
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalization and solubilization of polycyclic aromatic compounds by sulfoniumization†
Johannes E.
Erchinger‡
 a,
Tsubasa
Okumura‡
b,
Kanami
Nakata
b,
Daisuke
Shimizu
b,
Constanstin G.
Daniliuc
a,
Kazuma
Amaike
*c,
Frank
Glorius
a,
Kenichiro
Itami
*cd and
Hideto
Ito
*b
a,
Tsubasa
Okumura‡
b,
Kanami
Nakata
b,
Daisuke
Shimizu
b,
Constanstin G.
Daniliuc
a,
Kazuma
Amaike
*c,
Frank
Glorius
a,
Kenichiro
Itami
*cd and
Hideto
Ito
*b
aOrganisch-Chemisches Institut, University of Münster, Corrensstrasse 36, 48149 Münster, Germany
bDepartment of Chemistry, Graduate School of Science, Nagoya University, Nagoya 464-8602, Japan. E-mail: ito.hideto.p4@f.mail.nagoya-u.ac.jp
cMolecule Creation Laboratory, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: kazuma.amaike@riken.jp; kenichiro.itami@riken.jp
dInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Nagoya 464-8602, Japan
First published on 11th April 2025
Abstract
Despite their unique physical properties and diverse applications in materials science, poor solubility of polycyclic aromatic hydrocarbons (PAHs) limits further fine-tuning and investigation of these systems. Herein, we report a sulfoniumization strategy to solubilize and functionalize a diverse range of PAHs in a one-step protocol using a triethylene glycol ether-substituted diaryl sulfoxide. While mono-sulfoniumization is generally observed, modification of the reaction conditions to favor bis-sulfoniumization is shown. The downstream applicability of the resulting PAH sulfonium salts is validated through a series of post-functionalization reactions that include C–C and C–heteroatom bond formation, while their application in annulative π-extension (APEX) is showcased by the synthesis of tetra-tert-butylquaterrylene from perylene. The red-shifted absorption and fluorescence, along with high water solubility of the PAH sulfonium salts, enable their application in bio-imaging, where they demonstrate selective mitochondrial staining without cytotoxicity.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) represent an important cornerstone of materials science, fueling promising advances in the realm of organic semiconductors.1 In addition, the distinctive optical properties of PAHs offer significant potential for biological applications.2 As their electronic, photophysical and biological properties are directly influenced by the quantity and arrangement of sp2-hybridized carbons, the tailorability of PAHs, e.g., by late-stage functionalization such as annulative π-extension (APEX)3,4 has proven to be a vital tool to access novel PAHs and molecular nanocarbons4 or to establish structural libraries of target π-conjugated core structures.5 However, π-conjugation and rigidity come with the drawback of aggregation in solution due to strong π–π interactions,6 which may limit the synthesis and transformation of principally accessible PAHs, and various solution-based biological applications.For the easier synthesis and application of poorly soluble PAHs, the introduction of solubilizing groups or a solubility-independent synthetic method is strongly required. For example, Kubota, Ito and co-workers circumvented this issue by solid-state cross-coupling using ball-milling, introducing solubilizing substituents in the process (Fig. 1A).7 To minimize property changes upon the introduction of solubilizing groups in the core aromatic structure, a dendrimer support connected by a cleavable linker was utilized by Yagi, Itami and co-workers, granting access to main-chain-unsubstituted “bare” aromatic polymers.8 To effectively incorporate PAHs into biological studies, water soluble substituents, such as triethylene glycol (TEG) groups or carboxyl groups, are commonly introduced into PAHs. To date, several water-soluble PAHs, including a warped nanographene,9 a planar graphene containing 222 carbon atoms,10 and a dibenzo[hi,st]ovalene11 have been applied as imaging probes as well as in photothermal and photodynamic therapy. However, these strategies and molecules require functionalized groups such as halogens in starting materials, which is also problematic during the preparation step due to the low solubility of halogenated PAHs. Therefore, late-stage functionalization and transformation of bare PAH to afford soluble functionalized PAHs are highly demanded for further applications.
 | ||
| Fig. 1 Current strategies for PAH solubilization and regioselective arene functionalization. (A) Dissolution of PAHs. (B) Regioselective sulfoniumization of small arenes via thianthrenation. (C) This work. | ||
As one of the prominent examples of late-stage functionalization, the thianthrenation of small aromatic systems is an appealing methodology to pre-activate small aromatic systems with exceptional regioselectivity and independence from functional handles in the substrate (Fig. 1B).12,13 Among the broad variety of recently explored functionalization reactions of thianthrenium salts, amination14 or aryne formation followed by cycloaddition with furans or pyrroles15 highlights the opportunities for programmable lynchpin installations and annulations, respectively. However, sulfoniumization of bare PAHs remains underdeveloped16 and—to the best of our knowledge—there is no precedence for the transformation of bare PAHs and cross-coupling through thianthrenation/sulfoniumization in the literature.17 Furthermore, the introduction of highly organic solvent/water soluble sulfonium substituents to bare PAHs can also facilitate a handleability and cell permeability in organic and aqueous media, leading to further transformation of PAH-sulfonium salts such as cross-coupling and APEX, and biological applications. Therefore, the development of the sulfoniumization method for poorly soluble PAHs can provide a new tool in synthetic chemistry for nanographenes and biomolecular chemistry. Herein, we report a method for thianthrenation/sulfoniumization of various bare PAHs for solubilizing poorly soluble PAHs and enabling further applications. We aimed to combine the prospects of solubilization and functionalization of PAHs through sulfoniumization with newly developed sulfoxides bearing solubilizing side-chains (Fig. 1C). The thus-obtained highly soluble PAH-sulfoniums salts enabled further transformations such as regioselective halogenation, cross-coupling and APEX, and bio-imaging.
Results and discussion
Thianthrenation of PAHs
To probe whether the thianthrenation protocol can be extended from small aromatic systems12 to PAHs, we chose unsubstituted naphthalene as a model compound. Indeed, the thianthrenium salt 3a could be obtained in good yield using chlorinated solvents and 1.5 equiv. of acid and thianthrenium oxide 2a (Scheme 1, see the ESI† for optimization studies).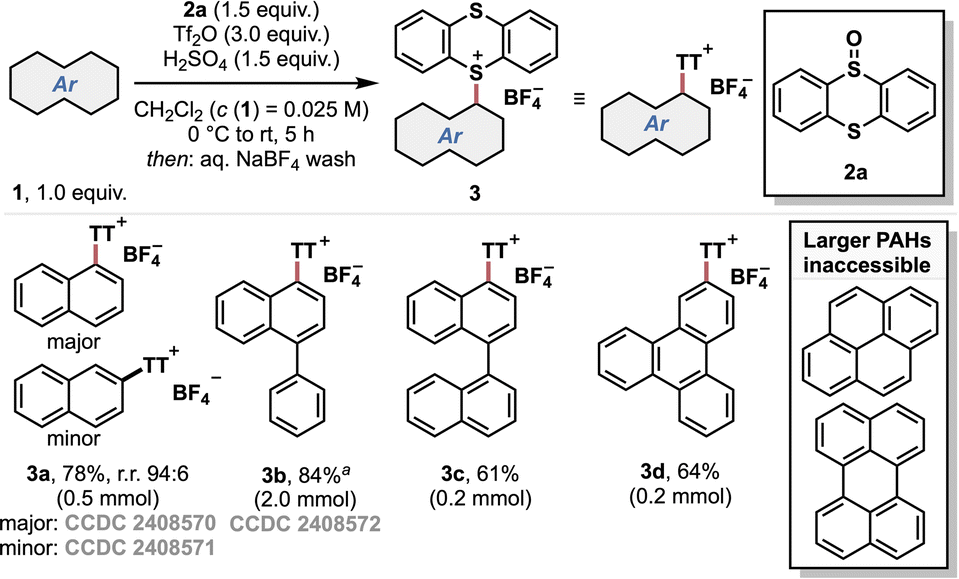 | ||
| Scheme 1 Thianthrenation of small aromatics and PAHs, and scope limitations. Isolated yields are given. Only one regioisomer was obtained, unless otherwise specified. a1.0 equiv. of thianthrene oxide 2a was used. | ||
The regioselectivity of the thianthrenation was corroborated by the X-ray crystal structure analysis of products 3a and 3b (see the ESI†). Interestingly, thianthrenation of 1-phenylnaphthalene occurred solely on the naphthalene moiety (3b). Furthermore, 1,1′-binaphthalene and triphenylene underwent efficient thianthrenation, affording thianthrenium salts 3c and 3d in good yield. However, it quickly became apparent that only small PAHs were suitable substrates for the thianthrenation protocol, as difficulties arose with both reactivity and—more generally—isolation of larger PAHs of interest, e.g., perylene (see the ESI†).
Sulfoniumization reagent screening
Along these lines, the pursuit of a broader sulfoniumization strategy clearly necessitates the use of solubilizing side-chains (Fig. 2A). We started our investigation with suitable PAH sulfoniumization reagents 2b–2f having a thianthrene core structure, screening possible tethers for their suitability under the naphthalene model reaction (Fig. 2B). While C(sp3)-, C(sp2)- and ether tethers on the thianthrene core exhibited poor performance (2b–2d, entries 1–3), silyl-based tethers showed promise but led to isolation issues (2e and 2f, entries 4–5, see the ESI†). The diaryl sulfoxide core bearing ortho-substitution did not yield the desired product (9a, entry 6), while the incorporation of electron-rich substituents at the para-position showed promising reactivity (9b, entry 7); additionally facile isolation was achieved with an ether tether (9c, entry 8). In comparison with installing long alkyl chains (9d, entry 9), the TEG motif was found to serve as a superior solubilization agent for both reactivity and the ease of isolation (see the ESI† for details), leading to the development of sulfoniumization reagent 9e (entry 10). Notably, simple but highly soluble sulfoxide 9e was easily prepared by etherification of 4,4′-sulfinyldiphenol with the corresponding alkyl bromide (see the ESI† for details). Furthermore, rotational freedom was found to be an important factor for sulfoniumization of naphthalene, as dibenzothiophene oxides 15a–c bearing a similar substitution pattern as their parent diaryl sulfoxides exhibited poor performance (entries 11–13). | ||
| Fig. 2 (A) Design and (B) screening of a sulfoniumization reagent for PAHs. | ||
PAH sulfoniumization scope
With reagent 9e in hand, the PAH scope was investigated (Scheme 2). To our delight—and in stark contrast to the thianthrenation protocol—a variety of larger PAHs including 1,1′-binaphthyl (1b), triphenylene (1c), perylene (1d), pyrene (1e), dibenzo[g,p]chrysene (1f), corannulene (1g) and coronene (1h) could undergo C–H sulfoniumization to yield the sulfonium salts 14b–h in good yields. Key to achieving high yields of products 14f–h was the solvent change from CH2Cl2 to 1,2-dichloroethane and prior solubilization of the PAHs under reflux before adding Tf2O and H2SO4 under ambient conditions to form the reactive cationic intermediate from reagent 9e. Interestingly, the bis-adduct 14d′ was observed as a side-product in the reaction with perylene, which could be favored by addition of an excess amount of 9e, Tf2O and H2SO4 to obtain 14d′ in 34% yield. Limitations include larger PAHs such as quaterrylene or hexabenzocoronene, which did not show reasonable solubility in refluxing 1,2-dichloroethane (see the ESI†). Notably, a series of PAH-sulfonium salts demonstrated exceptional solubility in MeOH, with values of >160 mg mL−1 for 14d and >297 mg mL−1 for 14h—over 1000 times greater than those of pristine perylene (0.16 mg mL−1) and coronene (0.09 mg mL−1) in MeOH. Additionally, 14d was found to dissolve even in deionized H2O with a solubility of 4.6 mg mL−1. | ||
| Scheme 2 PAH sulfoniumization scope. aConditions: 1 (1.0 equiv.), 9e (3.0 equiv.), Tf2O (9.0 equiv.), H2SO4 (4.6 equiv.), 1,2-dichloroethane, rt (24 °C), 5 h. bPAH was dissolved in refluxing 1,2-dichloroethane prior to the addition of Tf2O and H2SO4 (see the ESI†). | ||
Transformations of PAH sulfonium salts
Motivated by the multitude of functional group interconversions from thianthrenium/diarylsulfonium salts and driven by our interest in site-selective π-extension of the PAH systems, we applied several transformations to the functionalized PAHs according to the report by Ritter and co-workers.12 First, we examined sequential functionalization of triphenylene by consecutive thianthrenation with 2a followed by photo-induced iodination/cyanation, Suzuki–Miyaura cross-coupling and Sonogashira–Hagihara coupling reactions.18 We found that iodination, Suzuki–Miyaura coupling and Sonogashira–Hagihara coupling smoothly proceeded to afford 19a, 19c and 19d in overall 33%, 39% and 63% yields, respectively. On the other hand, photo-induced cyanation was somehow incompatible, affording 19b in only 12% yield.Next, we verified the transformability of the bis(4-alkoxyphenyl)sulfonium group on 14, as there is no precedent for the selective incorporation of acyclic triarylsulfonium salts in cross-coupling reactions in the literature (Scheme 3). Gratifyingly, Suzuki–Miyaura cross-coupling of bis(4-alkoxylphenyl)(perylene-3-yl)sulfonium salt 14d with phenylboronic acid preferentially gave 3-phenylperylene 20a in 78% yield. Motivated by this result, we also examined the two-fold cross-coupling between perylene bis-sulfonium salt 14d′ with phenylboronic acid and (3,6-di-tert-butylnaphthalen-1-yl)boronic acid pinacol ester, and diarylated perylenes 20b and 20c were obtained in high yields (77% and 83%). Dinaphthylperylene 20c was further extended to tetra-tert-butylquaterrylene 21 (ref. 19–21) by a FeCl3-promoted Scholl reaction, resulting in a high yield of 86%. Notably, 3,10-di-functionalized perylenes such as 3,10-dibromoperylene are an important structural motif as precursors for further cross-coupling for the synthesis of perylene-conjugated molecules as demonstrated here. However, the preparation of typically employed 3,10-dibromoperylene by bromination of bare perylene and its purification are often problematic due to the poor solubility of the products and the difficulty of separating them from other isomers such as 3,9-dibromoperylene.22 In this context, the present PAH-sulfoniumization strategy including solubilization and further functionalization offers an alternative synthetic tool for the efficient conversion of poorly soluble PAHs, bypassing the—oftentimes problematic—traditional bromination process.
 | ||
| Scheme 3 Transformation of PAH-sulfonium salts. Conditions for the synthesis of 19a: RuCl2(bpy)3·6H2O (3.3 mol%), CuBF4(MeCN)4 (1.0 equiv.), LiI (9.8 equiv.), MeCN/DMSO, blue LED, rt (23 °C), 21 h; conditions for the synthesis of 19b: RuCl2(bpy)3·6H2O (2.5 mol%), CuBF4(MeCN)4 (1.3 equiv.), NnBu4CN (2.5 equiv.), MeCN, blue LED, rt (24 °C), 21 h; conditions for the synthesis of 19c: PdCl2(dppf) (2.2 mol%), (4-tert-butylphenyl)boronic acid (1.4 equiv.), K3PO4 (2.0 equiv.), EtOH/1,4-dioxane, 50 °C, 48 h; conditions for the synthesis of 19d: PdCl2(dppf) (2.7 mol%), phenylacetylene (1.4 equiv.), CuI (21 mol%), N-methylmorpholine (2.1 equiv.), 1,4-dioxane, 40 °C, 48 h. | ||
The structure of 21 was elucidated by X-ray diffraction analysis for the first time (for further information, see the ESI†). Compared to a previous synthetic attempt for 21 from a naphthalene oligomer by Müllen and co-workers,19–21 our synthetic route to 21 offers several advantages in terms of step economy, the use of readily available pristine perylene as a starting material, the absence of aromatic ring rearrangement in the Scholl reaction, and a two-step APEX reaction with high yields.
Photophysical properties of perylene sulfonium salts
During the scope studies, we observed that the sulfoniumization strategy not only increased the solubility of PAH adducts in organic solvents, but also enabled the handling of these compounds in aqueous solution (vide supra). As the perylene core is well-known for its interesting photophysical properties, especially with regard to its diimide derivatives,23 we were interested in investigating the perylene mono- and bis-sulfonium salts further. Both the mono-adduct (14d) and bis-adduct (14d′) show red-shifted absorption maxima ( λabs (14d) = λabs (14d′) = 470 nm) compared to the parent perylene (λabs (perylene) = 437 nm) in CH2Cl2 (Fig. 3A). With regard to fluorescence, the emission maximum of mono-adduct 14d (λem (14d) = 518 nm) is red-shifted in comparison to perylene (λem (perylene) = 442 nm) due to charge separation (see density-functional theory (DFT) calculations and HOMO/LUMO representations in the ESI†), while the bis-adduct 14d′ shows a minor blue shift (λem (14d′) = 480 nm) with a small Stokes shift compared to 14d (Fig. 3B).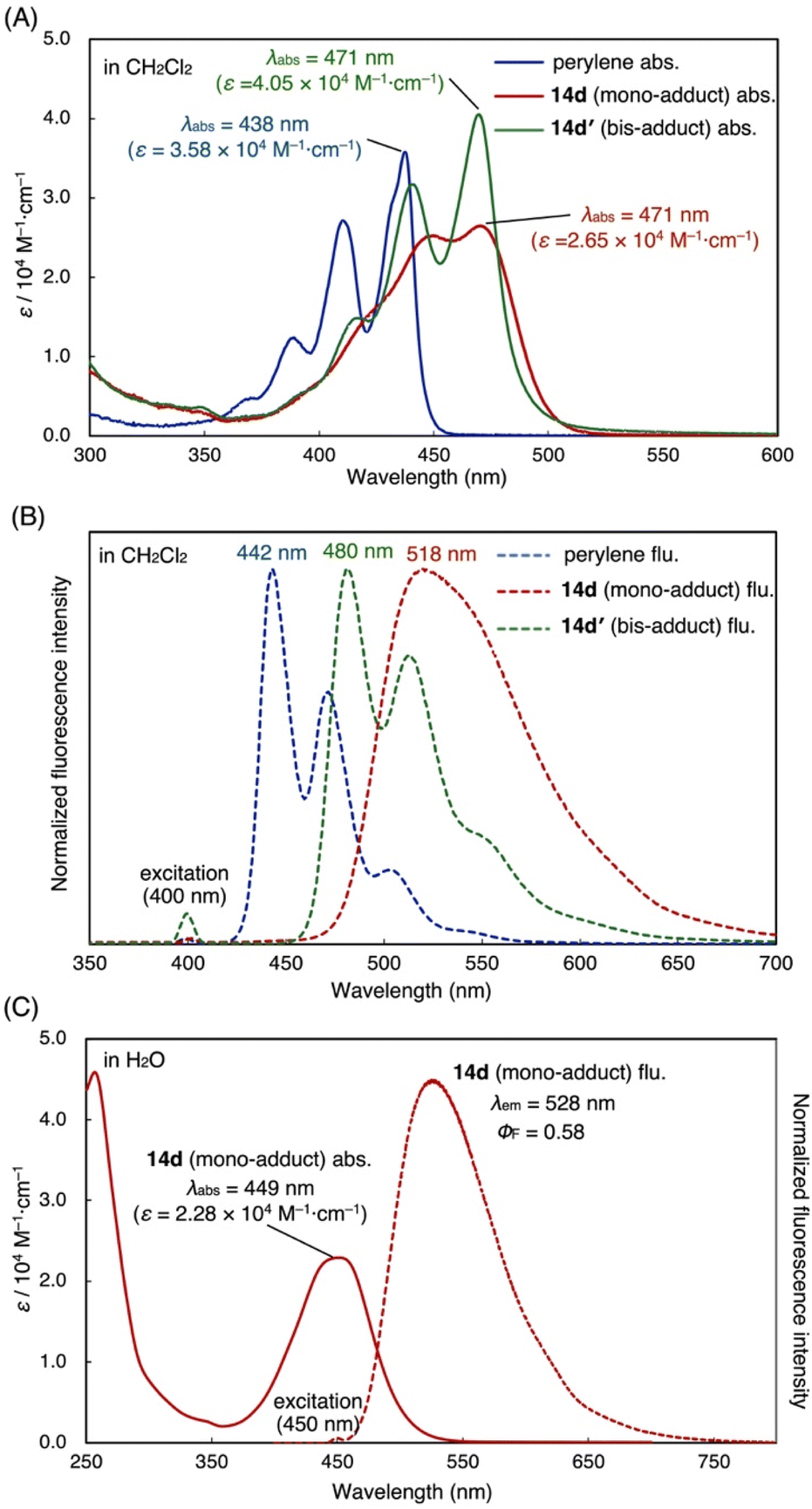 | ||
| Fig. 3 (A) Absorption and (B) emission spectra of 14d, 14d′ and perylene in CH2Cl2. (C) Absorption and emission spectra of 14d in H2O. | ||
This implies that the vibrational structure, as observed in rigid perylene, is largely restored in 14d′, whereas significant structural relaxation and charge transfer occur in 14d′. Furthermore, the water-soluble mono-adduct 14d shows a broad absorption band with a maximum at λabs = 449 nm, an emission with a maximum peak at λem = 528 nm and a fluorescence quantum yield of (ΦF) = 0.58 in H2O (Fig. 3C).
Mitochondria-selective bio-imaging
With better insight into the photophysical properties of the perylene sulfonium salt 14d and its good solubility in H2O, imaging of HeLa cells with 14d was performed (Fig. 4). | ||
| Fig. 4 Confocal microscopic imaging of HeLa cells with 14d. (A) Fluorescence of 14d and (B) mitochondrial marker. (C) Differential interference contrast (DIC). (D) Merged images of A, B and C. | ||
Notably, perylene derivatives have previously been identified as promising candidates for bio-imaging.24 Stimulated by this finding, 14d was incubated with HeLa cells for 30 min at 37 °C. Confocal microscopy images showed that its fluorescence was observed in the cells without any signs of cytotoxicity (Fig. 4A and S6†). Interestingly, it appeared to localize to specific organelles, which was validated using a mitochondrial marker. The Pearson correlation coefficients (r) for colocalization between each image were calculated to be 0.77 ± 0.02 (mean ± SE), supporting good correlation in mitochondria selective imaging (Fig S10†). This aligns well with the tendency of delocalized lipophilic cations (DLCs) to accumulate in mitochondria, as their lipophilic nature enables them to cross the membrane, drawn by its negative potential.25 To the best of our knowledge, this represents the first example of sulfonium salt localization in mitochondria.
Conclusions
Herein, a sulfoniumization strategy for the one-step solubilization and functionalization of PAHs is presented. Based on the disclosed scope limitations of the thianthrenation for PAHs, a tether-/core-based screening of plausible sulfoxide reagents identified diaryl sulfoxide bearing solubilizing TEG ether side-chains as a superb sulfoniumization reagent to enable the regioselective C–H functionalization of large PAH systems. The application of the PAH sulfonium salts is highlighted by various functional group interconversions. Upon sulfoniumization of the bare PAH, Suzuki–Miyaura cross-coupling followed by Scholl-type annulation offers a new, regioselective APEX pathway. The promising water solubility and photophysical properties of the perylene sulfonium salt were showcased in the bio-imaging of cells, demonstrating selective mitochondrial staining with no signs of cytotoxicity. We hope that this work sparks further interest in the selective functionalization and solubilization of PAHs to address current limitations in the characterization and modification of polyaromatic materials. Further detailed studies on the sulfoniumization of other nanocarbons, bio-imaging and the structure–function relationship with various PAH-sulfonium salts including 14d are currently underway.Data availability
Experimental and characterization data, including crystallo-graphic data [3a major regioisomer (CCDC No. 2408570), 3a minor regioisomer (CCDC No. 2408571), 3b (CCDC No. 2408572), and 21 (CCDC No. 2397243)], and NMR spectra, as well as computational investigations. The data supporting this article have been included as part of the ESI.†Author contributions
J. E. E. and T. O. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (GRK 2678–437785492, J. E. E.) and the Verband der Chemischen Industrie (VCI, Fonds der Chemischen Industrie, J. E. E.) for generous financial support. This study was supported by the Uehara Memorial Foundation (to K. I.), the Chugai Foundation for Innovative Drug Discovery Science (to K. I.), the Sumitomo Foundation (2300884, to H. I.), the Foundation of Public Interest of Tatematsu (22B025, to H. I.), the Kondo Memorial Foundation (2022-03, to H. I.), and the NAGAI Foundation of Science & Technology (to H. I.). We thank Dr Keigo Yamada and Mr Daiki Imoto for X-ray diffraction measurement, Dr Akiko Yagi (Nagoya University) for helpful discussion of this work and Prof. Masayasu Taki (Gifu University) for fruitful discussion about bio-imaging. The computations were performed at the Research Center for Computational Science, Okazaki, Japan (Project Nos. 23-IMS-C061 and 24-IMS-C059).Notes and references
- (a) H. Klauk, Chem. Soc. Rev., 2010, 39, 2643 RSC; (b) S.-I. Kawano, T. Hamazaki, A. Suzuki, K. Kurahashi and K. Tanaka, Chem.–Eur. J., 2016, 22, 15674 CrossRef CAS PubMed; (c) S. R. Forrest, Nature, 2004, 428, 911 CrossRef CAS; (d) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (e) L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J.-L. Brédas, K. N. Houk and A. L. Briseno, Acc. Chem. Res., 2015, 48, 500 CrossRef CAS PubMed.
- (a) N. Panwar, A. M. Soehartono, K. K. Chan, S. Zeng, G. Xu, J. Qu, P. Coquet, K.-T. Yong and X. Chen, Chem. Rev., 2019, 119, 9559 CrossRef CAS PubMed; (b) A. T. Krasley, E. Li, J. M. Galeana, C. Bulumulla, A. G. Beyene and G. S. Demirer, Chem. Rev., 2024, 124, 3085 CrossRef CAS PubMed.
- H. Ito, K. Ozaki and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 11144 CrossRef CAS PubMed.
- W. Matsuoka, H. Ito, D. Sarlah and K. Itami, Nat. Commun., 2021, 12, 3940 CrossRef CAS PubMed.
- W. Matsuoka, K. P. Kawahara, H. Ito, D. Sarlah and K. Itami, J. Am. Chem. Soc., 2023, 145, 658 CrossRef CAS.
- (a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS; (b) M. Rapacioli, F. Calvo, F. Spiegelman, C. Joblin and D. J. Wales, J. Phys. Chem. A, 2005, 109, 2487 CrossRef CAS PubMed; (c) N. J. Silva, F. B. C. Machado, H. Lischka and A. J. A. Aquino, Phys. Chem. Chem. Phys., 2016, 18, 22300 RSC; (d) K. Carter-Fenk and J. M. Herbert, Phys. Chem. Chem. Phys., 2020, 22, 24870 RSC.
- T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165 CrossRef CAS.
- S. Fujiki, K. Amaike, A. Yagi and K. Itami, Nat. Commun., 2022, 13, 5358 CrossRef CAS PubMed.
- H.-A. Lin, Y. Sato, Y. Segawa, T. Nishihara, N. Sugimoto, L. T. Scott, T. Higashiyama and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 2874 CrossRef CAS.
- X.-H. Ma, X. Gao, J.-Y. Chen, M. Cao, Q. Dai, Z.-K. Jia, Y.-B. Zhou, X.-J. Zhao, C. Chu, G. Liu and Y.-Z. Tan, J. Am. Chem. Soc., 2024, 146, 2411 CrossRef CAS PubMed.
- X. Zhu, Q. Chen, H. Zhao, Q. Yang, Goudappagouda, M. Gelléri, S. Ritz, D. Ng, K. Koynov, S. H. Parekh, V. K. Chetty, B. K. Thakur, C. Cremer, K. Landfester, K. Müllen, M. Terenzio, M. Bonn, A. Narita and X. Liu, J. Am. Chem. Soc., 2024, 146, 5195 CrossRef CAS.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS PubMed.
- F. Juliá, Q. Shao, M. Duan, M. B. Plutschack, F. Berger, J. Mateos, C. Lu, X.-S. Xue, K. N. Houk and T. Ritter, J. Am. Chem. Soc., 2021, 143, 16041 CrossRef.
- P. S. Engl, A. P. Häring, F. Berger, G. Berger, A. Pérez-Bitrián and T. Ritter, J. Am. Chem. Soc., 2019, 141, 13346 CrossRef CAS PubMed.
- R. A. Roberts, B. E. Metze, A. Nilova and D. R. Stuart, J. Am. Chem. Soc., 2023, 145, 3306 CrossRef CAS.
- J. Yan, A. P. Pulis, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2019, 58, 15675 CrossRef CAS PubMed.
- J. Sprachmann, A. Petrik, C. Douglas and O. Dumele, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-4nh7w-v2.
- X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang, Chem. Commun, 2020, 56, 5058 RSC.
- A. Bohnen, K.-H. Koch, W. Lüttke and K. Müllen, Angew. Chem., Int. Ed., 1990, 29, 525 CrossRef.
- K.-H. Koch and K. Müllen, Chem. Ber., 1991, 124, 2091 CrossRef CAS.
- U. Scherf and K. Müllen, Synthesis, 1992, 23 CrossRef CAS.
- (a) N. V. Korovina, C. H. Chang and J. C. Johnson, Nat. Chem., 2020, 12, 391 CrossRef CAS PubMed; (b) A. Matsumoto, M. Suzuki, H. Hayashi, D. Kuzuhara, J. Yuasa, T. Kawai, N. Aratani and H. Yamada, Chem.–Eur. J., 2016, 22, 14462 CrossRef CAS; (c) K. Hayashi and M. Inouye, Eur. J. Org Chem., 2017, 4334 CrossRef CAS.
- B. Zhang, H. Soleimaninejad, D. J. Jones, J. M. White, K. P. Ghiggino, T. A. Smith and W. W. H. Wong, Chem. Mater., 2017, 29, 8395 CrossRef CAS.
- (a) M. Sun, K. Müllen and M. Yin, Chem. Soc. Rev., 2016, 45, 1513 RSC; (b) M. Moniruzzaman, S. D. Dutta, K.-T. Lim and J. Kim, ACS Omega, 2022, 7, 37388 CrossRef CAS PubMed.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2408570, 2408571, 2408572 and 2397243. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01415h |
| ‡ J. E. E. and T. O. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |