
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeamless integration of CMOS microsensors into open microfluidic systems†
Raziyeh
Bounik
 a,
Alex E.
Landolt
a,
Jihyun
Lee
a,
Vijay
Viswam
ab,
Fernando
Cardes
a,
Mario M.
Modena
*a and
Andreas
Hierlemann
a
a,
Alex E.
Landolt
a,
Jihyun
Lee
a,
Vijay
Viswam
ab,
Fernando
Cardes
a,
Mario M.
Modena
*a and
Andreas
Hierlemann
a
aDepartment of Biosystems Science and Engineering, ETH Zürich, Basel, Switzerland. E-mail: mario.modena@bsse.ethz.ch
bMaxWell Biosystems AG, Zürich, Switzerland
First published on 27th March 2025
Abstract
As traditional two-dimensional (2D) cell cultures offer limited predictive capabilities for drug development, three-dimensional (3D) tissue models, such as spherical microtissues, have been introduced to better reproduce physiological conditions. The hanging-drop method, used to cultivate microtissues at an air–liquid interface, proves to be effective for microtissue formation and maintenance. Using that technology, it is possible to fluidically interconnect several hanging drops hosting different models of human organs to recapitulate relevant tissue interactions. Here, we combine microfluidics with microelectronics (i.e., complementary metal-oxide-semiconductor (CMOS) technology) and present a novel multifunctional CMOS microelectrode array (MEA) integrated into an open microfluidic system. The device can be used in hanging-drop mode for in situ microtissue readouts and in standing-drop mode like a conventional MEA. The CMOS-MEA chip features two reconfigurable electrode arrays with 1024 electrodes each, and enables electrophysiology, impedance spectroscopy, and electrochemical sensing to acquire a broad spectrum of biologically relevant information. We fabricated the chip using a 0.18 μm CMOS process and developed a strategy to integrate the CMOS-MEA chip into the open microfluidic system within a larger overall effort to incorporate discrete CMOS sensors into microfluidic devices. Proof-of-concept experiments demonstrate the capability to perform electrophysiology and impedance spectroscopy of human induced pluripotent stem cell (hiPSC)-derived cardiac microtissues, as well as electrochemical sensing of different analytes including hydrogen peroxide and epinephrine.
1. Introduction
Efficient in vitro assays are crucial for investigating fundamental biological questions, developing biotechnological products, and advancing the lengthy and expensive drug discovery process.1,2 This process features a high failure rate, so that significant improvements are necessary, including the development and utilization of reliable cellular and tissue models as well as of efficient screening methods.3 Conventional two-dimensional (2D) systems involve culturing cells as monolayers in pretreated or coated dishes or flasks using molecules that promote cell adhesion and growth. Although these systems have been valuable for studying biological phenomena and screening drugs, one major limitation includes their inability to faithfully replicate in vivo niches or pockets due to their planar monolayer structure.4 To better replicate physiological conditions, three-dimensional (3D) culture systems have been introduced that enable the recapitulation of important features like cell–cell interactions and density gradients.5–8 Among the most popular 3D culture systems are 3D microtissues, such as spheroids, which can be formed by using, e.g., low-attachment plates,9 or the hanging-drop method, where cells in suspension settle at the bottom, followed by spontaneous aggregation of the cells into microtissues.10–12Microfluidic hanging-drop network platforms, consisting of interconnected hanging drops, have been developed to form and culture 3D microtissues.13,14 These microfluidic systems provide precise control over the 3D culturing conditions and enable a better replication of the dynamic in vivo environment, e.g., with respect to perfusion rates and metabolite turnover.15 The open microfluidic system design ensures optimal gas exchange and facilitates easy loading and retrieval of microtissues for downstream assays. Additionally, this method helps to avoid common issues, such as bubbles that often lead to failure of microfluidics networks. Moreover, it offers the potential of combining different tissue models, supporting the concept of a “body-on-a-chip”.16,17
Microscopy and imaging play a crucial role in the analysis of 2D and 3D cell cultures. However, only limited information about the cellular state can be extracted in a culture environment. To gather more comprehensive information, the use of microsensor technology may be beneficial. Numerous microsensors have been introduced into 2D and 3D culture systems, operating under both static and dynamic conditions.18 In particular, label-free and noninvasive microsensors relying on electrical impedance spectroscopy (EIS), electrochemical biosensing, and electrophysiological measurements have attracted considerable interest for monitoring the status of cell cultures.19–23
In addition to single microsensors or a few electrodes, microelectrode-array (MEA) systems have been used in in vitro assays. MEA systems are primarily used for spatially resolved voltage recordings from electrogenic cells in vitro.24 Moreover, the use of MEAs enables two-dimensional “impedance imaging” of biological samples.19,25,26 Additionally, MEAs facilitate localized electrochemical measurements, including amperometry and voltammetry, which enables real-time two-dimensional electrochemical imaging.27–29 Finally, MEAs with multiple sensing modalities have been fabricated in CMOS technology.30–32
Several techniques have been developed to directly integrate microsensors into a hanging-drop platform. In one approach, glass inlays containing the electrodes were positioned in the ceilings of the hanging drops. This method has been used for EIS-based and electrochemical sensing systems with various electrode configurations; the electrodes were then connected to an off-chip external readout unit.16,33 In another work, to facilitate parallel measurements, multiple sets of electrodes were fabricated on a glass substrate, forming the ceiling of a microfluidic network containing eight hanging drops. Each hanging drop featured a set of electrodes, connected to an external readout, allowing for measuring the glucose concentration of all eight droplets in parallel.34
To augment the sensor system for a hanging-drop platform, several aspects are important: (i) the possibility to perform different types of measurements with a single system, (ii) an increase in measurement accuracy by improving the signal-to-noise ratio, and (iii) a reduction of the size of readout units to enable parallelization and seamless integration in dedicated microsystems. A promising approach to realize these features on a single device is to utilize CMOS technology and devise a microsystem, which then can be integrated into, e.g., a hanging-drop system.
This paper presents a novel device for in situ sensing in 3D microtissue cultures, featuring a multi-functional CMOS MEA system integrated into a microfluidic hanging-drop network platform (Fig. 1a top). In terms of functionality, the designed CMOS-MEA system includes electrophysiology, EIS, and electrochemical sensing modules, along with reconfigurable microelectrode arrays including a total of 1024 electrodes. The inclusion of a MEA as sensing unit for each hanging drop renders the device versatile and flexible and enables to flip the device upside down and operate it in a “standing-drop” configuration.35,36 In this configuration, the device works like a conventional high-density CMOS-MEA (Fig. 1a bottom).
 | ||
| Fig. 1 (a) Side view of the device in hanging-drop mode (top) and standing-drop mode (bottom). (b) Main components of the device, including the microfluidic system, the CMOS-MEA chip, and the glass substrate. (c) Block diagram of the CMOS-MEA chip, featuring two electrode arrays, each comprising 1024 electrodes that can – via a row-column selection scheme – be connected to 64 electrophysiology channels, eight impedance spectroscopy channels, eight electrochemical sensing channels, two current/voltage stimulation channels, and an on-chip waveform generator. A digital control unit receives user commands and transmits data from the functional units for off-chip post-processing. (d) Micrograph of the fabricated CMOS-MEA chip and a 3D image of a portion of the electrode array. (e) Three electrode array configuration options, where the gray squares represent the selected electrodes and the white squares represent the non-selected electrodes across different configurations: i. 64 pseudo-large electrodes formed by interconnecting groups of 16 neighboring electrodes, which were particularly useful for experiments in the hanging-drop mode; ii. 64 electrodes uniformly distributed across the array; and iii. a block of 64 electrodes for high-density recordings, the use of which was beneficial primarily in the standing-drop mode. | ||
2. Materials and methods
2.1. System design of the CMOS-MEA chip
Each electrode array comprises 1024 Pt microelectrodes, each with an area of 38 × 42 μm2, arranged at a center-to-center pitch of 50 μm. The electrodes cover a total area of 1.6 × 1.6 mm2. The reference electrode occupies an area of 0.52 mm2 and is positioned at the periphery of the array. The array dimensions were selected according to the droplet size in the microfluidic system (3 mm diameter). Additionally, considerations were made to ensure adequate space for stable bonding of the fluidics and to account for tolerances for slight misalignment during the assembly of the CMOS-MEA chip and the microfluidic system.The electrophysiology channels were designed to amplify low-amplitude extracellular signals from electrogenic cells. The circuitry of these channels was adapted from that published in ref. 30. The complete amplification chain comprises four stages with programmable gain and bandwidth. Following filtering and amplification, the signals were digitized using a 10-bit successive-approximation-register (SAR) analog-to-digital converter (ADC) with serial output. The ADC operated at 1.28 MS s−1, sampling each of the 64 channels at 20 kS s−1 (Fig. S1a†).
Impedance spectroscopy channels were designed to measure impedance magnitude and phase using a lock-in detection method19 within a frequency range of 1 Hz to 1 MHz. We used the circuitry published in ref. 30. A sinusoidal stimulus voltage, generated by the on-chip waveform generator, was applied between the reference electrode and the target working electrodes. Subsequently, the resulting currents were detected through eight low-noise transimpedance amplifiers (TIAs) and were mixed with the on-chip-generated synchronous in-phase (I) and quadrature (Q) square signals. Output signals from the mixers were then lowpass-filtered, multiplexed, and digitized by delta–sigma ADCs working at a sampling frequency of 1.28 MHz (Fig. S1b†). The output bitstreams of the ADCs were decimated by off-chip cascaded integrator-comb (CIC) filters to acquire the in-phase (ZI) and quadrature (ZQ) values with respect to the TIA feedback load. The extraction of impedance magnitude and phase values was then carried out as follows:

A waveform generator was implemented to produce the stepwise approximation of the required stimulus and I/Q mixing signals in the impedance spectroscopy module. The technique and circuitry of the analog components were adapted from ref. 30. To achieve different measurement frequencies, the 25.6 MHz system frequency was modified by using a digital frequency divider. The programmable sampled values of a full period of the desired waveform were stored as 10-bit data in the look-up table. The values in the look-up table were determined by the address indicated by a counter, operating at a frequency defined by the frequency divider. Subsequently, these values were converted to analog signals by a digital-to-analog converter (DAC) and buffered using an on-chip voltage buffer. Furthermore, a digital logic circuit was integrated to generate the I and Q signals of the impedance spectroscopy module, ensuring they were phase-locked to the stimulus sine-wave signal. The overall frequency range of the waveform generator was between 1 Hz and 1 MHz (Fig. S1c†).
The electrochemical sensing channels were designed to measure Faradaic currents resulting from oxidation and reduction reactions at the electrodes. The circuitry was adapted from the design reported in ref. 30 and specifically designed for fast-scan cyclic voltammetry (FSCV). A triangular scan voltage was applied between the sensing electrodes and the reference electrode by changing the potential of the sensing electrodes, while maintaining the reference electrode potential at the common-mode direct current (DC) voltage. In each channel, the scan voltage was buffered by an operational transconductance amplifier (OTA), configured in a unity-gain feedback loop, and the resulting current was detected by a resistively loaded transimpedance amplifier (TIA). The outputs of the eight channels, along with the applied scan voltage, were sampled and multiplexed at a rate of 20 kS s−1. Subsequently, they were digitized by a 10-bit successive-approximation-register (SAR) ADC with serial output (Fig. S1d†). Digitizing the scan voltage along with the current readout was essential to maintain timing information to obtain a precise cyclic voltammogram (CV). Additionally, amperometry can be performed by applying constant voltage and measuring the resulting current.
The digital controller consisted of one receiver and two transmitter blocks. The receiver block captured user commands via a serial peripheral interface (SPI) protocol. These commands were fetched by the receiver block, designed as a state machine, and subsequently transmitted to the system bus. The transmitter blocks continuously received the output data from the recording modules of each array, combined them into data frames, and generated serial output data associated with each array. Sending out the recorded data from each array in a serial format was crucial due to the constraints imposed by the integration of the CMOS-MEA chip and the microfluidic system, which limited the number of contact pads on the chip. The serial output data of each array was streamed out at a clock frequency of 25.6 MHz.
2.2. Fabrication and post processing of the CMOS-MEA chip
The CMOS-MEA chip was fabricated using 0.18 μm CMOS technology (1P6M) by X-FAB (Erfurt, Germany). The die size was 4.0 × 9.0 mm2. The chip included a shifted-electrode layout to ensure stable isolation of the CMOS circuitry from the liquid in contact with the electrodes.37 Various post-processing steps were applied, including the deposition of a multi-passivation layer of SiO2/Si3N4, reactive-ion etching to define the opening of electrodes and bond-wire pads, and platinum (Pt) deposition on the electrodes for biocompatibility and stability. The micrograph of the chip was taken with a scanning-laser 3D surface profiler (VK-X3000, Keyence, Itasca, US).2.3. Electrical characterization of the CMOS-MEA chip
To acquire the transfer function of the electrophysiology channels, a common sinusoidal signal with an amplitude of 2 mVpp was applied to all inputs, and the frequency was swept from 0.1 Hz to 10 kHz. The external load used to verify the operation of the impedance spectroscopy module, was implemented by off-chip resistors and a capacitor. The noise power spectral density of this module was measured by opening the input of the TIA and recording the output signal. To measure the gain of the electrochemical sensing module, a sinusoidal of 100 Hz and 100 nApp current was applied to the input of each channel, and the resulting voltage through the ADC was measured.2.4. Fabrication of the microfluidic system
The microfluidic system was fabricated in polydimethylsiloxane (PDMS) by standard soft lithography using a double-sided molding process. We used a bottom 3D-printed mold (ceramic-like Perform, Protolabs, Feldkirchen, Germany) and a top PDMS mold. The top mold was first plasma-treated (50 W, 25 s, 0.5 mbar O2, Diener Electronic, Ebhausen, Germany) and then silanized by 3.5 μL trichloro (1H,1H,2H,2H-perfluorooctyl)silane (Sigma-Aldrich, Buchs, Switzerland) for 1 h under vacuum to prevent the bonding of PDMS during the molding process. To fabricate the microfluidic system, we poured the liquid PDMS between the aligned top and bottom molds and degassed it under constant vacuum for ∼6 h to remove any bubbles. During this time, the molds were placed at an ∼20–30° angle to facilitate the flow of liquid PDMS between the molds. The PDMS was then cured in an oven at 80 °C for 3 h in a flat position. During the degassing and curing periods, a 1 kg metal weight was placed and fixed on the top mold to uniformly press the molds together and to guarantee uniform thickness of the chip. The microfluidic system was then obtained after detaching from top and bottom molds, respectively.2.5. Fabrication of the glass substrate
An 8-inch borosilicate glass wafer of 0.5 mm thickness was used to fabricate the glass substrate by using standard photolithography processes for Pt deposition and patterning. First, the lift-off resist (LOR3B, Microchem Corp., Newton, USA) was spin-coated for 5 s at 500 rpm and 45 s at 3700 rpm and soft-baked for 3 min at 200 °C to form a layer of 0.3–0.4 μm thickness. Then, the positive photoresist (S1813, Rohm-Haas, Schwalbach, Germany) was spin-coated for 5 s at 500 rpm and 45 s at 3700 rpm and soft-baked for 1 min at 116 °C to form a layer of 1.5 μm thickness. The Pt patterns were transferred into the resist by ultraviolet exposure through a transparency mask. The exposure was done by using a Hg vapor lamp I-line (365![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 nm wavelength) for 4 s (135 mJ cm−2) with hard contact (Mask Aligner Gen3, Karl Suess, München, Germany). The wafer was then developed in MF319 Developer (Rohm-Haas, Schwalbach, Germany) for 60 s. 0.2 μm-thick Pt was deposited with an adhesion layer of 20 nm WTi (10%) at 60 °C (Ionfab 300, Oxford Instruments, Abingdon, UK) through a sputtering process. The lift-off process was performed by using an ultrasonic bath for 4 min in Remover 1165 (Rohm-Haas, Schwalbach, Germany) solution, and the wafer was rinsed with isopropyl alcohol (IPA). In the next step, a 0.5 μm-thick Si3N4 passivation layer was deposited by using a plasma-enhanced chemical vapor deposition process (Plasmalab 80, Oxford Instruments). The passivation layer was re-opened at the sites of the pads for connection to CMOS-MEA chip and the pads for connection to the measurement setup. To reopen these sites, similar to the steps above, the S1813 positive resist was spin-coated for 5 s at 500 rpm and 45 s at 3700 rpm, followed by soft baking for 1 min at 116 °C. Exposure of the mask with opening patterns was then performed using a Hg vapor lamp I-line for 4 s with hard contact, followed by development for 60 s in MF319. The wafer was plasma-cleaned by O2 plasma for 20 min at 360 W. Reactive-ion etching (RIE; Plasmalab 100, Oxford Instruments) was then performed for 12 min. The resist was removed by using an ultrasonic bath for 4 min in Remover 1165, and the wafer was rinsed with IPA. Finally, the glass wafer was diced using a saw with a 200 μm-thick blade. For gold electroplating, a non-soluble Pt anode was employed and NB-Semiplate Au 100 (NB Technologies GmbH, Bremen, Germany) was used as the gold electrolyte. A 200 mA current was applied for 8 min to deposit a 1 μm-thick gold layer.
:4 nm wavelength) for 4 s (135 mJ cm−2) with hard contact (Mask Aligner Gen3, Karl Suess, München, Germany). The wafer was then developed in MF319 Developer (Rohm-Haas, Schwalbach, Germany) for 60 s. 0.2 μm-thick Pt was deposited with an adhesion layer of 20 nm WTi (10%) at 60 °C (Ionfab 300, Oxford Instruments, Abingdon, UK) through a sputtering process. The lift-off process was performed by using an ultrasonic bath for 4 min in Remover 1165 (Rohm-Haas, Schwalbach, Germany) solution, and the wafer was rinsed with isopropyl alcohol (IPA). In the next step, a 0.5 μm-thick Si3N4 passivation layer was deposited by using a plasma-enhanced chemical vapor deposition process (Plasmalab 80, Oxford Instruments). The passivation layer was re-opened at the sites of the pads for connection to CMOS-MEA chip and the pads for connection to the measurement setup. To reopen these sites, similar to the steps above, the S1813 positive resist was spin-coated for 5 s at 500 rpm and 45 s at 3700 rpm, followed by soft baking for 1 min at 116 °C. Exposure of the mask with opening patterns was then performed using a Hg vapor lamp I-line for 4 s with hard contact, followed by development for 60 s in MF319. The wafer was plasma-cleaned by O2 plasma for 20 min at 360 W. Reactive-ion etching (RIE; Plasmalab 100, Oxford Instruments) was then performed for 12 min. The resist was removed by using an ultrasonic bath for 4 min in Remover 1165, and the wafer was rinsed with IPA. Finally, the glass wafer was diced using a saw with a 200 μm-thick blade. For gold electroplating, a non-soluble Pt anode was employed and NB-Semiplate Au 100 (NB Technologies GmbH, Bremen, Germany) was used as the gold electrolyte. A 200 mA current was applied for 8 min to deposit a 1 μm-thick gold layer.
2.6. Materials for device assembly
To attach the CMOS-MEA chip to the glass substrate, a ∼25 μm layer of a two-component epoxy mixture (EPO-TEK H70E, Epoxy Technology Inc, MA, USA) was applied to the substrate. The CMOS-MEA chip was then aligned and placed, and the epoxy was cured for 1 hour at 150 °C. The glass substrate and the PDMS layers were plasma-activated for 25 s at 50 W in 0.5 mbar O2, before bonding. Wire-bonding was performed by using an automated bonding machine (ESEC WB 3100 plus, Zug, Switzerland), with 25 μm-diameter gold wires. A two-component epoxy (EPO-TEK 353ND, Epoxy Technology Inc) was employed to cover the bond wires. The two epoxy components were first mixed, and the mixture was degassed for 45 minutes under vacuum before being applied to cover the bond wires. Subsequently, the epoxy was cured in an oven at 80 °C for 12 hours. A bio-compatible glue (U307 UV-Curable Acrylate, H. B. Fuller Adhesives, Mannheim, Germany) was applied to fix the needles.2.7. Experimental setup
To perform the experiments, the device was plugged in a custom-made holder printed circuit board (PCB), which was designed to accommodate the device and to connect signals and supply lines from the device to a measurement PCB through a 75 mm flat flexible cable (FFC). In the hanging-drop mode, the holder PCB was clamped in a custom-made holder frame, which was placed in an OmniTray single-well plate (Thermo Fisher Scientific, Reinach, Switzerland) on the stage of an inverted microscope (Leica DMI6000B, Wetzlar, Germany). Precision syringe pumps (neMESYS, Cetoni GmbH, Korbussen, Germany) were connected to inlets and outlets of the device to initiate and control the fluidic flow. The pumps can be operated in pulse-free mode from 0.2 μL min−1, with a reported minimum flow rate of ∼1 nL min−1, which ensured a stable and continuous flow during operation. The open-source software YouScope38 was used to control the size of the droplets by means of a fluid-control feedback loop, which relied on continuously focusing on an SU-8 epoxy ring at the bottom of the control droplet and adjusting the flow rates to set and maintain the focus position. In the standing-drop mode, the syringes were controlled manually by the neMESYS User Interface software. To operate the CMOS-MEA chip, the control commands were generated by a custom-designed C# program and sent to the chip through the SPI interface to configure the chip. On the other end, the serial output data, produced by the chip, was acquired by using a data acquisition (DAQ) card (PXIe-6537, National Instruments, Austin, United States) and a custom-designed LabVIEW program. Data analysis was performed in MATLAB (MathWorks, Massachusetts, United States) (Fig. S2†). Hanging-drop experiments were conducted within a CO2-controlled microscope box, set to a temperature of 37 °C and a humidity level of 95%. Standing-drop experiments were carried out in an incubator, maintained at 37 °C with humidity regulated using a water bath.2.8. Preparation of cardiac microtissues
The human induced pluripotent stem cells (hiPSC) line, CW30318CC1 (Fujifilm Cellular Dynamics, Inc., Madison, WI, USA), derived from a healthy donor, was obtained from the California Institute of Regenerative Medicine (CIRM) hPSC Repository. This cell line was differentiated into cardiomyocytes, following the protocol in ref. 39. The spontaneously and synchronously beating cardiomyocytes were dissociated from the culture dish and were seeded into U-shaped-bottom, low-adhesion 96-well plates. The seeded cells were cultured in RPMI 1640 medium supplemented with B27, fetal bovine serum, and Y-27632 for 24 hours. Afterwards, the culture medium was replaced with RPMI 1640 medium, supplemented with B27, and refreshed every 2–3 days. After recovery in a humidified CO2-incubator, the cardiomyocytes started beating as early as two days after seeding. A beating microtissue was then transferred to a human fibronectin-coated array of the CMOS-MEA chip and put in the incubator for a minimum of two days of recovery before conducting the experiments.2.9. Device loading for electrophysiology recordings and impedance spectroscopy
At the start of each experiment, the tubes, inlet, and outlet syringes were pre-filled with 2.5 mL and 0.5 mL of the liquid of interest. Subsequently, the tubes were connected to the device's inlet and outlet, and the lateral droplets were filled with a volume of 10 μL. Pre-filling of the tubing and syringes was necessary to ensure bubble-free operation. The center droplets were then loaded with a volume of 8 μL through regular pipetting, and the microfluidic channels were connected by pulling the liquid between the droplets using a needle. Following the loading of the droplets, hiPSC-derived cardiac microtissues were directly transferred from a 96-well plate to the MEA surface using a 1 mL pipet tip, and the beating of the microtissue was confirmed using a digital microscope (AM5018MZTL, Dino-Lite, Almere, Netherlands). In hanging-drop experiments, a control 200 μm-thick SU-8 ring was placed inside the control drop, and the device was then flipped upside down to obtain the hanging drops.2.10. Fast-scan cyclic voltammetry (FSCV) of epinephrine
An off-chip-generated triangular waveform was scanned between −0.3 and 0.7 V at a rate of 400 V s−1 with a frequency of 10 Hz. Epinephrine solutions were freshly prepared from (±)-epinephrine hydrochloride (99% purity, Sigma-Aldrich, USA) in phosphate-buffered saline (PBS) (pH 7.4, Gibco, Thermo Fisher Scientific, USA). For measurements under static conditions, background measurements using PBS were first performed. Subsequently, epinephrine was diluted in the device's reservoir. After each dilution had been prepared, the solution in the device reservoir was mixed by using a pipet to ensure uniform distribution in the reservoir. Data was recorded after a stabilization time of 2 minutes. Up to sixteen electrodes were connected to form one pseudo-large electrode, varying the electrode surface area from 1596 μm2 to 25536 μm2. Data from 64 electrodes could be simultaneously recorded per condition.
Wash-out experiments of epinephrine were performed using the hanging-drop configuration. Two syringes were connected to the device inlet via a Y-splitter; one syringe was filled with PBS, while the other syringe was filled with 200 μM (±)-epinephrine hydrochloride in PBS. A third precision syringe pump was connected to the device outlet. The individual parts were connected using poly(ether–ether–ketone) (PEEK) tubing with an internal diameter of 0.5 mm. The experiment was carried out over a period of 3 hours, consisting of three stages: (i) during the first 30 minutes, a PBS baseline flow of 10 μL per minute was established by applying a positive/negative flow in the first/third syringe. (ii) Subsequently, the flow in the first syringe was stopped, and the flow in the second syringe was activated to flush the device with epinephrine solution for 90 minutes. (iii) The device was then flushed again with PBS for 80 minutes. Every 20 seconds, data from eight electrodes were recorded. Background subtraction was performed by averaging the data from the initial 15 minutes of the PBS recording and then subtracting this average value from all subsequent recordings.
2.11. Amperometry of hydrogen peroxide
Experiments to correlate the oxidation current with hydrogen peroxide concentration were conducted similarly to cyclic voltammetry experiments, except that a DC voltage of 0.65 V was applied instead of a triangular waveform. Before testing, the devices were plasma treated. Recordings were taken using up to 16 individual electrodes connected to form one pseudo-large electrode. Data were recorded from 64 pseudo-large electrodes per experimental condition.Fresh hydrogen peroxide solutions were prepared by diluting a 30% H2O2 solution for trace analysis (Art. 95321, Sigma Aldrich) in PBS (pH 7.4, Gibco, Thermo Fisher Scientific). Background measurements were taken using only PBS, followed by the preparation of dilutions directly in the device's reservoir. After each dilution was prepared, the solution was mixed by up-and-down pipetting.
Data were recorded for 1 second, and the obtained currents were averaged over the recording time. The background current, obtained from the initial PBS measurements, was subtracted from the value of each electrode for normalization. Linear models were fitted to the data in the range between 25 and 100 μM H2O2 for a single-electrode configuration (1596 μm2), between 1 and 100 μM for a pseudo-large electrode configuration with 2 × 2 (6384 μm2) or 3 × 3 (14364 μm2) connected electrodes, and between 1 and 250 μM H2O2 for a pseudo-large electrode configuration of 4 × 4 electrodes (25536 μm2). The detection limit was calculated by taking into account the standard deviation of the normalized values of all 64 electrodes at an H2O2 concentration of 0.01 μM.
3. Results
The overall system comprises three main components: (i) a CMOS-MEA chip housing two reconfigurable MEAs, (ii) the microfluidic system including two microfluidic networks, and (iii) a glass substrate to host the CMOS-MEA chip and the microfluidic system (Fig. 1b). We now describe the features and characterization results of the different components.3.1. Characterization of the CMOS-MEA chip
As described, the CMOS-MEA chip features two electrode arrays, each comprising 1024 electrodes that can be connected through a switch matrix to 64 electrophysiology, eight impedance spectroscopy, eight electrochemical sensing, and two current/voltage stimulation channels. Additionally, the chip incorporates two on-chip waveform generators and a digital controller unit for communication with the sensing units. The digital control unit receives user commands and transmits data for off-chip post-processing (Fig. 1c and d).The inclusion of two electrode arrays on a single chip enhances throughput and allows for concurrent testing of two experimental conditions using a single device. Compared to integrating two separate MEAs into the microfluidic system to achieve the same throughput, a co-integration of two MEAs on the same CMOS chip reduces the number of bond wires, as supply and reference voltages of the two electrode arrays can be shared, which effectively reduces the number of connections and setup complexity. Moreover, such co-integration serves as proof of concept for the realization of microfluidic networks with several CMOS-MEA chips. Additionally, measurement variations, originating from offset and process variations in CMOS components, are significantly lower for two MEAs on the same die as compared to two separate chips and dies. To ensure system flexibility, the system was designed to facilitate dynamic programming and adaptation of the sensing area and electrode configuration to the nature of the preparation and/or to record from regions of potential interest (Fig. 1e).
The overall power consumption of the CMOS-MEA chip with all modules in operation was measured to be 41 mW (Fig. 2a). The input-referred noise of an electrophysiology channel, integrated over the full band from 0.4 Hz to 10 kHz, was determined to be 4.9 ± 0.2 μVrms, which is in the range of similar state-of-the-art active MEAs.30,40 This value includes the noise contribution from amplifiers, filters, and ADC and excludes the noise generated by the electrodes. The integrated noise distribution indicated low levels across all channels (Fig. 2b). The transfer function of the electrophysiology channels is depicted in Fig. 2c, with a measured low-pass corner frequency of 3.19 ± 0.18 kHz and a high-pass corner frequency of 0.28 ± 0.04 Hz. The high-pass corner frequency can be adjusted to up to 84 Hz, which enables measurements of both action potentials (APs) and local field potentials (LFPs).40 The minimum and maximum measured gains were 29.2 dB and 75.4 dB, respectively, and all tested gain settings were within the expected range. A programmable gain is crucial to cope with the diverse signal levels generated by various electrogenic cells, ranging from small neural action potentials (tens of μVpp) to large cardiac action potentials (up to several tens of mVpp).41
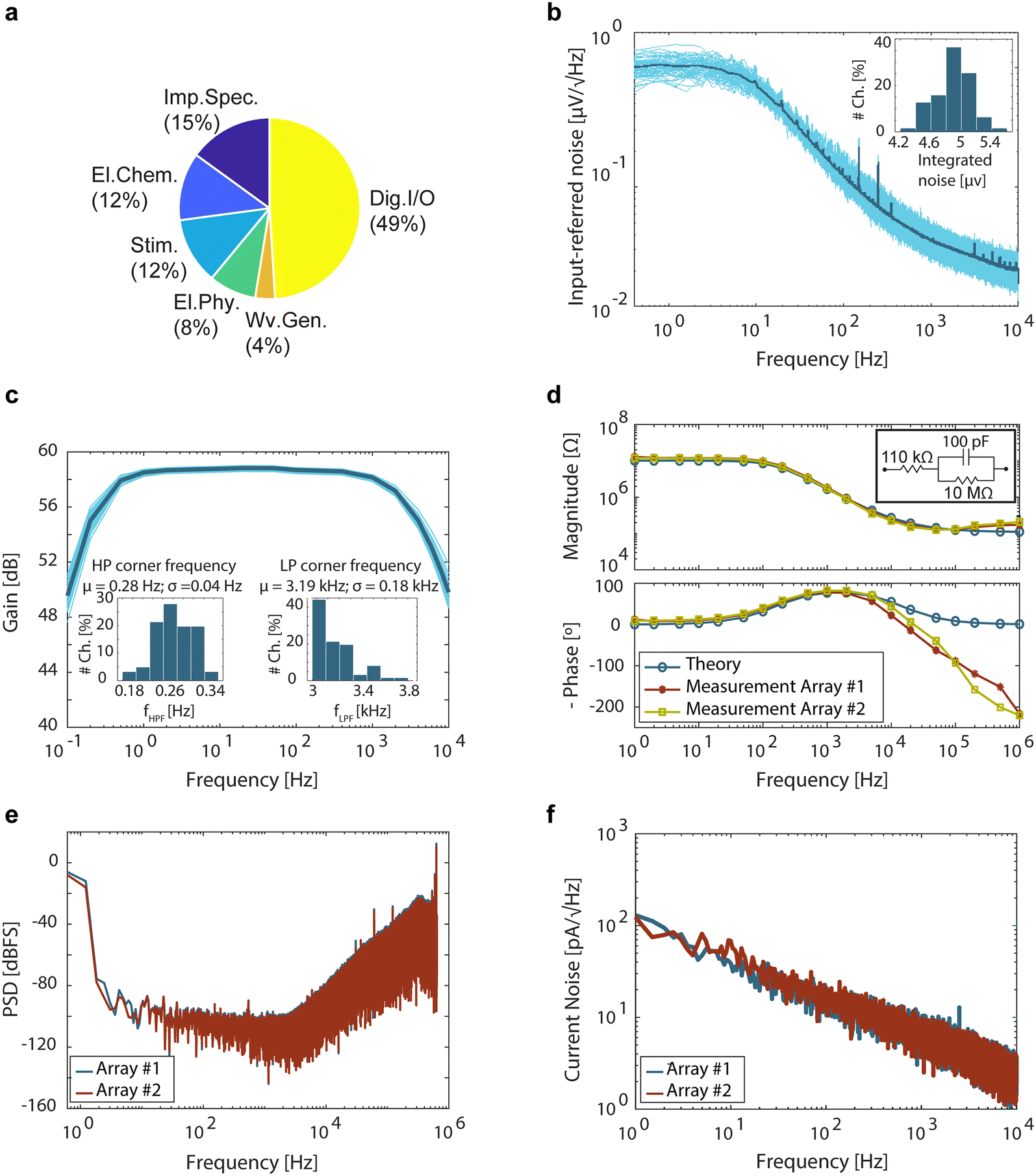 | ||
| Fig. 2 (a) Pie chart depicting the power consumption contributions of the various units of the CMOS-MEA chip, totaling 41 mW. (b) Noise measurement of the electrophysiology channels. The input-referred noise, integrated over the full band from 0.4 Hz to 10 kHz, was determined to be 4.9 ± 0.2 μVrms, measured across 64 channels. The subfigure shows the distribution of integrated noise across these 64 channels. (c) Gain transfer function of the 64 electrophysiology channels with the gain set to 59.3 dB, along with the respective average values. Subfigures illustrate the distribution of high-pass and low-pass corner frequencies across these channels. (d) Impedance spectroscopy of the external load measured by using the impedance spectroscopy module of both arrays. The subfigure displays the external load configured as a simple equivalent-circuit of the electrode-electrolyte interface impedance. (e) Noise power spectral density of the impedance spectroscopy channels measured without mixing and excluding the electrode, multiplexer, and CIC filter. (f) Noise power spectral density for the electrochemical channels. | ||
Impedance spectroscopy of an external load, configured to resemble the equivalent circuit of the electrode-electrolyte interface,42 was conducted across the entire frequency range to validate the impedance module's operation (Fig. 2d). At frequencies below 100 kHz, the recorded impedance aligned with the ideal calculated value. However, at higher frequencies, deviations from ideal values were observed, likely due to the parasitic capacitance and switch-on resistance of the lengthy probe-input leads, as well as the non-ideality of the components, which tends to be more noticeable at high frequencies. The noise power spectrum density of an impedance spectroscopy channel without mixing, excluding the electrode, multiplexer, and off-chip CIC filter, is illustrated in Fig. 2e, where the noise shaping characteristics of the delta sigma converter are clearly visible.
The integrated current noise for an electrochemical sensing channel was measured as 450 pArms and 554 pArms for the two arrays (Fig. 2f), falling within the range reported for state-of-the-art electrochemical sensors.30,43,44
3.2. Microfluidic system
The structure of the microfluidic system comprises patterns on two sides (Fig. S3†). On the top side, it features two parallel microfluidic networks, grooves designed to accommodate a potential lid for minimizing evaporation, and rectangular openings for the bond wires. At the bottom side, the microfluidic system was patterned to accommodate the CMOS-MEA chip. The design of the microfluidic networks was optimized to support the formation of stable hanging-drop networks, with drops of 3 mm diameter. We opted for a 3 mm-diameter aperture to ensure drop stability, simple integration with the CMOS-MEA, and ease of fabrication. Although prior reported studies on hanging-drop networks with integrated microsensors initially made use of 3.5 mm apertures,16,33 later designs shifted toward 3 mm droplets, as reducing the drop diameter provided increased system stability.8,35,45 Beyond stability, the 3 mm diameter offered a practical balance between experimental requirements and fabrication constraints. Compared to smaller diameters, it ensured sufficient area for accommodating the electrode array while reducing fabrication challenges associated with very small microfluidic structures entailing an increased risk of structural failure. Additionally, as the CMOS-MEA chip takes up a fraction of the droplet volume, further reducing the droplet size would have decreased the overall liquid volume, increasing the risk of medium evaporation.Each network comprised four circular compartments that were interconnected by channels with a cross-section of 0.3 × 0.3 mm2. One of the center droplets was designated to host the sample of interest and the CMOS-MEA, while the other was used to control the drop height in the hanging-drop mode, as explained in section 3.6. The two peripheral droplets could either provide extra medium volume for the samples, when the platform was placed in an incubator, or serve as connections to the fluidic inlet and outlet. Rim structures, 0.3 mm wide, surrounded the droplets and ensured their stability. The open microfluidic system facilitated gas exchange and oxygenation, continuous perfusion, and drug dosage. Continuous perfusion also supported tissue viability by providing continuous flow of fresh medium to the cells, prevented electrolyte and chemical up-concentration due to evaporation, and facilitated the removal of waste and secretory products. Additionally, the open microfluidic system allowed for a direct transfer to and harvesting of samples from the droplets. The droplet containing the CMOS-MEA chip featured a maximum volume of ∼8.5 μL, while the others had a maximum volume of ∼13.5 μL. The volume of the microfluidic system, including channels and the four droplets but excluding the inlet and outlet needles, amounted to ∼50 μL. The small volume required precise flow control in dynamic drug-testing applications. The overall system performance, determined by total volume, liquid exchange interface, system stability, and flow rate tolerance, also depended on the used external liquid handling system.
3.3. Glass substrate
The planar glass substrate served as a stable support for the CMOS-MEA chip and the microfluidic system, and featured Pt leads to establish connections between the CMOS-MEA chip and the setup measurement board. We chose glass as the substrate material due to its compatibility with plasma activation and its suitability for plasma bonding with the PDMS microfluidic system. Moreover, its transparency enabled optical access to the sample in the control droplet, a crucial factor for controlling and maintaining droplet size when using the device in the hanging-drop mode, as detailed in sections 3.5 and 3.6.Efficient wire bonding from the CMOS-MEA chip to the substrate necessitates a minimum thickness of 0.5 μm for the metal layer. Therefore, a 1 μm thick gold (Au) layer was deposited on the substrate pads through electroplating. Gold was chosen over Pt because electroplating of Au can be carried out at room temperature.
3.4. Device assembly
We devised an efficient method for assembling the various components of the system, which is suitable for system integration of CMOS sensor units and microfluidic systems. The primary challenge was to effectively protect the circuitry and contacts on the CMOS-MEA chips, preventing any contact with liquid, while keeping the CMOS electrode array surface open and accessible.To assemble the device, we initially affixed the CMOS-MEA chip on the glass substrate. Subsequently, both the microfluidic layer and the glass substrate with the CMOS-MEA chip underwent plasma activation. After careful alignment under an upright microscope, we bonded the microfluidic system to the substrate and the CMOS-MEA chip. In the following step, 1 mm-diameter holes were drilled into the substrate at the centers of the side droplets to establish connections for the fluidic inlet and outlets. Wire bonding was then performed to connect the pads on the CMOS-MEA chip with those on the substrate. To shield the bond wires from physical damage, a two-component biocompatible epoxy was applied. It was crucial to perform wire bonding at the latest possible stage to avoid any physical damage before applying epoxy protection. Subsequently, a PDMS layer with a thickness of 2 mm was aligned and plasma-bonded to the backside of the glass substrate, featuring 1 mm-diameter holes. This PDMS layer was utilized to accommodate inlet and outlet connections. 90 degree-bent metal needles with inner diameters of 0.6 mm and outer diameters of 0.9 mm were then inserted as inlets and outlets, and the needles were fixed by applying a biocompatible glue. The assembly steps of the device are summarized in Fig. 3.
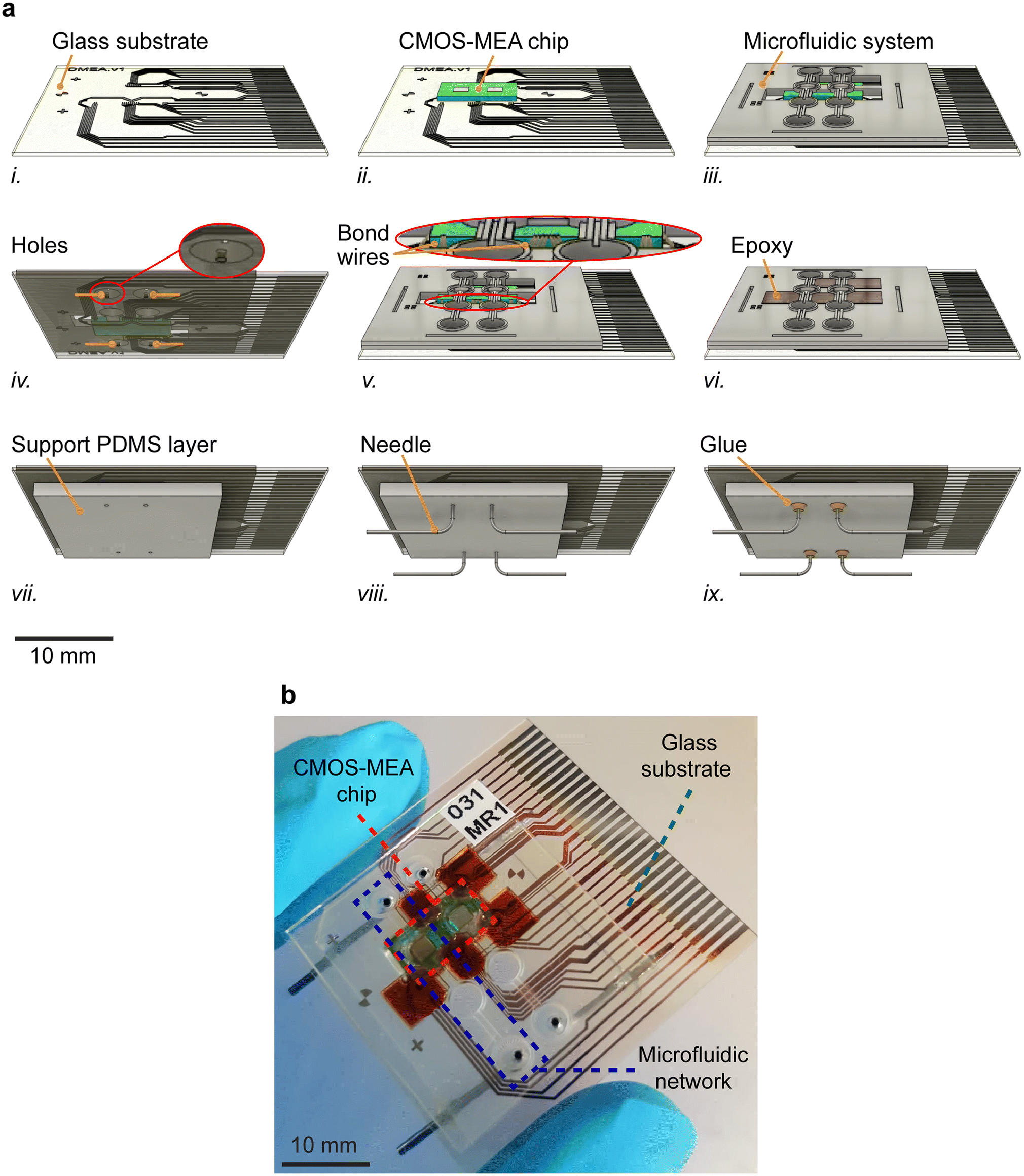 | ||
| Fig. 3 (a) Device assembly: i. glass substrate, ii. CMOS-MEA chip glued onto the glass substrate; iii. PDMS microfluidic system plasma-bonded onto the glass substrate and CMOS-MEA chip; iv. inlet and outlet holes drilled into the glass substrate; v. wire-bonding performed; vi. bond-wires covered with biocompatible epoxy; vii. back-side PDMS layer plasma-bonded to the bottom of the glass substrate; viii. inlet and outlet needles inserted; ix. needles affixed with biocompatible UV-curable glue. (b) Image of the final assembled device. | ||
The successful operation of the assembled system under liquid flow in the hanging-drop configuration (section 2.7) evidenced that the assembly process had properly worked. A tight sealing was particularly crucial, since liquid leakage would have disrupted device operation. Leakage would change the drop height or significantly disturb the operation of the CMOS-MEA chip. Out of 19 assembled devices, 13 were successfully fabricated and operated as intended, resulting in a success rate of approximately 68%. The main causes of failure included issues in the wire bonding process (Fig. 3av) and incomplete plasma bonding of the microfluidic system, particularly around the droplet structures, which led to epoxy partially covering the electrode array (Fig. 3aiii).
3.5. Electrophysiology recordings
We assessed the performance of the electrophysiology module through in vitro measurements. We recorded the electrical activity of hiPSC-derived cardiac microtissues in both hanging- and standing-drop modes (Fig. 4 and S4†). The average peak-to-peak amplitude map of a cardiac microtissue in the hanging-drop mode, featuring droplet height of approximately 700 μm, is depicted in Fig. 4a, which was acquired through electrophysiology recordings using 64 uniformly distributed pseudo-large electrodes consisting of 4 (configured as 2 × 2) electrodes. From this activity map, it is evident that the microtissue was somewhat displaced from the center of the droplet, as the droplet was rather shallow. In comparison to having only a single electrode at the center of the device, the presence of an electrode array ensured the detection of beating activity of the displaced microtissue. One of the high-amplitude signals is illustrated in Fig. 4b, where the different phases of cardiac beating activity, including the depolarization phase corresponding to Na+ influx and the repolarization phase corresponding to K+ efflux,46,47 can be clearly distinguished after band-pass filtering. The measured average beating rate was 44 beats per min. | ||
| Fig. 4 (a) Average peak-to-peak amplitude map of a hiPSC-derived cardiac microtissue in hanging-drop mode. This map was acquired through electrophysiology recordings using 64 uniformly distributed pseudo-large electrodes formed by interconnecting groups of 2 × 2 electrodes. Each pixel represents one pseudo-electrode. For clarity, only the exact position and configuration of the electrodes, not the entire CMOS-MEA chip, are shown in the schematic of the droplet. (b) One of the high-amplitude electrophysiology signals from part (a), showing depolarization and repolarization phases of cardiac beating activity after proper band-pass filtering. (c) Image of a hiPSC-derived cardiac microtissue on an electrode array in standing-drop mode under flow conditions (left), and the average peak-to-peak amplitude map obtained through high-resolution electrophysiology recordings from all 1024 electrodes (right). (d) Beating frequency of the microtissue in part (c) recorded from a 64-electrode block underneath the microtissue at different time points before and after the introduction of epinephrine through the integrated microfluidics. Signals A and B show the electrophysiological signal after band-pass filtering, immediately before the administration of epinephrine and after stabilization of the beating frequency, demonstrating the effect of epinephrine in increasing the beating frequency. | ||
In Fig. 4c, the average peak-to-peak amplitude map of a cardiac microtissue in the standing-drop mode under flow conditions is presented. This map was obtained through high-resolution electrophysiology recordings. The device's high gain and its low-noise characteristics allowed for the recording of the electrical activity of cardiac microtissues even when the microtissues did not adhere to the MEA surface and no adhesive coating was used. To showcase the drug delivery characteristics of the integrated fluidics, we administered 10 μM epinephrine and conducted electrophysiology recordings from a 64-electrode block underneath the microtissue. Recordings were conducted at various time points to observe the stability of the beating both before and after the introduction of epinephrine (Fig. 4d). The beating rate was approximately 21 beats per min before the arrival of epinephrine and increased to 54 beats per min around 4 minutes after dosage, confirming the expected effect of epinephrine in accelerating the beating rate.48–50 The outcome of this proof-of-concept experiment demonstrates the successful application of drugs through the integrated microfluidic system. In comparison to a manual application of the drug using traditional pipetting methods, drug administration through the integrated microfluidics allows for more precise and controllable dosage, and reduced operator-induced variations.
It is important to note that, although the recorded values for both hanging- and standing-drop mode reported in Fig. 4a and c are within the same range, the values cannot be directly compared. The two modes entail different measurement conditions, in addition to variations in the microtissue batch and morphology of sensing electrodes. Under identical conditions – in the standing mode – there is a direct contact between electrodes and the cell preparation, which also provides a sealing of the volume between cells and electrodes towards the droplet liquid phase. These effects would lead to higher signal amplitudes and a more pronounced “footprint” of the preparation, so that subcellular feature resolution can potentially be achieved, e.g., when the tissue is pressed onto or affixed to the electrode array. In contrast – in the hanging-drop configuration – there is no direct contact between tissue and microelectrodes, and a liquid medium film of considerable thickness is always present between the electrodes and the electrogenic-cell preparation. This liquid film and volume is directly connected to the rest of the droplet liquid and, therefore, entails diffusion of ions after being released by the cells, which attenuates and blurs the signals.
3.6. Impedance spectroscopy
The characterization of individual electrodes and pseudo-large electrodes was performed prior to any impedance measurement of the samples of interest (Fig. 5a). To explore the correlation between impedance and microtissue-to-electrode distance in hanging drops, an hiPSC-derived cardiac microtissue (330 μm diameter) was utilized in a hanging drop setup, featuring variations in droplet height (1200 μm, 1000 μm, and 800 μm), corresponding to volumes of ∼5 μL, ∼4.6 μL, and ∼4.3 μL, respectively. Data were recorded using three pseudo-large electrodes (2 × 2 electrodes) at different positions, each featuring an electrode surface area of 6384 μm2. An excitation wave of 200 mV amplitude at 100 kHz was applied. The data were normalized with respect to the average impedance value recorded with the largest droplet height (1200 μm). The resulting data revealed distinct impedance variations among the selected electrodes and an increase in impedance magnitude with decreasing droplet height. The presence of the microtissue within the hanging drop network could be successfully identified. The measured impedance values increased with decreasing drop height and microtissue-to-electrode distance, which is consistent with the fact that the microtissue is less conductive than the surrounding culture solution and blocks the respective current paths. Impedance values measured by the electrode closer to the reference electrode (El1) were less sensitive to variations in drop height, likely due to the existence of more low-impedance current paths between this electrode and the reference electrode (Fig. 5b). | ||
| Fig. 5 (a) Impedance spectroscopy of individual electrodes and pseudo-large electrodes formed by interconnecting 2 × 2 and 4 × 4 neighboring electrodes, averaged over 64 electrodes with error bars representing the standard deviation. (b) Correlation between impedance and microtissue-to-electrode distance in hanging drops containing a hiPSC-derived cardiac microtissue, recorded by using three pseudo-large electrodes (El1, El2, and El3) formed by interconnecting 4 × 4 neighboring electrodes. For better clarity, only the exact position and configuration of the electrodes, not the entire CMOS-MEA chip, are shown in the schematic of the droplet. (c) Impedance imaging of the same cardiac microtissue depicted in Fig. 4c, conducted at four distinct frequencies in standing-drop mode, acquired through high-density impedance measurements from all 1024 electrodes. | ||
Impedance imaging of the cardiac microtissue depicted in Fig. 4c was conducted at four distinct frequencies in the standing-drop mode. The acquired impedance images are presented in Fig. 5c. At 1 kHz, identification of the microtissue was not possible in the impedance image, as the electrode impedance obscured the impedance caused by the microtissue. With increasing frequency and the subsequent decrease in electrode impedance, microtissue-induced changes in impedance became more discernible. At 10 kHz, the presence and position of the microtissue were clearly visible in both magnitude and phase images. A greater portion of the microtissue became visible at 50 kHz, particularly in the impedance phase image, revealing areas with less tight contact to the electrodes in addition to the center of the microtissue, which always was in close contact. At 100 kHz, the impedance magnitude values yielded an image superior to those at 10 kHz and 50 kHz. The proof-of-concept experiments demonstrate the device's capability to monitor the presence and adhesion of tissues through impedance imaging. The technique is label-free, non-invasive, and holds potential for automated long-term measurements in an incubator.
Analyzing the impedance images obtained at 10 kHz, 50 kHz, and 100 kHz, it is evident that the center of the tissue yielded larger values, indicating a close contact between the microtissue and the electrodes, as can be anticipated due to the nearly spherical shape of the microtissue. Furthermore, in comparison to the optical image, the top-right part of the microtissue was not detected in any of the impedance images, indicating a relatively large tissue-to-electrode distance in that specific region. The large distance could also be the reason for the absence of any discernible electrical activity in the electrophysiology recordings of the same region (Fig. 4c): no beating activity was observed, presumably due to the tissue not being close enough to the electrodes. The proof-of-concept impedance measurements demonstrate the potential use of impedance imaging as a complementary technique to electrophysiology recordings. Impedance measurements can be employed to assess the location and adhesion of cells and tissues before conducting electrophysiology recordings.
3.7. Electrochemical sensing
We confirmed the feasibility of cyclic voltammetry using sulfuric acid (Fig. S5†). Thereafter, fast-scan cyclic voltammetry (FSCV) was utilized for qualitative catecholamine detection; increased non-Faradaic background currents at higher scan rates were handled through digital background subtraction. Background signals were recorded in PBS, and epinephrine solutions (1 μM to 200 μM) were measured with background subtraction under static conditions (Fig. 6a). The excitation potential varied from −0.4 V to 0.7 V at a scan rate of 400 V s−1, similar to that published by Yakushenko et al.51 The signals of 64 independent electrodes were sequentially analyzed and combining multiple electrodes to pseudo-large electrodes allowed for dynamic adjustments of the working electrode size from 1596 μm2 to 4788 μm2. The results showed a positive correlation between electrode surface area and measured current. Qualitative analyte detection down to 10 μM was achieved, with distinct voltammogram shapes observed at different concentrations. Furthermore, by using a larger electrode surface area, the voltammograms for different concentrations became more distinct. | ||
| Fig. 6 (a) FSCV of epinephrine solutions of different concentrations, ranging from 1 μM to 200 μM, and different electrode surface areas, ranging from 1596 μm2 to 4788 μm2. The used triangular potential was scanned from −0.3 V to 0.7 V at a scan rate of 400 V s−1. The individual colors represent different working electrodes of the microelectrode array. Data were background-subtracted using a PBS-only control measurement. (b) FSCV of epinephrine under flow in a wash-out experiment. Heat map visualizing the recorded FSCV voltammograms during the wash-out experiment in an experimental time point versus triangular-excitation voltage scan. The corresponding cyclic voltammograms were taken at individual time points. For the first 30 minutes, PBS was flushed through the device; the data recorded during the initial 15 minutes were used to establish the background current, which was then subtracted from the subsequent measurement data points. At time point E, we started with the perfusion of a 200 μM epinephrine solution. After 70 minutes, the dosage was stopped, and the medium was switched back to PBS without epinephrine (timepoint P). See Fig. S6† for a wash-out experiment with 100 μM epinephrine. (c) Background-subtracted oxidation current versus H2O2 concentration for different pseudo-large electrode sizes. The dashed trend lines for each pseudo-large electrode configuration were obtained through linear regression analysis, and the R2 values represent the goodness of the fit for each trend line. Data points represent the average of 64 independent electrodes, and error bars represent the standard deviation of these 64 independent electrodes in a single experiment. A zoomed-in view of the linear range is shown in Fig. S7.† | ||
The use of the MEA/microfluidic system combination enabled the execution of washout experiments directly in the droplets. In this type of experiments, cells were first exposed to a specific analyte, followed by its removal and a subsequent cultivation of the cells in analyte-free medium. This process provides valuable insights, especially when studying analytes that covalently bind to their targets. Through the integration of FSCV, we further enhanced our capability to qualitatively detect the presence of the analytes at high temporal resolution, which is not possible in traditional washout experiments. A baseline was first established by introducing PBS into the system, followed by a transition to the analyte solution (200 μM epinephrine) and subsequently returning to PBS (Fig. 6b). Alterations in peak characteristics were observed in the recorded voltammograms upon analyte presence, which returned to baseline levels in the presence of PBS during the washout. Importantly, a singular loop pattern in the voltammograms was observed when the experiments were conducted under flow conditions, which was in stark contrast to the previously observed “double-crossing” pattern under static conditions. This divergence can be attributed to the presence of a redox intermediate in the epinephrine reaction pathway52 that was effectively flushed away under flow conditions, resulting in a unipolar voltammogram. In contrast, under static conditions, the intermediate was found to accumulate, giving rise to a biphasic voltammogram. These experiments show the performance of the combined MEA/microfluidic system for determination of analyte presence using FSCV under static and flow conditions.
The feasibility of conducting quantitative measurements was subsequently explored through single-potential amperometric detection of hydrogen peroxide (H2O2), which plays an important role in cellular signaling and inflammation events. An external potential of 0.65 V was applied to the reference electrode. Various H2O2 concentrations (0.1 μM to 500 μM) were tested by using up to 16 electrodes grouped into pseudo-large electrodes with different surface areas (1596 μm2 to 25536 μm2). Background correction was performed by subtracting PBS-only measurements. The results displayed a linear relationship between oxidation current and H2O2 concentration (Fig. 6c). Linear regression models were applied to the 64 pseudo-large electrode configurations, revealing that larger surface areas increased the linear range (2.7 μM to 250 μM, R2 = 0.97) and improved the limit of detection (LOD) (3σ/slope, 1.9 μM). Compared to other nonenzymatic H2O2 biosensors, the results obtained with the MEA device demonstrate a detection limit and linear range that is on par with other state-of-the-art devices, as can be seen in Table 1.
| Electrode configuration | Linear range [M] | LOD [M] |
|---|---|---|
| a No indication on electrode surface area. | ||
| This work, 1 electrode (1596 μm2) | 2.5 × 10−5–1.0 × 10−4 | 7.9 × 10−6 |
| This work, 2 × 2 pseudo-large electrode configuration (6384 μm2) | 1.0 × 10−6–1.0 × 10−4 | 2.7 × 10−6 |
| This work, 3 × 3 pseudo-large electrode configuration (14364 μm2) |
2.5 × 10−6–1.0 × 10−4 | 1.9 × 10−6 |
| This work, 4 × 4 pseudo-large electrode configuration (25536 μm2) |
2.7 × 10−6–2.5 × 10−4 | 2.7 × 10−6 |
| Non-enzymatic hydrogen peroxide sensor based on platinum nanoparticle-modified nanoporous gold electrode53 (electrode area: 1.25 × 107 μm2) | 1.0 × 10−7–2.0 × 10−5 | 7.2 × 10−8 |
| Graphene–Pt nanocomposite for non-enzymatic detection of hydrogen peroxide54 (electrode area: 7.07 × 106 μm2) | 2.0 × 10−6–7.1 × 10−5 | 5.0 × 10−7 |
| Non-enzymatic hydrogen peroxide sensor based on a polypyrrole nanowire–copper nanocomposite-modified gold electrode55a | 7.0 × 10−6–4.3 × 10−3 | 2.3 × 10−6 |
| Mesocrystalline Cu2O hollow nanocubes: synthesis and application in non-enzymatic amperometric detection of hydrogen peroxide and glucose56 (electrode area ∼1.25 × 107 μm2) | 2.0 × 10−6–1.0 × 10−4 | 1.0 × 10−6 |
| Effects of microstructure of carbon nanofibers for amperometric detection of hydrogen peroxide57a | 1.8 × 10−4–2.6 × 10−3 | 4.0 × 10−6 |
4. Discussion and conclusion
This paper presents a novel multifunctional system featuring a CMOS-MEA chip integrated into an open microfluidic system, which is intended for biological applications. The system can be operated in two modes: a hanging-drop mode for in situ functional analysis of microtissues and a standing-drop mode that augments conventional MEAs with integrated fluidic tools. To the best of our knowledge, this is the first system that features a CMOS-MEA chip that was specifically designed for integration into a hanging-drop open microfluidic network. Compared to previous microsensor systems for hanging-drop platforms that offer only single functionality,16,58,59 this system includes modules for electrophysiology, impedance spectroscopy, and electrochemical sensing, all within a single setup. This integration enables dynamic analyses of microtissues and cell cultures. A new methodology was developed to co-integrate the CMOS-MEA chip with the open microfluidic networks, which opens a path to future integration of CMOS-based sensor systems into dynamic cell-culture platforms.Our open microfluidic CMOS-MEA system was specifically designed for in situ functional analysis of 3D microtissues, overcoming some of the limitations of conventional microfluidic and MEA platforms. While commercially available MEAs are optimized for planar cell cultures, our system enables real-time, label-free monitoring of the activity of 3D microtissues within a hanging-drop microfluidic network. Additionally, the operability in two modes – the hanging-drop mode for microtissue studies and the standing-drop mode for conventional MEA-like experiments – renders the system a powerful tool for a wide range of applications in biomedical research and bioengineering. The system integration with fluidics allows for continuous perfusion during recordings, so that experimental conditions better mimicking in vivo situations can be achieved. Moreover, the risk of contaminations is reduced, and more efficient drug delivery is possible in comparison to manual pipetting.16 The integration of the CMOS-MEA system enables non-invasive, comprehensive electrical data collection. For example, it allows for continuous monitoring of electrical activity of electrogenic cells through electrophysiology recordings, tracking tissue presence and/or adhesion via impedance sensing, and monitoring the presence and concentration changes of analytes in the medium through electrochemical detection.
The dual-mode feature of our device offers great flexibility. The hanging-drop configuration, one of the most widely used methods for forming and maintaining 3D microtissues, was the primary scenario for designing the device. The use of this mode preserves the physiological structure and integrity of microtissues by preventing direct tissue contact with any surface. Additionally, it allows for using the device from initial stages of microtissue formation, through culturing of formed tissues, to extended periods of drug exposure and wash-out experiments in long-term studies. However, as the microtissue is not in direct contact with the electrodes, the use of the system in the hanging-drop mode requires the usage of larger pseudo-electrodes, which entails lower spatial resolution. Measurement outcomes are influenced by droplet size and the microtissue-to-electrode distance, which adds complexity to the data interpretation. In contrast, the use of the standing-drop mode enables high-spatiotemporal-resolution measurements due to the direct contact between the tissue or cells and electrodes. However, such contact may gradually alter the morphology of 3D tissues and ultimately lead to tissue adhesion, rendering this mode less suitable for long-term culturing. Nonetheless, the standing-drop mode is well-suited for studying cell cultures and tissue slices.
One of the main features of the presented device is the open microfluidic system, which enables oxygenation, continuous perfusion, and precise drug dosing. Continuous perfusion better mimics in vivo conditions by providing a constant flow of fresh medium to the cell culture, helping to avoid up-concentration of waste products in small volumes. The open nature of the system allows for direct transfer of samples (cells, tissues, etc.) to and harvesting of tissues from the droplets. Additionally, different cell and tissue types can be hosted in fluidically connected droplets within a network in a “body-on-a-chip” configuration, enabling the study of multi-organ interactions.15,35
Another key feature of the device is the CMOS-MEA chip. CMOS technology enables the integration of signal conditioning (such as amplification and filtering) and digitization units on the same chip, providing high signal-to-noise ratio, compact system outline, and parallelization potential. CMOS technology also enables the integration of different functionalities in the device. The three main functionalities – electrophysiology, impedance spectroscopy, and electrochemical sensing – render it a versatile tool for experiments targeting multiple cellular parameters. Furthermore, the CMOS-MEA chip features two high-density microelectrode arrays that are reconfigurable and enable dynamic electrode configuration changes during experiments. The inclusion of two electrode arrays enables parallel recordings from two droplets with less efforts during the device assembly procedure and it provides more measurement options through the simultaneous use of two MEAs. The arrays were designed to allow for dynamic programming and adaptation of the sensing electrode configuration during a recording session. Additionally, groups of neighboring electrodes can be interconnected to form pseudo-large electrodes, which have proven useful, particularly in hanging-drop mode experiments. Compared to previous work featuring fixed-size electrodes, the option of creating pseudo-large electrodes with various effective sizes allows for better optimization for different applications and conditions.
Despite all advantageous features of the system, there is still room for improvements. The success rate of the assembly process was slightly lower than that for static CMOS arrays and can be improved. Reducing wire-bonding failures could be achieved by optimizing machine parameters and by adjusting the gold deposition thickness on the bond pads. Improving the alignment of components prior to plasma bonding could reduce the time and improve the success rates for this critical fabrication step, which would avoid issues, as epoxy infiltrating below the fluidic structure and covering the array. Moreover, the start-up of the device and the configuration of parameters in the hanging-drop setup is time-consuming and may be challenging for users, so that further optimization is necessary. There is currently no precise method for measuring the microtissue-to-electrode distance in the droplet; this distance is estimated based on the microtissue's spherical shape, size, and the droplet height, which may complicate the interpretation of results of different independent experiments. Furthermore, while our findings prove the utility of impedance sensing for monitoring microtissue-to-electrode distance and microtissue position, further investigations will be necessary to establish robustness in practical applications. Future studies will focus on optimizing electrode configurations and calibration procedures to enhance sensitivity of the system and reliability of the data. Impedance sensing in the hanging drop mode as a standalone readout method needs refinements. Finally, while small liquid volumes in the device offer advantages, they also bear the risk of evaporation, so that handling of the system under insufficient humidity could lead to rapid dehydration, which would then compromise microtissue viability.
Data availability
The data for this article, including .dat and .mat files, as well as MATLAB scripts for data analysis and figure generation, are available on Zenodo at https://doi.org/10.5281/zenodo.14947935.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation projects 205320_188910/1 and MHV 171267 (R. B.), and the Swiss State Secretariat for Education, Research and Innovation through the SwissChips initiative. The authors thank Peter Rimpf and Jonathan Schmidli at the Cleanroom Facility Basel (CRFB, ETH Zurich) for valuable support. The hiPSC line, CW30318CC1, was obtained from the CIRM hPSC Repository funded by the California Institute of Regenerative Medicine (CIRM).References
- R. C. Mohs and N. H. Greig, Drug discovery and development: Role of basic biological research, Alzheimer's Dementia, 2017, 3(4), 651–657 CrossRef.
- J. A. DiMasi, H. G. Grabowski and R. W. Hansen, Innovation in the pharmaceutical industry: New estimates of R&D costs, J. Health Econ., 2016, 47, 20–33 Search PubMed.
- D. Sun, W. Gao, H. Hu and S. Zhou, Why 90% of clinical drug development fails and how to improve it?, Acta Pharm. Sin. B, 2022, 12(7), 3049–3062 CrossRef PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. K. Stelzer, The third dimension bridges the gap between cell culture and live tissue, Nat. Rev. Mol. Cell Biol., 2007, 8(10), 839–845 Search PubMed.
- B. A. Justice, N. A. Badr and R. A. Felder, 3D cell culture opens new dimensions in cell-based assays, Drug Discovery Today, 2009, 14(1–2), 102–107 CrossRef PubMed.
- H. Baharvand, S. M. Hashemi, S. K. Ashtiani and A. Farrokhi, Differentiation of human embryonic stem cells into hepatocytes in 2D and 3D culture systems in vitro, Int. J. Dev. Biol., 2006, 50(7), 645–652 CrossRef PubMed.
- Y.-C. Tung, A. Y. Hsiao, S. G. Allen, Y. Torisawa, M. Ho and S. Takayama, High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array, Analyst, 2011, 136(3), 473–478 RSC.
- J. A. Boos, P. M. Misun, G. Brunoldi, L. A. Furer, L. Aengenheister and M. Modena, et al., Microfluidic Co-Culture Platform to Recapitulate the Maternal–Placental–Embryonic Axis, Adv. Biol., 2021, 5(8), 1–12 Search PubMed.
- A. Seifinejad, S. Li, C. Mikhail, A. Vassalli, S. Pradervand and Y. Arribat, et al., Molecular codes and in vitro generation of hypocretin and melanin concentrating hormone neurons, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(34), 17061–17070 Search PubMed.
- S. Rismani Yazdi, A. Shadmani, S. C. Bürgel, P. M. Misun, A. Hierlemann and O. Frey, Adding the ‘heart’ to hanging drop networks for microphysiological multi-tissue experiments, Lab Chip, 2015, 15(21), 4138–4147 RSC.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi and R. Polico, et al., 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained, Sci. Rep., 2016, 6, 1–11 Search PubMed.
- C. R. Cox, S. Lynch, C. Goldring and P. Sharma, Current Perspective: 3D Spheroid Models Utilizing Human-Based Cells for Investigating Metabolism-Dependent Drug-Induced Liver Injury, Front. Med. Technol., 2020, 2, 1–22 Search PubMed.
- R. Booth and H. Kim, Characterization of a microfluidic in vitro model of the blood-brain barrier (μBBB), Lab Chip, 2012, 12(10), 1784–1792 CAS.
- C. Blundell, E. R. Tess, A. S. R. Schanzer, C. Coutifaris, E. J. Su and S. Parry, A microphysiological model of the human placental barrier, Lab Chip, 2016, 16(16), 3065–3073 CAS.
- O. Frey, P. M. Misun, D. A. Fluri, J. G. Hengstler and A. Hierlemann, Reconfigurable microfluidic hanging drop network for multi-tissue interaction and analysis, Nat. Commun., 2014, 5, 4250 CAS.
- P. M. Misun, J. Rothe, Y. R. F. Schmid, A. Hierlemann and O. Frey, Multi-analyte biosensor interface for real-time monitoring of 3D microtissue spheroids in hanging-drop networks, Microsyst. Nanoeng., 2016, 2, 16022 CAS.
- S. N. Bhatia and D. E. Ingber, Microfluidic organs-on-chips, Nat. Biotechnol., 2014, 32(8), 760–772 CAS.
- J. Kieninger, A. Weltin, H. Flamm and G. A. Urban, Microsensor systems for cell metabolism-from 2D culture to organ-on-chip, Lab Chip, 2018, 18(9), 1274–1291 CAS.
- R. Bounik, F. Cardes, H. Ulusan, M. M. Modena and A. Hierlemann, Impedance Imaging of Cells and Tissues: Design and Applications, BME Front., 2022, 2022, 1–21 Search PubMed.
- A. Singh, A. Sharma, A. Ahmed, A. K. Sundramoorthy, H. Furukawa and S. Arya, et al., Recent advances in electrochemical biosensors: Applications, challenges, and future scope, Biosens. Bioelectron., 2021, 11(9), 1–31 Search PubMed.
- D. Özsoylu, T. Wagner and M. J. Schöning, Electrochemical Cell-based Biosensors for Biomedical Applications, Curr. Top. Med. Chem., 2022, 22(9), 713–733 CrossRef.
- C. Forro, D. Caron, G. Angotzi, V. Gallo, L. Berdondini and F. Santoro, et al., Electrophysiology Read-Out Tools for Brain-on-Chip Biotechnology, Micromachines, 2021, 12(2), 124 CrossRef PubMed.
- J. P. Piccini, A. M. Russo, P. S. Sharma, J. Kron, W. Tzou and W. Sauer, et al., Advances in Cardiac Electrophysiology, Circ.: Arrhythmia Electrophysiol., 2022, 15(12), e009911 Search PubMed.
- L. Xu, C. Hu, Q. Huang, K. Jin, P. Zhao and D. Wang, et al., Trends and recent development of the microelectrode arrays (MEAs), Biosens. Bioelectron., 2021, 175(88), 112854 CrossRef PubMed.
- V. Viswam, R. Bounik, A. Shadmani, J. Dragas, C. Urwyler and J. A. Boos, et al., Impedance Spectroscopy and Electrophysiological Imaging of Cells With a High-Density CMOS Microelectrode Array System, IEEE Trans. Biomed. Circuits Syst., 2018, 12(6), 1356–1368 Search PubMed.
- C. Laborde, F. Pittino, H. A. Verhoeven, S. G. Lemay, L. Selmi and M. A. Jongsma, et al., Real-time imaging of microparticles and living cells with CMOS nanocapacitor arrays, Nat. Nanotechnol., 2015, 10(9), 791–795 CrossRef PubMed.
- W. Tedjo, J. E. Nejad, R. Feeny, L. Yang, C. S. Henry and S. Tobet, et al., Electrochemical biosensor system using a CMOS microelectrode array provides high spatially and temporally resolved images, Biosens. Bioelectron., 2018, 114, 78–88 Search PubMed.
- A. Konno, K. Ino, Y. Utagawa and H. Shiku, Electrochemical imaging for cell analysis in microphysiological systems, Curr. Opin. Electrochem., 2023, 39, 101270 CrossRef.
- M. K. Zachek, J. Park, P. Takmakov, R. M. Wightman and G. S. McCarty, Microfabricated FSCV-compatible microelectrode array for real-time monitoring of heterogeneous dopamine release, Analyst, 2010, 135(7), 1556–1563 RSC.
- J. Dragas, V. Viswam, A. Shadmani, Y. Chen, R. Bounik and A. Stettler, et al., In Vitro Multi-Functional Microelectrode Array Featuring 59 760 Electrodes, 2048 Electrophysiology Channels, Stimulation, Impedance Measurement, and Neurotransmitter Detection Channels, IEEE J. Solid-State Circuits, 2017, 52(6), 1576–1590 Search PubMed.
- C. M. Lopez, H. S. Chun, S. Wang, L. Berti, J. Putzeys and C. Van Den Bulcke, et al., A multimodal CMOS MEA for high-throughput intracellular action potential measurements and impedance spectroscopy in drug-screening applications, IEEE J. Solid-State Circuits, 2018, 53(11), 3076–3086 Search PubMed.
- T. Chi, J. S. Park, J. C. Butts, T. A. Hookway, A. Su and C. Zhu, et al., A Multi-Modality CMOS Sensor Array for Cell-Based Assay and Drug Screening, IEEE Trans. Biomed. Circuits Syst., 2015, 9(6), 801–814 Search PubMed.
- Y. R. F. Schmid, S. C. Bürgel, P. M. Misun, A. Hierlemann and O. Frey, Electrical Impedance Spectroscopy for Microtissue Spheroid Analysis in Hanging-Drop Networks, ACS Sens., 2016, acssensors.6b00272 Search PubMed.
- N. Rousset, R. L. Sandoval, M. M. Modena, A. Hierlemann and P. M. Misun, Modeling and measuring glucose diffusion and consumption by colorectal cancer spheroids in hanging drops using integrated biosensors, Microsyst. Nanoeng., 2022, 8(1), 14 Search PubMed.
- J. A. Boos, P. M. Misun, A. Michlmayr, A. Hierlemann and O. Frey, Microfluidic Multitissue Platform for Advanced Embryotoxicity Testing In Vitro, Adv. Sci., 2019, 6(13), 1900294 Search PubMed.
- R. Bounik, J. Lee, V. Viswam, F. Cardes, M. M. Modena and A. Hierlemann, A CMOS Microelectrode Array Integrated into an Open, Continuously Perfused Microfluidic System, in 2022 IEEE Biomedical Circuits and Systems Conference (BioCAS), IEEE, 2022, pp. 491–494 Search PubMed.
- F. Heer, W. Franks, A. Blau, S. Taschini, C. Ziegler and A. Hierlemann, et al., CMOS microelectrode array for the monitoring of electrogenic cells, Biosens. Bioelectron., 2004, 20(2), 358–366 Search PubMed.
- M. Lang, F. Rudolf and J. Stelling, Use of youscope to implement systematic microscopy protocols, Curr. Protoc. Mol. Biol., 2012, 1(SUPPL.98), 1–23 Search PubMed.
- M. Zhao, Y. Tang, Y. Zhou and J. Zhang, Deciphering Role of Wnt Signalling in Cardiac Mesoderm and Cardiomyocyte Differentiation from Human iPSCs: Four-dimensional control of Wnt pathway for hiPSC-CMs differentiation, Sci. Rep., 2019, 9(1), 1–15 Search PubMed.
- X. Yuan, A. Hierlemann and U. Frey, Extracellular Recording of Entire Neural Networks Using a Dual-Mode Microelectrode Array with 19 584 Electrodes and High SNR, IEEE J. Solid-State Circuits, 2021, 2466–2475 Search PubMed.
- M. Ballini, J. Muller, P. Livi, Y. Chen, U. Frey and A. Stettler, et al., A 1024-channel CMOS microelectrode array with 26,400 electrodes for recording and stimulation of electrogenic cells in vitro, IEEE J. Solid-State Circuits, 2014, 49(11), 2705–2719 Search PubMed.
- W. Franks, I. Schenker, P. Schmutz and A. Hierlemann, Impedance characterization and modeling of electrodes for biomedical applications, IEEE Trans. Biomed. Eng., 2005, 52(7), 1295–1302 Search PubMed.
- S. Lu and Y. Liao, A 19 μW, 50 kS/s, 0.008-400 V/s Cyclic Voltammetry Readout Interface With a Current Feedback Loop and On-Chip Pattern Generation, IEEE Trans. Biomed. Circuits Syst., 2021, 15(2), 190–198 Search PubMed.
- M. Jamalzadeh, E. Cuniberto and D. Shahrjerdi, A Framework for Benchmarking Emerging FSCV Neurochemical Sensors, Adv. Phys. Res., 2024, 3(2), 2300079 Search PubMed.
- N. Rousset, C. Lohasz, J. A. Boos, P. M. Misun, F. Cardes and A. Hierlemann, Circuit-Based Design of Microfluidic Drop Networks, Micromachines, 2022, 13(7), 1124 Search PubMed.
- L. Sala, D. Ward-Van Oostwaard, L. G. J. Tertoolen, C. L. Mummery and M. Bellin, Electrophysiological analysis of human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs) using multi-electrode arrays (MEAs), J. Visualized Exp., 2017, 2017(123), 1–15 Search PubMed.
- D. Millard, Q. Dang, H. Shi, X. Zhang, C. Strock and U. Kraushaar, et al., Cross-site reliability of human induced pluripotent stem cell-derived cardiomyocyte based safety assays using microelectrode arrays: Results from a blinded cipa pilot study, Toxicol. Sci., 2018, 164(2), 550–562 CAS.
- O. Sirenko, C. Crittenden, N. Callamaras, J. Hesley, Y. W. Chen and C. Funes, et al., Multiparameter in vitro assessment of compound effects on cardiomyocyte physiology using iPSC cells, J. Biomol. Screening, 2013, 18(1), 39–53 CAS.
- X. Fan, X. Gu, R. Zhao, Q. Zheng, L. Li and W. Yang, et al., Cardiac 2-Adrenergic receptor phosphorylation at Ser355/356 Regulates receptor internalization and functional resensitization, PLoS One, 2016, 11(8), 1–15 Search PubMed.
- C. Prajapati, M. Ojala and K. Aalto-Setala, Divergent effects of adrenaline in human induced pluripotent stem cell-derived cardiomyocytes obtained from hypertrophic cardiomyopathy, Dis. Models Mech., 2018, 11(2), dmm032896 CrossRef PubMed.
- A. Yakushenko, V. Schöps, D. Mayer, A. Offenhäusser and B. Wolfrum, On-chip fast scan cyclic voltammetry for selective detection of redox active neurotransmitters, Phys. Status Solidi A, 2014, 211(6), 1364–1371 Search PubMed.
- A. Cristian, A. Dobre, I. Sandu, A. Lungu and C. Mihailciuc, Electrochemical study of epinephrine at platinum electrode, Rev. Roum. Chim., 2010, 55(4), 249–254 Search PubMed.
- G. Yin, L. Xing, X.-J. Ma and J. Wan, Non-enzymatic hydrogen peroxide sensor based on a nanoporous gold electrode modified with platinum nanoparticles, Chem. Pap., 2014, 68(4), 435–441 CAS.
- F. Xu, Y. Sun, Y. Zhang, Y. Shi, Z. Wen and Z. Li, Graphene–Pt nanocomposite for nonenzymatic detection of hydrogen peroxide with enhanced sensitivity, Electrochem. Commun., 2011, 13(10), 1131–1134 Search PubMed.
- T. Zhang, R. Yuan, Y. Chai, W. Li and S. Ling, A Novel Nonenzymatic Hydrogen Peroxide Sensor Based on a Polypyrrole Nanowire-Copper Nanocomposite Modified Gold Electrode, Sensors, 2008, 8(8), 5141–5152 CrossRef CAS PubMed.
- Z. Gao, J. Liu, J. Chang, D. Wu, J. He and K. Wang, et al., Mesocrystalline Cu2O hollow nanocubes: synthesis and application in non-enzymatic amperometric detection of hydrogen peroxide and glucose, CrystEngComm, 2012, 14(20), 6639 RSC.
- Z. Li, X. Cui, J. Zheng, Q. Wang and Y. Lin, Effects of microstructure of carbon nanofibers for amperometric detection of hydrogen peroxide, Anal. Chim. Acta, 2007, 597(2), 238–244 CrossRef PubMed.
- R. Bounik, M. Gusmaroli, P. M. Misun, V. Viswam, A. Hierlemann and M. M. Modena, Integration of Discrete Sensors and Microelectrode Arrays into Open Microfluidic Hanging-Drop Networks, Proc. IEEE Int. Conf. Micro Electro Mech. Syst., 2019, 2019, 441–444 Search PubMed.
- S. C. Bürgel, L. Diener, O. Frey, J.-Y. Kim and A. Hierlemann, Automated, Multiplexed Electrical Impedance Spectroscopy Platform for Continuous Monitoring of Microtissue Spheroids, Anal. Chem., 2016, 88(22), 10876–10883 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc01000k |
| This journal is © The Royal Society of Chemistry 2025 |