
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArginine-functionalised hydrogels as a novel atmospheric water-harvesting material†
Moki K.
Thanusing
 ,
Brett L.
Pollard
and
Luke A.
Connal
*
,
Brett L.
Pollard
and
Luke A.
Connal
*
Australian National University, Australia. E-mail: luke.connal@anu.edu.au
First published on 24th February 2025
Abstract
Atmospheric water harvesting is a versatile but underutilised source of potable water. In this study, a poly(HEMA-co-PEGMA) linear copolymer and PEGDMA-crosslinked hydrogel were post-functionalised using Steglich esterification to attach L-arginine onto HEMA side chains. The water-harvesting properties of the resulting polymers were then tested. The functionalised polymers had a water uptake of 130–150 mg g−1 water after 24 hours. The thermal phase transitions were around 60–80 °C, however this can be easily adjusted by varying composition and degree of functionalisation. Notably, there was a significant decrease in the rate of water uptake after 2–3 hours. This property was further explored with a rapid cycling test, in which 70-minute water-harvesting cycles yielded 2 g water per gram of polymer after 24 hours. The data presented in this body of work showcases the water-harvesting potential of guanidinium moieties, as well as highlighting the broad scope of materials and synthetic methods that could be used for developing water-harvesting polymeric materials.
Introduction
Polymer desiccants are an interesting and diverse class of atmospheric water harvesting (AWH) materials. Atmospheric water is found in all climates, notably arid climates with few other accessible water sources, making it a promising avenue for alleviating water scarcity. The large scope of functionalities and synthetic methods available allows for the fine-tuning of desired properties. The rate of water uptake is generally the bottleneck of the water-harvesting process, however, and as a result many existing polymeric materials use hygroscopic additives to enhance uptake.1–5 Lithium chloride is the most commonly used in current literature given its high water-uptake ability.6 Such materials harvest water at low humidities and can be renewed at low temperatures (and low energy inputs), visibly improving upon current AWH technologies. However, there are clear drawbacks, especially when considering future commercial and industrial use. There is a growing industrial demand for LiCl, most notably for the production of lithium-ion batteries.7 Another more general drawback for additives in water-harvesting systems is leaching over time, effectively decreasing the material efficiency. Scaling up the production of a LiCl-dependent water-harvesting material is therefore impractical.Identifying hygroscopic functionalities which can be covalently bonded to the polymer backbone therefore represents an attractive target. Although the relationship between chemical structure and hygroscopicity (or water sorption) is complex and not precisely understood, charge is a rough indicator for hygroscopic behaviour and can help direct the identification of promising AWH material candidates.8–12 This was well demonstrated with the poly([2-(acryloyloxy)ethyl]trimethylammonium acetate) (PAETA-Ac) system of Wu et al., where the polymer network's intrinsic charge was used for water harvesting to capture ∼0.53 g g−1 day−1.13 Natural polymers have also been used for AWH, like in the Park et al. alginate/acrylamide/carbon nanotube composite system which gave a daily yield of 1.81 g g−1 day−1.14 However, this material used CaCl2 as a hygroscopic additive, meaning leaching (however minimal) would result in reduced performance over time. Even though polymeric AWH materials have only been studied in earnest within the last two decades, there are already a wide variety of approaches published to literature, including using MOFs, COFs, and more bioinspired systems.15–17 The ubiquity of atmospheric water compared to other water sources allows for AWH to fill environmental niches not available to other water harvesting methods. Given its relative novelty, we feel drawn to AWH focusing on expanding the breadth of chemical structures and functionalities, synthetic methods, and harvesting environments rather than the optimisation of known systems.
One well-known hygroscopic moiety that has not yet been utilised for AWH materials is the guanidinium moiety, sustainably (and affordably) available in the form of arginine. Arginine, being an amino acid, also offers convenient routes for functionalisation that do not interfere with the guanidinium side chain. In this work, we report our efforts to design, prepare and characterise an additive-free water-harvesting polymer system using hygroscopic guanidinium moieties attached by esterification of L-arginine.
Results and discussion
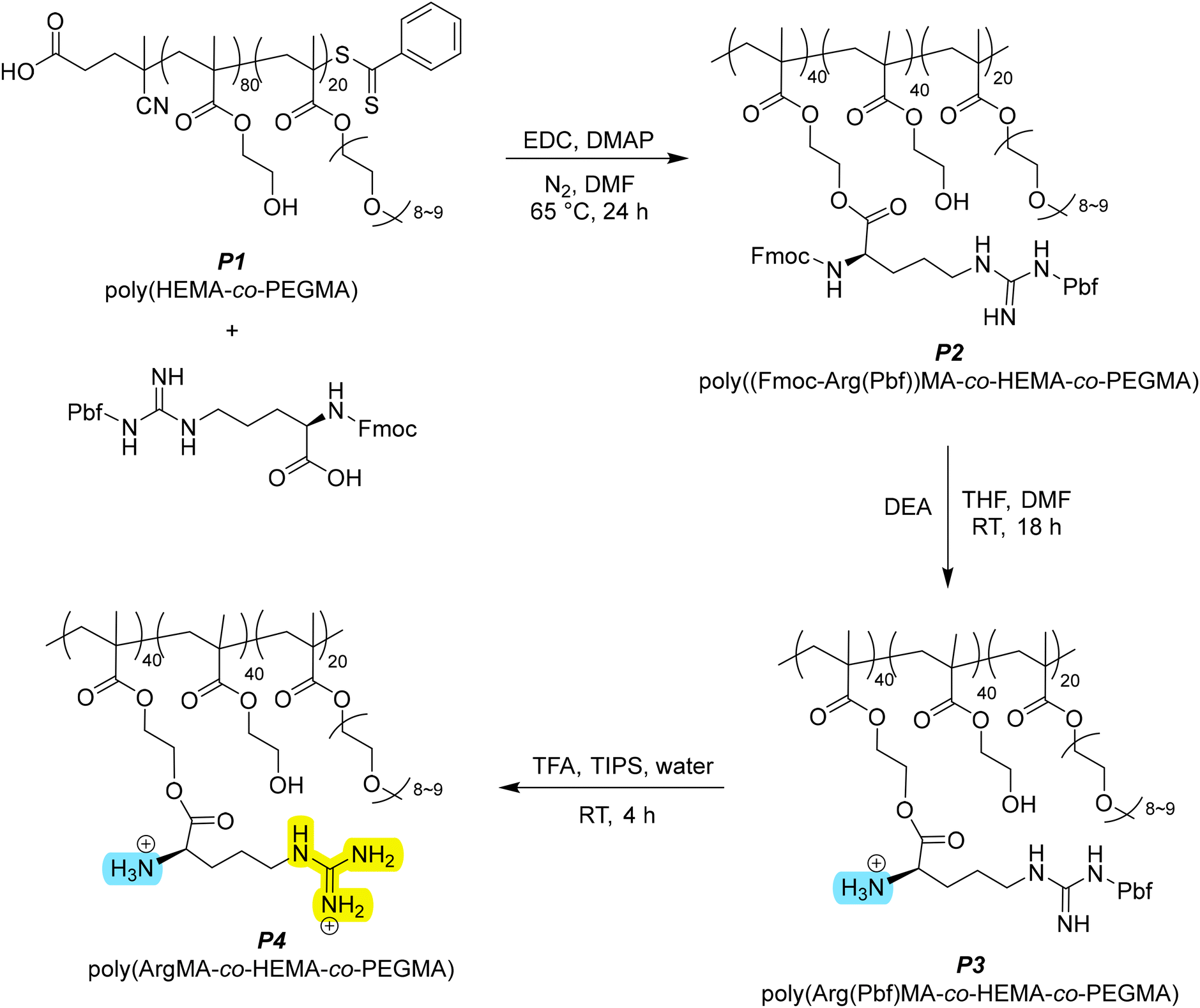
We designed a synthetic pathway to understand polymer structure–property relationships by copolymer functionality, specifically with guanidinium (via arginine) in this study. To form a parent polymer for functionalisation with arginine groups, we first prepared poly(2-hydroxyethyl methacrylate-co-poly(ethylene glycol) methyl ether methacrylate) (poly(HEMA-co-PEGMA)) (P1, Scheme S1†). This was done using reverse addition fragmentation transfer (RAFT) polymerisation to control dispersity. HEMA is thermoresponsive, hydrophilic, and can be readily modified through the side chain alcohol. The poly(HEMA) homopolymer is, however, brittle and insoluble in common solvents, rendering further processing impractical. The addition of PEGMA allows for a more soluble and flexible material, as well as maintaining thermoresponsivity. The final molar composition of the polymer, as calculated by 1H nuclear magnetic resonance (1H NMR) spectroscopy, was found to be 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 HEMA:PEGMA with a theoretical molecular weight of around 21 kDa. Gel permeation chromatography (GPC) data obtained indicated a Mn of 58 kDa and a dispersity around 6. Polymeric inter- and intramolecular hydrogen-bonding interactions can inflate dispersity measurements; this is indicated by FTIR data and a broad, multimodal GPC trace (Fig. S5 and S14†).18 Dispersity is significant when considering size-dependent properties such as thermal phase transitions; a narrow molecular weight distribution allows for a faster phase transition over a smaller temperature range.19 However, the effects of molecular weight (and dispersity) on other water harvesting properties is unclear, and so in the context of this study we performed measurements using a singular batch of bulk sample so as to retain molecular weight as a controlled variable.
:19 HEMA:PEGMA with a theoretical molecular weight of around 21 kDa. Gel permeation chromatography (GPC) data obtained indicated a Mn of 58 kDa and a dispersity around 6. Polymeric inter- and intramolecular hydrogen-bonding interactions can inflate dispersity measurements; this is indicated by FTIR data and a broad, multimodal GPC trace (Fig. S5 and S14†).18 Dispersity is significant when considering size-dependent properties such as thermal phase transitions; a narrow molecular weight distribution allows for a faster phase transition over a smaller temperature range.19 However, the effects of molecular weight (and dispersity) on other water harvesting properties is unclear, and so in the context of this study we performed measurements using a singular batch of bulk sample so as to retain molecular weight as a controlled variable.
Steglich esterification was then conducted to attach N-terminus and side group protected-arginine to the HEMA sidechain (P2, Scheme 1). The amine was protected with fluorenylmethoxycarbonyl (Fmoc) and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) protected the guanidine group. N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) was selected as the byproduct 1-(3-(dimethylamino)propyl)-3-ethylurea (EDU) is soluble in water and enables facile purification. Functionalisation was confirmed via1H NMR analysis which indicated that ca. 50% of HEMA groups had been converted. The overall composition of the functionalised polymer P2 was hence determined to be poly((Fmoc-Arg(Pbf))MA40-co-HEMA40-co-PEGMA20). The mild conditions of Steglich esterification were expected to minimise the undesired deprotection of Fmoc during the reaction, though we still observed some cleavage by 1H NMR analysis. A change in the material colour also indicated that the RAFT chain transfer agent end groups were removed during the esterification. P2 was also insoluble in water, likely due to the presence of the bulky protecting groups.
 | ||
| Scheme 1 Scheme depicting functionalisation of P1via Steglich esterification (top) and sequential deprotection of side chain Fmoc-Arg(Pbf) (right and bottom). Highlighted are side-chain arginine primary amine (blue) and guanidine (yellow) exposed after deprotection. | ||
The orthogonality of the deprotection chemistry enables us to reveal the polar groups sequentially. Initially, we removed Fmoc to liberate the primary amine. Some Fmoc had already been removed during the esterification and so the complete removal of Fmoc was conducted (P3, Scheme 1). We next removed the Pbf group to liberate the guanidine group (P4, Scheme 1). Characterisation using 1H NMR spectroscopy confirmed the successful removal of the protecting groups. As expected, the polymer composition stayed consistent to that calculated from P2, indicating that the deprotection conditions did not cause any unwanted side reactions or degradation.
With our controlled deprotection we had 4 polymers with varying polarity and functional groups: the parent polymer (P1), with HEMA and PEGMA; P2, with fully protected arginine; P3 with free primary amine and protected guanidine; and P4 with free amine and guanidine. We next set about measuring the water uptake for products P1–P4 over a period of 24 hours (Fig. 1a). The fully deprotected arginine-functionalised polymer (P4) had increased water uptake at 24 hours from 92 mg g−1 to 134 mg g−1 water to polymer; a 46% increase compared to the base polymer P1. Considering that approximately 50% of HEMA groups were functionalised (i.e. 40% of the polymer by molar ratio), this shows arginine has around twice the water uptake capacity as HEMA. In terms of sequential deprotection, there was a 150% increase in water uptake after 24 hours from P2 to P3, then a 77% increase from P3 to P4. While the guanidine group is known to be hygroscopic and was expected to be the main contributor to increasing water uptake, the first deprotection of Fmoc (exposing the primary amine) appeared more impactful by percentage increase in uptake. However, the sequential improvements in water uptake are contributed to by both the removal of bulky protecting groups and the exposure of hydrophilic/hygroscopic functional groups on the arginine side chain.
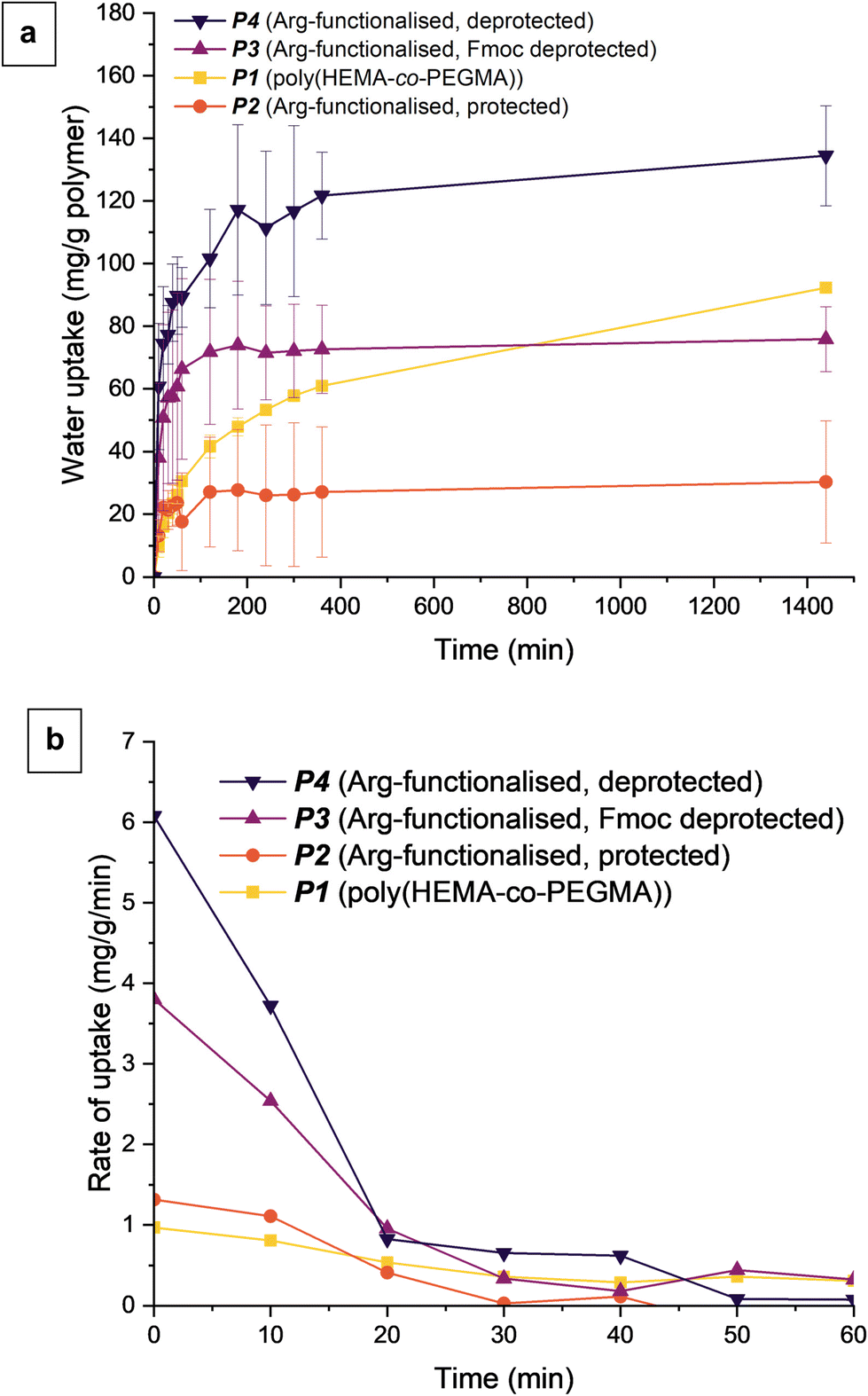 | ||
| Fig. 1 (a) Water uptake over 24 hours, showing the effects of arginine functionalisation and sequential deprotection. (b) First derivative of water uptake over 1 hour, highlighting the different rates of change in water uptake with functionalisation and sequential deprotection. | ||
When comparing P1 and P4, the arginine side chain greatly increased the initial rate of uptake compared to HEMA (Fig. 1b). The rapid water uptake within the first 3 hours for P4 highlights the possibility of applications tailored toward more rapid cycling. P2 and P3 showed similar changes of rate to P4, however this is more likely due to bulky protecting groups hindering water uptake. The bulky and hydrophobic protecting groups reduce the adsorption capacity of the polymer. Interestingly, functionalised samples P2–P4 showed a sudden decrease in the rate of water uptake at around 120–180 minutes, contrasted with a more gradual attenuation for P1. Rapid surface adsorption of water vapour followed by a relatively slow diffusion of water into the polymer network are the main mechanisms of water uptake in these materials.20,21 The decrease in water uptake rate likely indicates saturation of surface hygroscopic groups, with further water uptake depending on the rate of diffusion. The relative rates of diffusion are unclear from the data, though over short time periods this would be less relevant.
While the linear, soluble polymer is useful for characterisation, it would be solubilised in water-harvesting applications and hence contaminate the harvested water. Crosslinking eliminates any polymer solubility in water, alleviating these concerns. A hydrogel system based on the arginine-functionalised polymers above is shown in Fig. 2. This was synthesised with a 75:5:20 molar feed ratio of HEMA, poly(ethylene glycol) dimethacrylate (PEGDMA) 750 crosslinker and PEGMA (Scheme S2†). PEGDMA 750 was selected because of its similarity in structure to PEGMA and its positive contributions to water uptake observed in our past work.22 Functionalisation and deprotection were conducted using the same methods employed for the linear polymer to afford base gel G1 and arginine-functionalised gel G2 (Scheme S2†). Given the insolubility of hydrogel products, characterisation of the resulting gel products was performed using FTIR spectroscopy. Disappearance of the free HEMA alcohol O–H stretch at 3670 cm−1, the splitting of the alkane C–H stretch at around 2900 cm−1, as well as the appearance of amine peaks between 3100 cm−1 and 3350 cm−1, indicated that functionalisation was successful (Fig. S9 and S10†). After lyophilisation, G2 takes on a powdery appearance, and although the porosity is minimal (as shown by SEM imaging, Fig. 2c), the polymer was still found to have a large surface area. While the powdery, crumbly nature of G2 was not amenable to mechanical testing, its high surface area rendered it particularly suitable for water-harvesting applications.
 | ||
| Fig. 2 (a) Graphical representation and chemical structure of the functionalised hydrogel G2, showing its single-network structure, PEGDMA 750 crosslinking, and interspersion of the arginine-functionalised sidechain. (b) Photo of G2 after lyophilisation. (c) SEM image of G2 (scale bar 20 μm). | ||
Because the fully deprotected polymer was the lead candidate in terms of water uptake, we went straight to investigating the fully deprotected gel G2. The gel, like its linear counterparts, showed rapid water uptake which plateaued after 2 hours. After 24 hours, G2 absorbed a total of 149 mg g−1 water compared to P4's 134 mg g−1, showing that, as in our previous study, the addition of PEGDMA crosslinker did not negatively affect water uptake (Fig. 3).22
 | ||
| Fig. 3 (a) Water uptake over 24 hours of G2 and P4, showing that crosslinking with PEGDMA 750 had no negative effects on water uptake. (b) First derivative of water uptake over 1 hour of G2 and P4, showing the minimal impact of crosslinking. | ||
We next turned our attention to water release. Thermoresponsive polymers allow for the energy-efficient harvesting of water at low temperatures. HEMA and PEGMA homopolymers are both thermoresponsive, as well as the copolymer P1, with a cloud point close to 56 °C at a concentration of 10 mg mL−1 in water, and a 69.5 °C lower critical solution temperature (LCST) after swelling.23–25 Differential scanning calorimetry (DSC) was used to determine phase transition temperatures to account for varying solubility of products and allow for direct comparison. We determined P4 to have a LCST of 81.9 °C, meaning that the incorporation of arginine greatly increases the temperature required for water release. Arginine is cationic at neutral pH, hindering the thermal transition change and increasing the LCST.23 In contrast to its linear counterpart, G2 has a volume phase transition temperature (VPTT) of 62.2 °C, which is more amenable to the desired application of water harvesting. While we were satisfied with our measured VPTT, it is well known that this property is dependent on the specific hydrophilic/hydrophobic balance of a system and so could be further tuned by adjusting the polymer composition and the degree of arginine functionalisation.26,27
A cycling experiment was then conducted on G2 to evaluate the combined uptake and release abilities over time (Fig. 4). G2 lends itself to short cycle times as the initial rapid adsorption slows at around 120 minutes. After calculating total output based on uptake and cycle times, we decided that a 60-minute uptake and 10-minute release cycle would be most optimal. Gratifyingly, after conducting 25 cycles we observed no loss of uptake or release capacity. The scale of these experiments was in the sub-gram range, hence some water loss would be attributed to evaporation. However, the ability to renew the material quickly at low temperatures nonetheless is promising for future water harvesting applications.
 | ||
| Fig. 4 Water harvesting cycling data of G2, showing minimal/no decay of performance. Uptake cycles were conducted at 70% RH at around 25 °C for 1 hour, while release cycles were conducted in a 70 °C oven for 10 minutes. | ||
Conclusions
In summary, we have demonstrated the water-harvesting potential of L-arginine-functionalised polymers and a novel use of the hygroscopic guanidine moiety. Notably, arginine-functionalised polymers exhibited around 140 mg g−1 of uptake after 24 hours at 70% RH without any additives. We demonstrated that the primary amine group has positive effects on water harvesting but an even greater effect when combined with the guanidine group. Rapid cycling behaviour has also been demonstrated, with 70-minute cycles potentially yielding 2 g g−1 of water after 24 hours. Water uptake and release properties were retained over an extended period of time. Side-chain functionalisation was also demonstrated to affect physical properties such as thermal phase transitions, allowing another avenue for the material tunability required for specialised applications. Additional studies on this work would include changes in copolymerisation ratios and arginine conversion, as well as investigations into more efficient post-functionalisation methods. These outcomes highlight the expansive potential for new hygroscopic moieties and underutilised synthetic methods in the water-harvesting material space, in particular, the further study of arginine and other guanidinium-containing molecules.Experimental section
Fmoc-Arg(Pbf)-OH was purchased from GL Biochem. All other chemicals were purchased from Sigma-Aldrich and were used as purchased with the following exceptions: poly(ethylene glycol) methyl ether methacrylate (PEGMA, average Mn 500, containing 200 ppm butylated hydroxytoluene (BHT), 100 ppm 4-methoxyhenol (MEHQ) as inhibitor), 2-hydroxyethyl methacrylate (HEMA, containing ≤50 ppm MEHQ as inhibitor), and poly(ethylene glycol) dimethacrylate (PEGDMA average Mn 750, containing 80–120 ppm MEHQ as inhibitor, 270–330 ppm BHT as inhibitor). For these, basic-alumina chromatography was conducted to remove inhibitors immediately prior to polymerisation.Gel permeation chromatography (GPC) was performed with an Agilent 1260 Infinity II, using N,N-dimethylacetamide (DMAc) as solvent with a 1.00 mL min−1 flow rate and a column temperature of 30 °C. Molecular weights were calibrated against polyethylene oxide/glycol EasiVirals® (Agilent Technologies, U.K.) standards that covered an average molecular mass range from 1576000 to 106 g mol−1. 1H NMR spectra were obtained at 298 K on a Bruker Ascend 400 at 400 MHz using DMSO-d6 as solvent. FTIR spectroscopy was conducted on a PerkinElmer Spectrum 2 using lyophilised samples. Field emission electron scanning microscopy (FESEM) images were taken at 3 kV using a Zeiss Crossbeam 550 FIB-SEM. Samples were desiccated and coated with platinum before analysis. Humidity was measured inside a custom set-up as shown in Fig. S15.†22 Humidity and ambient temperature were measured using a RS-1260 humidity temperature meter. DSC experiments were conducted using a PerkinElmer DSC8500 apparatus.
Synthesis of poly(HEMA-co-PEGMA) copolymer
CTP chain transfer agent (4-cyano-4-(phenyl-carbonothioylthio)pentanoic acid) (27.9 mg, 0.10 mmol), HEMA (2.08 g, 16.00 mmol), PEGMA 500 (2.00 g, 4.00 mmol), 2,2′-azobis(2-methylpropionitrile) initiator (AIBN) (1.64 mg, 0.010 mmol) and 5 mL of anhydrous N,N-dimethylformamide (DMF) were charged into a 25 mL round bottom flask. Once dissolved, the solution was sealed with a rubber septum, magnetically stirred and maintained at 0 °C while being degassed with nitrogen for 20 minutes. The flask was then magnetically stirred at 65 °C in an oil bath for 24 hours. The resultant mixture was then precipitated into vigorously stirred cold diethyl ether (80 mL) to afford a pink, solid mass. Further purification was conducted by dialysis in water over 3 days. The polymer product was lyophilised for 24 hours to afford a flexible pink film P1 (poly(HEMA80-co-PEGMA20), 84%).1 H NMR poly(HEMA 80 -co-PEGMA 20 ) ( P1 ), 400 MHz, DMSO-d 6 δ 4.79 (s, 202 × 1H), 4.01 (s, 47 × 2H, signal overlapping), 3.90 (s, 202 × 2H, signal overlapping), 3.58–3.51 (s, 47 × 38H, signal overlapping), 3.44, 3.43, 3.42 (s, 202 × 2H), 3.24 (s, 47 × 3H), 2.53 (s, 47 × 2H), 1.78 (b, 202 × 2H, signal overlapping), 1.46 (b, 47 × 2H, signal overlapping), 0.94 (s, 47 × 3H, signal overlapping), 0.78 (202 × 3H, signal overlapping).
Synthesis of poly(HEMA-co-PEGDMA-co-PEGMA) hydrogel
CTP chain transfer agent (27.9 mg, 0.10 mmol), HEMA (1.95 g, 15.00 mmol), PEGMA 500 (2.00 g, 20.00 mmol), PEGDMA 750 (0.750 g, 1.00 mmol), AIBN initiator (1.64 mg, 0.010 mmol) and 6 mL of anhydrous DMF were charged into a 25 mL round bottom flask. The resulting solution was seal with a rubber septum and magnetically stirred at 0 °C while being degassed with nitrogen for 20 minutes, with stirring. The flask was then magnetically stirred at 65 °C in an oil bath for 24 hours, resulting in a solid pink gel. This product was broken into small chunks with a spatula. Excess monomer and other impurities were removed by soaking in water over 3 days. The polymer product was lyophilised for 24 hours to afford a soft pink solid G1 (poly(HEMA75-co-PEGDMA5-co-PEGMA20)).Arginine functionalisation procedure
Steglich esterification was conducted with poly(HEMA-co-PEGMA) polymer (P1) or poly(HEMA-co-PEGDMA-co-PEGMA) gel (G1), Fmoc-Arg(Pbf)-OH, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and 4-dimethylaminopyridine (DMAP) catalyst in anhydrous DMF. The magnetically stirred solution was allowed to react at room temperature for 18 hours before the reaction vessel was exposed to air. Products P2 and G1* were afforded from this procedure.A detailed procedure for poly(Fmoc-Arg(Pbf)MA-co-HEMA-co-PEGMA) (P2) is as follows: P1 (303 mg, 43 wt% HEMA, 1.00 mmol HEMA), Fmoc-Arg(Pbf)-OH (649 mg, 1.00 mmol), EDC initiator (186 mg, 1.20 mmol), DMAP catalyst (36.7 mg, 0.30 mmol) and 7 mL of anhydrous DMF were charged into a 25 mL round bottom flask. The flask was sealed and magnetically stirred at room temperature overnight, with the solution going from clear, bright yellow to colourless. The resultant mixture was then precipitated into vigorously stirred cold water (80 mL) to afford an off-white precipitate. The precipitate was lyophilised for 24 h to afford a beige powder P2 (473 mg, approx. 50% HEMA conversion as determined by 1H NMR).
1 H NMR poly(Fmoc-Arg(Pbf)MA 40 -co-HEMA 40 -co-PEGMA 20 ) ( P2 ), 400 MHz, DMSO-d 6 δ 7.89 (dt), 7.72 (d), 7.62 (dd), 7.43 (dtd), 4.75 (b), 4.29–4.19 (m, signal overlapping), 4.00 (s), 3.89 (s), 3.47 (s, signal overlapping), 3.21 (s, signal overlapping), 3.02–2.90 (m, signal overlapping), 2.48 (s, signal overlapping), 2.42 (s), 2.00, 1.99, 1.97 (t + s, signal overlapping), 1.65 (s, signal overlapping), 1.56 (s, signal overlapping), 1.45 (s, signal overlapping), 1.41, 1.39, 1.35 (t + s, signal overlapping), 0.92 (s, signal overlapping), 0.78 (s, signal overlapping).
Deprotection procedure
Fmoc protecting groups on Arg-functionalised products (P2 and G1*) were removed with diethylamine (DEA) in an equal volume of anhydrous tetrahydrofuran (THF). For G1*, 1 mL DMF was added to keep the gel swollen and to allow DEA to penetrate the network for deprotection. The mixture was stirred at room temperature for 18 hours, after which the contents were concentrated under reduced pressure and washed with cold diethyl ether. A tan solid was afforded after gravity filtration and air drying. Pbf protecting groups were removed with a mixture of trifluoroacetic acid, water and triisopropylsilane (95:2.5:2.5). The mixture was stirred at room temperature for 4 hours, after which the contents were washed with cold diethyl ether. A beige solid was afforded after lyophilisation.
Fmoc was removed from P2 to afford P3 (poly(Arg(Pbf)MA40-co-HEMA40-co-PEGMA20)), and Pbf was removed from P3 to afford P4 (poly(ArgMA40-co-HEMA40-co-PEGMA20)).
Both Fmoc and Pbf groups were removed from G1* (poly(HEMA-co-Fmoc-Arg(Pbf)MA-co-PEGDMA-co-PEGMA)) to afford G2 (poly(HEMA-co-ArgMA-co-PEGDMA-co-PEGMA)).
1 H NMR poly(Arg(Pbf)MA 40 -co-HEMA 40 -co-PEGMA 20 ) ( P3 ), 400 MHz, DMSO-d 6 δ 4.78 (b), 3.99, 3.90 (m + d, signal overlapping), 3.5 (s, signal overlapping), 3.23 (s, signal overlapping), 3.04 (s, signal overlapping), 2.96 (s, signal overlapping), 2.48 (s, signal overlapping), 2.42 (s, signal overlapping), 2.01 (s), 1.57 (m, signal overlapping), 1.41 (s, signal overlapping), 0.97 (s, signal overlapping), 0.79 (s, signal overlapping).
1 H NMR poly(ArgMA 40 -co-HEMA 40 -co-PEGMA 20 ) ( P4 ), 400 MHz, DMSO-d 6 δ 4.79 (b), 4.05, 3.90 (m + s, signal overlapping), 3.51 (s, signal overlapping), 3.44 (s, signal overlapping), 3.24 (s, signal overlapping), 2.95, 2.93, 2.92, 2.89 (q, signal overlapping), 1.72, 1.54 (m + s + s, signal overlapping), 0.93 (s, signal overlapping), 0.78 (s, signal overlapping).
Water uptake measurement
Samples were lyophilised overnight right before water uptake experiments were conducted to minimise residual water. The humidity in the box was adjusted to approximately 70% RH. Room temperature was consistent at 23 °C. Plastic Petri dishes containing samples were placed into the humidity box shown in Fig. S15.† Samples were removed, weighed and returned to the box as quickly as possible. Water content was determined as the mass of water per gram of polymer. For the first hour, measurements were taken every ten minutes, then every hour for the next five hours, then once more 24 hours after the experiment was commenced. Mean values were taken from at least three trials for each sample.LCST and VPTT measurement
The LCST of polymer products and VPTT of hydrogel products were determined using DSC. Water-swollen samples were blotted with filter paper and a small portion (∼10 mg) was loaded into a hermetically sealed aluminium pan. One cycle was run from ambient temperature to 100 °C. Endothermic phase transitions were defined by the peak of the endotherm.24Cycling experiments
G2 was heated at 70 °C for several hours before this experiment was commenced. The water uptake method described above was followed, with measurements taken after 1 hour. After this, the sample was placed into an oven maintained at 70 °C. After 10 minutes, the sample was weighed then returned to the humidity box. This uptake-release cycle was repeated for another 24 cycles.Author contributions
M. T., B. P. and L. C. conceived and designed the experiments. L. C. supervised the project. M. T. performed the experiments, analysed experimental results, and wrote the manuscript. B. P. and L. C. edited and revised the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by an Australian Research Council (DP230100679) and through an Australian Government Research Training Program (RTP) Scholarship.The authors acknowledge Microscopy Australia at the Centre for Advanced Microscopy, The Australian National University, a facility enabled by NCRIS and university support.
References
- F. Zhao, X. Zhou, Y. Liu, Y. Shi, Y. Dai and G. Yu, Super Moisture-Absorbent Gels for All-Weather Atmospheric Water Harvesting, Adv. Mater., 2019, 31 (10), 1806446, DOI:10.1002/adma.201806446.
- Y. Guo, W. Guan, C. Lei, H. Lu, W. Shi and G. Yu, Scalable super hygroscopic polymer films for sustainable moisture harvesting in arid environments, Nat. Commun., 2022, 13(1), 2761, DOI:10.1038/s41467-022-30505-2.
- D. Maity, A. P. Teixeira and M. Fussenegger, Hydratable Core–Shell Polymer Networks for Atmospheric Water Harvesting Powered by Sunlight, Small, 2023, 19(47), 2301427, DOI:10.1002/smll.202301427.
- Q. Luo, M. Chen, D. Yu, T. Zhang, J. Zhao, L. Zhang, X. Han, M. Zhou, Y. Hou and Y. Zheng, An Atmospheric Water-Harvester with Ultrahigh Uptake-Release Efficiency at Low Humidity, ACS Nano, 2024, 18(22), 14650–14660, DOI:10.1021/acsnano.4c02866.
- J. Xu, T. Li, T. Yan, S. Wu, M. Wu, J. Chao, X. Huo, P. Wang and R. Wang, Ultrahigh solar-driven atmospheric water production enabled by scalable rapid-cycling water harvester with vertically aligned nanocomposite sorbent, Energy Environ. Sci., 2021, 14(11), 5979–5994, 10.1039/D1EE01723C.
- A. G. Tereshchenko, Deliquescence: Hygroscopicity of Water-Soluble Crystalline Solids, J. Pharm. Sci., 2015, 104(11), 3639–3652, DOI:10.1002/jps.24589.
- Fortune Business Insights. Lithium Chloride Market Size, Share & Industry Analysis, By Application (Batteries, Ceramics & Glass, Lubricants, Polymer Production, Air Treatment, and Others) and Regional Forecast, 2024–2032; FBI108063, 2024. https://www.fortunebusinessinsights.com/lithium-chloride-market-108063 Search PubMed.
- E. Hsiao, A. L. Barnette, L. C. Bradley and S. H. Kim, Hydrophobic but Hygroscopic Polymer Films – Identifying Interfacial Species and Understanding Water Ingress Behavior, ACS Appl. Mater. Interfaces, 2011, 3(11), 4236–4241, DOI:10.1021/am200894h.
- Y. Cheng, X. Zhang, J. Zhang, Z. He, Y. Wang, J. Wang and J. Zhang, Hygroscopic hydrophobic coatings from cellulose: Manipulation of the aggregation morphology of water, Chem. Eng. J., 2022, 441, 136016, DOI:10.1016/j.cej.2022.136016.
- J. E. Coughlin, A. Reisch, M. Z. Markarian and J. B. Schlenoff, Sulfonation of polystyrene: Toward the “ideal” polyelectrolyte, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(11), 2416–2424, DOI:10.1002/pola.26627.
- P. Schweng, F. Mayer, D. Galehdari, K. Weiland and R. T. Woodward, A Robust and Low-Cost Sulfonated Hypercrosslinked Polymer for Atmospheric Water Harvesting, Small, 2023, 19(50), 2304562, DOI:10.1002/smll.202304562.
- L. J. Mauer, Deliquescence of crystalline materials: mechanism and implications for foods, Curr. Opin. Food Sci., 2022, 46, 100865, DOI:10.1016/j.cofs.2022.100865.
- M. Wu, R. Li, Y. Shi, M. Altunkaya, S. Aleid, C. Zhang, W. Wang and P. Wang, Metal- and halide-free, solid-state polymeric water vapor sorbents for efficient water-sorption-driven cooling and atmospheric water harvesting, Mater. Horiz., 2021, 8(5), 1518–1527, 10.1039/D0MH02051F.
- H. Park, I. Haechler, G. Schnoering, M. D. Ponte, T. M. Schutzius and D. Poulikakos, Enhanced Atmospheric Water Harvesting with Sunlight-Activated Sorption Ratcheting, ACS Appl. Mater. Interfaces, 2022, 14(1), 2237–2245, DOI:10.1021/acsami.1c18852.
- C. Lei, W. Guan, Y. Zhao and G. Yu, Chemistries and materials for atmospheric water harvesting, Chem. Soc. Rev., 2024, 53(14), 7328–7362, 10.1039/D4CS00423J.
- P. Schweng and R. T. Woodward, Challenging POPular opinion: Porous organic polymers for atmospheric water harvesting, React. Funct. Polym., 2024, 203, 106014, DOI:10.1016/j.reactfunctpolym.2024.106014.
- F. Ni, N. Qiu, P. Xiao, C. Zhang, Y. Jian, Y. Liang, W. Xie, L. Yan and T. Chen, Tillandsia-Inspired Hygroscopic Photothermal Organogels for Efficient Atmospheric Water Harvesting, Angew. Chem., Int. Ed., 2020, 59(43), 19237–19246, DOI:10.1002/anie.202007885 , (acccessed 2025/01/25).
- B. Diggle, Z. Jiang, R. W. Li and L. A. Connal, Self-Healing Polymer Network with High Strength, Tunable Properties, and Biocompatibility, Chem. Mater., 2021, 33 (10), 3712–3720, DOI:10.1021/acs.chemmater.1c00707.
- C. Ramkissoon-Ganorkar, L. Feng, M. Baudys and S. W. Kim, Effect, of molecular weight and polydispersity on kinetics of dissolution and release from pH/temperature-sensitive polymers, J. Biomater. Sci., Polym. Ed., 1999, 10(10), 1149–1161, DOI:10.1163/156856299X00739.
- A. C. Newns, Sorption and Diffusion in Polymers, Nature, 1968, 218(5139), 355–356, DOI:10.1038/218355b0.
- X. Zhou, H. Lu, F. Zhao and G. Yu, Atmospheric Water Harvesting: A Review of Material and Structural Designs, ACS Mater. Lett., 2020, 2(7), 671–684, DOI:10.1021/acsmaterialslett.0c00130.
- M. K. Thanusing, P. Shen, B. L. Pollard and L. A. Connal, Rational design of water-harvesting hydrogels, Mol. Syst. Des. Eng., 2024, 9(1), 63–72, 10.1039/D3ME00132F.
- W. S. Ng, E. Forbes, G. V. Franks and L. A. Connal, Xanthate-Functional Temperature-Responsive Polymers: Effect on Lower Critical Solution Temperature Behavior and Affinity toward Sulfide Surfaces, Langmuir, 2016, 32(30), 7443–7451, DOI:10.1021/acs.langmuir.6b00211.
- R. Fei, J. T. George, J. Park, A. K. Means and M. A. Grunlan, Ultra-strong thermoresponsive double network hydrogels, Soft Matter, 2013, 9 (10), 2912–2919, 10.1039/C3SM27226E.
- D. Singh, D. Kuckling, V. Choudhary, H.-J. Adler and V. Koul, Synthesis and characterization of poly(N-isopropylacrylamide) films by photopolymerization, Polym. Adv. Technol., 2006, 17(3), 186–192, DOI:10.1002/pat.713.
- Y. Yuan, K. Raheja, N. B. Milbrandt, S. Beilharz, S. Tene, S. Oshabaheebwa, U. A. Gurkan, A. C. S. Samia and M. Karayilan, Thermoresponsive polymers with LCST transition: synthesis, characterization, and their impact on biomedical frontiers, RSC Appl. Polym., 2023, 1(2), 158–189, 10.1039/D3LP00114H.
- R. Singh, S. A. Deshmukh, G. Kamath, S. K. R. S. Sankaranarayanan and G. Balasubramanian, Controlling the aqueous solubility of PNIPAM with hydrophobic molecular units, Comput. Mater. Sci., 2017, 126, 191–203, DOI:10.1016/j.commatsci.2016.09.030.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00373j |
| This journal is © The Royal Society of Chemistry 2025 |