
Tough and adhesive conductive hydrogels with fast gelation from a polyphenol–aluminium ion dual self-catalysis system for wearable strain sensors and triboelectric nanogenerators†
Maolin
Yu
,
Yuecong
Luo
,
Qiannian
Yang
,
Tengfei
Duan
,
Zengmin
Tang
,
Lijian
Xu
,
Na
Li
and
Jianxiong
Xu
 *
*
Hunan Key Laboratory of Biomedical Nanomaterials and Devices, College of Life Sciences and Chemistry, Hunan University of Technology, Zhuzhou 412007, P. R. China. E-mail: xujianxiong8411@163.com
First published on 18th September 2024
Abstract
Hydrogels have been widely used as flexible electrodes for the construction of strain sensors and triboelectric nanogenerators (TENGs) with high performance owing to their attractive flexibility and conductivity. However, traditional fabrication methods of hydrogels involve time-consuming synthesis and/or use of external stimuli (i.e., heat and light). Herein, a tough and adhesive conductive double network hydrogel (PVA/PHEAA–TA–Al3+ gel) was prepared via rapid in situ room temperature gelation processes (25 °C, 215 s) in a tannic acid–aluminium ion (TA–Al3+) dual self-catalysis system. This involved the collaborative use of TA–Al3+ to induce the decomposition of ammonium persulfate (APS), which generated abundant free radicals to trigger the polymerization of the HEAA monomer within a polyvinyl alcohol/N-(2-hydroxyethyl)acrylamide/tannic acid (PVA/HEAA/TA) aqueous solution. The obtained hydrogel showed excellent mechanical properties (tensile stress/strain of 240 kPa/920%), adhesion, and self-healing ability. Benefitting from the ultra-wide sensing range (1–600%), high sensing sensitivity (GF = 2.7) and long-term stability (500 cycles), the PVA/PHEAA–TA–Al3+ gel was used to construct a strain sensor, which can accurately identify and distinguish the changes in human expression and joint movement. Furthermore, the PVA/PHEAA–TA–Al3+ gel was used to fabricate TENGs (named PT-TENGs). PT-TENGs with an area of 2 × 2 cm2 exhibited attractive electrical output properties (VOC = 109 V, ISC = 1.3 μA, and QSC = 35 nC at a fixed frequency of 2.0 Hz), which can power 22 LED arrays. This TA–Al3+ dual self-catalysis system is expected to provide a new way for the fabrication of tough and adhesive conductive hydrogels toward health monitoring sensors and energy supply.
Introduction
Flexible and stretchable electronic devices have received a great deal of attention due to their potential applications in human–machine interaction, electronic skin, and energy storage devices.1–5 However, their energy supply problems are also becoming increasingly prominent.6 Triboelectric nanogenerators (TENGs), based on the triboelectric effect and electrostatic coupling effect, have been extensively studied as promising future power supply devices due to their exceptional power generation performance, simple structure, and lightweight design.7–9 Although there are many electrode materials for the construction of TENGs (such as gold, aluminum, and copper), these rigid electrodes often lack stretchability, thus hindering their applications in wearable electronics.10–12Conductive hydrogels combine the characteristics of outstanding electrical conductivity and flexibility, making them ideal materials for flexible TENGs.13–15 In practical application, high electrical conductivity and excellent flexibility are the key factors to realize their functionalization. A qualified flexible electronic device needs to have favorable ductility, adhesion, and sensing sensitivity simultaneously.16 In order to design hydrogels that meet the above-mentioned demands, the synthesis process becomes complex and time-consuming, even requiring external energy (radical polymerization initiated by ultraviolet light or heating, etc.), which greatly hinders the large-scale production and practical application of the hydrogels.17–19 Therefore, it is of great significance to build versatile hydrogels that can be rapidly gelated at room temperature.
Using redox initiators to reduce the energy barrier for initiator decomposition, rather than utilizing external energy to overcome the barrier, is known as a more convenient method.20 Metal ions play a double role in the enhancement of conductivity and the construction of reversible coordination bonds with active groups.21–23 On the one hand, these metal coordination bonds act as sacrificial bonds, effectively dissipating energy to enhance the mechanical properties of the hydrogels.24–28 On the other hand, variable valence metal ions (Fe3+, Cu2+, etc.), as a common oxidant, formed a redox pair with ammonium persulfate (APS) while the reduced variable valence metal ions participated in the single electron transfer reaction as electron donors.29–34 Lu et al. proposed a dual self-catalysis system consisting of variational metal ions (Fe3+, Co3+, Ni3+, and Zr3+) and catechol-containing small molecules (dopamine, tea polyphenol, and tannic acid).35 The introduction of catechols can generate the oxidation products of catechol, semiquinone and quinone, which also formed a catalytic system and promoted the decomposition of APS to produce free radicals. This self-polymerized hydrogel showed low tensile strength (ca. 15 kPa), which may be due to the single network structure with insufficient energy dissipation. To improve this problem, Wei et al. reported a double network (DN) conductive hydrogel (0.3 Fe/TA–CNF–PAA) with relatively high tensile strain (121 kPa) by using tannic acid (TA)-modified cellulose nanofibers and poly(acrylic acid).36 However, most of the hydrogels based on a dual self-catalysis system exhibited unsatisfactory mechanical properties, mainly due to the following two reasons. (i) Incomplete polymerization. Excessive intermediates derived from semiquinone/quinone groups and subvalent valence metal ions consumed a lot of free radicals in the redox-active process. (ii) Weak metal coordination. The metal coordination interaction was weakened because of reduced high valence of variable metal ions in the self-catalytic reaction.
Recently, a few works reported that nonvariable valence metal ions (for example, Ag+, Li+) can trigger free radical polymerization of vinyl monomers without requiring external energy.37–39 Yang et al. used a TA–silver dual catalysis strategy to induce rapid polymerization of poly(acrylamide)@cellulose nanocrystal composite hydrogels, which showed unsatisfactory tensile stress (128.12 kPa).40 Liu et al. fabricated a lignin–nonvariable alkali metal ion self-catalytic system to form poly(AM–DLx–LiCly) hydrogels, where catechols can coordinate with alkali metal ions to form complexes. The poly(AM–DL0.1–LiCl3) hydrogel showed a high tensile stress of 0.4 MPa at a strain of 1125%.41 Compared with these alkali metal ions, the oxidation ability of nonvariable metal ions in the high valence state is relatively stronger, as well as their metal coordination capacity, but it hasn’t been reported yet. Therefore, utilizing a nonvariable valence metal ion-induced self-catalytic strategy to perform highly efficient and rapid polymerization of tough conductive hydrogels in self-catalytic systems is essential and challenging.
In this work, we developed a polyphenol–nonvariable metal ion dual self-catalysis strategy consisting of TA and Al3+, which was used to initiate the polymerization of the monomer (N-(2-hydroxyethyl)acrylamide, HEAA), resulting in the rapid gelation of the poly(vinylalcohol) (PVA), HEAA and TA mixture at room temperature to produce a PVA/PHEAA–TA–Al3+ gel. The resultant hydrogels exhibited outstanding mechanical properties, strong adhesion, and rapid self-healing ability. In addition, the strain sensor constructed with the PVA/PHEAA–TA–Al3+ gel showed excellent sensitivity that can effectively distinguish different joint movements of human body and transmit encrypted messages. Meanwhile, triboelectric nanogenerators based on the hydrogel exhibited excellent electrical output performances, which can be applied as an external power source to power a 22 light-emitting diode (LED) array without the capacitor.
Experimental
Materials
Poly(vinyl alcohol) (PVA, Mn ≈ 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was provided by Acros Organics. N-(2-hydroxyethyl)acrylamide (HEAA) and tannic acid (TA) were purchased from Alfa Aesar. Ecoflex00-10 was obtained from Smooth-on. Sodium tetraborate decahydrate (Na2B4O7·10H2O), aluminium trichloride (AlCl3), methylene bisacrylamide (MBA), and ammonium persulfate (APS) were purchased from Aladdin. Unless otherwise stated, all reagents were used without further purification. Distilled water was purified with a Milli-Q system in all experiments.
000) was provided by Acros Organics. N-(2-hydroxyethyl)acrylamide (HEAA) and tannic acid (TA) were purchased from Alfa Aesar. Ecoflex00-10 was obtained from Smooth-on. Sodium tetraborate decahydrate (Na2B4O7·10H2O), aluminium trichloride (AlCl3), methylene bisacrylamide (MBA), and ammonium persulfate (APS) were purchased from Aladdin. Unless otherwise stated, all reagents were used without further purification. Distilled water was purified with a Milli-Q system in all experiments.
Preparation of the hydrogels
Typically, APS (0.023 g), MBA (0.0015 g), TA (0.04 g), 0.08 wt% borax solution (1 mL), and 10 wt% PVA solution (0.6 g) were mixed with 1.33 mL of water. Then, HEAA (4.0 g) and 0.1 mol L−1 AlCl3 solution (1 mL) were added to the above mixture under stirring to obtain a homogeneous precursor solution. The solution was injected into a 1 mm thickness glass mold and reacted at 25 °C to obtain a targeted hydrogel (PVA/PHEAA–TA–Al3+ gel). The control (PHEAA) was prepared as follows: HEAA (4.0 g) and APS (0.023 g) were added into a deionized water vial sequentially and stirred. Then the mixture was injected into a 1 mm thickness glass mold and was further reacted for 2 h at 60 °C to obtain the sample.Characterization
Fourier transform infrared (FTIR) spectroscopy (Nicolet Is10, USA) was performed to detect the chemical components of the hydrogels. The microstructure of the gels was observed with an emission scanning electron microscope (SEM, JSM-6380LV, Tokyo, Japan). Before observation, the samples were frozen with liquid nitrogen and then freeze-dried in a vacuum freeze dryer. The surface morphology of the hydrogel after gold plating was investigated. Rheology measurements were done on an Anton Paar MCR 92. Electron spin resonance (ESR) spectra were recorded on an ESR spectrometer (Bruker EMX PLUS). Density functional theory (DFT) calculations were carried out using the DMol3 module in Materials Studio.42,43 The exchange–correlation function was treated with generalized gradient approximation with the Perdew–Burke–Ernzerhof (GGA-PBE).44 Considering unpaired single electrons, spin unrestricted calculations were performed. The DFT semi-core pseudopotential method was adopted for core treatment. The double numeric with polarization (DNP) 3.5 method was selected as the basis set for expanding the electronic functions.45 During the electronic step optimization process, the convergence criterion of the self-consistent field (SCF) was set at 10−6 Ha and the thermal orbital occupancy was realized by using smearing (0.005 Ha). The energy, maximum force and maximum displacement of convergence tolerance were set at 10−5 Ha, 0.002 Ha Å−1, 0.005 Å, respectively. An X-ray photoelectron spectrometer (Thermo Scientific K-Alpha, USA) was used. The electrical output performances of the TENG were measured with an electrometer (Keithley-6514), and real-time data acquisition was realized using a software platform, which was constructed based on LabView.Results and discussion
Synthesis and characterization
As shown in Fig. 1a, the rapid gelation process of the PVA/PHEAA–TA–Al3+ gel is as follows. All raw materials including PVA, borax, HEAA, TA, AlCl3, APS, and MBA were mixed in water and stirred uniformly to form a precursor solution, which was further injected into a mold and polymerized at 25 °C. In this process, borax was easily dissociated into tetrahedral borate ions (B(OH)4−), which formed a central complex with cis-diols on the PVA chain as the first network, resulting in cross-linking and tanglement with PVA chains. Simultaneously, the TA–Al3+ catalytic system induced APS to produce sulfate radicals and further formed hydroxyl radicals, which initiated monomer HEAA polymerization to construct the second network, realizing rapid gelation at room temperature. In this system, TA as a natural plant polyphenol contains a large number of phenolic hydroxyl groups. Based on this, TA facilitated the formation of hydrogen bonds with PHEAA or PVA chains. Meanwhile, phenolic hydroxyl groups within TA generate metal coordination with Al3+, which further enhanced the cross-linking density of the hydrogel network. This elaborate design could improve the mechanical properties and provide excellent adhesion to the hydrogel, broadening the application range of self-catalytic hydrogels in the field of wearable electronic devices. | ||
| Fig. 1 (a) The synthesis of the PVA/PHEAA–TA–Al3+ gel. (b) The FTIR spectra of the PVA, PHEAA, and PVA/PHEAA–TA–Al3+ gel. (c) The SEM photograph and (d) corresponding elemental mapping images of the PVA/PHEAA–TA–Al3+ gel. (e) Digital photographs of the PVA/PHEAA–TA–Al3+ gel under the twisted stretching (top) and knotted stretching (bottom) states. | ||
The successful preparation of the PVA/PHEAA–TA–Al3+ gel was proved by comparing the Fourier transform infrared (FTIR) spectra of PVA, PHEAA, and PVA/PHEAA–TA–Al3+ gels (Fig. 1b). The results showed several characteristic peaks of PVA at about 3380 cm−1 (the stretching vibration from –OH) and PHEAA at about 1548 cm−1 (the bending vibration from N–H) and 1059 cm−1 (the stretching vibration from C–OH). Notably, all the characteristic peaks of the PVA/PHEAA–TA–Al3+ gel were compared with individual PVA and PHEAA could be detected and slightly shifted from their original positions, indicating the successful construction of the targeted hydrogel.
Scanning electron microscopy (SEM) images and the corresponding energy dispersive X-ray spectroscopy (EDS) elemental mapping were used to analyze the internal pore size and elemental types of the PVA/PHEAA–TA–Al3+ gel (Fig. 1c and d). The results proved that the hydrogel showed a uniform porous structure and the average pore size was observed to be about 2.5 μm. Meanwhile, multiple elements (C, N, O and Al) were uniformly distributed in the hydrogel. Besides, the PVA/PHEAA–TA–Al3+ gel can withstand various deformations (twisted or knotted stretching), revealing outstanding stretchability due to the intense interactions between TA, PHEAA, PVA network and Al3+ (Fig. 1e).
Rapid gelation process
To validate the rapid gelation process under mild conditions enabled by the synergistic effect of AlCl3 and APS, three kinds of hydrogels were fabricated. The gelation temperature and rheological properties of these hydrogel precursors (without Al3+ but containing TA, Al3+ but without TA, and both Al3+ and TA) were measured using a thermal imager and rheometer. The starting time of gelation is defined as the time node in which the storage modulus of the precursor solution was equal to the loss modulus (G′ = G′′). Fig. 2a and b show that G′ < G′′ under the whole process in the presence of TA but without Al3+, indicating that gelation was not observed in the precursor solution. Interestingly, the precursor solution including Al3+ and the absence of TA showed obvious gelation (328 s, G′ = G′′, Fig. 2c and d). The solution temperature exhibited an increasing trend during the gelation process without external energy input, which suggested that the rapid gelation was triggered by AlCl3. To shorten the gelation time, TA as a polyphenol was introduced to build a dual self-catalysis system. Compared with the absence of TA, the precursor solution containing both Al3+ and TA displayed a shorter gelation time (215 s) (Fig. 2e and f). The above results indicated that the presence of Al3+ is essential for the rapid gelation of the PVA/PHEAA–TA–Al3+ gel at room temperature and the addition of TA can expedite the polymerization process. | ||
| Fig. 2 Digital pictures, rheological tests and thermal imaging photos of the gel (a) and (b) with TA but without Al3+, (c) and (d) without TA but with Al3+, and (e) and (f) with TA and Al3+ for the rapid gelation process. | ||
Density functional theory (DFT) calculations were performed to obtain more insights into the Al3+-induced self-catalysis system. In the presence of Al3+, the bond length of the peroxyl bond (O–O) in (S2O8–Al)+ system was calculated as 1.53 Å, which was longer than that of the Al3+-absence system (S2O8–2NH4, 1.47 Å) (Fig. 3a). This indicates that the O–O bond tends to break with the addition of Al3+. Moreover, the energy barrier of S2O82− decomposing into SO4−˙ with the catalysis of Al3+ (22.46 kJ mol−1) was much lower than it without (190.86 kJ mol−1) (Fig. 3b). The homolysis of S2O82− was endothermic with the energy change value (ΔE) of 120.60 kJ mol−1, while the redox reaction of S2O82− and Al3+ was exothermic with ΔE of −39.10 kJ mol−1. Therefore, the reaction between S2O82− and Al3+ was favorable both kinetically (energy barrier) and thermodynamically. Finally, the reaction energies of the Al3+–catechol complex (C6H4(O2Al)+ and C6H4(OHAl)2+) were calculated as −16.53 eV (quinone state) and −20.18 eV (semiquinone state), implying that the reaction more readily occurred in the TA–Al3+ dual self-catalysis system (Fig. 3c). In conclusion, the spontaneous reaction can be achieved in the presence of an Al3+ system and the addition of TA can accelerate the polymerization.
 | ||
| Fig. 3 (a) The molecular structure and bond length of O–O bonds of the S2O8–2NH4 system and the (S2O8–Al)+ system. (b) DFT calculation of energy changes of S2O82− decomposed into SO4−˙ with/without the catalysis of Al3+. (c) DFT calculation of relative energies of the reduced compounds (C6H4(O2Al)+ and C6H4(OHAl)2+). | ||
To further verify the mechanism of rapid gelation of the PVA/PHEAA–TA–Al3+ gel at room temperature, the hydroxyl radical indicator Coomassie brilliant blue G250 (BB) was used to qualitatively analyse the hydroxyl radicals in the system (Fig. S1a, ESI†). The solution exhibited a blue color and displayed its highest absorption peak at 595 nm (Fig. S1b, ESI†). Upon addition of AlCl3 and APS at 25 °C, the solution turned completely colorless, indicating the formation of hydroxyl radicals. Consequently, the absorption peak of BB at 595 nm disappeared. Notably, the color of the BB solution remained unchanged upon the separate addition of each component, suggesting the absence of hydroxyl radicals. It was observed that the absorption intensity slightly decreased after introducing AlCl3 and APS at 25 °C, possibly due to their influence on pH value of the solution, leading to the color change of the BB solution. Further heating to 60 °C under APS and BB mixture, the sample turned transparent due to the generation of hydroxyl radicals from the thermal homolysis of APS. To exclude the impact of heating on BB solution's color, the BB solution was heated to 60 °C and revealed no change in color. Furthermore, the free radicals produced by the TA–Al3+ dual self-catalysis system were identified through 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-trapped electron spin-resonance (ESR) experiments (Fig. S1c, ESI†). The results displayed a quartet of signals from the DMPO–OH adducts with relative intensities of 1:2:2:1 during the reaction, confirming the presence of hydroxyl radicals in the hydrogel polymerization process. Furthermore, high-resolution XPS spectra show peaks at 284.80 eV, 286.00 eV, and 287.75 eV, which are ascribed to C–C bonds, C–O bonds and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the hydrogel, respectively (Fig. S2, ESI†). Meanwhile, metal Al0 and Al3+ appeared at 74.45 eV and 72.55 eV, indicating the presence of the dual self-catalysis system (TA–Al3+).
O bonds in the hydrogel, respectively (Fig. S2, ESI†). Meanwhile, metal Al0 and Al3+ appeared at 74.45 eV and 72.55 eV, indicating the presence of the dual self-catalysis system (TA–Al3+).
Mechanical properties
The network composition of the hydrogel is the key factor affecting the mechanical properties of the PVA/PHEAA–TA–Al3+ gel. Here, the effects of the content of PVA, the mass of HEAA, and the concentration of AlCl3 on the mechanical properties of the PVA/PHEAA–TA–Al3+ gel were studied systematically. Firstly, the effect of PVA content on the mechanical properties of the PVA/PHEAA–TA–Al3+ gel was studied. As shown in Fig. 4a with the increase of the PVA content from 10 wt% to 15 wt%, the tensile stress and the elastic modulus of the PVA/PHEAA–TA–Al3+ gel increases from 241/67 kPa to 325/92 kPa (Fig. 4b and c). This is due to the rigidity of PVA and the increase of PVA content increases the toughness of the hydrogel. With the increase of PVA content, the PVA/PHEAA–TA–Al3+ gel formed a denser network structure, which endowed the gel with higher ability to disperse stress, thus enhancing the mechanical strength of the gel. | ||
| Fig. 4 (a), (d) and (g) The stress–strain curves of the PVA/PHEAA–TA–Al3+ gel with different PVA concentrations, HEAA mass and AlCl3 concentration. (b), (e) and (h) Corresponding tensile stresses and (c), (f) and (i) elastic modulus values. | ||
Then, the stress–strain curves of the PVA/PHEAA–TA–Al3+ gel prepared under different HEAA masses were shown in Fig. 4d. When the weight of HEAA increased from 2.4 g to 4.4 g, the tensile stress and elastic modulus of the PVA/PHEAA–TA–Al3+ gel increased from 44/17 kPa to 296/97 kPa (Fig. 4e and f). The main reason was the stronger hydrogen bond interaction with the –OH on the PVA chain and the phenolic hydroxyl groups on the TA during the increase of the mass of HEAA, the –CONH– and –OH on the HEAA chain. Finally, the effect of AlCl3 concentration on the mechanical properties of the PVA/PHEAA–TA–Al3+ gel was investigated. Fig. 4g showed that the tensile stress and elastic modulus of the PVA/PHEAA–TA–Al3+ gel increase from 241/67 kPa to 288/75 kPa (Fig. 4h and i) with Al3+ concentration in the range of 0.1–0.7 mol L−1. This is due to the metal coordination between phenolic hydroxyl groups on TA and Al3+, which increased the crosslinking density and rigid modulus of the hydrogel. To sum up, the PVA/PHEAA–TA–Al3+ gel consisting of 10 wt% PVA, 4.0 g HEAA and 0.1 mol L−1 AlCl3 showed the best mechanical properties, which is one of the best among the state-of-the-art self-catalytic hydrogels (Table S1, ESI†). The tensile stress under this ratio is 240 kPa, the tensile strain is 920%, and the hydrogel with this ratio is used for all subsequent tests and applications.
In order to evaluate the fatigue resistance of the PVA/PHEAA–TA–Al3+ gel, cyclic tensile test was performed. As shown in Fig. S3a (ESI†), the PVA/PHEAA–TA–Al3+ gel was tested through 10 continuous loading–unloading cycles under 200% strain. After 10 continuous stretching cycles, the hydrogel recovery rate remained basically unchanged at 55% (Fig. S3b, ESI†). In addition, we also investigated the fatigue resistance of the PVA/PHEAA–TA–Al3+ gel under 80% compressive strain. As shown in Fig. S3c and d (ESI†), the hydrogel recovery rate remained basically unchanged up to 59% after 10 continuous compression cycles. In each cycle, the hydrogel showed an obvious hysteresis loop, indicating that there is a mechanism of energy dissipation. The energy dissipation mechanism was attributed to the coordination bond between the phenolic hydroxyl group of TA and Al3+ as the main sacrificial bond, which can effectively consume mechanical stress and dissipate energy in the process of large deformation. When the double network structure was destroyed, the non-covalent interactions between polymer chains, including metal coordination, hydrogen bonding and hydrophobic interactions, effectively dispersed energy. Because these non-covalent bonds were dynamic and reversible, it can be reformed after removing the external force endowing the hydrogel with excellent flexibility and fatigue resistance.
Self-healing and adhesive performances
The PVA/PHEAA–TA–Al3+ gel exhibited not only self-healing ability (stored at 25 °C for 6 h), but also withstood bending or stretching after self-healing (Fig. S4a and b, ESI†). Then, the PVA/PHEAA–TA–Al3+ gel was connected as a conductor to a closed circuit with a LED lamp (Fig. S4c, ESI†). When the hydrogel is completely cut, the circuit was broken and the LED light went out. After self-healing of the hydrogel, the LED lamp was lit again. In order to quantify the self-healing effect of the PVA/PHEAA–TA–Al3+ gel, the mechanical properties of the healed hydrogel were tested (Fig. S4d, ESI†). The tensile stress of the healed hydrogel was 93 kPa and tensile strain was 206%. According to the formula of self-healing rate, the self-healing rate of the PVA/PHEAA–TA–Al3+ gel at 25 °C was determined to be 38.7%.In the development of practical applications such as triboelectric nanogenerators, the proper fitting of the hydrogel with the friction material is crucial. Poor fitting can lead to separation under significant force, jeopardizing the structural integrity of the nanogenerator. Therefore, the adhesion of the hydrogel is vital for optimizing the electrical output performance of triboelectric nanogenerators. TA contains numerous phenolic hydroxyl groups that exhibit polyphenolic properties similar to mussel proteins, allowing them to mimic the adhesion mechanism observed in mussels. Due to the interaction of phenolic hydroxyl groups introduced by TA, –CONH– on the HEAA chain and a large number of free –OH on the PVA chain, the hydrogel can quickly form non-covalent bonds (such as hydrogen bonds and metal coordination bonds) with other groups on different substrates. As shown in Fig. 5a, the PVA/PHEAA–TA–Al3+ gel possessed excellent adhesion to a variety of surfaces, including steel, plastic, ceramic, glass, and skin. The good adhesion strength can be explained by multiple interactions (hydrogen bond and metal coordination) between the hydrogels and substrate surface (Fig. 5b). The lap shear testing showed that the adhesive strength of the PVA/PHEAA–TA–Al3+ gel to Ecoflex, paper, copper, polyethylene terephthalate (PET), and porcine skin was measured at 28 kPa, 14 kPa, 9 kPa, 8 kPa, and 4 kPa, respectively (Fig. S5, ESI†). Subsequently, the effect of shear rate on the shear strength of the PVA/PHEAA–TA–Al3+ gel on glass was investigated via the lap shear test (Fig. 5c). The shear strength of the PVA/PHEAA–TA–Al3+ gel increased from 47 kPa to 108 kPa when the shear rate ranged from 5 mm min−1 to 100 mm min−1 (Fig. 5d and e). This phenomenon suggested that the low shear rate can give the gel enough time to restore the relaxation state and maximize the energy dissipation caused by mechanical hysteresis.
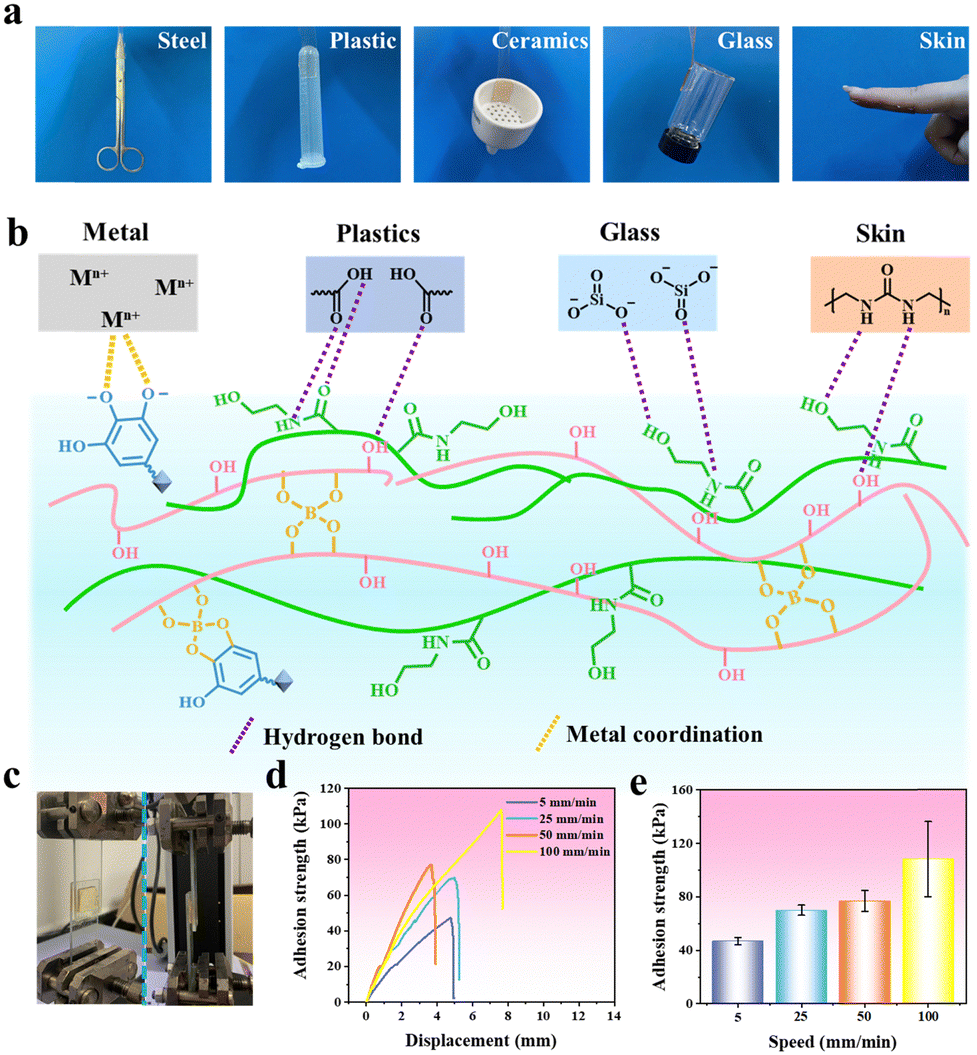 | ||
| Fig. 5 (a) The photographs of the PVA/PHEAA–TA–Al3+ gel adhering to various matrixes including steel, plastic, ceramic, glass and skin. (b) Schematic diagram showing the adhesion mechanism between hydrogels and different substrates. (c) Experimental apparatus for shearing tests of the PVA/PHEAA–TA–Al3+ gel. The adhesive (d) curves and (e) strength of the hydrogels at different shear rates. | ||
Strain sensing properties
Because of the excellent comprehensive performance of the PVA/PHEAA–TA–Al3+ gel, the hydrogel was first used to construct a strain sensor, and its strain sensing performance was tested. As shown in Fig. 6a and b, the resistance change rates of PVA/PHEAA–TA–Al3+ gel-based sensors are measured under small strain (1%, 5%, 10%, 20%, 30%, 40%, and 50%) and large strain (100%, 200%, 300%, 400%, 500%, and 600%). Under small strain (1–50%), the resistance change rate increased from 2% to 52%, indicating that the PVA/PHEAA–TA–Al3+ gel strain sensor can recognize small deformation and can be used to detect muscle or facial expression changes. Under large strain (100–600%), the resistance change rate increases from 120% to 1485%, so the hydrogel sensor can also monitor large deformation. In order to further evaluate the sensitivity of the sensor, the gauge coefficients (GF) in different strain ranges are calculated. As shown in Fig. 6c, the GF was 1.3 in the 200% strain range, while the GF increased to 2.7 in the 200% to 600% strain range. As shown in Fig. 6d, the response times of the PVA/PHEAA–TA–Al3+ gel strain sensor during tension and release processes were accurately measured, and its excellent sensing responsiveness was verified. The response times of the PVA/PHEAA–TA–Al3+ gel-based strain sensor during stretching and release were 0.15 s and 0.30 s, respectively. In addition, the durability and stability of hydrogel-based flexible sensors will directly affect their application prospects in the field of wearable electronics. Therefore, the durability and stability of the PVA/PHEAA–TA–Al3+ gel strain sensor were investigated by measuring the resistance change rate during the tensile cycle. As shown in Fig. S6 (ESI†), the resistance change rate of the hydrogel strain sensor remained stable during 500 tensile cycles from 0% to 50% strain, indicating that the hydrogel sensor can maintain good sensing stability in the long-term tensile cycles. | ||
| Fig. 6 Relative resistance changes of the PVA/PHEAA–TA–Al3+ gel-based strain sensor at different strains in the range of (a) 1–50% and (b) 100–600%. (c) Relative resistance variation–tensile strain curve of the PVA/PHEAA–TA–Al3+ gel. (d) Response time of the hydrogel-based sensor. | ||
Applications
Due to the excellent sensing characteristics of the PVA/PHEAA–TA–Al3+ gel, it was used to monitor the expression changes of human body via building a sensor. The results showed that the strain sensor can accurately identify and distinguish the facial expression changes of smiling, looking up and opening mouth (Fig. 7a–c). Meanwhile, the PVA/PHEAA–TA–Al3+ gel-based strain sensor was attached to the finger joint, and outputs electrical signals of the sensor with different angles according to the bending angle of the finger (Fig. 7d–f) and showed similar results even after self-healing (Fig. 7g–i). Therefore, the sensor based on the PVA/PHEAA–TA–Al3+ gel can be used to monitor joint curvature, which revealed its application potential in the field of joint rehabilitation training. | ||
| Fig. 7 Monitoring of facial expression changes for (a) smiling, (b) lifting and (c) mouthing. Response to 30°, 60°, and 90° bending of the strain sensor attached on the index finger (d)–(f) before and (g)–(i) after self-healing. Various encrypted messages of (j) “SOS”, (k) “HELP”, and (l) “HUT” via Morse code. | ||
To further verify the superior sensing characteristics of the PVA/PHEAA–TA–Al3+ gel strain sensor, it was further used to transmit encrypted information in real time. The strain sensor was attached to the index finger joint, and the output of the electrical signal was controlled by controlling the bending time of the finger. Various distress signals (“SOS” and “HELP”) and word abbreviations (“HUT”) can be transmitted through Morse code, respectively (Fig. 7j–l). This implied that the PVA/PHEAA–TA–Al3+ gel can further expand its application range in information encryption and transmission by adjusting various codes of the electrical signal output.
Triboelectric nanogenerators (PT-TENG) based on the PVA/PHEAA–TA–Al3+ gel is constructed by using Ecoflex 00-10 as the negative friction material, PI as the positive friction material and the PVA/PHEAA–TA–Al3+ gel as the flexible electrode. As a flexible friction nano-generator, PT-TENG can return to its original state after being subjected to various deformations (folding, twisting, twisting stretching and normal stretching) under the action of external force (Fig. 8a). The contact separation of the positive and negative friction materials moved the anions and cations inside the hydrogel, causing electrons inside the copper wire to move under electrostatic interaction, resulting in continuous alternating current in the external circuit (Fig. 8b).
 | ||
| Fig. 8 (a) The digital photos of the PT-TENG in the original, folded, twisted, twisted stretching and normal stretching states. (b) Schematic diagram of the power generation mechanism. (c) VOC, (d) ISC, and (e) QSC of the PT-TENG with an area of 2 × 2 cm2. (f) Open-circuit voltage of the stretchable PT-TENG under different strains. (g) Performance stability of the PT-TENG for 10000 cycles at a frequency of 2.0 Hz. (h) Configuration of the circuit with external loads. (i) Charging behavior of PT-TENG to different commercial capacitors (1–22 μF) at 2.0 Hz. (j) Configuration of the circuit and digital image (the inset) of the LEDs powered by tapping PT-TENG. | ||
The power generation characteristics of the friction nano-generator based on the PVA/PHEAA–TA–Al3+ gel with an area of 2 × 2 cm2 were studied quantitatively for evaluating the output performance of PT-TENG. By optimizing the testing conditions, the PT-TENG device achieved a VOC of 109 V, an ISC of 1.3 μA, and a QSC of 35 nC under 2.0 Hz contact separation motion frequency and 30 N tapping pressure (Fig. S7 (ESI†) and Fig. 8c–e). Then, the output voltage of PT-TENG under different tensile strains was further investigated. VOC showed an increased trend with the increase of tensile strain at different tensile strains in the range of 0–120% (Fig. 8f). After continuous contact for 10000 cycles, PT-TENG can still maintain a stable output voltage during long-term use, indicating excellent durability (Fig. 8g).
To better illustrate the charging capability of the device as a practical sustainable power supply, we first measured the charging speed of PT-TENG on commercial capacitors with different capacities (1 μF, 4.7 μF, 10 μF, and 22 μF) (Fig. 8h and i). With the increase of the capacity of commercial capacitors, the charging speed decreases accordingly. Furthermore, 22 LED arrays are powered directly using PT-TENG (Fig. 8j). The results showed that PT-TENG can continuously illuminate the LED array under the contact separation motion.
Conclusions
In summary, we proposed a polyphenol–aluminium ion (TA–Al3+) induced initiator to generate free radicals. A flexible, adhesive, self-healing DN conductive hydrogel (PVA/PHEAA–TA–Al3+ gel) was synthesized by in situ rapid gelation of the hydrogel at room temperature. The PVA/PHEAA–TA–Al3+ gel showed excellent stretchability and self-healing ability. Then, the hydrogel electrode was used to construct a flexible strain sensor and a triboelectric nanogenerator. The strain sensor assembled with the PVA/PHEAA–TA–Al3+ gel exhibited an ultra-wide sensing range (1–600%), high sensing sensitivity (GF = 2.7) and long-term stability, which accurately identified and distinguished the changes of human expression and joint movement. Additionally, the triboelectric nanogenerator constructed with the PVA/PHEAA–TA–Al3+ gel as the electrode exhibited outstanding electrical output performance (VOC of 109 V, ISC of 1.3 μA, and QSC of 35 nC), which was enough to light 22 LEDs in series. These multifunctional properties broaden the application range of the PVA/PHEAA–TA–Al3+ gel in the field of wearable electronics. This study provides a potential multi-functional material for energy harvesting, health monitoring and electronic skin.Author contributions
Maolin Yu: visualization, methodology, software, funding acquisition, and writing – original draft. Yuecong Luo: visualization and data curation. Qiannian Yang: data curation; Tengfei Duan: software. Zengmin Tang: resources. Lijian Xu: funding acquisition and resources; Na Li: funding acquisition and resources. Jianxiong Xu: funding acquisition, supervision, and writing – review & editing.Data availability
All data for this study are available in this main text or are included in the ESI,† which is also available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52474326, 52477213, and 52374387), the Joint Funds of the National Natural Science Foundation of China (U23A20138), the Hunan Natural Science Foundation for Distinguished Young Scholars (2024JJ2029), the Hunan Provincial Natural Science Foundation of China (2023JJ40264), and the Scientific Research Fund of Hunan Provincial Education Department (23A0430).References
- R. Yin, D. Wang, S. Zhao, Z. Lou and G. Shen, Adv. Funct. Mater., 2021, 31, 2008936 CrossRef CAS.
- Z. Xu, R. Li, H. Li, G. Gao and T. Chen, Soft Matter, 2022, 18, 7103–7111 RSC.
- T. Gao, N. Li, Y. Yang, J. Li, P. Ji, Y. Zhou and J. Xu, J. Energy Chem., 2024, 92, 63–73 CrossRef CAS.
- W.-Y. Guo and M.-G. Ma, J. Mater. Chem. A, 2024, 12, 9371–9399 RSC.
- T. Gao, W. Luo, Y. Yang, Y. Zhou, J. Xu, N. Li, J. Li and Z. Liu, Colloids Surf., A, 2024, 684, 133057 CrossRef CAS.
- Z. L. Wang, J. Chen and L. Lin, Energy Environ. Sci., 2015, 8, 2250–2282 RSC.
- L. Xu, Y. Chen, M. Yu, M. Hou, G. Gong, H. Tan, N. Li and J. Xu, Nano Energy, 2023, 107, 108119 CrossRef CAS.
- Y. Qin, J. Mo, Y. Liu, S. Zhang, J. Wang, Q. Fu, S. Wang and S. Nie, Adv. Funct. Mater., 2022, 32, 2201846 CrossRef CAS.
- M. Hou, M. Yu, W. Liu, H. Zhang, Z. Wang, J. Du, L. Xu, N. Li and J. Xu, Chem. Eng. J., 2024, 483, 149299 CrossRef CAS.
- J.-h Son, K. Cha, S.-H. Chung, D. Heo, S. Kim, M. Choi, I. S. Park, J. Hong and S. Lee, Adv. Sci., 2023, 10, 2301609 CrossRef CAS PubMed.
- Y. Wu, Y. Luo, T. J. Cuthbert, A. V. Shokurov, P. K. Chu, S.-P. Feng and C. Menon, Adv. Sci., 2022, 9, 2106008 CrossRef CAS PubMed.
- Z. Zhao, L. Zhou, S. Li, D. Liu, Y. Li, Y. Gao, Y. Liu, Y. Dai, J. Wang and Z. L. Wang, Nat. Commun., 2021, 12, 4686 CrossRef CAS PubMed.
- B. Ying, R. Zuo, Y. Wan and X. Liu, ACS Appl. Electron. Mater., 2022, 4, 1930–1938 CrossRef CAS.
- Z. Li, C. Li, W. Sun, Y. Bai, Z. Li and Y. Deng, ACS Appl. Mater. Interfaces, 2023, 15, 12787–12796 CrossRef CAS PubMed.
- R. Li, Z. Xu, L. Li, J. Wei, W. Wang, Z. Yan and T. Chen, Chem. Eng. J., 2023, 454, 140261 CrossRef CAS.
- T. Zhu, Y. Ni, G. M. Biesold, Y. Cheng, M. Ge, H. Li, J. Huang, Z. Lin and Y. Lai, Chem. Soc. Rev., 2023, 52, 473–509 RSC.
- Y. Xu, B. Ding, Z. Huang, L. Dai, P. Liu, B. Li, W. Cai, H.-M. Cheng and B. Liu, Light: Sci. Appl., 2023, 12, 1 CrossRef CAS PubMed.
- H. Zhang, H. Shen, J. Lan, H. Wu, L. Wang and J. Zhou, Carbohydr. Polym., 2022, 295, 119848 CrossRef CAS PubMed.
- W. Zhang, P.-L. Wang, L.-Z. Huang, W.-Y. Guo, J. Zhao and M.-G. Ma, Nano Energy, 2023, 117, 108875 CrossRef CAS.
- A. S. Sarac, Prog. Polym. Sci., 1999, 24, 1149–1204 CrossRef CAS.
- Q. Yang, M. Li, R. Chen, D. Gao, Z. Wang, C. Qin, W. Yang, H. Liu and P. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 51774–51784 CrossRef CAS PubMed.
- L. Zhou, Y. Li, J. Xiao, S.-W. Chen, Q. Tu, M.-S. Yuan and J. Wang, Anal. Chem., 2023, 95, 3811–3820 CrossRef CAS PubMed.
- Y. Xin, J. Liang, L. Ren, W. Gao, W. Qiu, Z. Li, B. Qu, A. Peng, Z. Ye, J. Fu, G. Zeng and X. He, Biomacromolecules, 2023, 24, 1287–1298 CrossRef CAS PubMed.
- J. Shen, Y. Dai, F. Xia and X. Zhang, Prog. Polym. Sci., 2022, 135, 101622 CrossRef CAS.
- J. Xu, H. Zhang, Z. Guo, C. Zhang, H. Tan, G. Gong, M. Yu and L. Xu, Int. J. Biol. Macromol., 2023, 230, 123195 CrossRef CAS PubMed.
- M. Gao, R. Zhao, B. Kang, Z. Zhao and S. Song, Colloids Surf., A, 2023, 663, 131051 CrossRef CAS.
- J. Yang, J. Cheng, G. Qi and B. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 17163–17174 CrossRef CAS PubMed.
- H. Zhang, Q. Yang, L. Xu, N. Li, H. Tan, J. Du, M. Yu and J. Xu, Nano Energy, 2024, 125, 109521 CrossRef CAS.
- M. Li, H. Wang, J. Hu, J. Hu, S. Zhang, Z. Yang, Y. Li and Y. Cheng, Chem. Mater., 2019, 31, 7678–7685 CrossRef CAS.
- Q. Wang, X. Pan, C. Lin, X. Ma, S. Cao and Y. Ni, Chem. Eng. J., 2020, 396, 125341 CrossRef CAS.
- D. Sun, N. Li, J. Rao, S. Jia, Z. Su, X. Hao and F. Peng, J. Mater. Chem. A, 2021, 9, 14381–14391 RSC.
- N. Li, D. Sun, Z. Su, X. Hao, M. Li, J. Ren and F. Peng, Carbohydr. Polym., 2021, 269, 118306 CrossRef CAS PubMed.
- Y. Jiang, Y. Chen, W. Feng, X. Zhong, D. Yu and W. Wang, Chem. Eng. J., 2024, 484, 149460 CrossRef CAS.
- D. Sun, Y. Feng, S. Sun, J. Yu, S. Jia, C. Dang, X. Hao, J. Yang, W. Ren, R. Sun, C. Shao and F. Peng, Adv. Funct. Mater., 2022, 32, 2201335 CrossRef CAS.
- M. Li, H. Wang, J. Hu, J. Hu, S. Zhang, Z. Yang, Y. Li and Y. Cheng, Chem. Mater., 2019, 31, 7678–7685 CrossRef CAS.
- Y. Chen, D. Wang, A. Mensaha, Q. Wang, Y. Cai and Q. Wei, J. Colloid Interface Sci., 2022, 606, 1457–1468 CrossRef CAS PubMed.
- S. Hao, C. Shao, L. Meng, C. Cui, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 56509–56521 CrossRef CAS.
- M. Zhang, Q. Yang, T. Hu, L. Tang, Y. Ni, L. Chen, H. Wu, L. Huang and C. Ding, ACS Appl. Mater. Interfaces, 2022, 14, 8728–8742 CrossRef CAS PubMed.
- Y. Yang, Y. Zhu, A. Yang, T. Liu, Y. Fang, W. Wang, Y. Song and Y. Li, Int. J. Biol. Macromol., 2024, 260, 129378 CrossRef CAS PubMed.
- S. Hao, C. Shao, L. Meng, C. Cui, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 56509–56521 CrossRef CAS.
- H. Su, Q. Guo, C. Qiao, X. Ji, L. Gai and L. Liu, Adv. Funct. Mater., 2024, 34, 2316274 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, J. Phys. Chem., 1996, 100, 6107–6110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc02897j |
| This journal is © The Royal Society of Chemistry 2024 |