–11477 CrossRef CAS.
- S. Hughes, V. Jenkins, M. J. Dar, A. Engelman and P. Cherepanov, J. Biol. Chem., 2010, 285, 541–554 CrossRef CAS.
- A. Yokoyama and M. L. Cleary, Cancer Cell, 2008, 14, 36–46 CrossRef CAS.
- J. T. Ernst, J. Becerril, H. S. Park, H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2003, 42, 535–539 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, Y. J. Reddy and K. Praneeth, Synthesis, 2009, 1520–1524 CrossRef CAS.
- A. Loupy and L. Perreuxin Microwave in Organic Synthesis (ed. A. Loupy), Wiley-VCH, Weinheim, 2006, pp. 134–218 Search PubMed.
- E. Van der Eycken and C. O. Kappein Topics in Heterocyclic Chemistry, ed. G. Springer, Berlin, 2006, pp. 267–304 Search PubMed.
- B. K. Singh, C. Cavalluzzo, M. De Maeyer, Z. Debyser, V. S. Parmar and E. Van der Eycken, Synthesis, 2009, 2725–2728 CAS.
- T. Okauchi, M. Itonaga, T. Minami, T. Owa, K. Kitoh and H. Yoshino, Org. Lett., 2000, 2, 1485–1487 CrossRef CAS.
- P. Delhaise, M. Bardiaux, M. De Maeyer, M. Prevost, D. Vanbelle, J. Donneux, I. Lasters, E. Vancustem, P. Alard and S. Wodak, J. Mol. Graph. Model., 1988, 6, 219 Search PubMed.
- P. Cherepanov, Z. Y. Sun, S. Rahman, G. Maertens, G. Wagner and A. Engelman, Nat. Struct. Mol. Biol., 2005, 12, 526–532 CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- R. Pauwels, J. Balzarini, M. Baba, R. Snoeck, D. Schols, P. Herdewijn, J. Desmyter and E. De Clercq, J. Virol. Methods, 1988, 20, 309–321 CrossRef CAS.
- A. Adachi, H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson and M. A. Martin, J. Virol., 1986, 59, 284–291 CAS.
- K. Busschots, J. Vercammen, S. Emiliani, R. Benarous, Y. Engelborghs, F. Christ and Z. Debyser, J. Biol. Chem., 2005, 280, 17841–17847 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00582k |
| ‡ the authors equally contributed to the paper. |
| This journal is © The Royal Society of Chemistry 2012 |