
Efficient synthesis of polylactide and copolymers under industrial conditions by multinuclear β-ketoimide zinc complexes†
Yu
Cheng
abc,
Zihe
Zhao
a,
Xiaowei
Xu
a,
Chunxiao
Ren
a,
Xiaohui
Wei
a,
Yanxiang
Yang
a,
Jin
Li
d,
Daqiang
Jiang
b,
Kunyu
Zhang
*a,
Bin
Wang
 *e and
Yi
Luo
a
*e and
Yi
Luo
a
aAdvanced Materials Research Center, Petrochemical Research Institute, PetroChina Company Limited, Beijing, 102206, China. E-mail: zhangkunyu010@petrochem.com.cn
bCollege of New Energy and Materials, China University of Petroleum Beijing, No. 18 Fuxue Rd., Beijing, 102249, China
cNational Elite Institute of Engineering, CNPC, Beijing, 100096, China
dPetroChina Guangxi Petrochemical, Company, China
eSchool of Materials Science and Engineering, Tianjin University, 135 Yaguan Road, Tianjin, 300350, China. E-mail: binwang@tju.edu.cn
First published on 20th May 2025
Abstract
Polylactic acid (PLA), a representative degradable aliphatic polyester, has the advantages of biodegradability, biocompatibility, and good thermal and mechanical properties. The industrial production of high-molecular-weight PLA is achieved via the ring-opening polymerization (ROP) of L-lactide (L-LA) catalyzed by Sn(II)-2-ethyl-hexanoate (Sn(Oct)2) under melt and bulk conditions. Although huge efforts have been made to develop organometallics with low toxicity, and many catalysts that are highly active under mild laboratory conditions have been found, very few candidates can compete with Sn(Oct)2 under industrially relevant conditions. Here, we report novel multinuclear β-ketoiminate zinc complexes as efficient catalysts for L-LA polymerization under industrially relevant conditions, with a turnover frequency as high as 5880 h−1. The catalysts exhibited good stability and activity and compete well with Sn(Oct)2 under industrial conditions, affording colorless PLLA with high crystallinity. Preliminary copolymerization experiments suggested that the zinc catalyst can also catalyze the random copolymerization of L-LA with ε-caprolactone and dioxanone under melt and bulk conditions.
1. Introduction
The prevalence of plastics in modern life has driven an exponential increase in their production, leading to significant challenges pertaining to energy, the environment, and climate change.1–6 The transition from current petroleum-based plastics to sustainable alternatives represents a pivotal yet formidable challenge for modern society.7–9 Biodegradable polymers derived from renewable resources, such as polylactide (PLA) and polycaprolactone (PCL), along with their copolymers, offer a promising avenue for addressing these concerns. PLA, derived from renewable sources, such as corn sugar and sugar cane, is the most well-known commercially implemented aliphatic polyester, with a global production exceeding 300 kt per year. It has been applied in the fields of packaging, electronics, and biomedicine because of its renewability, biodegradability, biocompatibility, and good thermomechanical properties.10–12 The ring-opening polymerization (ROP) of cyclic esters using metal-based complexes,13–16 organic catalysts,17–20 and cooperative catalysis21–23 is the most promising method for synthesizing PLA and its copolymers, as ROP allows for good control over molecular weight and its distribution, and stereoregularity of the polymer.The current industrial production of high-molecular-weight PLA and copolymers is still based on the ROP of L-lactide by Sn(II)-2-ethyl-hexanoate (SnOct2) in the presence of excess alcohol at high temperatures (170–190 °C) and under solvent-free conditions, as Sn(Oct)2 has good performance in terms of activity and low racemization rate. Even though Sn(Oct)2 is FDA-approved and is considered safe at residual levels below 20 ppm, concerns remain over issues, such as cellular inflammation caused by toxic metal residues, as it is not possible to completely eliminate catalyst residues from polymers. For this reason, organometallics based on non-toxic metals, including Mg,24 Ca,25 Zn,26 Fe,27 and Al,28 have been developed for the ROP of LA. These complexes typically contain a kinetically-stabilizing organic ligand, an initiator group, and a free coordination site at the metal center, which allows the coordination of the monomer. Although there are many examples of catalysts that perform well in terms of activity, control, and stereoselectivity, most of the reported catalysts have been tested under mild conditions, namely, in solution at temperatures ranging from 25 °C to 100 °C. While some studies have reported polymerizations under solvent-free conditions, the typical temperatures range from130 °C to150 °C, and the conditions would be impractical for industrial application. In addition, while technical-grade monomer is used in industrial processes, the monomer is purified by recrystallization according to most literature reports.
Among the various catalysts, the classical β-diketiminate and β-ketoiminate metal catalysts have drawn our attention. The β-ketoiminate ligands have both oxygen and nitrogen coordination atoms, which allows them to bind well with the metal centers and placing a tunable and bulky substituent on one side can protect the metal center while leaving the other side open, enhancing the activity of the metal complexes. Zinc β-diketiminate/β-ketoiminate complexes have attracted great attention due to their low toxicity, high catalytic activities, and low price. Coates et al. first reported the heteroleptic zinc complexes L1ZnOiPr containing an N,N-β-diketiminato ligand (I, Scheme 1).29 These complexes showed good activity and high selectivity for the ROP of rac-lactide ([M]0/[C]0 = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 95% conv. in 20 min, 25 °C). Following this elegant work, various modifications of the β-diketiminato ligand have been conducted in the search for zinc complexes with high catalytic activity and thermal stability.30–34 Many ligands including bidentate N,O ketiminato and tridentate N,N,N,35 and O,N,N ligands with pendent coordination donors have been designed and synthesized.35–37 The easy modification of the electronic and steric properties in the β-diketiminato ligand allows the synthesis of tailor-made catalysts. Herres-Pawlis et al. reported an ultra-high-activity bisguanidine zinc complex (II, Scheme 1) for the ROP of LA. The polymerization rate constant (Kapp) of the catalyst was 1.432 L mol−1 s−1, which was nearly one order of magnitude greater than that of the industrial catalyst Sn(Oct)2.38 Ma et al. reported a series of oxazoline-based aminophenolate zinc complexes (III, Scheme 1) that showed unprecedentedly excellent activity with a turnover frequency (TOF) of up to 44000 h−1 under solvent-free immortal conditions with catalyst loadings as low as 0.005 mol% (relative to monomer), which has potential for industrial application.39 Pellecchia et al. reported a family of Zn(II) complexes bearing variously substituted monoanionic [N,O−] (imidazole[1,5-a]pyrid-3-yl)phenolate ligands (IV, Scheme 1), as catalysts for the ROP of L-LA under industrially relevant (190 °C, in the melt, technical grade unpurified monomer) conditions with a TOF of 29700 h−1.40 Jones et al. reported a series of Zn(II)-complexes for the ROP of rac-LA under industrial melt conditions at 180 °C with a [rac-LA]:[Init]:[BnOH] ratio of 3000:1:10, and the highest activity of Zn2(2)2Et2 (V, Scheme 1) leads to a high monomer conversion of up to 94% within one minute.41
:1; 95% conv. in 20 min, 25 °C). Following this elegant work, various modifications of the β-diketiminato ligand have been conducted in the search for zinc complexes with high catalytic activity and thermal stability.30–34 Many ligands including bidentate N,O ketiminato and tridentate N,N,N,35 and O,N,N ligands with pendent coordination donors have been designed and synthesized.35–37 The easy modification of the electronic and steric properties in the β-diketiminato ligand allows the synthesis of tailor-made catalysts. Herres-Pawlis et al. reported an ultra-high-activity bisguanidine zinc complex (II, Scheme 1) for the ROP of LA. The polymerization rate constant (Kapp) of the catalyst was 1.432 L mol−1 s−1, which was nearly one order of magnitude greater than that of the industrial catalyst Sn(Oct)2.38 Ma et al. reported a series of oxazoline-based aminophenolate zinc complexes (III, Scheme 1) that showed unprecedentedly excellent activity with a turnover frequency (TOF) of up to 44000 h−1 under solvent-free immortal conditions with catalyst loadings as low as 0.005 mol% (relative to monomer), which has potential for industrial application.39 Pellecchia et al. reported a family of Zn(II) complexes bearing variously substituted monoanionic [N,O−] (imidazole[1,5-a]pyrid-3-yl)phenolate ligands (IV, Scheme 1), as catalysts for the ROP of L-LA under industrially relevant (190 °C, in the melt, technical grade unpurified monomer) conditions with a TOF of 29700 h−1.40 Jones et al. reported a series of Zn(II)-complexes for the ROP of rac-LA under industrial melt conditions at 180 °C with a [rac-LA]:[Init]:[BnOH] ratio of 3000:1:10, and the highest activity of Zn2(2)2Et2 (V, Scheme 1) leads to a high monomer conversion of up to 94% within one minute.41
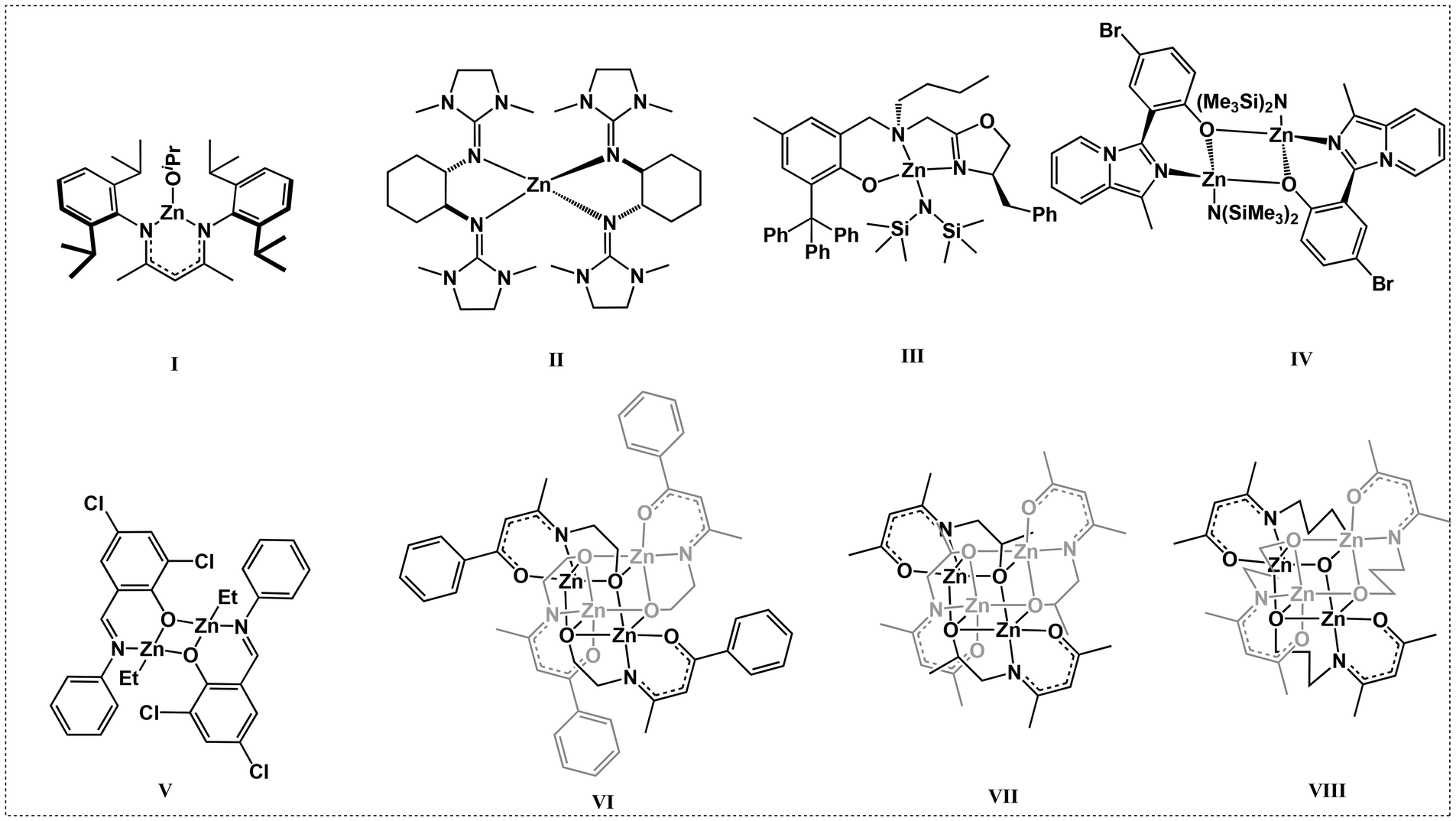 | ||
| Scheme 1 L1ZnOiPr and typical zinc complexes. | ||
It has been reported that tridentate N,N,N and O,N,N-chelating β-ketoiminate ligands containing a modestly π-electron rich framework are known to effectively stabilize metal centers. Jones et al. introduced an active functional group –OH on the aliphatic N-substitutes of the ligands and observed a multidentate chelation phenomenon upon coordination with the metal center to produce multinuclear zinc complexes (VI, Scheme 1).42 The most active zinc complexes could promote the ROP of rac-lactide in the absence of alcohol, and the monomer conversion reached 88% in 10 min with a monomer feed ratio LA:Zn = 1000:1 at 130 °C. Peng et al. also synthesized two tetranuclear zinc N-alkoxide ketoiminate complexes (VII and VIII, Scheme 1) for the ROP of rac-lactide, and the TOF was as high as 19000 h−1 at 130 °C under melt and bulk conditions.43 These elegant works suggested that the pendent coordination donor strongly affected the stability and impurity tolerance of the catalyst, as well as its subsequent polymerization behavior. Nonetheless, the catalyst performances remain far from industrial standards. It was assumed that these multinuclear zinc complexes would be decomposed at high temperatures because the flexible pendent –OH could not encapsulate the zinc center very well. Against this background, we designed and synthesized a tridentate N,N,O-ligand with rigid pendent –OH groups. The thermostability and impurity tolerance of the corresponding zinc complexes are further improved, as the zinc center is embedded entirely in the ligand pincer. As expected, these novel β-ketoimine zinc complexes exhibited higher catalytic performance than most currently reported zinc-based complexes. They could promote L-LA polymerization under industrially relevant conditions (in bulk and melt, 150–190 °C), producing colorless semi-crystalline PLLA with high efficiency. In addition, technical grade L-LA without further purification could also be polymerized rapidly due to the good tolerance to impurities at high temperature. Finally, these zinc complexes could effectively catalyze the copolymerization of L-LA with other cyclic esters.
2. Results and discussion
2.1 Catalyst design, synthesis and characterization
The design concept for the zinc catalysts originated from previous findings that zinc complexes bearing β-ketoiminate ligands bearing a flexible pendent coordination donor are highly active in catalyzing the ROP of rac-LA, but their thermal stability and impurity resistance are poor. Therefore, to further improve the catalytic activity, thermal stability, and impurity resistance, we used rigid β-benzocyclohexanone imines as a backbone and introduced rigid pendent –OH groups (L1–L6, Scheme 2) containing aryl and aliphatic rings. We further employed classical acetylacetone to synthesize the β-ketoiminate ligands (L7–L12) to investigate the steric effect on the catalytic activity. The β-ketoiminate ligands were prepared according to the synthetic routes in Scheme 2A. The corresponding zinc complexes (1–12, Scheme 2B) were obtained by reacting the ligands with diethylzinc in a glove box at room temperature (see ESI† for detailed procedures).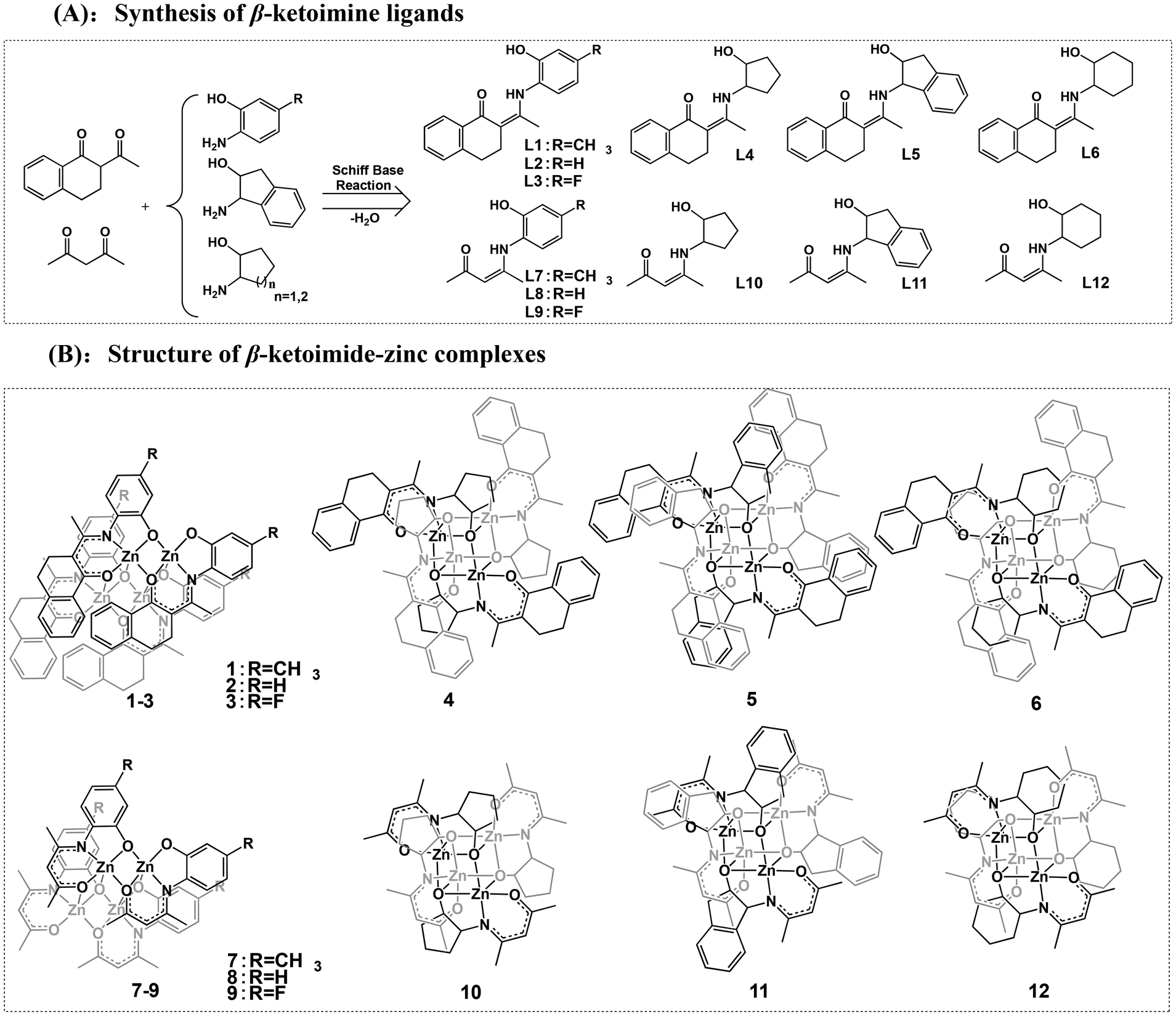 | ||
| Scheme 2 Synthesis of the β-ketoimine ligands (A) and structures of the β-ketoimide-zinc complexes (B). | ||
The purities and identities were confirmed by 1H and 13C NMR (Fig. S1–S36†). The molecular structures of L7, 1, and 6 were further confirmed by single-crystal X-ray diffractions (Fig. 1 and Fig. S37, Tables S1, S2†). The core of complex 1 is a planar Zn2O2 ring, and the coordination environment of each zinc atom consists of the atoms of the tridentate phenoxide-ketoiminate ligand. Each zinc atom is also coordinated to the oxygen atom in another dimeric complex. Finally, the five-coordinated environment of each zinc atom is completed by the phenoxide oxygen atom of the adjacent subunit. In contrast, complex 6 adopts a tetranuclear Zn4O4 heterocubane-type structure in the solid state, with four zinc atoms and four bridging alkoxy oxygen atoms. Each zinc atom is penta-coordinate, surrounded by one ketoiminate ligand and two additional oxygen atoms of the pendent N-alkoxide arm from two other adjacent ligands, which results in a highly distorted square-based pyramidal coordination geometry. The basal plane consists of one nitrogen and two oxygen atoms from one ligand and a third oxygen atom from another ligand. The apical position is occupied by an oxygen atom from a third ligand. From the structures, we clearly observed that the ketoiminate ligands are fully deprotonated and act as tridentate dianionic ligands in their corresponding zinc complexes. Based on zinc analogs reported in the literature, the structure of the complex is determined by the pendent –OH groups.43,44 If the side-arm donors are aromatic N-substitutes, the core of the complex is a Zn2O2 ring. When aliphatic N-substitutes are used, the core of the complex is a tetranuclear Zn4O4 heterocubane-type structure. Based on the previous work of Jones and Peng42,43 and the molecular structures of 1 and 6, we deduced that complexes 1, 2, 3, 7, 8, and 9 bearing an aryl group are structurally similar, and that those of complexes 4, 5, 6, 10, 11, and 12 with an alicyclic group are also similar to one another (Scheme 2B).
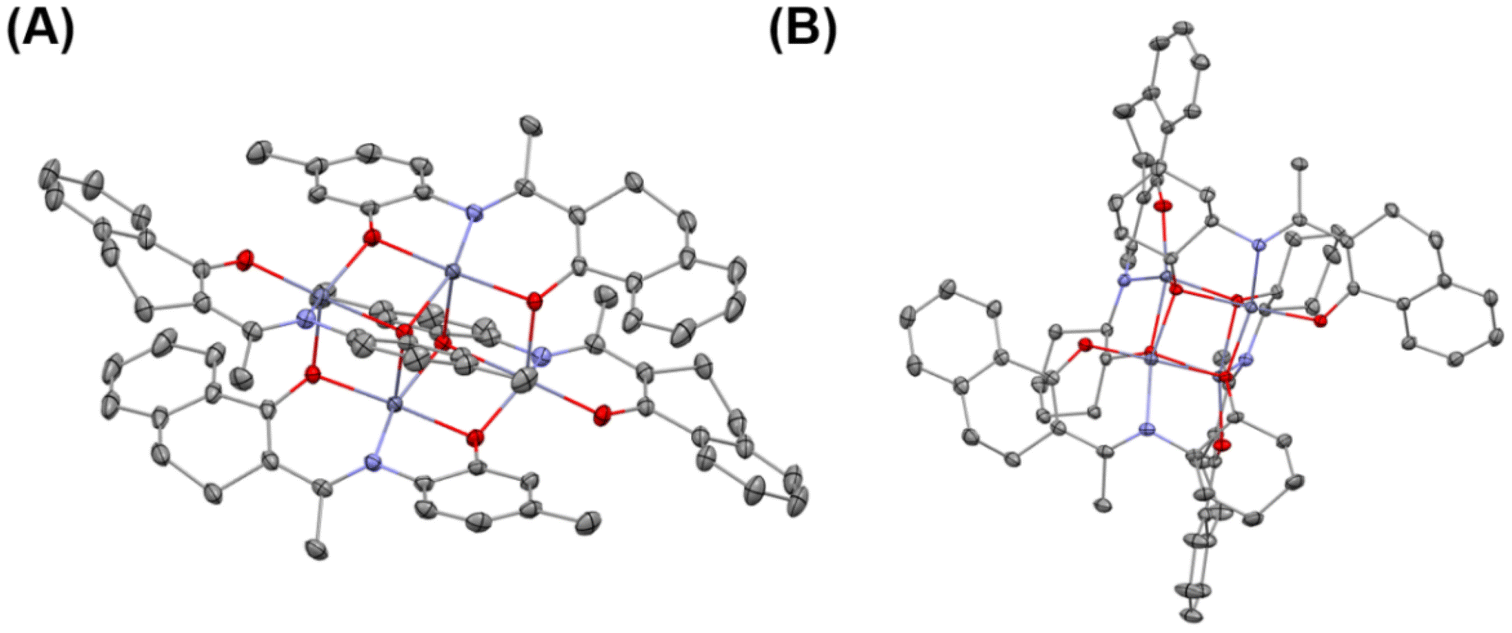 | ||
| Fig. 1 Molecular structures of complexes 1 (A) and 6 (B). The single crystals were developed by the slow evaporation method in dichloromethane. The thermal ellipsoids are plotted at the 30% probability level. H atoms are omitted for clarity. | ||
2.2 Catalyst evaluation in solution polymerization
After successfully synthesizing zinc β-ketoiminate complexes with various structures, we first evaluated their catalytic properties for L-LA polymerization in solution to clarify the structure–property relationships (Table 1). Complex 1 with R = CH3 converted ca. 18.0% of L-LA within 3 h, and the TOF was 6 h−1. The gradual increase in the activity of complexes 2 (R = H) and 3 (R = F) suggests that the side-arm donors of the ligand influence the catalytic activity through electronic effects, and introducing an electron-withdrawing group improves the catalytic activity. We reasoned that the electron-withdrawing groups increased the Lewis acidity of the zinc center, which promotes monomer activation. Replacing the aromatic donor with an aliphatic one significantly increased the catalytic activity (complexes 4–6). When the rigid β-benzocyclohexanone imine backbone was replaced with acetylacetone, the catalytic activities of the resulting complexes 7–12 showed the same trend. Complexes 10–12 bearing aliphatic side-arm donors showed higher catalytic activities than 7–9 bearing aromatic side-arm donors. In addition, the steric effects of the β-ketoiminate ligands also significantly affect the catalytic activities. The activities of complexes 10, 11, and 12 were much higher than those of 4, 5, and 6, probably because the reduced steric hindrance of their ligands allows easier access of the L-LA monomer to the metal center.| Runa | Cat. | t (min) | Conv.b (%) | TOF (h−1) |
M
n,calcc (kDa) |
M
n,GPCd (kDa) |
Đ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a The reactions were carried out with [M]0 = 1 M in toluene, [L-LA]:[M]:BnOH = 100:1:1 at 100 °C.
b The conversion was determined by 1H NMR.
c
Mn, calc = 144.14 × ([L-LA]0/[BnOH]0) × conv. + (%) + 108.13.
d Determined by SEC analysis vs. PS standards, and the Mn of PLA was corrected by Mn = 0.58Mn,GPC.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1 | 180 | 18 | 6 | 2.70 | 2.08 | 1.08 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 2 | 210 | 47 | 13 | 6.88 | 2.42 | 1.09 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 3 | 60 | 15 | 15 | 2.27 | 1.27 | 1.09 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 4 | 45 | 54 | 72 | 7.89 | 7.78 | 1.09 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 5 | 15 | 92 | 368 | 13.37 | 12.72 | 1.08 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 6 | 10 | 98 | 587 | 14.23 | 13.89 | 1.10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 7 | 360 | 45 | 7 | 6.59 | 5.64 | 1.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 8 | 180 | 25 | 8 | 3.71 | 3.37 | 1.15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 9 | 120 | 20 | 10 | 2.99 | 2.02 | 1.10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 10 | 30 | 95 | 190 | 13.80 | 10.89 | 1.09 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 11 | 8 | 98 | 735 | 14.23 | 10.96 | 1.11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 12 | 5 | 81 | 972 | 11.78 | 10.79 | 1.10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Complexes 1–12 showed a high degree of control toward L-LA polymerization in toluene, as evidenced by the close theoretical and experimental number-averaged molecular weights, as well as the narrow polydispersity indices (Đ = 1.08–1.15) (Table 1). Additionally, the molecular weights linearly increased with the monomer conversions, and the molecular weight distribution always remained monomodal and narrow (Fig. 2A and Fig. S38A†). The kinetic studies indicated that the L-LA polymerizations exhibited first-order dependence on both monomer concentration (Fig. 2B and Fig. S38B, S39†) and catalyst concentration (Fig. 2C and D), namely, d[PLLA]/dt = k[L-LA][Cat.].
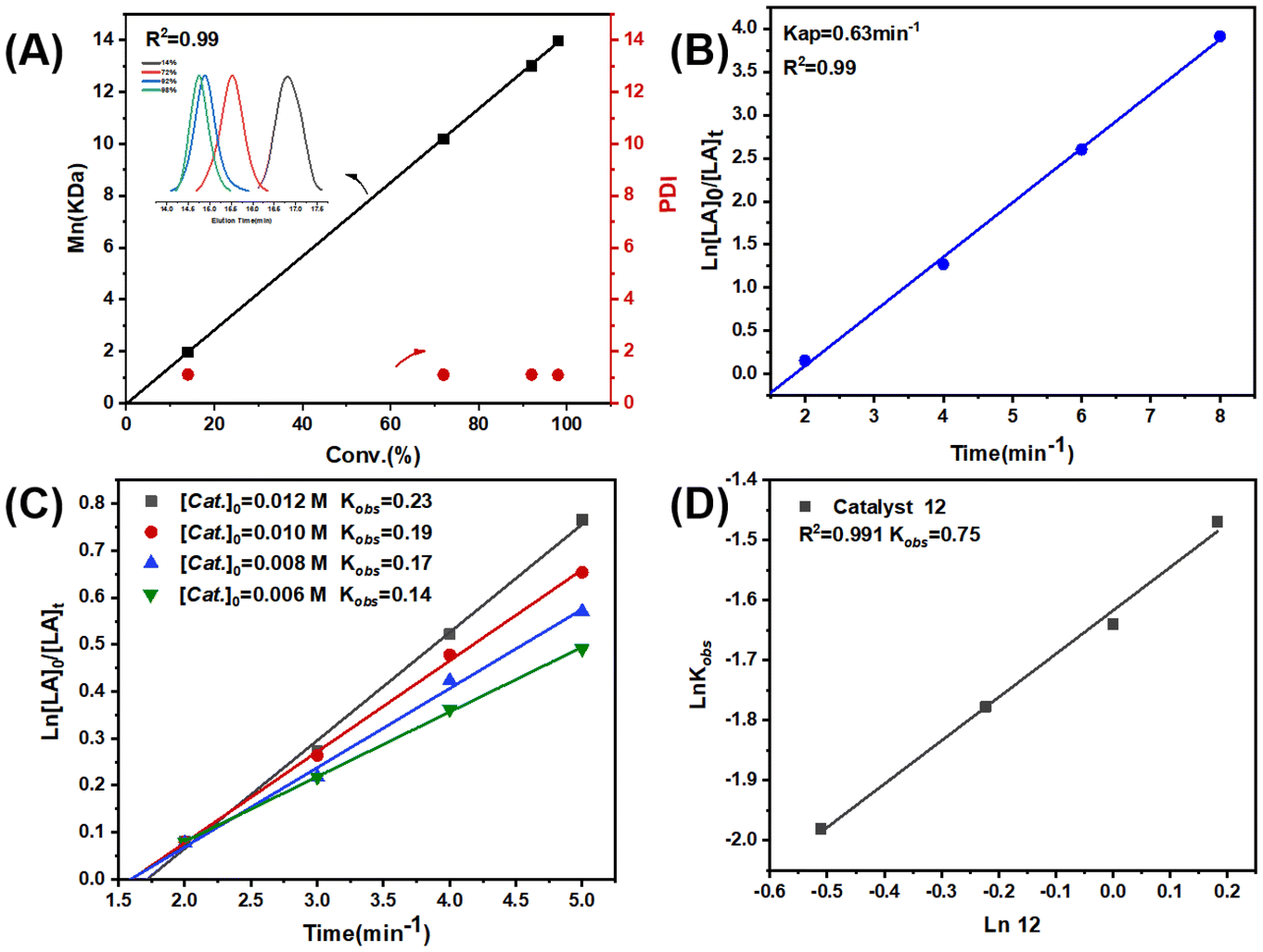 | ||
| Fig. 2 (A) Dependence of Mn,GPC and Đ on L-LA conversion. Inset: GPC traces of PLLA. (B) Kinetic plot for L-LA polymerization using complex 12 as the catalyst ([L-LA]:[Zn]:[BnOH] = 100:1:1, [L-LA] = 1 mol L−1, in toluene, 85 °C). (C) Kinetic plot of L-LA polymerization for varying catalyst concentrations ([L-LA]:[Zn]:[BnOH] = 100:1:1, [L-LA] = 1 mol L−1, toluene, 75 °C). (D) Double-logarithmic plot of lnKobsversus ln[12], suggesting that the L-LA polymerizations exhibited first-order dependence on the catalyst concentration. | ||
In order to compare the catalytic performance of the highly active complexes, the polymerization kinetics of 11 and 12 were determined in toluene at 85 °C. The apparent rate constants (k) were obtained from the slopes of the semi-log plots of the L-LA conversions versus time (Fig. 3A). The results showed that 12 was the most active catalyst. The ROP of L-LA by the β-ketoiminate zinc complexes showed an obvious induction period at 85 °C.44 The β-ketoiminate zinc complex may require a certain activation time when catalyzing the ROP of L-lactide (L-LA). We conducted in situ high-resolution mass spectrometry tests on the reaction between the complex and benzyl alcohol (BnOH) (Fig. S40†). The experimental m/z values agree well with the theoretical ones for [12 + H]+. These results show that the complex still maintains a tetranuclear structure in the solution and does not form a new structure by reacting with BnOH. This finding weakens the hypothesis of the coordination insertion mechanism. Analysis of the polymers by MALDI-TOF mass spectrometry shows the presence of BnO/H end groups (Fig. 3B). Therefore, we proposed the activated monomer mechanism; this process leads to the appearance of an induction period. In addition, the transesterification side reaction may occur, based on the presence of fragments with a molecular weight interval of 72.
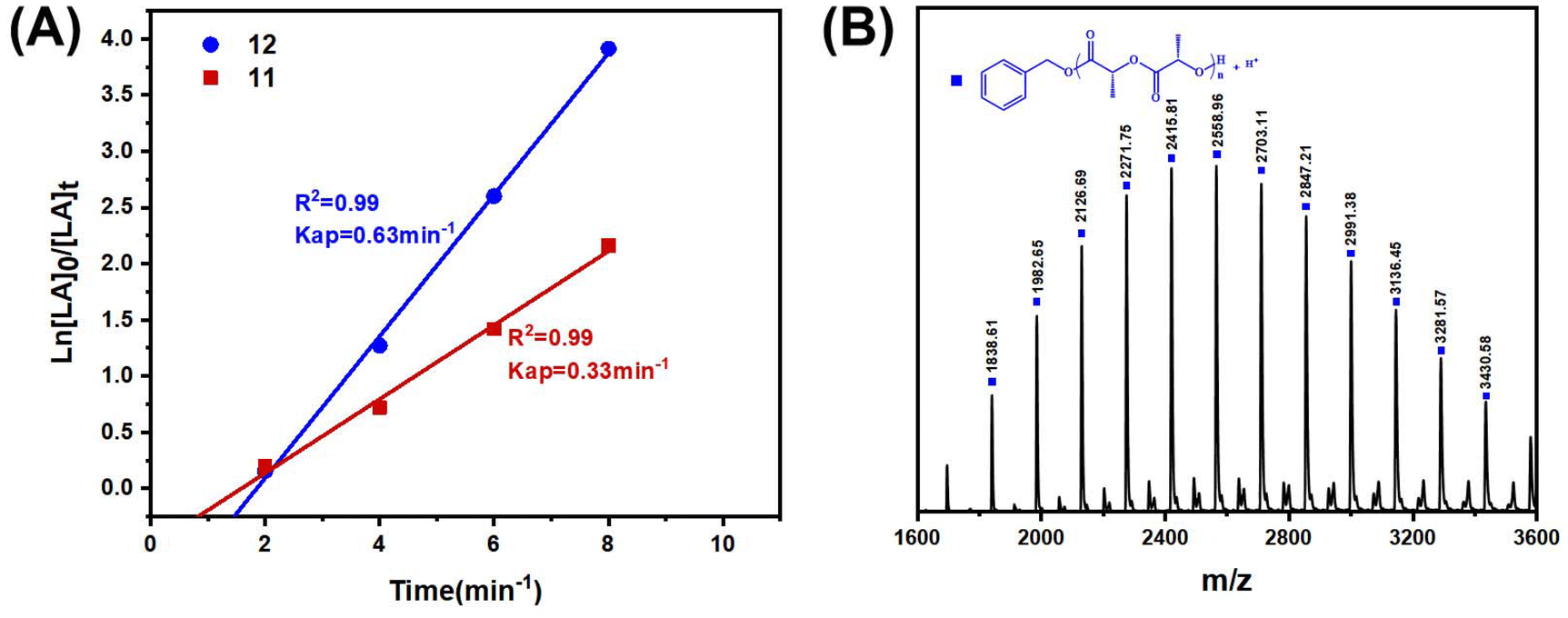 | ||
| Fig. 3 (A) The kinetics of L-LA polymerization by complexes 11 and 12 ([L-LA]:[Zn]:[BnOH] = 100:1:1, [L-LA] = 1 mol L−1, toluene, 85 °C). (B) MALDITOF mass spectrum of PLLA ([L-LA]:[Zn]:[BnOH] = 100:1:1, [L-LA] = 1 mol L−1, toluene, 85 °C, 2 min). | ||
To elucidate the origin of the different catalytic activities, density functional theory (DFT) studies were performed. The Mulliken charges of the zinc atoms (Table 2) in four catalysts (complexes 8 and 10–12) were calculated at the level of TPSSh-D3BJ/def2-TZVP//TPSSTPSS-D3BJ/6-31G* (non-metal atoms)/lanl2dz (Zn). According to the theoretical calculations, as expected, the zinc center of 12, with an average Mulliken charge (Qaver) of +0.860, exhibits greater electropositivity than that of 11 (Qaver: +0.845). Given the nucleophilic tendency of L-LA towards electron-deficient zinc centers, complex 12 exhibits significantly higher catalytic activity during L-LA polymerization compared to 11 (entries 11 vs. 12, Table 1). The same tendency is observed for 11 and 10 in terms of both the average Mulliken charge on their zinc atoms (+0.845 vs. +0.840) and their catalytic activities in L-LA polymerization (entries 10 vs. 11, Table 1). Moreover, the zinc center in 8 displays a notably less-positive average Mulliken charge of +0.778 compared to that of 12 (+0.860). This diminished electropositivity anticipates a markedly lower activity of 8 in catalyzing LA polymerization, which is in the agreement with experimental observations (entries 8 vs. 12, Table 1). Therefore, the average Mulliken charge (Qaver) of the zinc atoms seems to be a reliable and effective descriptor for designing polymerization catalysts with enhanced performance.
| Run | Cat. | Q (Zn1) | Q (Zn2) | Q (Zn3) | Q (Zn4) | Q aver |
|---|---|---|---|---|---|---|
| 1 | 8 | 0.811 | 0.81 | 0.746 | 0.745 | 0.778 |
| 2 | 10 | 0.878 | 0.805 | 0.805 | 0.873 | 0.840 |
| 3 | 11 | 0.835 | 0.855 | 0.855 | 0.835 | 0.845 |
| 4 | 12 | 0.782 | 0.925 | 0.790 | 0.943 | 0.860 |
Subsequently, we conducted DFT calculations to explore the reaction pathway for complex 12. Considering that the experimental observations suggest that a monomer activation mechanism is operative in the current system, the DFT calculations were performed according to the monomer activation mechanism. As shown in Fig. 4, the catalytic cycle initiates with the coordination of the first L-LA molecule to the dual Zn centers, accompanied by the formation of a crucial hydrogen bond between benzyl alcohol and a ligand oxygen atom, resulting in the stable ternary intermediate 2. Subsequently, the benzyloxy oxygen nucleophilically attacks the L-LA carbonyl carbon through transition state TS2-3 with an activation enthalpy (ΔH‡) of 28.6 kcal mol−1, leading to intermediate 3. This intermediate then undergoes spontaneous isomerization to afford intermediate 4, which is exergonic by 0.9 kcal mol−1, followed by a barrierless and hydrogen-bond-assisted ring-opening process to yield intermediate 5. A potential energy surface scan (Fig. S41†) also indicates that the conversion of 4 to 5 is a barrierless process, which also explains why the transition state for this process was not found. The catalytic cycle is completed by the facile coordination of the second L-LA molecule with the catalyst and benzyl alcohol to regenerate the active ternary complex 7, demonstrating the efficient and continuous nature of the catalytic process.
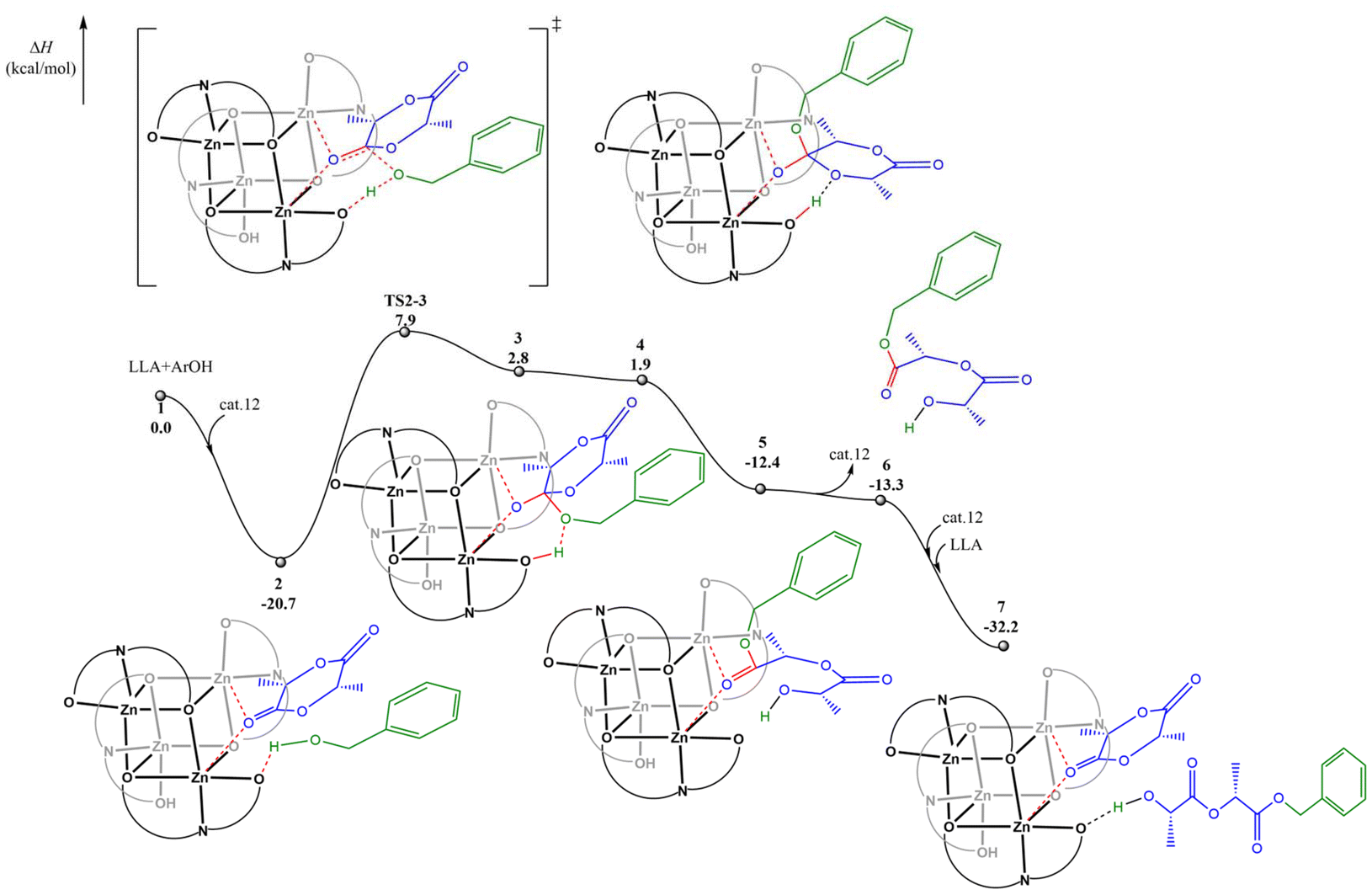 | ||
| Fig. 4 The calculated reaction pathway for L-LA polymerization catalyzed by complex 12 with benzyl alcohol. | ||
A comparative analysis of complexes 10 and 12 indicates a correlation between Mulliken charge and coordination energy. The more positive charge on Zn in 12 (0.860 vs. 0.840 in complex 10) correlates with stronger monomer coordination (−20.7 vs. −20.1 kcal mol−1) and the observed higher catalytic activity (TOF 972 vs. 190 h−1).
2.3 Evaluation of catalyst ROP performance under industrial conditions
As mentioned in the introduction, while most studies have focused on optimization of performance under mild conditions (room temperature, recrystallized monomer at low monomer/catalyst ratios, using solvents such as dichloromethane, THF, or toluene), industrial processes work under much harsher conditions, i.e., in the melt at T = 150–200 °C, using unpurified technical-grade monomer at very high [LA]:[Cat.] molar ratios (1000–200000) and an excess of alcohol with respect to Sn(Oct)2. Therefore, we further investigated the catalytic performance of complexes 1–12 for the L-LA bulk polymerizations at 150–190 °C under industrially relevant conditions with [L-LA]:[Zn]:[BnOH] = 1000:1:1 (Table S3†). The structure–activity relationships in the bulk were same as those in solution polymerization. PLLA with a high molecular weight and narrow distribution was readily prepared by β-ketoiminate/BnOH at a high monomer/catalyst feed ratio (Table 3). The results showed that the catalysts maintained high catalytic activity in all cases, and the catalytic activity continued to increase by increasing temperature (entries 1, 2, and 3, Table 3). Compared with the optimal catalyst complex 12, although catalyst I (entries 4, Table 3) exhibits high activity for the polymerization of rac-LA, it is still limited to solution polymerization. As the most active catalyst reported to date, catalyst II (entry 5, Table 3) exhibited a remarkably high TOF of 57662 h−1 and PLA with a high crystallinity. It represents a highly successful catalyst; however, it is noteworthy that the authors only conducted experiments below 150 °C and did not attempt polymerization at higher temperatures. Catalysts III, IV, and V (entries 6, 7, and 8, Table 3) all demonstrated good activity, but were tested at low monomer/initiator ratios (<300), failing to achieve high-molecular-weight PLA. Furthermore, catalyst V showed poor control over the molecular weight distribution (PDI = 1.91). Catalysts VI, VII, and VIII (entries 9, 10, and 11, Table 3) also demonstrate remarkable polymerization activity under the bulk melting conditions at 130 °C. However, the molecular weight distributions of the obtained polymers are relatively broad. Taking catalyst VIII with the highest activity as an example, the polymer molecular weight is only 8.7 kDa, and the molecular weight distribution is relatively wide (Đ = 1.72).
| Runa |
Cat. | [L-LA]:[M]:BnOH |
T ( °C) | t (min) | Conv.b (%) |
TOF (h−1) |
M
n,calcc (kDa) |
M
n,GPCd (kDa) |
Đ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Unless otherwise stated, the L-LA polymerizations were performed in the bulk and melt without solvents.
b Determined using 1H NMR spectroscopy.
c
M
n, calc = 144.14 × ([L-LA]0/[BnOH]0) × conv. (%) + 108.13 (runs 1–8), Mn, calc = 144.14 × ([L-LA]0/[DODE]0) × conv. (%) + 186.34 (run 9) or Mn, calc = 144.14 × ([L-LA]0/[DODE]0) × conv. (%) + 90.12 (run 10).
d Determined using gel permeation chromatography in THF with polystyrene standards and corrected by a coefficient of 0.58.
e Cited from ref. 29.
f Cited from ref. 38.
g Cited from ref. 39.
h Cited from ref. 40.
i Cited from ref. 41.
j Cited from ref. 42.
k Cited from ref. 43.
l Cited from ref. 43.
m 1-Dodecanol was used as the initiator.
n L(+)-Lactic acid was used the initiator.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 12 | 1000:1:1 |
150 | 20 | 97 | 2910 | 139.92 | 75.76 | 1.50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 12 | 1000:1:1 |
170 | 15 | 97 | 3880 | 139.92 | 60.53 | 1.40 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 12 | 1000:1:1 |
190 | 10 | 98 | 5880 | 141.37 | 55.61 | 1.56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4e | I | 200:1:0(rac-LA, 0.4 M, CH2Cl2) |
20 | 20 | 95 | 582 | 27.96 | 21.98 | 1.10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5f | II | 5000:1:1(rac-LA) |
150 | 3.8 | 74 | 57662 |
533.43 | 84.68 | 1.70 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6g | III | 10000:1:100 |
110 | 12 | 88 | 44000 |
12.70 | 8.70 | 1.69 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7h | IV | 1000:1:5 |
190 | 2 | 99 | 29700 |
28.40 | 9.74 | 1.50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8i | V | 3000:1:10(rac-LA) |
180 | <1 | 94 | >16920 |
40.70 | 31.64 | 1.91 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9j | VI | 1000:1:0 (rac-LA) |
130 | 10 | 88 | 5380 | 126.95 | 59.19 | 1.68 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10k | VII | 1000:1:0 (rac-LA) |
130 | 3 | 96 | 19000 |
138.48 | 52.89 | 1.94 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11l | VIII | 1000:1:0 (rac-LA) |
130 | 3 | 99 | 19800 |
142.81 | 8.70 | 1.72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 12 | 2000:1:1 |
150 | 45 | 96 | 2560 | 276.97 | 103.31 | 1.53 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 12 | 5000:1:1 |
150 | 120 | 92 | 2287 | 663.15 | 211.10 | 1.51 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 12 | 10000:1:1 |
150 | 180 | 93 | 2818 | 1340.61 | 297.99 | 1.50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | Sn(Oct) 2 | 10000:1:1 |
150 | 300 | 72 | 1440 | 1037.92 | 287.42 | 1.54 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | 12 | 100000:1:1 |
190 | 4320 | 97 | 1347 | 13981.69 |
60.90 | 1.56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17m | 12 | 1000:1:1 |
150 | 20 | 98 | 2940 | 141.44 | 110.04 | 1.51 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18n | 12 | 1000:1:1 |
150 | 20 | 93 | 2790 | 134.14 | 67.54 | 1.54 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
At higher monomer/catalyst feed ratios, the L-LA bulk polymerizations also proceed smoothly. As the monomer/catalyst feed ratio was increased, the catalyst remained highly active, and the molecular weight significantly increased (entries 1, 12, 13, and 14, Table 3). It is noteworthy that the TOF of 12 was as high as 2818 h−1, whereas the reaction of Sn(Oct)2 showed relatively low activity under the same conditions (TOF = 1440 h−1). Apparently, complex 12 exhibited higher catalytic activity than Sn(Oct)2, generating PLLA with a higher molecular weight and narrower distribution under the same conditions (entries 14 and 15, Table 3). 97% of the L-LA was converted after 72 h (three days) by complex 12 with a catalyst concentration as low as 0.001 mol% (entry 16, Table 3). The monomer conversion increased continuously during polymerization, suggesting that the active species were not thermally decomposed. These results clearly indicated that the β-ketoimide zinc catalysts exhibited excellent thermostability. The molecular weights of the resultant PLLAs did not significantly increase at ultra-high monomer/catalyst feed ratio (100000:1:1). We assumed that the dosage of trace protonic impurities (such as water and residual lactic acid) would also increase at high monomer loading, and that severe chain transfer reaction would reduce the molecular weight. However, the number-average molecular weights were in the valuable range (Mn,GPC = 60.90 kDa). These results indicated that the zinc complex showed much higher activity than the tin catalyst under the industrially relevant conditions. For industrial applications, the colors of the catalyst and polymers are also important. Although complexes 1–12 showed dark colors (Fig. S42†), the resulting PLLAs were white solids and were not affected by the color of the catalyst (Fig. S43†).
In practical applications, the properties of PLA largely depend on the nature of the end groups,45 and commercially available PLA products also provide different capping options to meet the needs of different applications. Therefore, we combined complex 12 with two different initiators (n-dodecanol and lactic acid) to catalyze the ROP of L-LA (entries 17 and 18, Table 3) Additionally, we carried out the polymerization with these initiators at low monomer equivalents (Table S4†). The results demonstrated the successful synthesis of alkyl-capped and acid-capped PLLA (Fig. S44–S48†).
The 4-core β-ketoiminate zinc catalysts also showed good tolerance to chain transfer agents (CTAs) under bulk and melt conditions (up to 100 equivalents relative to Zn catalysts) (Fig. 5A). By increasing CTA loading, the molecular weight of PLLA gradually decreased, and the polymer polymerization rate increased owing to the increased number of polymer chains. These results suggest that the catalyst loading can be further reduced without sacrificing the catalytic performance, and that the molecular weight can be further regulated by the loading dose of CTA. The melting temperatures of the PLLA decreased, probably because of the decreased molecular weight (Fig. 5B). These results clearly indicated that the zinc complexes can serve as alternatives to Sn(Oct)2 for industrial PLA production.
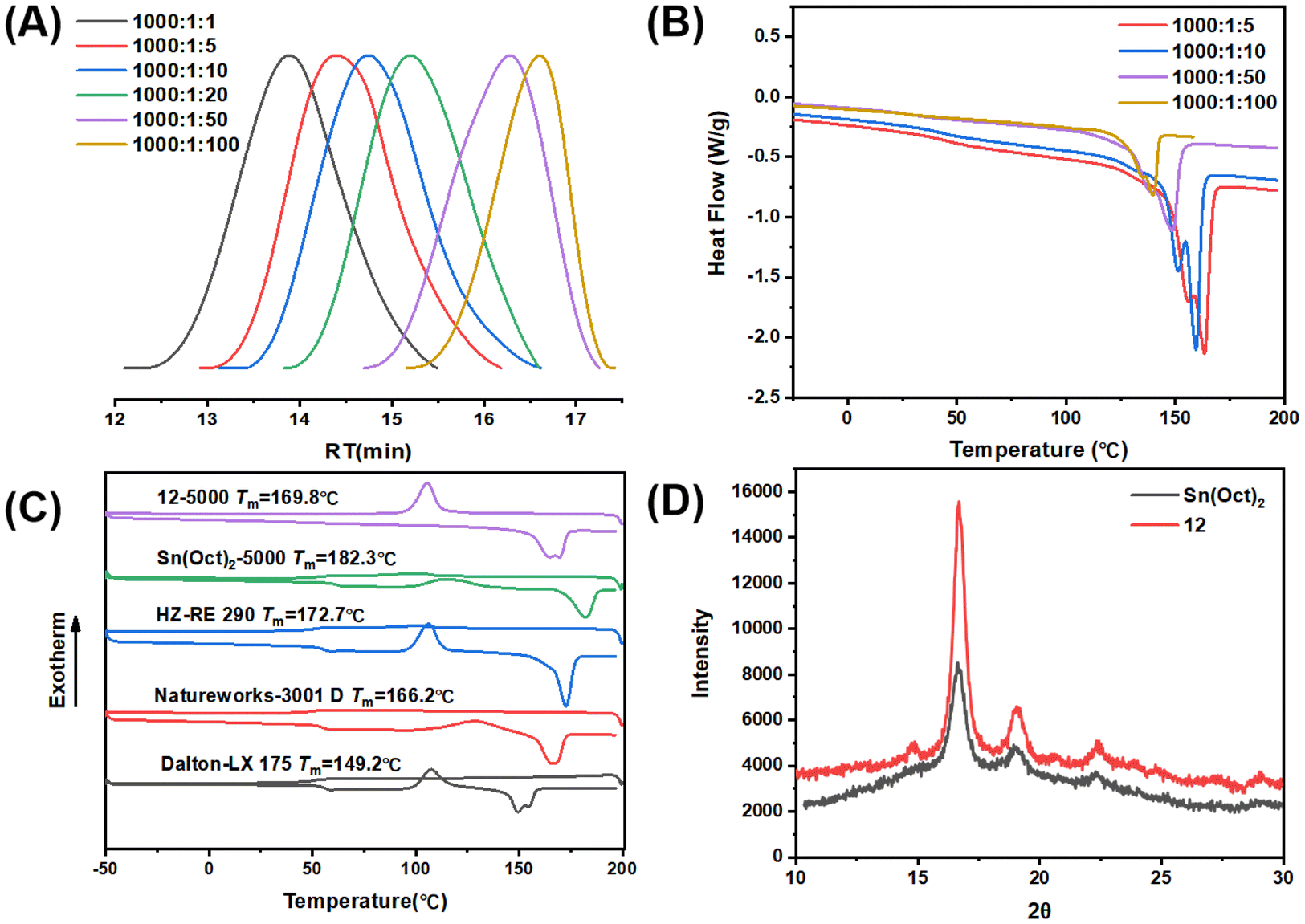 | ||
| Fig. 5 (A) GPC elution curves with different CTA contents. (B) DSC curves of PLLA samples obtained under different L-LA/BnOH feed ratios. (C) DSC curves of the PLLA sample obtained using complex 12 and other commercial PLLAs. (D) XRD profiles of the PLLA synthesized from 12 and Sn(OCt)2. | ||
The zinc catalyst not only exhibited high activity in the bulk and melt polymerization, it also produced polymeric materials with a higher degree of crystallinity, which avoids additional post-processing to obtain the desired properties. The typical DSC curves of the different PLLA samples are shown in Fig. 5C. It clearly indicated that the PLLA obtained by complex 12 showed an obvious crystallization peak in the first cooling cycle, and there was no cold crystallization peak in the second heating cycle. In contrast, the PLLA synthesized using Sn(OCt)2 and other commercial PLLAs showed no crystallization peak but displayed cold crystallization peaks. These results clearly indicate that the PLLA synthesized by zinc complex possesses stronger crystallinity. Wide-angle XRD also indicated that the PLLA sample obtained by zinc catalyst exhibited stronger crystallinity than the Sn(OCt)2 samples (Fig. 5D); the crystallinity of PLLA synthesized using Sn(OCt)2 was 15.6%, while the crystallinity of PLLA synthesized using complex 12 was 37.1%. The degree of isotacticity (Pm) of the two PLLA samples was characterized using 1H NMR with homo-nuclear decoupling; under the same conditions, the Pm of PLLA synthesized using Sn(OCt)2 was 0.93, while the Pm of PLLA synthesized using complex 12 was 0.98 (Fig. S49†). In combination with the results of the single-crystal diffraction, it can be concluded that the unique multinuclear spatial structure of the polymers causes their metal centers to be surrounded by the ligands and thus suppresses the transesterification side reactions from occurring. Second, it effectively reduces the occurrence of epimerization at high temperatures, which gives the polymer excellent crystalline properties. The high crystallization ability will greatly improve the heat deflection temperature and the mechanical strength, which will greatly expand the application prospects.
2.4 Ring-opening copolymerization catalyzed by zinc β-ketoiminium complexes
PLLA is known to have good mechanical strength, but its poor toughness and slow degradation rate have hindered its wide applications. The physical properties of PLLA and PCL/PPDO are quite different and somewhat complementary. PCL possesses notable elasticity and thermal properties, and PPDO has fast hydrolysis, excellent mechanical strength and ideal thermal processing properties. Obviously, the copolymerization of ε-CL/PDO and L-LA could enable the preparation of materials with improved features in comparison with the parent homopolymers. The ideal advantage would be to exploit the flexibility (low Tg) of PCL/PPDO for the improvement of the slow degradation rate and elasticity of PLLA and thus obtain copolymers with diverse mechanical and thermal properties.The homopolymerization of ε-CL and PDO catalyzed by zinc β-ketoiminium exhibited higher catalytic activity under melt conditions (entries 1 and 2, Table 4), so P(LLA-co-CL) and P(LLA-co-PDO) random copolymers were prepared using the one-pot method. As expected, complex 12 could effectively catalyze the copolymerization of L-LA with ε-CL/PDO, producing copolymers with high molecular weights and narrow molecular weight distributions (entries 3–6, Table 4 and Fig. S50†). When Sn(Oct)2 was used as the catalyst, the molecular weight distribution of the P(LLA-co-CL) copolymer was broad (Đ = 1.81) (entry 7, Table 4). The reactivity of PDO in the copolymerization reaction was relatively low, and the content of PPDO in the random copolymer was much lower than the administered dose (entry 4, Table 4). In contrast, when complex 12 was used as a catalyst, P(LLA-co-PDO) random copolymers with high PDO incorporation were readily synthesized in 60 min (entries 5 and 6, Table 4). To further analyze the sequence structures of the P(LLA-co-CL) and P(LLA-co-PDO) copolymers, we synthesized P(LLA-co-CL) and P(LLA-co-PDO) copolymers with a feed ratio of [L-LA]:[M]:[Cat.]:BnOH = 500:500:1 (entries 4 and 6, Table 4). Subsequently, the copolymers were characterized using 1H and 13C NMR (Fig. S51–S54†). The carbonyl carbon signals in the P(LLA-co-CL) copolymer are mainly concentrated between 165 ppm and 175 ppm. Based on the signal intensity, the average sequence lengths of the LA and CL structural units in the copolymer (LLA = LLL) and caprolactone (LCL = LC) were calculated according to the literature.46,47 For the copolymer obtained by the catalysis of complex 12, the average sequence segment lengths are LLL = 1.93 and LC = 1.38, which is a typical random copolymer. In the carbonyl region of the P(LLA-co-PDO) copolymer (169–170.5 ppm), the peaks at 169.59 ppm and 170.00 ppm were assigned to continuous LA–LA and PDO–PDO sequences, respectively. Additionally, strong characteristic peaks corresponding to LA–LA–PDO, PDO–PDO–LA, LA–PDO–LA and PDO–LA–PDO sequences were tentatively assigned with the assistance of theoretical simulation (Fig. S54†), further supporting the random copolymerization of LA and PDO. These results indicated that the zinc complex is also a promising catalyst for synthesis of PLA copolymers under industrial conditions.
| Runa | Cat. | [L-LA]:[M]:[Cat.]:BnOH |
T (°C) | t (min) | Conv.b [LA] (%) |
Conv.b [M] (%) |
M
n,calcc (kDa) |
M
n,GPCd (kDa) |
Đ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Unless otherwise stated, the L-LA polymerizations were performed in bulk and melt without solvents. b Determined by 1H NMR spectroscopy. c M n, calc = 114.14 × ([ε-CL]0/[BnOH]0) × conv. (%) + 108.13 (entry 1) or Mn, calc = 102.09 × ([PDO]0/[BnOH]0) × conv. (%) + 108.13 (entry 2) or Mn, calc = 114.14 × ([ε-CL]0/[BnOH]0) × conv. (%) + 144.14 × ([L-LA]0/[BnOH]0) × conv. (%) + 108.13 (entries 3 and 5) or Mn, calc = 102.09 × ([PDO]0/[BnOH]0) × conv. (%) + 144.14 × ([L-LA]0/[BnOH]0) × conv. (%) + 108.13 (entries 4 and 6). d Determined by gel permeation chromatography in THF using polystyrene standards. e Copolymer monomer M = ε-CL. f Copolymer monomer M = PDO. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
12e |
0:1000:1:1 |
150 | 10 | — | 99 | 114.11 | 104.77 | 1.45 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
12f |
0:1000:1:1 |
80 | 20 | — | 88 | 89.86 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
12e |
700:300:1:1 |
150 | 60 | 99 | 77 | 109.68 | 84.99 | 1.50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |
12e |
500:500:1:1 |
150 | 90 | 99 | 98 | 128.11 | 108.25 | 1.53 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
12f |
700:300:1:1 |
120 | 40 | 99 | 72 | 122.93 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |
12f |
500:500:1:1 |
120 | 60 | 99 | 80 | 113.01 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |
Sn(Oct)
2
e |
700:300:1:1 |
150 | 720 | 99 | 94 | 133.73 | 135.72 | 1.81 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 |
Sn(Oct)
2
f |
700:300:1:1 |
120 | 1440 | 94 | 8 | 97.31 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3. Conclusions
Multinuclear β-ketoimide zinc complexes were proven to be a promising alternative to Sn(Oct)2 for L-LA polymerization. The catalytic activity was affected by the electronic and steric effects of the substituents; the introduction of an electron-withdrawing group or the smaller steric effects of acetylacetone could further increase the catalytic activity. The results of computational studies show that the activity of the catalyst is directly proportional to the average Mulliken charge on the zinc atoms. These novel zinc complexes exhibit good thermal stability and can catalyze the polymerization of L-LA to obtain semi-crystalline PLLA under industrially relevant conditions. Compared with Sn(Oct)2, the optimal zinc complex mediates the ROP of L-LA to afford PLLA with higher crystallinity. In addition, complex 12 can catalyze the copolymerization of L-LA with ε-caprolactone /dioxanone to produce P(LLA-co-CL)/P(LLA-co-PDO) random copolymers with high comonomer incorporation.Data availability
The experimental details, polymerization data, synthesis of monomers, NMR spectra, and GPC elution curves of the polymers. These data are available in the ESI.† The datasets supporting this article have been uploaded as part of the ESI.† The single crystal data for ligand 7 (CCDC 2413820†) and complexes 1 (CCDC 2413821†) and 6 (CCDC 2413824†) were deposited at The Cambridge Crystallographic Data Centre (CCDC).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is financially supported by the National Key Research and Development Program of China (2022YFB3704900), National Natural Science Foundation of China (Grant no. 52222302) and Sponsor Program of China National Petroleum Corporation (23-LH-59-05).References
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. De Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS PubMed.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- R. W. Clarke, T. Sandmeier, K. A. Franklin, D. Reich, X. Zhang, N. Vengallur, T. K. Patra, R. J. Tannenbaum, S. Adhikari, S. K. Kumar, T. Rovis and E. Y. X. Chen, Nature, 2023, 616, 731–739 CrossRef CAS PubMed.
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272–276 CrossRef CAS PubMed.
- Z. Li, Y. Shen and Z. Li, Macromolecules, 2024, 57, 1919–1940 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- M. S. Kim, H. Chang, L. Zheng, Q. Yan, B. F. Pfleger, J. Klier, K. Nelson, E. L. W. Majumder and G. W. Huber, Chem. Rev., 2023, 123, 9915–9939 CrossRef CAS PubMed.
- E. F. Fiandra, L. Shaw, M. Starck, C. J. McGurk and C. S. Mahon, Chem. Soc. Rev., 2023, 52, 8085–8105 RSC.
- J.-M. Raquez, Y. Habibi, M. Murariu and P. Dubois, Prog. Polym. Sci., 2013, 38, 1504–1542 CrossRef CAS.
- A. Michalski, M. Brzezinski, G. Lapienis and T. Biela, Prog. Polym. Sci., 2019, 89, 159–212 CrossRef CAS.
- I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. Lopez-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS.
- S. Gesslbauer, G. Hutchinson, A. J. P. White, J. Burés and C. Romain, ACS Catal., 2021, 11, 4084–4093 CrossRef CAS.
- L.-J. Wu, W. Lee, P. K. Ganta, Y.-L. Chang, Y.-C. Chang and H.-Y. Chen, Coord. Chem. Rev., 2023, 475, 214847 CrossRef CAS.
- X.-L. Chen, B. Wang, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 3502–3512 CrossRef CAS.
- E. Fazekas, P. A. Lowy, M. Abdul Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- W. Lv, Y. Wang, M. Li, X. Wang and Y. Tao, J. Am. Chem. Soc., 2022, 144, 23622–23632 CrossRef CAS PubMed.
- C. Li, Y.-F. Dang, B. Wang, L. Pan and Y.-S. Li, Macromolecules, 2021, 54, 6171–6181 CrossRef CAS.
- Z. Ding, M. Wang, B. Wang, L. Pan and Y. Li, Macromolecules, 2024, 57, 98–109 CrossRef.
- B. Lin and R. M. Waymouth, Macromolecules, 2018, 51, 2932–2938 CrossRef CAS.
- M. Hong, J. Chen and E. Y. X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS PubMed.
- H.-Y. Ji, D.-P. Song, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2019, 21, 6123–6132 RSC.
- H.-Y. Ji, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 641–648 RSC.
- F. Santulli, G. Gravina, M. Lamberti, C. Tedesco and M. Mazzeo, Mol. Catal., 2022, 528, 112480 CrossRef CAS.
- N. Liu, D. Liu, B. Liu, H. Zhang and D. Cui, Polym. Chem., 2021, 12, 1518–1525 RSC.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2020, 59, 21778–21784 CrossRef CAS PubMed.
- J. A. Stewart, P. McKeown, O. J. Driscoll, M. F. Mahon, B. D. Ward and M. D. Jones, Macromolecules, 2019, 52, 5977–5984 CrossRef CAS.
- Z.-B. Wang, M. Wang, Z. Ding, X.-S. Zhang and B. Wang, Polym. Sci. Technol., 2024 DOI:10.1021/polymscitech.4c00020.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785–2794 CrossRef CAS PubMed.
- A. P. Dove, V. C. Gibson, E. L. Marshall, A. J. P. White and D. J. Williams, Dalton Trans., 2004, 4, 570–578 RSC.
- M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2005, 44, 8004–8010 CrossRef CAS PubMed.
- T. J. J. Whitehorne and F. Schaper, Chem. Commun., 2012, 48, 10334–10336 RSC.
- S. Ghosh, E. Glöckler, C. Wölper, A. Tjaberings, A. H. Gröschel and S. Schulz, Organometallics, 2020, 39, 4221–4231 CrossRef CAS.
- S. Ghosh, P. M. Schäfer, D. Dittrich, C. Scheiper, P. Steiniger, G. Fink, A. N. Ksiazkiewicz, A. Tjaberings, C. Wölper, A. H. Gröschel, A. Pich, S. Herres-Pawlis and S. Schulz, ChemistryOpen, 2019, 8, 951–960 CrossRef CAS PubMed.
- C. Scheiper, C. Wölper, D. Bläser, J. Roll and S. Schulz, Z. Naturforsch., B, 2014, 69, 1365–1374 CrossRef CAS.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., 2020, 132, 21962–21968 CrossRef.
- C. Kan, J. Hu, Y. Huang, H. Wang and H. Ma, Macromolecules, 2017, 50, 7911–7919 CrossRef CAS.
- M. C. D'Alterio, I. D'Auria, L. Gaeta, C. Tedesco, S. Brenna and C. Pellecchia, Macromolecules, 2022, 55, 5115–5122 CrossRef.
- J. Payne, P. McKeown, M. F. Mahon, E. A. C. Emanuelsson and M. D. Jones, Polym. Chem., 2020, 11, 2381–2389 RSC.
- C. Di Iulio, M. Middleton, G. Kociok-Köhn, M. D. Jones and A. L. Johnson, Eur. J. Inorg. Chem., 2013, 2013, 1541–1554 CrossRef CAS.
- C. Yang, Y. Peng, J. Wang, P. Chen and X. Gong, Inorg. Chem. Commun., 2020, 119, 108136 CrossRef CAS.
- Y. Huang, X. Kou, Y.-L. Duan, F.-F. Ding, Y.-F. Yin, W. Wang and Y. Yang, Dalton Trans., 2018, 47, 8121–8133 RSC.
- B. Kost, M. Basko, M. Bednarek, M. Socka, B. Kopka, G. Łapienis, T. Biela, P. Kubisa and M. Brzeziński, Prog. Polym. Sci., 2022, 130, 101556 CrossRef CAS.
- J. Kasperczyk and M. Bero, Makromol. Chem., 1993, 194, 913–925 CrossRef CAS.
- J. Kasperczyk and M. Bero, Makromol. Chem., 1991, 192, 1777–1787 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2413820, 2413821 and 2413824. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4py01486c |
| This journal is © The Royal Society of Chemistry 2025 |