
Ribosome-targeting antibiotics and resistance via ribosomal RNA methylation
Learnmore
Jeremia
a,
Benjamin E.
Deprez
a,
Debayan
Dey
 b,
Graeme L.
Conn
*bc and
William M.
Wuest
*ac
b,
Graeme L.
Conn
*bc and
William M.
Wuest
*ac
aDepartment of Chemistry, Emory University, 1515 Dickey Dr., Atlanta, GA 30322, USA. E-mail: wwuest@emory.edu
bDepartment of Biochemistry, Emory University School of Medicine, 1510 Clifton Rd., Atlanta, GA 30322, USA. E-mail: gconn@emory.edu
cEmory Antibiotic Resistance Center, Emory University School of Medicine, 1510 Clifton Rd., Atlanta, GA 30322, USA
First published on 1st March 2023
Abstract
The rise of multidrug-resistant bacterial infections is a cause of global concern. There is an urgent need to both revitalize antibacterial agents that are ineffective due to resistance while concurrently developing new antibiotics with novel targets and mechanisms of action. Pathogen associated resistance-conferring ribosomal RNA (rRNA) methyltransferases are a growing threat that, as a group, collectively render a total of seven clinically-relevant ribosome-targeting antibiotic classes ineffective. Increasing frequency of identification and their growing prevalence relative to other resistance mechanisms suggests that these resistance determinants are rapidly spreading among human pathogens and could contribute significantly to the increased likelihood of a post-antibiotic era. Herein, with a view toward stimulating future studies to counter the effects of these rRNA methyltransferases, we summarize their prevalence, the fitness cost(s) to bacteria of their acquisition and expression, and current efforts toward targeting clinically relevant enzymes of this class.
Introduction
Treatment of infections caused by pathogenic bacteria has been complicated by the emergence of multi-drug resistant (MDR) “superbugs”. Overuse and abuse of antibiotics as cure-all therapeutics in healthcare and their widespread use as growth-promoting supplements in animal husbandry have fueled resistance through evolutionary pressure and resuscitation of ancient resistance-conferring genes from long before the human antibiotic era.1–4 The rapid development of resistance against commonly used antibiotics has left few treatment options unaffected, necessitating the use of drugs of last resort which may have serious side effects, such as polymyxins,5 or alternative strategies such as phage therapy.6Antimicrobial resistance (AMR) is estimated to cause at least 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year globally and is projected to be the leading cause of death by the year 2050.7 With healthcare resources recently diverted to curbing the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, efforts to mitigate AMR through surveillance and stewardship policies have been severely diminished,8,9 with potentially long-term consequences. Cases of bacterial coinfection with the coronavirus disease also led to the extensive use of antibiotics during the pandemic, likely exacerbating the development of bacterial resistance to these drugs.10 From 2019 to 2020, the Centers for Disease Control (CDC) reports a 15% increase in hospitalizations as a result of drug-resistant infections.9 Reports estimate that the United States currently spends over $4.6 billion annually on combating vancomycin-resistant Enterococcus (VRE), carbapenem-resistant Acinetobacter, carbapenem-resistant Enterobacterales (CRE), extended-spectrum β-lactamase (ESBL)-producing Enterobacterales, MDR Pseudomonas aeruginosa, and methicillin-resistant Staphylococcus aureus (MRSA), six infections that have been identified as the most alarming resistance threats.9,11 Despite declining investment in antibacterial discovery and development by pharmaceutical companies,12,13 these factors have served as motivation for continued antibiotic research and innovation.
000 deaths per year globally and is projected to be the leading cause of death by the year 2050.7 With healthcare resources recently diverted to curbing the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, efforts to mitigate AMR through surveillance and stewardship policies have been severely diminished,8,9 with potentially long-term consequences. Cases of bacterial coinfection with the coronavirus disease also led to the extensive use of antibiotics during the pandemic, likely exacerbating the development of bacterial resistance to these drugs.10 From 2019 to 2020, the Centers for Disease Control (CDC) reports a 15% increase in hospitalizations as a result of drug-resistant infections.9 Reports estimate that the United States currently spends over $4.6 billion annually on combating vancomycin-resistant Enterococcus (VRE), carbapenem-resistant Acinetobacter, carbapenem-resistant Enterobacterales (CRE), extended-spectrum β-lactamase (ESBL)-producing Enterobacterales, MDR Pseudomonas aeruginosa, and methicillin-resistant Staphylococcus aureus (MRSA), six infections that have been identified as the most alarming resistance threats.9,11 Despite declining investment in antibacterial discovery and development by pharmaceutical companies,12,13 these factors have served as motivation for continued antibiotic research and innovation.
Ongoing areas of study aim to minimize the effects of AMR by identifying novel therapeutics and improving the activity of existing antibiotics either through the use of drug combinations, drug functionality modifications, or drug adjuvant co-treatments. For example, the odilorhabdins are a recently discovered class of antibacterial secondary metabolites produced by a non-ribosomal peptide synthetase gene cluster of Xenorhabdus nematophila, a nematode symbiont.14 These peptide natural products were demonstrated to target a unique site on the ribosome small subunit and to interfere with protein synthesis. Together with the natural products darobactin and dynobactin, produced by Photorhabdus, the discovery of these antibiotics has revealed bacterial symbionts of invertebrates to be reservoirs of potent antibacterial metabolites.15–17 Additionally, improved culturing approaches like the use of isolation chip (iChip) technology, which led to the discovery of teixobactin, allow for the culturing of previously unculturable bacterial antibiotic producers (Fig. 1).18,19 Application of the synergistic effects of oxacillin and an erythromycin derivative (SIPI-8294) against MRSA, and a tetracycline/nalidixic acid combination against hospital-acquired MDR strains of Acinetobacter baumannii and Escherichia coli demonstrates that drug combinations are an expanding avenue of treatment.20,21 In addition to the common structure–activity relationship studies (SAR) that result in improved antibacterials,22,23 a set of physicochemical qualities that dictate drug accumulation in Gram-negative bacteria have recently been established (eNTRY rules; Fig. 1) and, as an example, have since been applied to the transformation of a Gram-positive FabI inhibitor, Debio-1452, into an extended-spectrum antibiotic.24,25 Finally, antibiotic potentiators like β-lactamase inhibitors, drug permeability enhancers, and efflux pump inhibitors are presently being used in clinical settings while others are advancing in the drug approval pipeline.26 The revitalization of widely prescribed broad-spectrum antibiotic regimens, mostly β-lactams,27 has been a useful strategy in countering the effects of resistance.
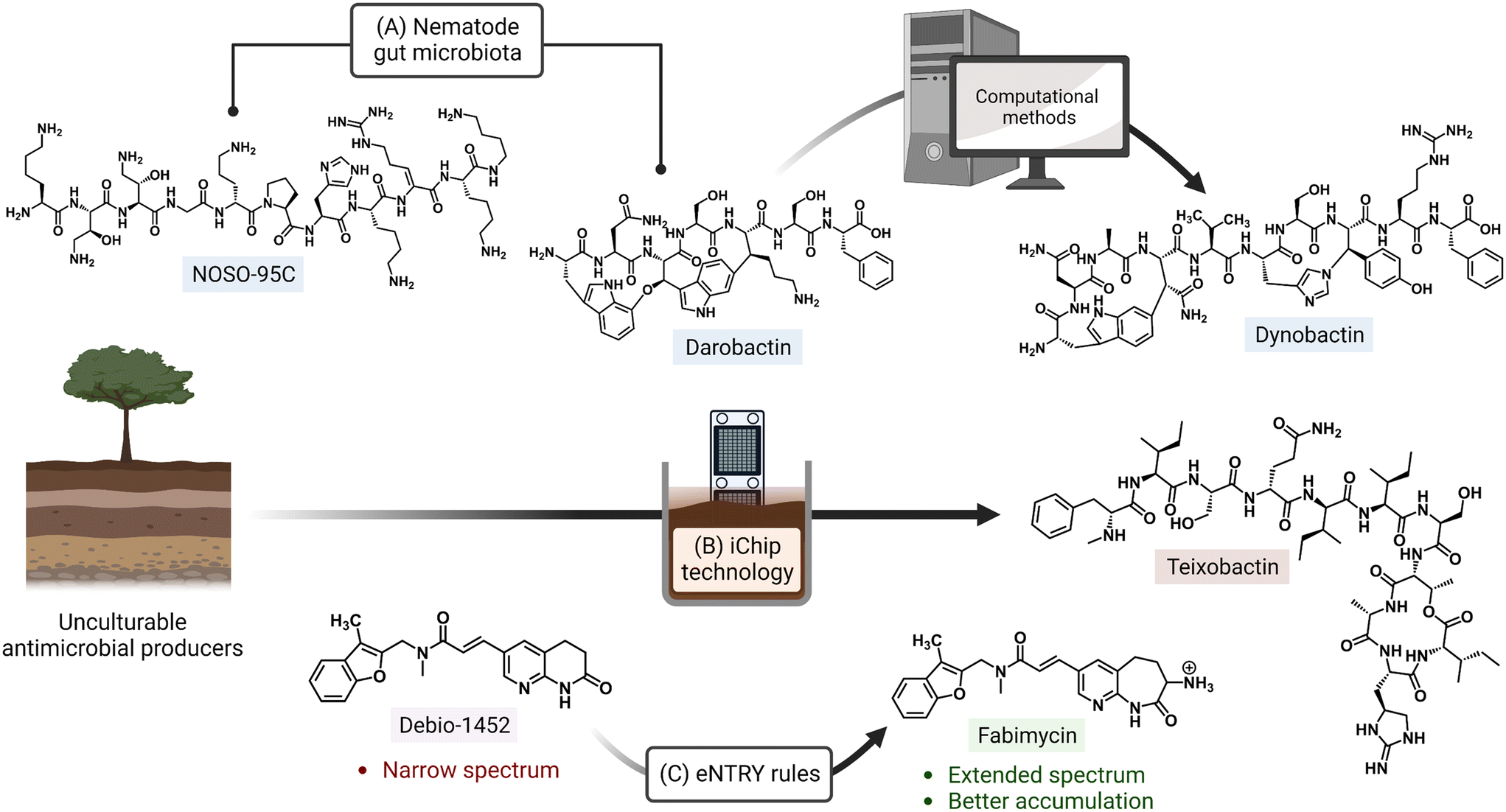 | ||
| Fig. 1 Approaches for discovery of antibiotics with novel mechanisms of action and improving activity of existing drugs. (A) The odilorhabdin (NOSO-95C) and darobactin were both isolated from nematode symbionts. Search for biosynthetic gene clusters similar to that of darobactin led to another inhibitor, dynobactin A. (B) Non-traditional culturing methods (e.g. iChip) have also enabled the culturing of bacterial species with currently untapped natural product repositories. (C) Implementation of physicochemical guidelines (eNTRY rules) has expanded the spectrum of originally Gram-positive-only antibiotics. Created with https://BioRender.com. | ||
Ribosome-targeting antibiotics
Ribosome-targeting antibiotics comprise a diverse group molecules, including many widely prescribed broad-spectrum antibiotics. The ribosome serves a critical function in all cells by translating messenger RNA (mRNA) into proteins necessary for life. To accurately decode mRNA, two conserved A-site residues A1492 and A1493 (note: E. coli rRNA numbering is used throughout) in the 30S (small) subunit flip out from helix 44 (h44) to probe the minor groove of a newly formed mRNA codon-transfer RNA (tRNA) anticodon duplex.28,29 While wobble base pairing is permitted for the base pair formed by the third codon nucleotide, non-Watson–Crick pairing for the first two positions is unfavorable and results in the rejection of non-cognate tRNA.30 Early studies using model A-site rRNA fragments and, later, intact 30S subunits revealed that deoxystreptamine (DOS) aminoglycosides directly promote A1492 and A1493 base flipping to stabilize the mRNA-tRNA interaction, even when non-cognate, and thus induce translational inaccuracy.29,31–34 More recently, single particle high-resolution cryo-electron microscopy (cryo-EM) structures of amikacin and paromomycin bound to the A. baumannii and E. coli 70S ribosomes, respectively, confirmed that both 4,6- and 4,5-disubstituted 2-DOS (4,6-DOS and 4,5-DOS, respectively) aminoglycosides interact directly with A1493 and A1492, in addition to other residues within their h44 A-site pocket, in this biologically most relevant context (Fig. 2).35,36 Cocrystal structures of other aminoglycosides, including gentamicins and plazomicin (4,6-DOS) and neomycins (4,5-DOS), with the Thermus thermophilus 70S ribosome reveal high conservation of the observed networks of drug-rRNA interactions.37,38 Apramycin, a monosubstituted 4,6-DOS with a bicyclic core, also has ribosome binding modes similar to disubstituted 4,6-DOS aminoglycosides for the deoxystreptamine ring.39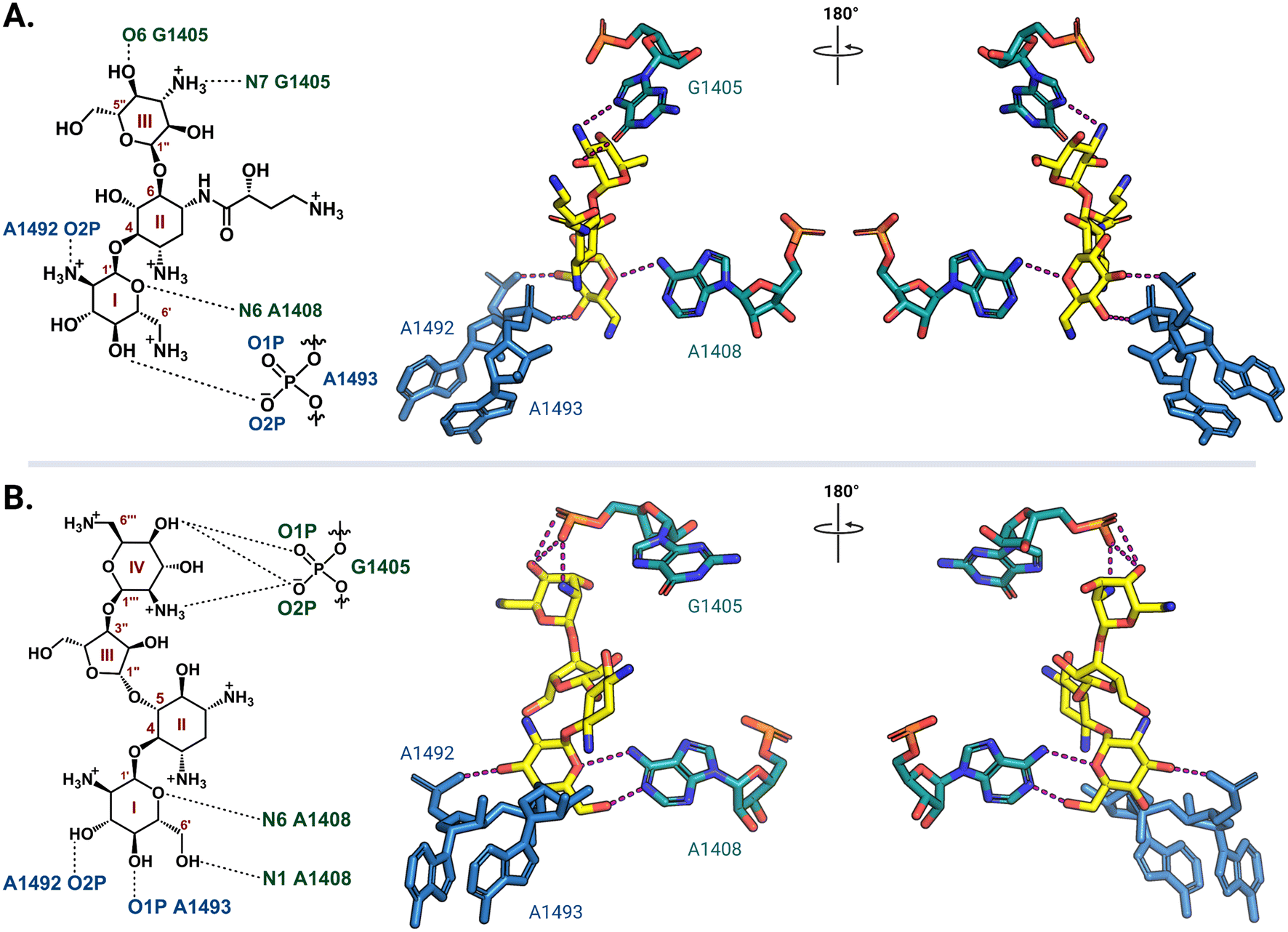 | ||
| Fig. 2 Aminoglycoside interaction networks with h44 residues in the decoding center. (A) The 4,6-DOS aminoglycoside amikacin (PDB: 6YPU) and (B) 4,5-DOS aminoglycoside paromomycin (PDB: 7K00) bind in the A site on the 30S ribosomal subunit, and they both interact directly with conserved decoding nucleotides (A1492 and A1493) and the targets of the aminoglycoside-resistance 16S rRNA methyltransferases (G1405 and A1408). Interaction with G1405 is different for the two DOS aminoglycoside groups, whereas A1408 is close to the neamine core for both. For clarity, only the noted residues of interest and their direct interactions (dashed lines) with the aminoglycosides (yellow) are shown. All structure images were prepared using PyMol. | ||
On the 50S (large) ribosomal subunit, macrolide antibiotics target a distinct functional site to impede protein synthesis by reversibly binding and occluding the nascent peptide exit tunnel (NPET). Macrolides thus block the extension of the growing peptide chain and cause premature peptidyl-tRNA drop-off.40 Drug binding along the NPET involves interactions with both 23S rRNA nucleotides and ribosomal proteins that line the interior of the tunnel's narrowest section. The cryo-EM structure of the macrolide erythromycin bound to the T. thermophilus ribosome reveals a water-mediated intermolecular bonding network crucial for blocking the channel and establishes the C5 desosamine sugar as an essential moiety for macrolide binding (Fig. 3A).41 These ribosome-macrolide interactions are consistent with other structures for second- (azithromycin),42 third- (telithromycin),41 and fourth-generation (solithromycin)43 macrolides. Additionally, the binding sites for macrolides with an extended C5 saccharide chain, such as tylosin (Fig. 3B), and the lincosamides extend from the NPET towards the peptidyl transferase center (PTC) positioning these antibiotics near highly-conserved PTC nucleotides.44,45 Several other classes of structurally unrelated antibiotics also share a common binding site at the PTC, including the phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A, collectively labeled PhLOPSA (Fig. 3C and D). At this binding site, these antimicrobials are positioned to sterically disturb the orbital positioning of the nucleophile-electrophile pair involved in the peptide elongation reaction.46,47 Alignment of X-ray crystal structures of ribosome-bound clindamycin,48 chloramphenicol,48 linezolid,49 tiamulin,50 and dalfopristin51 (Fig. 3E) illustrates the overlapping binding site of these molecules in the ribosome.
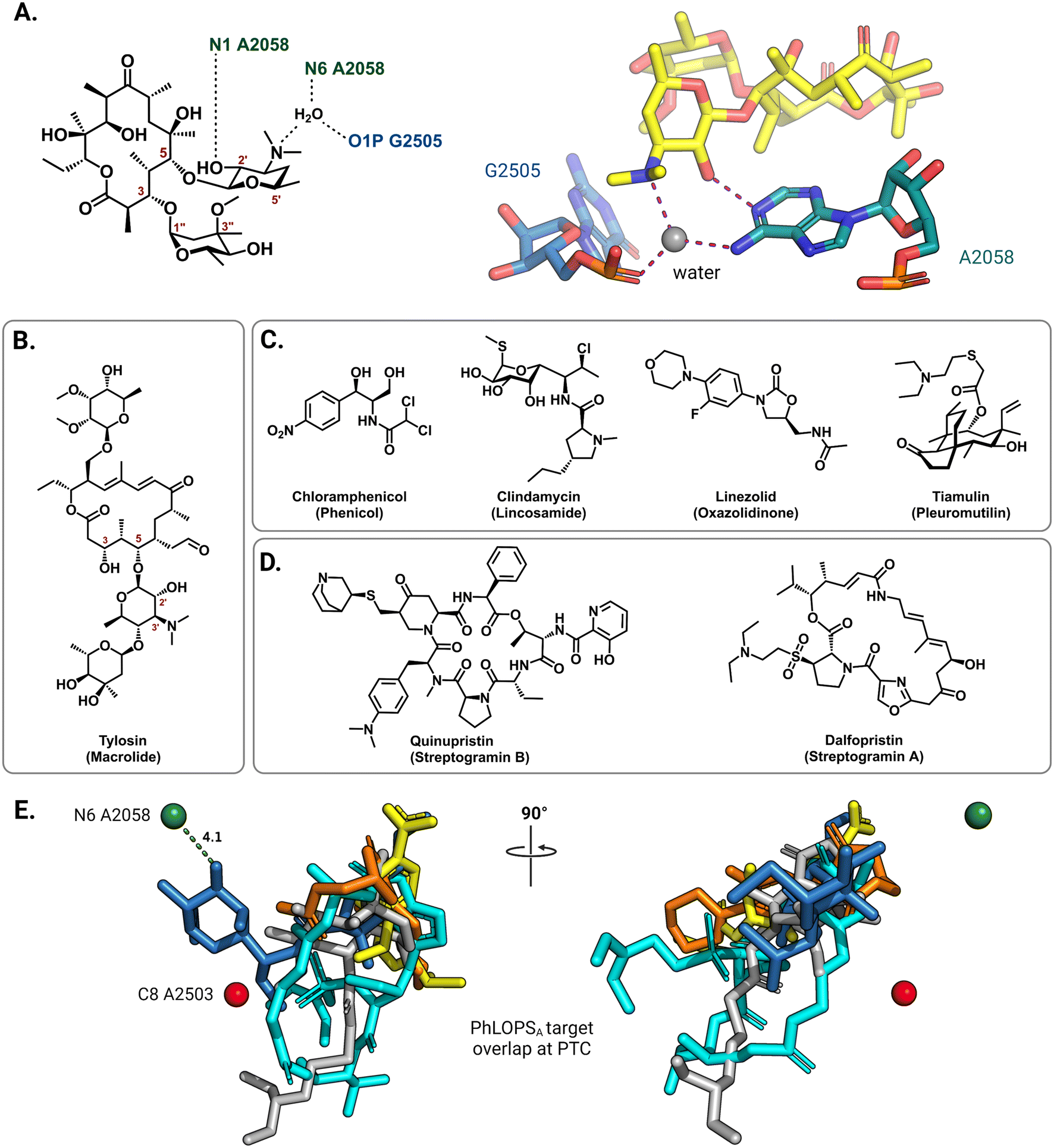 | ||
| Fig. 3 Interactions of antibiotics with the 50S ribosome. (A) The macrolide desosamine ring directly interacts with A2058 (N1) and forms water-bridged interactions with the indicated NPET residues (PDB: 6XHX). A2058 (N6) is the target site of macrolide-resistance 23S rRNA methyltransferases. For clarity, only residues of interest are shown; erythromycin is shown as yellow sticks. (B) Chemical structure of the 16-member macrolide tylosin. (C) Chemical structures of other PTC targeting antibiotics noted in the main text. (D) Chemical structure of streptogramin antibiotics. (E) Superposition of PTC binding antibiotics clindamycin (blue, PDB: 1JZX), chloramphenicol (yellow, PDB: 1K01), tiamulin (grey, PDB: 1XBP), linezolid (orange, PDB: 3CPW) and dalfopristin (light blue, PDB: 1SM1) from the respective ribosome-antibiotic cocrystal structures. | ||
Several oligomeric antibiotics that target the 50S subunit are widely used in veterinary medicine and have been considered for human use in the treatment of MDR pathogens. Orthosomycins are a class of oligosaccharide antibiotics that act by blocking the action of specific aminoacyl-tRNAs at the A site of a translating ribosome.52 CryoEM structures of the orthosomycins, evernimicin and avilamycin, each bound to the E. coli ribosome show that they have a unique binding site and validate the lack of cross-resistance with other ribosome-targeting antibiotics.53,54 Thiostrepton is a cyclic oligopeptide antibiotic belonging to the thiopeptide class of ribosome-targeting antibiotics that bind at the “GTPase center”. Binding of these drugs prevents GTP hydrolysis and thus essential action in translocation of the translation factors elongation factor G (EF-G) and elongation factor 4 (EF4; also known as LepA).55
In response to selective pressure, bacteria can acquire or develop resistance toward ribosome-targeting antibiotics through one or more of several mechanisms (Fig. 4). These mechanisms of resistance include limiting drug uptake, increasing drug efflux, chemically modifying the drug scaffolds, degrading the drug, the action of ribosome protection proteins, or altering the drug target by mutation or chemical modification.56
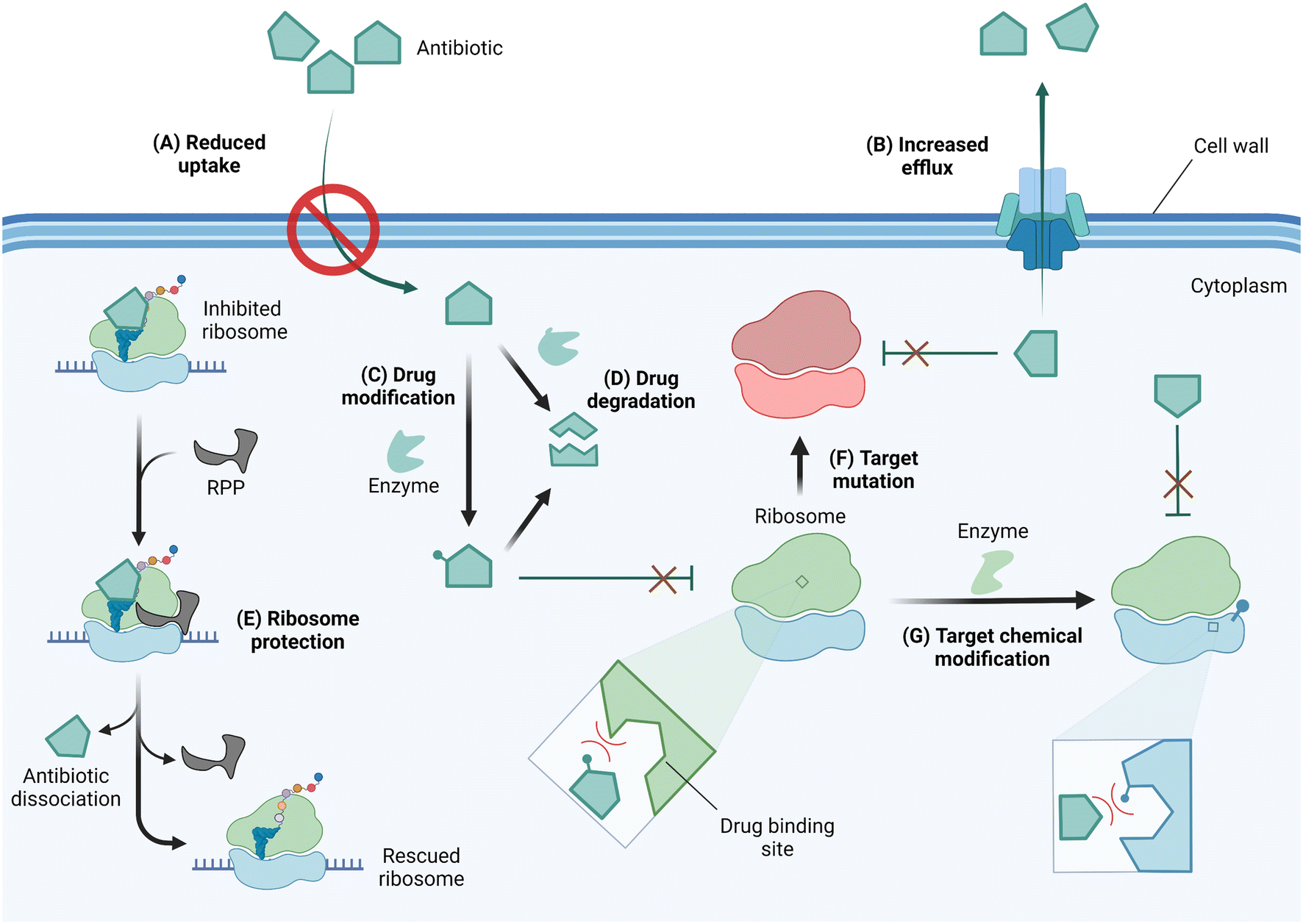 | ||
| Fig. 4 Mechanisms of resistance against ribosome-targeting antibiotics. (A) To prevent antibiotic entry, drug-resistant bacteria can alter the expression level or pore structure of porins. (B) The expression of efflux pumps facilitates drug extrusion from the cell thereby reducing intracellular drug concentrations. (C) As antibiotics reach the cytoplasm, they may be deactivated by drug-modifying enzymes or (D) degraded in their native form or after modification to enhance degradation. (E) Through the use of ribosome protection proteins (RPP), bacteria can resist inhibition by dislodging antibiotics from their ribosomal binding site. Resistant bacteria can also alter the drug target site via (F) mutation of residues critical for drug-binding or (G) chemical modification of target residues. Drug or target site chemical modification reduces binding affinity due to steric clash and/or electrostatic repulsion. Created with https://BioRender.com. | ||
Antibiotics with an intracellular target face the challenge of reaching the cytoplasm in high enough concentration to inhibit bacterial growth. As a resistance strategy, bacteria can therefore lower drug accumulation by reducing or eliminating the expression of porins, or narrowing the porin channel via adaptation or mutation.57,58 Drug-resistant strains of A. baumannii, P. aeruginosa, and Mycobacterium tuberculosis have also been reported to increase the expression of multidrug efflux pumps, thereby similarly limiting the intracellular accumulation of antibiotics.56,59–61
Once in the cytoplasm, antibiotics can be modified and/or degraded. For example, aminoglycosides can be deactivated by acetylation, phosphorylation, or adenylation of the amino and hydroxyl functional groups via aminoglycoside modifying enzymes (AMEs) to reduce their affinity for their h44 binding site,62 and macrolides are hydrolyzed through esterase-mediated cleavage.63 Ribosome protection proteins such as TetM/TetO64 and members of the ATP-binding cassette F (ABCF) family65,66 can confer resistance to ribosome-targeting antibiotics by dislodging them from their binding sites. Target site alterations via mutation or posttranscriptional modification sterically and/or electrostatically reduce drug binding affinity. An A2058G mutation in the NPET renders macrolides, lincosamides, and streptogramin B (MLSB) antibiotics inactive.67 Chemical modification at the binding sites of ribosome-targeting antibiotics occurs on strategic rRNA residues such that a compromise between protein synthesis efficiency and maximum protection is reached. Enzymes responsible for target alteration through methylation of rRNA residues are increasingly becoming a cause for alarm, and their features, prevalence, fitness cost, and inhibition efforts are the focus of this review.
Resistance-conferring rRNA methyltransferases
S-Adenosyl-L-methionine (SAM)-dependent methyltransferases are ubiquitous enzymes across all domains of life that facilitate the delivery of a methyl group to a wide range of substrates, including proteins, nucleic acids, and small molecules.68 In the process of substrate modification, the SAM cosubstrate is demethylated to form S-adenosylhomocysteine (SAH). Methyl transfer mediated by these enzymes proceeds via a second-order nucleophilic substitution (SN2) or a radical mechanism (Fig. 5A and B).69 As the two clinically most relevant examples, resistance-conferring 16S or 23S rRNA methyltransferases modify conserved rRNA residues in the A site or NPET/PTC to render bacteria insensitive to aminoglycosides and MLSB/PhLOPSA, respectively.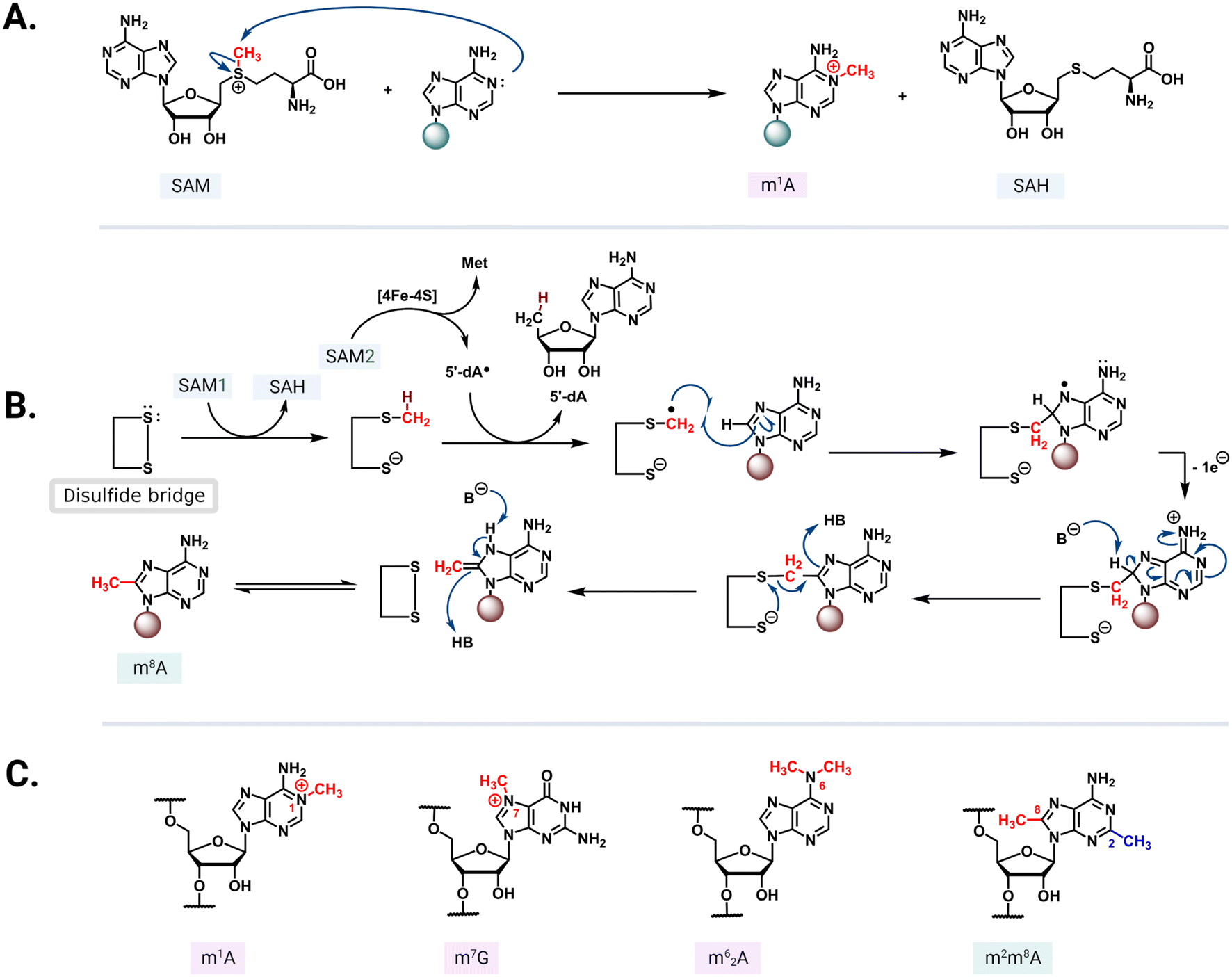 | ||
| Fig. 5 Methyl transfer mechanisms and modified residues. (A) In the SN2 mechanism, the nucleophilic nitrogen from the nucleotide base attacks the electrophilic methyl group of a positively charged SAM. (B) Radical mechanism mediated by an iron–sulfur cluster that reductively cleaves the second SAM (SAM2) to methionine and a 5′-dA radical. The radical abstracts hydrogen from a methylcysteine intermediate formed via an SN2 mechanism between SAM1 and a cysteine residue. The resulting radical then reacts with the target adenine base and the conserved active site disulfide bridge reforms to complete the transfer followed by an enamine/imine tautomerization mediated by the undefined base B- and acid/conjugate base HB. (C) Select structures of methylated rRNA residues: m1A, m7G, m62G, and m2m6A. | ||
In the 30S subunit, methylation of G1405 at the N7 position by ArmA or RmtA-H causes the modified residue to carry a positive charge (Fig. 5C) that results in charge repulsion with an ammonium functionality on ring III of 4,6-DOS aminoglycosides in addition to steric hindrance. The longer distance between the 4,5-DOS aminoglycosides and G1405 means that interactions between these drugs and the A-site residues are impacted less by the methylation of G1405. Apramycin does not interact directly with G1405 and thus its activity is unaffected by methylation of this residue. The neamine core (rings I and II) present in both 4,6- and 4,5-DOS aminoglycosides is proximal to the N1 position of a second methylated nucleotide, A1408 (Fig. 2), such that modification at this site by NpmA-B introduces steric clash for both drug scaffolds. Non-DOS compounds like streptomycin do not interact with the same A-site binding pocket and these modifications therefore do not affect their activity.29
In the 50S subunit, mono- or dimethylation of the N6 position on A2058 by Erm 23S rRNA methyltransferases introduces a steric clash with a required water molecule at the NPET.41 This prevents the formation of a hydrogen bonding network that bridges the macrolide desosamine ring to the NPET residues, thus significantly reducing the affinity of this class of drugs for their binding site (Fig. 3A). Only dimethylation of N6 A2058 confers resistance to the bactericidal, slow-dissociating third- and fourth-generation macrolides (ketolides).70,71 Similarly, C8 methylation of A2503 by Cfr introduces a steric clash of the PhLOPSA antibiotics with the modified residue, again significantly affecting their binding.72 The efficacy of lincosamide antibiotics and the synergistic streptogramin drug combinations (streptogramins A and B, Fig. 3C) is notably affected by both A2058 and A2503 methylations. Tylosin and other macrolides that extend into the PTC due to their extended sugar side chains are also sensitive to both A2058 and A2503 modification.73 Additionally, m1G748 and m6A2058 methylations have been shown to confer resistance specifically against tylosin and another 5- and 14-position glycosylated macrolactone, mycinamycin.74 Methylation of these residues is performed by TlrB (Erm32) and TlrD (ErmN) resistance-conferring 23S rRNA methyltransferases.74,75
Though less clinically relevant, modifications at several other 23S rRNA residues confer resistance to 50S subunit binding antibiotics. The oligosaccharide antibiotics, evernimicin and avilamycin, are also impacted by resistance-rRNA methylations. Base methylation of G2535 in h91 by AviRa and the 2′-hydroxyl of U2479 in h89 by AviRb have been demonstrated to render orthosomycins inactive.76 Molecular modeling suggests that m7G2535 is the most likely modification conferring resistance, and a similar model has confirmed the effect of methylation by AviRb.53 The methylation of domain V residue, G2470, by EmtA also renders bacteria insensitive to orthosomycins.77 Finally, an Am1067 methylation performed by the thiostrepton-resistance methyltransferase TsnR confers high-level resistance against thiostrepton and other related thiopeptide antibiotics which bind at the ribosome GTPase Center.78
Since most essential, clinically used chemotherapeutics are bacterial natural products, antibiotic-producing actinomycetes like Micromonospora spp. and Streptomyces spp. possess resistance mechanisms against these compounds as a method to prevent self-inhibition.79 Commonly, in these drug-producing bacteria, genes encoding resistance rRNA methyltransferases are embedded in the chromosomal biosynthetic gene cluster. Examples of such proteins include Sgm, KamB, and PikR1/PikR2 which incorporate 16S rRNA m7G1405, 16S rRNA m1A1408, and 23S rRNA m62A2058 modifications, respectively.80,81 In pathogenic bacteria, genes that encode resistance-conferring rRNA methyltransferases are more often located on mobile genetic elements and can thus be transferred horizontally between bacterial species.82,83 The acquired resistance-conferring rRNA methyltransferases are likely to have evolved from their drug-producer homologs though in many cases significant differences are found in gene and protein sequences for the two sets of extant enzymes. Other resistance rRNA methyltransferases may have originated from enzymes responsible for housekeeping rRNA methylations. This idea is supported by the mechanistic and sequence similarities between the radical SAM enzymes, RlmN (formerly YfgB) and Cfr, responsible for m2A2503 and m8A2503 modifications, respectively, the latter of which renders bacteria resistant to PhLOPSA.84,85 Furthermore, evidence shows that Cfr can perform both C2 and C8 methylations on A2503, but preferentially modifies the C8 position.86
The Arm/Rmt, Npm, and Erm family enzymes deliver methyl groups to their target residues via an SN2 mechanism. These three families are class I Rossmann-fold methyltransferases that possess a core seven-stranded β-sheet with GXG(XG) SAM-binding motif sandwiched between α-helices (Fig. 6A–C). G1405 enzymes have a large N-terminal domain with two bundles of three α-helices that appear to be critical for 30S subunit binding (Fig. 6A).87–89 In contrast, despite recognizing a similar region of the 30S subunit, NpmA has internal extensions between strands β5/β6 and β6/β7 that are important for rRNA recognition and binding (Fig. 6B),88,90 while Erm family methyltransferases have a unique C-terminal domain (Fig. 6C(ii)).91 In contrast, RlmN/Cfr proteins are class VII methyltransferases as they bear a partial α6/β6 TIM barrel fold that accommodates an iron–sulfur cluster ([4Fe–4S]) bound by a conserved CX3CX2C motif.92 While rRNA methyltransferase structures built on these two cores are unique for each family, many structural features required for methylation activity are universal and can be exploited in the design of inhibitors. For all the rRNA methylating enzymes, a positive electrostatic potential surface is essential for substrate recognition and binding (Fig. 6).
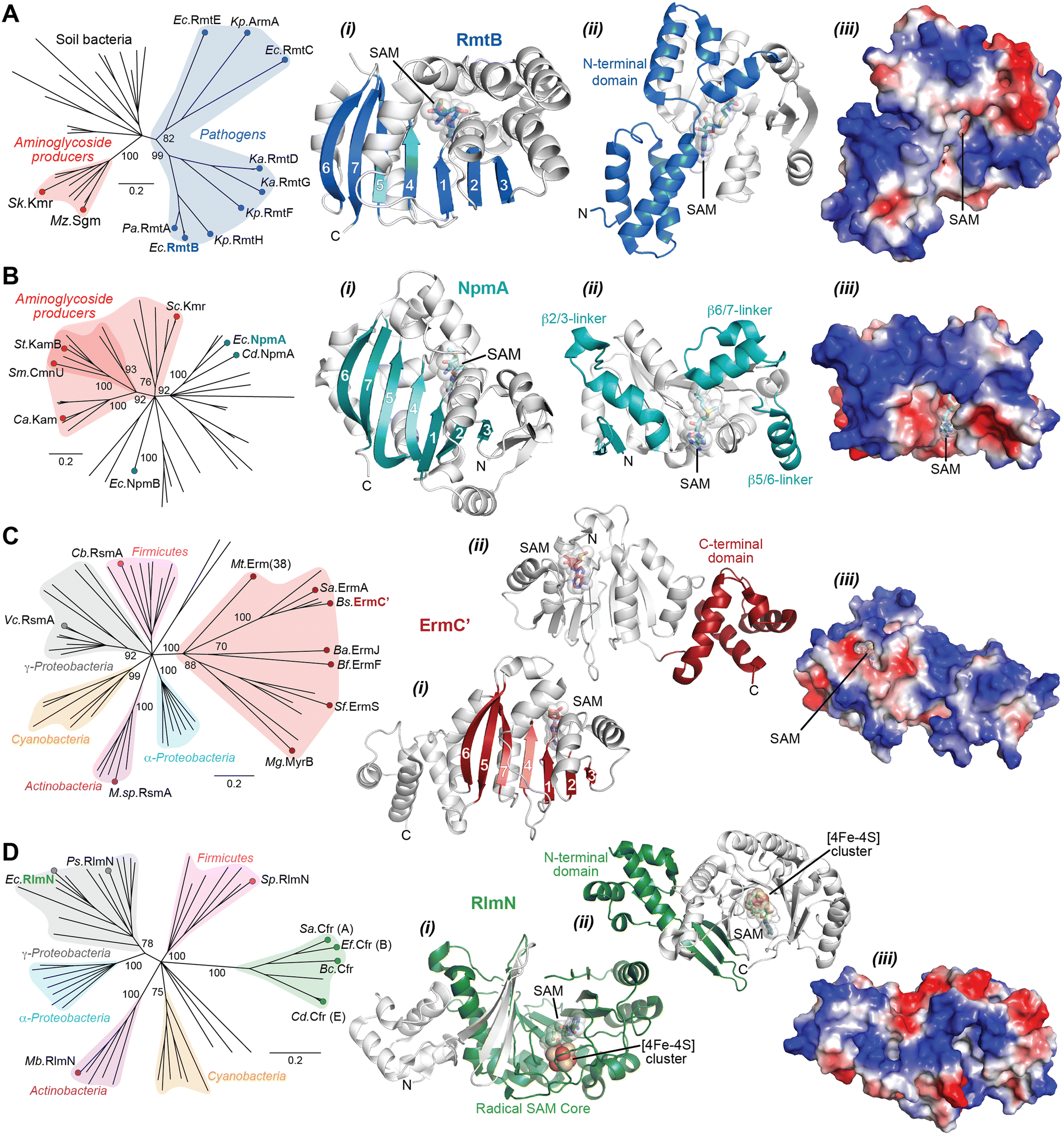 | ||
| Fig. 6 Phylogenetic and structural comparison of antibiotic-resistance methyltransferase families. (A) Neighbor joining phylogenetic tree created with 100 steps of bootstrapping (left) and views of the RmtB structure (PDB 3FRH) as an example of the m7G1405 methyltransferase family. Structure views (left to right) highlight: (i) the seven β-strand core with the SAM binding pocket, (ii) appended region with critical determinants for rRNA interaction, and (iii) the same orientation but shown as an electrostatic surface. (B) and (C) As for panel A, but for the aminoglycoside-resistance m1A1408 (with example of NpmA; PDB 3MTE) and macrolide-resistance Erm (with example structure of ErmC′; PDB 1QAO) and related methyltransferases, respectively. (D) Phylogentic analysis of Cfr and the related RlmN enzymes. Example RlnM structure (4PL1) highlights (left to right): (i) the radical SAM core fold with bound SAM and [Fe4–S4] cluster, (ii) appended N-terminal domain, and (iii) the same orientation but shown as an electrostatic surface. Species abbreviations: Ba, Bacillus anthracis; Bc, Bacillus clausii; Bf, Bacteroides fragillis; Bs, Bacillus subtilis; Ca, Catenulisporales acidiphilia; Cb, Clostridium botulinum; Cd, Clostridioides difficile; Ec, Escherichia coli; Ef, Enterococcus faecium; Ka, Klebsiella aerogenes; Kp, Klebsiella pneumoniae; Mb, Mycobacterium bovis; Mg, Micromonospora griseorubida; Mt, Mycobacterium tuberculosis; Mz, Micromonospora zionesis; Pa, Pseudomonas aeruginosa; Ps, Pseudomonas stutzeri; Sa, Staphylococcus aureus; Sc, Sorangium cellulosum So ce56; Sf, Streptomyces fradiae; Sk, Streptomyces kanamyceticus; Sm, Saccharothrix mutabilis subsp. capreolus; Sp, Streptococcus pyogenes; St, Streptoalloteichus tenebrarius. | ||
In 2014, Dunkle et al. reported the structural basis for 30S subunit recognition by NpmA and proposed a model for methyl transfer catalysis.93 The X-ray crystal structure of the T. thermophilus 30S subunit complexed with NpmA and sinefungin (a SAM analog) revealed the large interaction surface between NpmA and the 16S rRNA, with which it interacts exclusively. NpmA recognizes the unique structure of the rRNA sugar-phosphate backbone of four rRNA helices which are spatially adjacent only in the mature 30S, explaining this and related enzymes' requirement for the full subunit as their substrate. Despite the large interaction surface, the only rRNA sequence-specific interaction is a hydrogen bond between the backbone carbonyl of NpmA residue F105 and the N6 amine of target base A1408. Upon binding, additional residues in the NpmA regions liking β5/β6 and β6/β7 stabilize an rRNA conformational change in which A1408 flips out of h44, and is sandwiched between two active site tryptophan residues via π–π stacking interactions. This arrangement positions A1408 in a desirable orientation for catalysis with N1 positioned adjacent to the bound SAM analog (Fig. 7). The SAM analog sits in a negatively charged pocket, and this feature is conserved among SAM-dependent methyltransferases.89,93
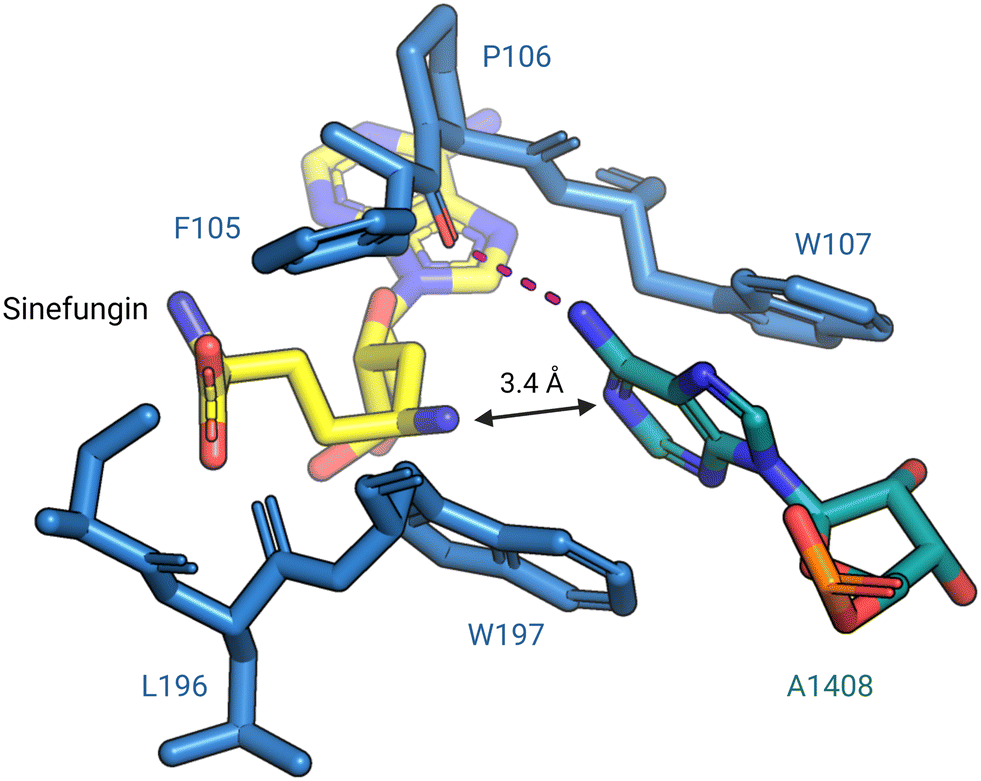 | ||
| Fig. 7 Base flipping positions A1408 for N1-methylation by NpmA. During methylation, A1408 is flipped from its helix by NpmA and the adenosine nucleotide is stabilized by π–π stacking interactions with two tryptophan residues. The backbone carbonyl of F105 forms a hydrogen bond (red dashed line) with N6 of A1408, positioning the N1 for methylation (PDB: 4OX9). Structure image was prepared using PyMol. | ||
Prevalence of acquired antibiotic-resistance rRNA methyltransferases
Resistance-conferring rRNA methyltransferase genes are named based on amino acid sequence identities between the enzymes they encode. In the nomenclature proposed by Doi and colleagues,94 a gene that displays an established aminoglycoside resistance profile and has either an amino acid sequence identity lower than 50% or methylates a different nucleotide of the 16S rRNA is given a new name (e.g., rmt, npm). Similarly, those with an amino acid homology in the range 50 – 95% are appended with a new alphabet letter (e.g., rmtB and rmtC), whereas those that are more than 95% identical to a known rRNA methyltransferase receive a variant number (e.g., armA2). Likewise, the nomenclature for MLSB resistance determinants was initiated by Roberts et al. in 1999: genes encoding a pair of rRNA methyltransferases with more than 80% amino acid identity are classified with the same letter designation whereas those with less than 80% homology are labeled with different letters or numbers e.g. ermB or erm(35).95 To encompass all 23S rRNA methyltransferases, the nomenclature is applied to the cfr family and a living document enlisting these MLSB and PhLOPSA resistance rRNA methyltransferases is curated by the Nomenclature Center for MLS Resistance Genes (https://faculty.washington.edu/marilynr/). The arm/rmt, npm, erm, and cfr families of acquired resistance-conferring ribosomal methyltransferase genes are, to the best of our knowledge, the most globally clinically relevant and are thus the focus of the following sections.Arm/Rmt aminoglycoside-resistance methyltransferases (m7G1405)
The group comprising the![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) minoglycoside-
minoglycoside-![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) esistance
esistance ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) ethylase (Arm) and ibosomal ethyl
ethylase (Arm) and ibosomal ethyl![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif) ransferase (Rmt) 16S rRNA m7G1405 methyltransferases is a prevalent family of aminoglycoside resistance-conferring rRNA modifiers. Phylogenetic analysis of the Arm/Rmt family suggests a common ancestor of these proteins with methyltransferases of aminoglycoside-producing bacteria which are present to inhibit self-intoxication (Fig. 6A). Enzymes acquired by pathogens including E. coli, Klebsiella pneumoniae, and P. aeruginosa, are divided into two early branching subclades containing the most widespread of these enzymes, to date, ArmA (along with RmtC and RmtE), while the other contains RmtB and the other Rmt enzymes. Notably, these genes are also present in other non-drug producing soil bacteria which might act as reservoirs for horizontal gene transfer (HGT).
ransferase (Rmt) 16S rRNA m7G1405 methyltransferases is a prevalent family of aminoglycoside resistance-conferring rRNA modifiers. Phylogenetic analysis of the Arm/Rmt family suggests a common ancestor of these proteins with methyltransferases of aminoglycoside-producing bacteria which are present to inhibit self-intoxication (Fig. 6A). Enzymes acquired by pathogens including E. coli, Klebsiella pneumoniae, and P. aeruginosa, are divided into two early branching subclades containing the most widespread of these enzymes, to date, ArmA (along with RmtC and RmtE), while the other contains RmtB and the other Rmt enzymes. Notably, these genes are also present in other non-drug producing soil bacteria which might act as reservoirs for horizontal gene transfer (HGT).
ArmA was first discovered in 1996 in a clinical isolate of Citrobacter freundii from Poland.96 Through rigorous surveillance efforts, genes that encode ArmA have since been identified in Gram-negative multidrug-resistant pathogens from most regions of the world. In Europe, a 2016 six-month-long study involving 14 UK hospital laboratories identified armA in 54.4% (43 of 79) of high-level 4,6-DOS aminoglycoside resistant clinical isolates (amikacin minimum inhibitory concentration (MIC) >256 μg mL−1), possessing at least one 16S rRNA methyltransferase gene.97 Of even greater concern, the ArmA-encoding gene is often not the sole resistance determinant identified. For example, in a Belgian study, 18 of 22 pathogenic isolates of E. coli, K. pneumoniae, Enterobacter aerogenes, Enterobacter cloacae, and Citrobacter amalonaticus exhibiting high-level resistance to amikacin, gentamicin, and tobramycin (MIC >512 μg mL−1) were found harboring the armA gene alongside aminoglycoside modifying enzymes and extended-spectrum β-lactamases.98
Immunocompromised individuals who require critical care and prolonged hospitalization are at a much higher risk of contracting nosocomial infections with bacteria possessing ArmA. For example, Ghafoor and coworkers found ArmA-encoding genes in strains of K. pneumoniae, P. aeruginosa, and E. coli isolated from twelve cancer patients with pneumonia in a Pakistan hospital.99 The worldwide abundance of bacteria possessing armA is additionally demonstrated by their occurrence in food and domesticated animals. In Spain, surveillance efforts conducted by González-Zorn and coworkers identified armA in an E. coli isolate from the fecal matter of a diarrheic pig100 and in seven K. pneumoniae isolates from infected house cats and dogs.101 The sequencing of armA in the Salmonella enterica isolated from chicken meat sold at a supermarket in La Réunion Island brings to light the role played by food-borne pathogens in the distribution of resistance-conferring rRNA methyltransferases.102 ArmA genes have also been found in Africa,103,104 Oceania,105 and North and South America,106 and a subvariant of ArmA (designated ArmA2) was recently sequenced in Myanmar from an MDR A. baumannii strain.107
Functionally identical enzymes with the alternative Rmt designation (RmtA-H) have also been widely reported, with RmtB being the second most widespread aminoglycoside-resistance m7G1405 16S rRNA methyltransferase. RmtB has been identified in bacteria isolated from farm animals,108 companion animals,109 and clinical settings since its first identification in 2002 from a patient in Japan.106,110 Although RmtB and ArmA rRNA methyltransferases modify the same residue, strains co-harboring genes that encode both enzymes have recently been identified. Two carbapenem-resistant K. pneumoniae strains exhibiting high-level aminoglycoside insensitivity were isolated from bloodstream infections of patients in China, and ArmA jointly with RmtB methyltransferase genes were located on separate plasmids.111 RmtA was isolated in Japan from a clinical sample of P. aeruginosa in 1997, yet reports of its occurrence are relatively uncommon.82 However, it is not only the most common subclass genes that are found in food animals: the genetic elements for the first two RmtE variants, rmtE and rmtE2, were both first identified in different E. coli strains found in cattle from USA and swine in China, respectively.112,113
In recent years, the value of extensive surveillance efforts has been showcased by the surging identification of less common Rmt methylases coupled with the discovery of novel subclass variants. A UK/Ireland-wide study of 550 A. baumannii clinical isolates collected between 2004 and 2015 reported the first occurrence of rmtE3 although this gene was in only one out of the 531 isolates possessing 16S rRNA methyltransferase genes.114 In 2011 and 2013, the genes encoding RmtF, RmtG, and RmtH were isolated from K. pneumoniae strains found in France,115 Brazil,116 and Iraq,117 respectively. These genes have since been identified in other drug-resistant Gram-negative bacteria, including E. coli and P. aeruginosa.118 Collectively, the global dissemination of established Rmt family genes and the emergence of novel subclass rRNA methyltransferases is an indication of a continually developing and globally expanding resistance mechanism.
Npm aminoglycoside-resistance methyltransferases (m1A1408)
The![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif) ovel
ovel ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) lasmid-ediated 16S rRNA m1A1408 methyltransferase (Npm) family confers high-level resistance to structurally diverse aminoglycosides, including some 4,6-DOS, 4,5-DOS and others, a resistance profile dissimilar to ArmA/RmtA-H. However, as for the m7G1405 enzymes, phylogenetic analysis of wider group of m1A1408 methyltransferases suggests a common ancestor with enzymes from aminoglycoside-producing soil bacteria (Fig. 6B), but not one that is shared between the two groups of aminoglycoside-resistance enzymes. While more distant from enzymes of known drug-producing bacteria, the pathogen associated enzymes NpmA and NpmB1/B2 are most closely related to soil bacteria which might have acted as the source for acquisition by HGT.
lasmid-ediated 16S rRNA m1A1408 methyltransferase (Npm) family confers high-level resistance to structurally diverse aminoglycosides, including some 4,6-DOS, 4,5-DOS and others, a resistance profile dissimilar to ArmA/RmtA-H. However, as for the m7G1405 enzymes, phylogenetic analysis of wider group of m1A1408 methyltransferases suggests a common ancestor with enzymes from aminoglycoside-producing soil bacteria (Fig. 6B), but not one that is shared between the two groups of aminoglycoside-resistance enzymes. While more distant from enzymes of known drug-producing bacteria, the pathogen associated enzymes NpmA and NpmB1/B2 are most closely related to soil bacteria which might have acted as the source for acquisition by HGT.
In comparison to ArmA and RmtB, NpmA is currently significantly less prevalent. The gene encoding NpmA was first isolated in 2003 from a drug-resistant E. coli strain infecting a patient in a general hospital in Japan.119 In the 2007 report, Wachino et al. proposed that methylation by NpmA conferred insensitivity to 4,6-DOS (MIC >256 μg mL−1 for both kanamycin and gentamicin) and 4,5 DOS (MIC >256 μg mL−1 for neomycin), but not streptomycin or spectinomycin (non-DOS) in E. coli ARS3. However, the use of gentamicin in surveillance to determine the resistance profile of A1408 methyltransferases has been a subject of debate as this molecule has been observed to have reduced sensitivity to m1A1408 methylation compared to other drugs (e.g. kanamycin).120 NpmA genes have also been identified in K. pneumoniae and Enterobacter spp. (E. aerogenes and E. cloacae) strains in a study from Saudi Arabia, with four strains of the 218 isolates analyzed in the study found to harbor both npmA and β-lactamase genes.121 Until Marsh et al. identified npmA and npmA2 in clinical isolates of Clostridioides difficile using whole genome sequencing,122 the existence of acquired 16S rRNA methyltransferases in Gram-positive bacteria was unknown. Previously, it had only been demonstrated that Bacillus subtilis with an rmtC-bearing recombinant plasmid conferred high-level resistance to kanamycin and gentamicin.123 The C. difficile strains hosting the NpmA genes were isolated from humans, swine, and bovine in four countries on three different continents: USA, Japan, Canada, and Australia.122 The npmA gene has also been found in P. aeruginosa in Iraq, and nearly 44% of the isolates examined in this surveillance effort possessed the gene.124 Other subclasses and variants in the Npm family are emerging in pathogenic bacteria and some appear more distantly related to known pathogenic enzymes than to many sequences from soil bacteria (Fig. 6B). In particular, recently, two aminoglycoside-resistant E. coli strains from the UK were found to harbor npmB1 and npmB2 after a search for NpmA-like methyltransferases in the National Center for Biotechnology Information (NCBI) database.125
Erm macrolide-resistance methyltransferase family
While 16S rRNA methyltransferases are predominantly found in Gram-negative pathogens, enzymes that methylate the A2058 nucleobase on the 23S rRNA have been widely identified in Gram-positive bacteria. In 1956, Chabbert reported S. aureus strains exhibiting erythromycin resistance,126 and later, Griffith et al. noted antagonism between the macrolide and lincomycin in the isolates.127 The Weisblum group demonstrated that this bacterium was also insensitive to streptogramin B which binds an overlapping binding site with macrolides and lincosamides.128 Subsequently, the gene that encodes ErmA, the first member of the![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) rythromycin ibosome ethylation (Erm) family of enzymes, was sequenced in 1973 from a clinical isolate of S. aureus and the MLSB resistance phenotype was attributed to this methyltransferase. Several subclasses in the Erm family alongside their variants have since been reported including ErmB–ErmZ and Erm(30)–Erm(55).
rythromycin ibosome ethylation (Erm) family of enzymes, was sequenced in 1973 from a clinical isolate of S. aureus and the MLSB resistance phenotype was attributed to this methyltransferase. Several subclasses in the Erm family alongside their variants have since been reported including ErmB–ErmZ and Erm(30)–Erm(55).
Phylogenetic analysis suggests that the Erm family methyltransferases share a common ancestor with RsmA, an rRNA methyltransferase which dimethylates two adjacent adenosines (A1518 and A1519) of 16S rRNA in the 30S ribosomal subunit. The RsmA group itself is divided into several clades corresponding to different bacterial phyla. In the Erm methyltransferase clade, this enzyme group is divided in to four main subclades containing ErmA/ErmC, ErmF/ErmJ, ErmS and MyrB (also known as ErmW), along with the more divergent Erm38 from Mycobacterium tuberculosis (Fig. 6C).
The presence of Erm resistance-conferring methyltransferase genes in gut-dwelling bacteria accelerates their global dissemination. In India, ErmB genes were found in the non-pathogenic bacteria Streptococcus pneumoniae and Enterococcus faecalis sampled from sewage water branching into River Ganga,129 the main water source in the region. In the USA, outflowing water from midwestern land reserved for animal husbandry and treated with swine manure was observed to have ermB and ermF-harboring bacteria.130 The existence of rRNA methyltransferases in bacteria found in food complicates the treatment of infections like Campylobacteriosis. In Tunisia, ermB genes were isolated in the Gram-negative food-borne pathogens, Campylobacter jejuni and Campylobacter coli, co-harboring other resistance determinants.131 Recently, the Erm(54) gene was located on a plasmid from livestock-acquired MRSA originating from Germany,132 and Erm(53) genes were isolated from E. coli found in Canada and USA.133 Despite the erm genes having been identified first in the 20th century, earlier MLSB resistance-conferring rRNA methyltransferases continue to disseminate worldwide and the discovery of new variants has not ceased.
Cfr radical SAM methyltransferase family
The Cfr methyltransferase family shares a common ancestor with the housekeeping RlmN methyltransferases (Fig. 6D), which incorporate an m2A2503 in 23S rRNA and m2A37 in tRNAs. The![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) hloramphenicol-
hloramphenicol-![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) lorfenicol esistance (cfr) gene was first discovered on a plasmid in Staphylococcus sciuri from a calf suffering a respiratory infection.134 In 2005, a MRSA strain coexpressing cfr and ermB, thus resistant to most clinically relevant 50S subunit targeting antibiotics, was isolated from a patient with a fatal case of pneumonia in Colombia.135 Recently, a study in Portugal identified cfr in livestock-acquired MRSA strains of swine origin alongside other resistance determinants like fexA (encodes florfenicol-chloramphenicol exporter), blaZ (encodes β-lactamase), and tetK (encodes a tetracycline efflux pump).136 In another surveillance effort, cfr was identified in clinical isolates of MRSA collected between 2018 and 2019 in hospitals in Pakistan.137 Aside from staphylococcal strains, cfr has also been detected in E. coli, E. faecalis, and Streptococcus suis in Europe and Asia.138–140 Four other homologs of cfr have been identified in diverse genetic contexts and geographical locations. In New Orleans, USA, Cfr(B) genes were first isolated from E. faecium recovered from patients at a medical center.141 In 2017, Cfr(C) was isolated from the food-borne Campylobacter pathogens in the USA and later that year in C. difficile.142,143 Cfr(D) genes first appeared in E. faecium human isolates from France, and recently, the genes were identified in another E. faecium isolate from a Western Australian man suffering from bacteremia.144,145 In both cases, the cfr(D) was detected alongside optrA, an emerging transferable gene that encodes an ABCF ribosome protection protein conferring resistance to oxazolidinones and phenicols.146 The co-existence of cfr and optrA has also been observed in E. faecalis in Brazil.147 Presence of transferable resistance determinants in commensal bacteria unveils the gut and nasal microbiota as a hub for rRNA methyltransferase gene exchange in humans and animals. Since these bacteria are non-pathogenic, this accelerates worldwide resistance-gene dissemination as nothing warrants investigation. Lastly, the latest Cfr-like rRNA methyltransferase, Cfr(E), was identified in 2015 from clinical C. difficile isolates in Mexico.148 The transcontinental spread of Cfr and Cfr-like rRNA methyltransferases poses a significant threat as it considerably limits antibiotic treatment options.
lorfenicol esistance (cfr) gene was first discovered on a plasmid in Staphylococcus sciuri from a calf suffering a respiratory infection.134 In 2005, a MRSA strain coexpressing cfr and ermB, thus resistant to most clinically relevant 50S subunit targeting antibiotics, was isolated from a patient with a fatal case of pneumonia in Colombia.135 Recently, a study in Portugal identified cfr in livestock-acquired MRSA strains of swine origin alongside other resistance determinants like fexA (encodes florfenicol-chloramphenicol exporter), blaZ (encodes β-lactamase), and tetK (encodes a tetracycline efflux pump).136 In another surveillance effort, cfr was identified in clinical isolates of MRSA collected between 2018 and 2019 in hospitals in Pakistan.137 Aside from staphylococcal strains, cfr has also been detected in E. coli, E. faecalis, and Streptococcus suis in Europe and Asia.138–140 Four other homologs of cfr have been identified in diverse genetic contexts and geographical locations. In New Orleans, USA, Cfr(B) genes were first isolated from E. faecium recovered from patients at a medical center.141 In 2017, Cfr(C) was isolated from the food-borne Campylobacter pathogens in the USA and later that year in C. difficile.142,143 Cfr(D) genes first appeared in E. faecium human isolates from France, and recently, the genes were identified in another E. faecium isolate from a Western Australian man suffering from bacteremia.144,145 In both cases, the cfr(D) was detected alongside optrA, an emerging transferable gene that encodes an ABCF ribosome protection protein conferring resistance to oxazolidinones and phenicols.146 The co-existence of cfr and optrA has also been observed in E. faecalis in Brazil.147 Presence of transferable resistance determinants in commensal bacteria unveils the gut and nasal microbiota as a hub for rRNA methyltransferase gene exchange in humans and animals. Since these bacteria are non-pathogenic, this accelerates worldwide resistance-gene dissemination as nothing warrants investigation. Lastly, the latest Cfr-like rRNA methyltransferase, Cfr(E), was identified in 2015 from clinical C. difficile isolates in Mexico.148 The transcontinental spread of Cfr and Cfr-like rRNA methyltransferases poses a significant threat as it considerably limits antibiotic treatment options.
Fitness cost and regulation of rRNA methyltransferase genes
The rate of global transmission of antibiotic-resistance rRNA methyltransferase genes relies, in part, on their overall efficacy as agents of resistance for the host bacteria. However, carrying and expressing resistance genes often comes with a fitness cost. The E. coli 16S rRNA is decorated with a total of eleven well-conserved nucleotide modifications, with several proximal to the A site: m4Cm1402, m5C1407, and m3U1498 on h44 (Fig. 8A).149 These four methylations are incorporated by different SAM-dependent housekeeping rRNA methyltransferases (RsmH, -I, -F, and -E) and are crucial for translational fidelity.149–151 In 2006, Andersen and Douthwaite established that a fitness cost is associated with the lack of an m5C1407 modification by RsmF (formerly YebU) in E. coli149 and Čubrilo et al. subsequently showed that m7G1405 modification by the aminoglycoside resistance-methyltransferase Sgm interferes with the ability of RsmF to introduce the functionally important m5C1407 modification into the E. coli 30S subunit.152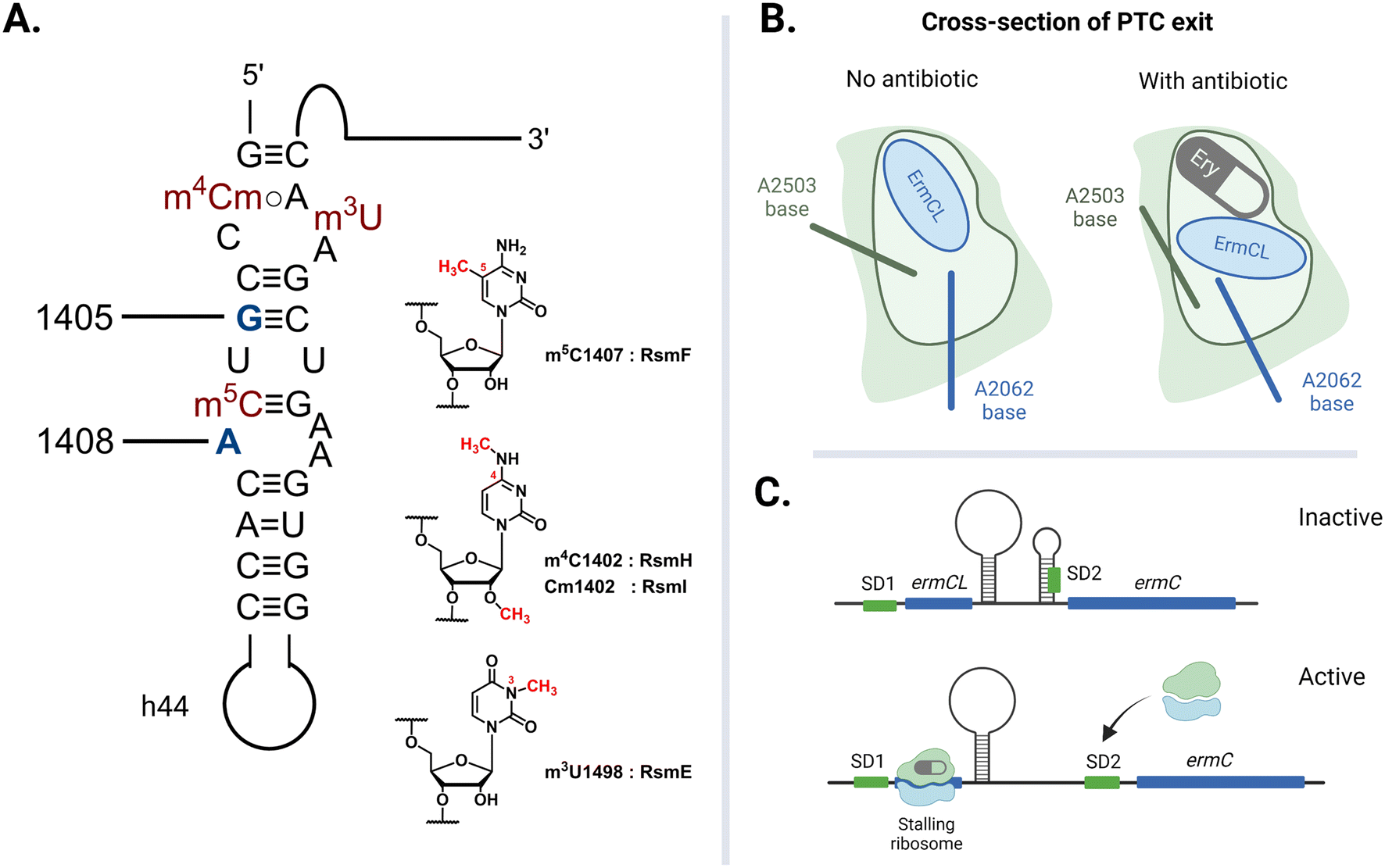 | ||
| Fig. 8 Factors that dictate the fitness cost of bacteria hosting resistance rRNA methyltransferase genes. (A) Locations of E. coli housekeeping methylations in h44 impacted by adjacent resistance modifications at G1405 and A1408. (B) The exiting nascent peptide does not interact with either A2062 or A2503 when the tunnel is clear. The A2503 base changes conformation to relay nascent peptide information from the NPET to the PTC when the channel is blocked by erythromycin (Ery). (C) Ribosome-erythromycin-ErmCL complex stalling at ermCL triggers the mRNA to release a second ribosome binding sequence (SD2). Created with https://BioRender.com. | ||
While similar impacts on translational efficiency could be expected for the Sgm homologs ArmA and RmtA-H, detailed studies of these pathogen-derived enzymes have revealed some unexpected differences. In 2012, Gutierrez and colleagues showed that ArmA, RmtB, and RmtC indeed interfered with RsmF, but rmtC-harboring E. coli surprisingly had an insignificant fitness cost in the absence of antibiotic challenge.153 In further contrast, in 2014 Lioy et al. reported that ArmA only interfered with 2′-O-methylation (Cm1402) by RsmI and not with the m5C1407 methylation.154 The same group also established that npmA-expressing cells had only a minor fitness cost despite m1A1408 modification by NpmA also impeding the adjacent m5C1407 methylation.154 Subsequently, in 2016, Yang and colleagues used E. coli strains harboring rmtB to demonstrate that there is a substantial fitness cost affiliated with carrying this resistance gene.155 Furthermore, competition and growth rate experiments reported by Ishizaki et al. in 2018 involving npmA and armA in the Gram-negative pathogenic strains, E. coli, P. aeruginosa, and K. pneumoniae, revealed the instability of npmA in the presence of insertion sequence (IS) elements.156 This study suggested that npmA expression is highly unfavorable and disadvantageous for bacterial growth and survival, consistent with this gene's relatively low acquisition and dissemination rate. The contrasting findings from these works call for more research in this arena using consistent experimental standards. While 16S rRNA modifying methyltransferase genes may decrease bacterial fitness in an antibiotic-free environment, compensatory mutations can also develop to restore fitness while maintaining the benefit of possessing the resistance determinant.
The expression of acquired resistance-conferring rRNA methyltransferase genes to modify domain V nucleotides of the ribosomal 50S subunit leads to interesting behavior in bacteria. From growth rate and competition studies conducted by LaMarre and coworkers, a low fitness cost was observed for a laboratory strain of S. aureus harboring a plasmid encoding the cfr resistance gene.157 A follow-up experiment in which cells with an active and a non-active Cfr protein were cocultured showed that the fitness cost was a result of the bacteria producing the protein rather than C8 methylation on A2503.157 The A2503 nucleotide, via a syn–anti base conformational transition, is reported to relay nascent peptide chain information from the NPET to the PTC during antibiotic-induced ribosome stalling (Fig. 8B).158 While C2 methylation facilitates this conformational change,159 it is probable that a second methylation at the C8 position does not alter the cell's viability as A2503 is not directly involved in protein synthesis. The low fitness burden reported for the S. aureus strain harboring cfr can explain the capacity of cfr and cfr-like genes to be rapidly disseminated. In contrast, the report by LaMarre et al. also revealed that expression of ermB alongside cfr significantly decreased bacterial survival157 This result is consistent with other findings that expression of Erm (MLSB resistance genes) are deleterious for bacteria,160 and are thus closely regulated.
Extensive research has been done to elucidate the regulation system of Erm-based MLSB resistance. In 1980, studies using the ermCL-ermC operon established that erythromycin induces the translation of ErmC rRNA methyltransferases.161,162 Ribosomes with macrolides blocking the NPET are stalled at the ermCL leader peptide sequence positioned upstream of the ermC open-reading frame. The macrolide-mRNA-ribosome complex thus induces a conformational change on the ermCL-ermC mRNA which then exposes a second Shine–Dalgarno (SD2) sequence, previously sequestered within a stem-loop structure allowing for the translation of ErmC (Fig. 8C). Recent cryo-EM structures solved by Beckert et al. of erythromycin- and telithromycin-bound ribosomes stalled by ermDL unveil how the nascent ErmDL peptide residues cause the conserved PTC nucleotides to change conformation and initiate translational arrest.163 These findings are consistent with reports made by Dzyubak and Yap studying the S. aureus ermBL-ermB operon as they showed that the clinically prevalent constitutive expression of ermB results from complete loss of the regulatory sequence or impairment of the ermBL region.164 Selective pressure imposed by the use of non-inducing macrolides like tylosin as growth promoters in animal husbandry is attributed to the increased occurrence of constitutively expressed Erm genes.165 In the absence of MLSB drugs, erm mRNA reverts to the inactive conformation. Since the production of the resistance-conferring proteins and the unnecessary methylation of rRNA in an antibiotic-free context leads to reduced fitness, controlled expression and regulation of rRNA methyltransferase-encoding genetic material underpins the continued global spread of these determinants.
Circumventing resistance-conferring rRNA methylations
Apramycin and its derivatives (apralogs) have garnered attention as potential broad-spectrum treatment options against strains of MDR bacteria harboring m7G1405 resistance-conferring rRNA methyltransferases. Current research efforts are aimed at lowering apramycin ototoxicity and increasing its activity against pathogenic bacteria.166,167 Recent reports have demonstrated that apralogs with a furanosyl moiety at the 5 position (Fig. 9A) have reduced selectivity for the human A site, therefore, providing a starting point for the next generation of nontoxic aminoglycosides.168,169 In addition to improved activity against bacteria encoding ArmA, RmtB, RmtC, and RmtF the resulting 4,5-DOS apralogs are less amenable to inactivation by AMEs as compared to other DOS aminoglycosides due to the unique bicyclic scaffold.170,171 Other 4,5-DOS analogs like the paromomycin derivative, propylamycin, are also promising leads.172 These compounds are however still susceptible to resistance mediated by NpmA/B as they directly bind to A1408.171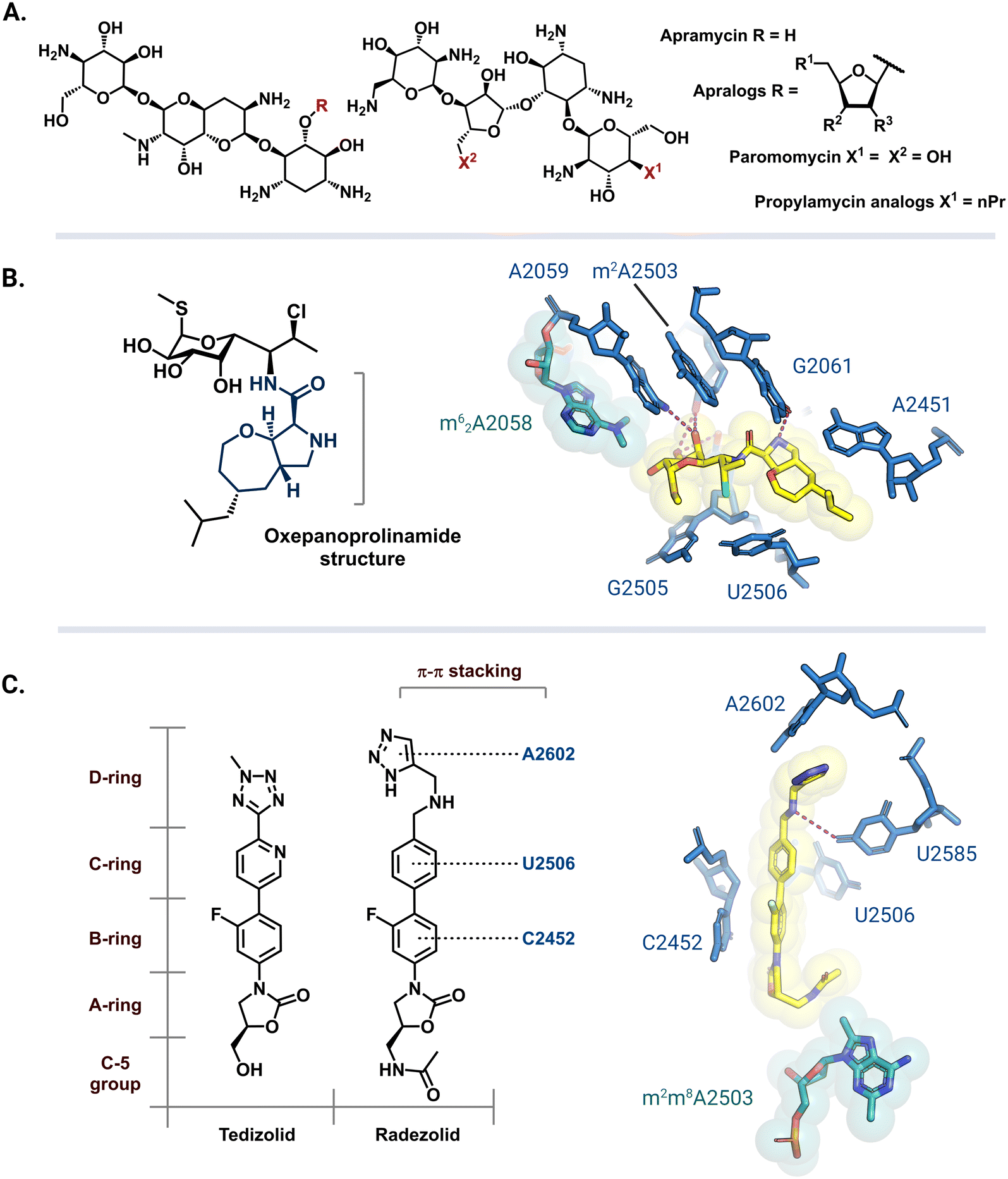 | ||
| Fig. 9 Antibiotic analogs that are active against bacteria with resistance rRNA methyltransferase genes. (A) Chemical structure of apramycin and 5-furanosyl appended apralogs. Studies have explored different substituents at R1, R2, and R3. (B) Chemical structure of iboxamycin and the structural basis for its binding in an Erm-modified ribosome (PDB: 7RQ9). (C) Chemical structures of tedizolid and radezolid and structure of radezolid (yellow) bound to Cfr-modified ribosome (PDB: 7S1K). Structure images were prepared using PyMol. | ||
The exploration of more than 500 lincosamide derivatives by Mitcheltree and colleagues led to an oxepanoprolinamide scaffold that aids iboxamycin evasion of both Erm and Cfr-based resistance.173 A cocrystal structure of iboxamycin and an m62A2058 modified T. thermophilus ribosome (Fig. 9B) shows that the antibiotic binds in the NPET/PTC site, and extends into the A-site pocket where it forms new hydrophobic interactions believed to counteract drug-m62A2058 clashes that would otherwise dislodge clindamycin and lincomycin.173 Similarly, since the discovery of Cfr mediated resistance against the oxazolidinone antibiotic linezolid, extensive SAR studies have led to more potent second generation derivatives like tedizolid and radezolid (Fig. 9C).174 Structures of ribosome-bound tedizolid and radezolid demonstrate additional interactions including π–π stacking with binding site residues as a result of aryl CD ring additions.175,176 A cryoEM structure of radezolid bound to a Cfr-modified ribosome shows that the m2m8A2503 residue shifts to avoid clashing with the C-5 acetamide.176 This conformation change is not expected for tedizolid with a hydroxymethyl at the C-5 position, which explains its increased affinity as compared to radezolid.174
Antibiotic derivatives that retain efficacy against bacteria harboring resistance-conferring rRNA methyltransferases do so by having additional binding contacts and/or evading clashes with the methylated residues. While the synthesis and biological evaluation of antibiotic analogs is a challenging and time-consuming task, the studies involving apramycin, tedizolid and radezolid, and iboxamycin show that more potent analogs can be discovered using this approach.
Targeting resistance-conferring rRNA methyltransferases
Several strategies have been employed to identify and develop small-molecule rRNA methyltransferase inhibitors to curb Erm-based MLSB resistance in bacteria. In 1995, Clancy and coworkers used high-throughput screening (HTS) to identify potential ErmC inhibitors with half maximal inhibitory concentrations (IC50) of 0.45 to 22.1 μM.177 To test for selectivity, inhibitory activity of the compounds was also tested against the E. coli methyltransferase EcoRI and catechol-O-methyltransferase (COMT) derived from rat liver. Their most promising hit, UK-105730 (Fig. 10A), was selective for ErmC and lowered the MIC of azithromycin in synergy assays against MLSB-resistant bacteria, but failed to protect mice from a clinical strain of ermC-harboring S. aureus. The hit compounds in the study lacked similarities in chemical structure but had remarkable selectivity and synergy profiles overall and could therefore be used as starting points in future searches.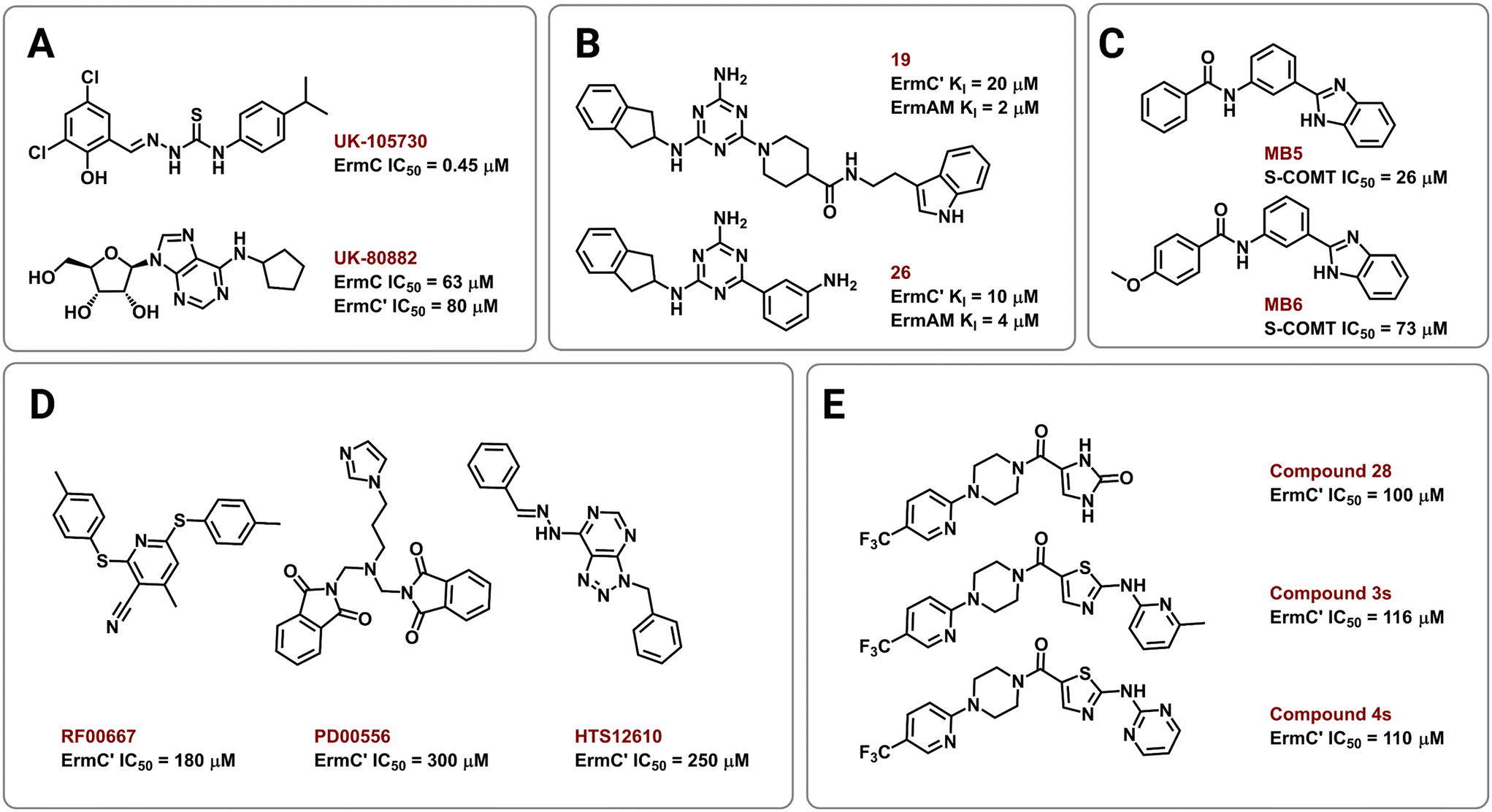 | ||
| Fig. 10 Chemical structures of Erm methyltransferase inhibitors. Erm inhibitors identified using (A) high throughput screening (HTS), (B) NMR based screening followed by an extensive SAR campaign, (C) in silico docking on ErmC′ and in vitro assays using COMT, (D) virtual screening using ErmC′ 3D structure, and (E) virtual screening then SAR of the lead compound after in vitro studies. | ||
In other efforts, Hajduk and colleagues identified 2,4,6-trisubstituted triazine-containing hits using an NMR-based screening method against the ErmAM protein.178 Subsequent SAR studies explored the utility of each substitution through the parallel synthesis of more than 600 analogs. The best compounds, 19 and 26 (Fig. 10B), had inhibitory constants (Ki) of 2 μM and 4 μM against ErmAM, and 20 μM and 10 μM against ErmC′, respectively. The leads identified using this technique target the conserved methyltransferase enzyme active site yet, interestingly, the compounds were reported to be ineffective against EcoRI methyltransferase hinting at selectivity for the Erm rRNA methyltransferases.
The availability of high-resolution crystal structures of rRNA methyltransferase enzymes has streamlined the computer-aided development of novel inhibitors. In 2006, Kreander and coworkers docked 200000 compounds in the SAM binding pocket of ErmC′ and selected 49 molecules for additional screening through in vitro assays using a soluble version of COMT eventually leading to two hits, MB5 and MB6 (Fig. 10C).179 However, the use of human methyltransferases for experimental validation could potentially select for cytotoxic compounds through targeting of conserved SAM-binding sites.
Subsequently, Feder et al. performed an in silico screen of 58679 small molecules using an ErmC′ crystal structure and identified 14 compounds, out of the 77 selected for in vitro and in vivo studies, predicted to bind at the rRNA binding site.180 The remaining 63 were predicted to bind in the pocket meant for the adenine substrate, and this criterion was met by the three most potent inhibitors, RF00667, PD00556, and HTS12610 (Fig. 10D). These three compounds lowered the MIC of erythromycin against E. coli harboring an ermC recombinant gene and had in vitro activity between 180 μM and 300 μM (IC50 against purified ErmC′). RF00667 and PD00556 were non-competitive with SAM while HTS12610 was competitive with the cosubstrate as it was modeled extending into the SAM binding pocket. The docking of UK-80882 (Fig. 10A) identified by Clancy et al. showed similar binding modes to the three hits; the modeled inhibitors were observed to interact with a crucial tyrosine residue via π–π stacking at the catalytic site.177,180 Following a similar strategy, the group again used virtual screening to identify inhibitors that bind both the SAM and the adenine binding pockets to allow for better in vitro activity while maintaining high ErmC′ selectivity.181 These new lead compounds (Fig. 10E) exhibited different inhibition modes, including: SAM-competitive (compound 4s), non-competitive (compound 28), and uncompetitive (compound 3s), and exhibited a greater binding affinity for the target site than the previously identified inhibitors.180,181
Collectively, these works set the stage for the rational design of Erm and other rRNA methyltransferase inhibitors, while also highlighting some of the challenges faced in targeting these enzymes. To the best of our knowledge, to date, no inhibitors of any 16S rRNA resistance methyltransferases or the Cfr and Cfr-like proteins have been identified, and none of the inhibitors reported so far have demonstrated efficacy in murine infection models. Thus, while a promising approach to revitalizing several key antibiotic classes, much work remains to be done.
Conclusion
As quickly as bacteria have evolved and broadened their arsenal of resistance mechanisms since the introduction of antibiotics into clinical practice, we have been correspondingly slow to mobilize efforts to counter such resistance despite its almost immediate identification. As part of these efforts, surveillance and identification of rRNA methyltransferases must be expanded; for example, data from Asia, Europe, and North America is ample as compared to Africa. Although there is a notable distribution trend for the rRNA methyltransferase families and their ever-expanding number of variants, it is also apparent that their rate of prevalence is still low compared to many other resistance mechanisms that affect some antibiotic classes. Thus, while new approaches are urgently needed to counter the effects of these resistance determinants, the window of opportunity remains open, for now, to mitigate their aminoglycoside, MLSB, and PhLOPSA resistance phenotypes.With improved technology and the availability of high-resolution crystal structures of the rRNA methyltransferases, the computer-aided design of inhibitors that can be used as drug adjuvants should be possible. The high sequence homogeneity between variants as well as the mechanistic and structural similarity between rRNA methyltransferase classes and families increases our chances to develop universal inhibitors. Although bacteria remain capable of developing resistance to adjuvants, the clinical success of β-lactamase inhibitors demonstrates that targeting resistance enzymes is a viable strategy in the ongoing fight against antimicrobial resistance.182
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding support provided by the National Institute of General Medical Sciences (R35 GM119426 to W. M. W.), National Institute of Allergy and Infectious Diseases (R01 AI088025 to G. L. C.), and Emory University through an Accelerator Grant from the Biological Discovery through Chemical Innovations initiative (to G. L. C. and W. M. W.).References
- L. Armand-Lefèvre, C. Angebault, F. Barbier, E. Hamelet, G. Defrance, E. Ruppé, R. Bronchard, R. Lepeule, J. C. Lucet, A. El Mniai, M. Wolff, P. Montravers, P. Plésiat and A. Andremont, Antimicrob. Agents Chemother., 2013, 57, 1488–1495 CrossRef PubMed.
- T. P. Van Boeckel, C. Brower, M. Gilbert, B. T. Grenfell, S. A. Levin, T. P. Robinson, A. Teillant and R. Laxminarayan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5649–5654 CrossRef CAS PubMed.
- M. Barlow and B. G. Hall, J. Mol. Evol., 2002, 55, 314–321 CrossRef CAS PubMed.
- J. Larsen, C. L. Raisen, X. Ba, N. J. Sadgrove, G. F. Padilla-González, M. S. J. Simmonds, I. Loncaric, H. Kerschner, P. Apfalter, R. Hartl, A. Deplano, S. Vandendriessche, B. Černá Bolfíková, P. Hulva, M. C. Arendrup, R. K. Hare, C. Barnadas, M. Stegger, R. N. Sieber, R. L. Skov, A. Petersen, Ø. Angen, S. L. Rasmussen, C. Espinosa-Gongora, F. M. Aarestrup, L. J. Lindholm, S. M. Nykäsenoja, F. Laurent, K. Becker, B. Walther, C. Kehrenberg, C. Cuny, F. Layer, G. Werner, W. Witte, I. Stamm, P. Moroni, H. J. Jørgensen, H. de Lencastre, E. Cercenado, F. García-Garrote, S. Börjesson, S. Hæggman, V. Perreten, C. J. Teale, A. S. Waller, B. Pichon, M. D. Curran, M. J. Ellington, J. J. Welch, S. J. Peacock, D. J. Seilly, F. J. E. Morgan, J. Parkhill, N. F. Hadjirin, J. A. Lindsay, M. T. G. Holden, G. F. Edwards, G. Foster, G. K. Paterson, X. Didelot, M. A. Holmes, E. M. Harrison and A. R. Larsen, Nature, 2022, 602, 135–141 CrossRef CAS PubMed.
- F. Shahbazi and S. Dashti-Khavidaki, Expert Rev. Clin. Pharmacol., 2015, 8, 423–448 CrossRef CAS PubMed.
- G. F. Hatfull, R. M. Dedrick and R. T. Schooley, Annu. Rev. Med., 2022, 73, 197–211 CrossRef PubMed.
- J. O'Neill, The Review on Antimicrobial Resistance, 2016, pp. 1–84 Search PubMed.
- B. D. Huttner, G. Catho, J. R. Pano-Pardo, C. Pulcini and J. Schouten, Clin. Microbiol. Infect., 2020, 26, 808–810 CrossRef CAS PubMed.
- CDC A, Centers Dis. Control Prev Search PubMed.
- S. Soltani, S. Faramarzi, M. Zandi, R. Shahbahrami, A. Jafarpour, S. Akhavan Rezayat, I. Pakzad, F. Abdi, P. Malekifar and R. Pakzad, New Microbes New Infect., 2021, 43, 100910 CrossRef CAS PubMed.
- R. E. Nelson, K. M. Hatfield, H. Wolford, M. H. Samore, R. D. Scott, S. C. Reddy, B. Olubajo, P. Paul, J. A. Jernigan and J. Baggs, Clin. Infect. Dis., 2021, 72, S17–S26 CrossRef PubMed.
- A. Towse, C. K. Hoyle, J. Goodall, M. Hirsch, J. Mestre-Ferrandiz and J. H. Rex, Health Policy, 2017, 121, 1025–1030 CrossRef PubMed.
- K. Outterson, J. H. Powers, G. W. Daniel and M. B. McClellan, Health Aff., 2015, 34, 277–285 CrossRef PubMed.
- L. Pantel, T. Florin, M. Dobosz-Bartoszek, E. Racine, M. Sarciaux, M. Serri, J. Houard, J. M. Campagne, R. M. de Figueiredo, C. Midrier, S. Gaudriault, A. Givaudan, A. Lanois, S. Forst, A. Aumelas, C. Cotteaux-Lautard, J. M. Bolla, C. Vingsbo Lundberg, D. L. Huseby, D. Hughes, P. Villain-Guillot, A. S. Mankin, Y. S. Polikanov and M. Gualtieri, Mol. Cell, 2018, 70, 83–94.e7 CrossRef CAS.
- J. M. Crawford and J. Clardy, Chem. Commun., 2011, 47, 7559–7566 RSC.
- R. D. Miller, A. Iinishi, S. M. Modaresi, B.-K. Yoo, T. D. Curtis, P. J. Lariviere, L. Liang, S. Son, S. Nicolau, R. Bargabos, M. Morrissette, M. F. Gates, N. Pitt, R. P. Jakob, P. Rath, T. Maier, A. G. Malyutin, J. T. Kaiser, S. Niles, B. Karavas, M. Ghiglieri, S. E. J. Bowman, D. C. Rees, S. Hiller and K. Lewis, Nat. Microbiol., 2022, 7, 1661–1672 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS PubMed.
- D. Nichols, N. Cahoon, E. M. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewis and S. S. Epstein, Appl. Environ. Microbiol., 2010, 76, 2445–2450 CrossRef CAS PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS.
- X. Liu, P. J. Pai, W. Zhang, Y. Hu, X. Dong, P. Y. Qian, D. Chen and H. Lam, Sci. Rep., 2016, 6, 1–12 CrossRef CAS PubMed.
- A. Gaurav, V. Gupta, S. K. Shrivastava and R. Pathania, Commun. Biol., 2021, 4, 1–12 CrossRef PubMed.
- S. F. Askari Rizvi, Biomed. J. Sci. Tech. Res., 2018, 7, 001–010 Search PubMed.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, MedChemComm, 2019, 10, 1719–1739 RSC.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- E. N. Parker, B. S. Drown, E. J. Geddes, H. Y. Lee, N. Ismail, G. W. Lau and P. J. Hergenrother, Nat. Microbiol., 2020, 5, 67–75 CrossRef CAS PubMed.
- H. Douafer, V. Andrieu, O. Phanstiel and J. M. Brunel, J. Med. Chem., 2019, 62, 8665–8681 CrossRef CAS PubMed.
- L. A. Hicks, M. G. Bartoces, R. M. Roberts, K. J. Suda, R. J. Hunkler, T. H. Taylor and S. J. Schrag, Clin. Infect. Dis., 2015, 60, 1308–1316 Search PubMed.
- N. Demeshkina, L. Jenner, E. Westhof, M. Yusupov and G. Yusupova, Nature, 2012, 484, 256–259 CrossRef CAS PubMed.
- A. P. Carter, W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Nature, 2000, 407, 340–348 CrossRef CAS PubMed.
- N. Demeshkina, L. Jenner, E. Westhof, M. Yusupov and G. Yusupova, Nature, 2012, 484, 256–259 CrossRef CAS PubMed.
- B. François, R. J. M. Russell, J. B. Murray, F. Aboul-ela, B. Masquida, Q. Vicens and E. Westhof, Nucleic Acids Res., 2005, 33, 5677–5690 CrossRef.
- Q. Vicens and E. Westhof, Structure, 2001, 9, 647–658 CrossRef CAS.
- Q. Vicens and E. Westhof, Chem. Biol., 2002, 9, 747–755 CrossRef CAS.
- Q. Vicens and E. Westhof, J. Mol. Biol., 2003, 326, 1175–1188 CrossRef CAS PubMed.
- D. Nicholson, T. A. Edwards, A. J. O'Neill and N. A. Ranson, Structure, 2020, 28, 1087–1100.e3 CrossRef CAS PubMed.
- Z. L. Watson, F. R. Ward, R. Méheust, O. Ad, A. Schepartz, J. F. Banfield and J. H. D. Cate, eLife, 2020, 9, 1–62 Search PubMed.
- M. A. Borovinskaya, R. D. Pai, W. Zhang, B. S. Schuwirth, J. M. Holton, G. Hirokawa, H. Kaji, A. Kaji and J. H. D. Cate, Nat. Struct. Mol. Biol., 2007, 14, 727–732 CrossRef CAS PubMed.
- T. Golkar, A. V. Bassenden, K. Maiti, D. P. Arya, T. M. Schmeing and A. M. Berghuis, Commun. Biol., 2021, 4, 1–8 CrossRef.
- T. Matt, C. L. Ng, K. Lang, S. H. Sha, R. Akbergenov, D. Shcherbakov, M. Meyer, S. Duscha, J. Xie, S. R. Dubbaka, D. Perez-Fernandez, A. Vasella, V. Ramakrishnan, J. Schacht and E. C. Böttger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10984–10989 CrossRef CAS.
- D. N. Wilson, Nat. Rev. Microbiol., 2013, 12, 35–48 CrossRef PubMed.
- M. S. Svetlov, E. A. Syroegin, E. V. Aleksandrova, G. C. Atkinson, S. T. Gregory, A. S. Mankin and Y. S. Polikanov, Nat. Chem. Biol., 2021, 17, 412–420 CrossRef CAS PubMed.
- F. Schlünzen, J. M. Harms, F. Franceschi, H. A. S. Hansen, H. Bartels, R. Zarivach and A. Yonath, Structure, 2003, 11, 329–338 CrossRef PubMed.
- B. Llano-Sotelo, J. Dunkle, D. Klepacki, W. Zhang, P. Fernandes, J. H. D. Cate and A. S. Mankin, Antimicrob. Agents Chemother., 2010, 54, 4961–4970 CrossRef CAS PubMed.
- J. L. Hansen, J. A. Ippolito, N. Ban, P. Nissen, P. B. Moore and T. A. Steitz, Mol. Cell, 2002, 10, 117–128 CrossRef CAS PubMed.
- M. R. Tirumalai, M. Rivas, Q. Tran and G. E. Fox, Microbiol. Mol. Biol. Rev., 2021, 85(4), e00104-21 CrossRef PubMed.
- K. H. Kaminska, E. Purta, L. H. Hansen, J. M. Bujnicki, B. Vester and K. S. Long, Nucleic Acids Res., 2009, 38, 1652–1663 CrossRef PubMed.
- L. S. Mccoy, Y. Xie and Y. Tor, Wiley Interdiscip. Rev.: RNA, 2011, 2, 209–232 CrossRef CAS PubMed.
- F. Schlünzen, R. Zarivach, J. Harms, A. Bashan, A. Tocilj, R. Albrecht, A. Yonath and F. Franceschi, Nature, 2001, 413, 814–821 CrossRef PubMed.
- J. A. Ippolito, Z. F. Kanyo, D. Wang, F. J. Franceschi, P. B. Moore, T. A. Steitz and E. M. Duffy, J. Med. Chem., 2008, 51, 3353–3356 CrossRef CAS PubMed.
- F. Schlünzen, E. Pyetan, P. Fucini, A. Yonath and J. M. Harms, Mol. Microbiol., 2004, 54, 1287–1294 CrossRef PubMed.
- J. M. Harms, F. Schlünzen, P. Fucini, H. Bartels and A. Yonath, BMC Biol., 2004, 2, 1–10 CrossRef PubMed.
- K. Mangano, J. Marks, D. Klepacki, C. K. Saha, G. C. Atkinson, N. Vázquez-Laslop and A. S. Mankin, Nat. Chem. Biol., 2022, 18, 1277–1286 CrossRef CAS PubMed.
- S. Arenz, M. F. Juette, M. Graf, F. Nguyen, P. Huter, Y. S. Polikanov, S. C. Blanchard and D. N. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7527–7532 CrossRef CAS PubMed.
- P. V. Adrian, C. Mendrick, D. Loebenberg, P. McNicholas, K. J. Shaw, K. P. Klugman, R. S. Hare and T. A. Black, Antimicrob. Agents Chemother., 2000, 44, 3101–3106 CrossRef CAS.
- J. D. Walter, M. Hunter, M. Cobb, G. Traeger and P. C. Spiegel, Nucleic Acids Res., 2012, 40, 360–370 CrossRef CAS PubMed.
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- L. Armand-Lefèvre, V. Leflon-Guibout, J. Bredin, F. Barguellil, A. Amor, J. M. Pagès and M. H. Nicolas-Chanoine, Antimicrob. Agents Chemother., 2003, 47, 1165–1168 CrossRef.
- L. Fernández and R. E. W. Hancock, Clin. Microbiol. Rev., 2012, 25, 661–681 CrossRef PubMed.
- S. Coyne, N. Rosenfeld, T. Lambert, P. Courvalin and B. Périchon, Antimicrob. Agents Chemother., 2010, 54, 4389–4393 CrossRef CAS PubMed.
- D. M. Daigle, L. Cao, S. Fraud, M. S. Wilke, A. Pacey, R. Klinoski, N. C. Strynadka, C. R. Dean and K. Poole, J. Bacteriol., 2007, 189, 5441–5451 CrossRef CAS PubMed.
- O. Danilchanka, C. Mailaender and M. Niederweis, Antimicrob. Agents Chemother., 2008, 52, 2503–2511 CrossRef CAS PubMed.
- G. D. Wright, Curr. Opin. Microbiol., 1999, 2, 499–503 CrossRef CAS PubMed.
- M. Zieliński, J. Park, B. Sleno and A. M. Berghuis, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- S. Arenz, F. Nguyen, R. Beckmann and D. N. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5401–5406 CrossRef CAS PubMed.
- C. R. Fostier, L. Monlezun, F. Ousalem, S. Singh, J. F. Hunt and G. Boël, FEBS Lett., 2021, 595, 675–706 CrossRef CAS PubMed.
- L. K. R. Sharkey, T. A. Edwards and A. J. O'Neill, MBio, 2016, 7, e01975-15 CrossRef PubMed.
- D. Tu, G. Blaha, P. B. Moore and T. A. Steitz, Cell, 2005, 121, 257–270 CrossRef CAS PubMed.
- A. W. Struck, M. L. Thompson, L. S. Wong and J. Micklefield, ChemBioChem, 2012, 13, 2642–2655 CrossRef CAS PubMed.
- T. L. Grove, J. S. Benner, M. I. Radle, J. H. Ahlum, B. J. Landgraf, C. Krebs and S. J. Booker, Science, 2011, 332, 604–607 CrossRef CAS PubMed.
- M. Liu and S. Douthwaite, Antimicrob. Agents Chemother., 2002, 46, 1629–1633 CrossRef CAS PubMed.
- M. S. Svetlov, N. Vázquez-Laslop and A. S. Mankin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13673–13678 CrossRef CAS PubMed.
- K. S. Long, J. Poehlsgaard, C. Kehrenberg, S. Schwarz and B. Vester, Antimicrob. Agents Chemother., 2006, 50, 2500–2505 CrossRef CAS PubMed.
- L. K. Smith and A. S. Mankin, Antimicrob. Agents Chemother., 2008, 52, 1703–1712 CrossRef CAS PubMed.
- M. Liu and S. Douthwaite, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14658–14663 CrossRef CAS PubMed.
- I. Lebars, C. Husson, S. Yoshizawa, S. Douthwaite and D. Fourmy, J. Mol. Biol., 2007, 372, 525–534 CrossRef CAS PubMed.
- I. Treede, L. Jakobsen, F. Kirpekar, B. Vester, G. Weitnauer, A. Bechthold and S. Douthwaite, Mol. Microbiol., 2003, 49, 309–318 CrossRef CAS PubMed.
- P. A. Mann, L. Xiong, A. S. Mankin, A. S. Chau, C. A. Mendrick, D. J. Najarian, C. A. Cramer, D. Loebenberg, E. Coates, N. J. Murgolo, F. M. Aarestrup, R. V. Goering, T. A. Black, R. S. Hare and P. M. McNicholas, Mol. Microbiol., 2001, 41, 1349–1356 CrossRef CAS PubMed.
- E. G. Kuiper and G. L. Conn, J. Biol. Chem., 2014, 289, 26189–26200 CrossRef CAS PubMed.
- G. P. Dinos, Br. J. Pharmacol., 2017, 174, 2967–2983 CrossRef CAS PubMed.
- E. Cundliffe, Annu. Rev. Microbiol., 1989, 43, 207–233 CrossRef CAS PubMed.
- M. M. Almutairi, S. R. Park, S. Rose, D. A. Hansen, N. Vázquez-Laslop, S. Douthwaite, D. H. Sherman and A. S. Mankin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12956–12961 CrossRef CAS PubMed.
- K. Yokoyama, Y. Doi, K. Yamane, H. Kurokawa, N. Shibata, K. Shibayama, T. Yagi, H. Kato and Y. Arakawa, Lancet, 2003, 362, 1888–1893 CrossRef CAS PubMed.
- M. Galimand, S. Sabtcheva, P. Courvalin and T. Lambert, Antimicrob. Agents Chemother., 2005, 49, 2949–2953 CrossRef CAS PubMed.
- F. Yan, J. M. Lamarre, R. Röhrich, J. Wiesner, H. Jomaa, A. S. Markin and D. G. Fujimori, J. Am. Chem. Soc., 2010, 132, 3953–3964 CrossRef CAS PubMed.
- K. H. Kaminska, E. Purta, L. H. Hansen, J. M. Bujnicki, B. Vester and K. S. Long, Nucleic Acids Res., 2010, 38, 1652–1663 CrossRef CAS PubMed.
- A. M. B. Giessing, S. S. Jensen, A. Rasmussen, L. H. Hansen, A. Gondela, K. Long, B. Vester and F. Kirpekar, RNA, 2009, 15, 327–336 CrossRef CAS PubMed.
- M. Nosrati, D. Dey, A. Mehrani, S. E. Strassler, N. Zelinskaya, E. D. Hoffer, S. M. Stagg, C. M. Dunham and G. L. Conn, J. Biol. Chem., 2019, 294, 17642–17653 CrossRef CAS PubMed.
- N. Husain, S. Obranić, L. Koscinski, J. Seetharaman, F. Babić, J. M. Bujnicki, G. Maravić-Vlahoviek and J. Sivaraman, Nucleic Acids Res., 2011, 39, 1903–1918 CrossRef CAS PubMed.
- E. Schmitt, M. Galimand, M. Panvert, P. Courvalin and Y. Mechulam, J. Mol. Biol., 2009, 388, 570–582 CrossRef CAS PubMed.
- R. MacMaster, N. Zelinskaya, M. Savic, C. R. Rankin and G. L. Conn, Nucleic Acids Res., 2010, 38, 7791–7799 CrossRef CAS PubMed.
- G. Schluckebier, P. Zhong, K. D. Stewart, T. J. Kavanaugh and C. Abad-Zapatero, J. Mol. Biol., 1999, 289, 277–291 CrossRef CAS PubMed.
- A. K. Boal, T. L. Grove, M. I. McLaughlin, N. H. Yennawar, S. J. Booker and A. C. Rosenzweig, Science, 2011, 332, 1089–1092 CrossRef CAS PubMed.
- J. A. Dunkle, K. Vinal, P. M. Desai, N. Zelinskaya, M. Savic, D. M. West, G. L. Conn and C. M. Dunham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6275–6280 CrossRef CAS PubMed.
- Y. Doi, J. I. Wachino and Y. Arakawa, Antimicrob. Agents Chemother., 2008, 52, 2287–2288 CrossRef CAS PubMed.
- M. C. Roberts, J. Sutcliffe, P. Courvalin, L. B. Jensen, J. Rood and H. Seppala, Antimicrob. Agents Chemother., 1999, 43, 2823–2830 CrossRef CAS PubMed.
- M. Gniadkowski, I. Schneider, A. Pałucha, R. Jungwirth, B. Mikiewicz and A. Bauernfeind, Antimicrob. Agents Chemother., 1998, 42, 827–832 CrossRef CAS PubMed.
- E. Taylor, A. M. Bal, I. Balakrishnan, N. M. Brown, P. Burns, M. Clark, M. Diggle, H. Donaldson, I. Eltringham, J. Folb, N. Gadsby, M. Macleod, N. V. D. V. Ratnaraja, C. Williams, M. Wootton, S. Sriskandan, N. Woodford and K. L. Hopkins, J. Antimicrob. Chemother., 2021, 76, 2428–2436 CrossRef CAS PubMed.
- P. Bogaerts, M. Galimand, C. Bauraing, A. Deplano, R. Vanhoof, R. De Mendonca, H. Rodriguez-Villalobos, M. Struelens and Y. Glupczynski, J. Antimicrob. Chemother., 2007, 59, 459–464 CrossRef CAS PubMed.
- S. Ghafoor, B. Zeshan, M. Khan and S. Kabir, Pak. J. Pharm. Sci., 2021, 34, 1737–1741 CAS.
- B. González-Zorn, T. Teshager, M. Casas, M. C. Porrero, M. A. Moreno, P. Courvalin and L. Domínguez, Emerging Infect. Dis., 2005, 11, 954–956 CrossRef PubMed.
- L. Hidalgo, B. Gutierrez, C. M. Ovejero, L. Carrilero, S. Matrat, C. K. S. Saba, A. Santos-Lopez, D. Thomas-Lopez, A. Hoefer, M. Suarez, G. Santurde, C. Martin-Espada and B. Gonzalez-Zorn, Antimicrob. Agents Chemother., 2013, 57, 4532–4534 CrossRef CAS PubMed.
- S. A. Granier, L. Hidalgo, A. S. Millan, J. A. Escudero, B. Gutierrez, A. Brisabois and B. Gonzalez-Zorn, Antimicrob. Agents Chemother., 2011, 55, 5262–5266 CrossRef CAS PubMed.
- Z. Belbel, H. Chettibi, M. Dekhil, A. Ladjama, S. Nedjai and J. M. Rolain, Microb. Drug Resist., 2014, 20, 310–315 CrossRef CAS PubMed.
- M. A. E. G. El-Sayed-Ahmed, M. A. Amin, W. M. Tawakol, L. Loucif, S. Bakour and J. M. Rolain, Antimicrob. Agents Chemother., 2015, 59, 3602–3605 CrossRef CAS PubMed.
- S. Shoma, M. Kamruzzaman, A. N. Ginn, J. R. Iredell and S. R. Partridge, Diagn. Microbiol. Infect. Dis., 2014, 78, 93–97 CrossRef CAS PubMed.
- T. R. Fritsche, M. Castanheira, G. H. Miller, R. N. Jones and E. S. Armstrong, Antimicrob. Agents Chemother., 2008, 52, 1843–1845 CrossRef CAS PubMed.
- T. Tada, H. Uchida, T. Hishinuma, S. Watanabe, M. Tohya, K. Kuwahara-Arai, S. Mya, K. N. Zan, T. Kirikae and H. H. Tin, J. Global Antimicrob. Resist., 2020, 22, 122–125 CrossRef PubMed.
- L. Chen, Z. L. Chen, J. H. Liu, Z. L. Zeng, J. Y. Ma and H. X. Jiang, J. Antimicrob. Chemother., 2007, 59, 880–885 CrossRef CAS PubMed.
- J. Xia, L. X. Fang, K. Cheng, G. H. Xu, X. R. Wang, X. P. Liao, Y. H. Liu and J. Sun, Front. Microbiol., 2017, 8, 529 Search PubMed.
- Y. Doi, K. Yokoyama, K. Yamane, J. I. Wachino, N. Shibata, T. Yagi, K. Shibayama, H. Kato and Y. Arakawa, Antimicrob. Agents Chemother., 2004, 48, 491–496 CrossRef CAS PubMed.
- Y. Zhou, W. Ai, Y. Guo, X. Wu, B. Wang, Y. Xu, L. Rao, H. Zhao, X. Wang and F. Yu, Microbiol. Spectrum, 2022, 10, e0237121 CrossRef PubMed.
- M. A. Davis, K. N. K. Baker, L. H. Orfe, D. H. Shah, T. E. Besser and D. R. Call, Antimicrob. Agents Chemother., 2010, 54, 2666–2669 CrossRef CAS PubMed.
- J. Xia, J. Sun, L. Li, L. X. Fang, H. Deng, R. S. Yang, X. P. Li, X. P. Liao and Y. H. Liu, Antimicrob. Agents Chemother., 2015, 59, 7921–7922 CrossRef CAS PubMed.
- E. Taylor, E. Jauneikaite, S. Sriskandan, N. Woodford and K. L. Hopkins, J. Med. Microbiol., 2022, 71, 001531 CrossRef CAS PubMed.
- M. Galimand, P. Courvalin and T. Lambert, Antimicrob. Agents Chemother., 2012, 56, 3960–3962 CrossRef CAS PubMed.
- M. F. C. Bueno, G. R. Francisco, J. A. O'Hara, D. De Oliveira Garcia and Y. Doi, Antimicrob. Agents Chemother., 2013, 57, 2397–2400 CrossRef CAS PubMed.
- J. A. O'Hara, P. McGann, E. C. Snesrud, R. J. Clifford, P. E. Waterman, E. P. Lesho and Y. Doi, Antimicrob. Agents Chemother., 2013, 57, 2413–2416 CrossRef PubMed.
- L. Hidalgo, K. L. Hopkins, B. Gutierrez, C. M. Ovejero, S. Shukla, S. Douthwaite, K. N. Prasad, N. Woodford and B. Gonzalez-Zorn, J. Antimicrob. Chemother., 2013, 68, 1543–1550 CrossRef CAS PubMed.
- J. I. Wachino, K. Shibayama, H. Kurokawa, K. Kimura, K. Yamane, S. Suzuki, N. Shibata, Y. Ike and Y. Arakawa, Antimicrob. Agents Chemother., 2007, 51, 4401–4409 CrossRef CAS PubMed.
- M. Savic, J. Lovrić, T. I. Tomic, B. Vasiljevic and G. L. Conn, Nucleic Acids Res., 2009, 37, 5420–5431 CrossRef CAS PubMed.
- Y. A. Al Sheikh, M. A. M. Marie, J. John, L. G. Krishnappa and K. H. M. Dabwab, Libyan J. Med., 2014, 9, 24432 CrossRef PubMed.
- J. W. Marsh, M. P. Pacey, C. Ezeonwuka, S. L. Ohm, D. Snyder, V. S. Cooper, L. H. Harrison, Y. Doi and M. M. Mustapha, J. Antimicrob. Chemother., 2019, 74, 521–523 CrossRef CAS PubMed.
- J. I. Wachino, K. Shibayama, K. Kimura, K. Yamane, S. Suzuki and Y. Arakawa, FEMS Microbiol. Lett., 2010, 311, 56–60 CrossRef CAS PubMed.
- A. A.-A. Abdul-Wahid and A. M. Almohana, Kufa J. Nurs. Sci., 2015, 5, 156–165 Search PubMed.
- A. Kawai, M. Suzuki, K. Tsukamoto, Y. Minato and Y. Doi, Antimicrob. Agents Chemother., 2021, 65, e01009-21 CrossRef PubMed.
- Y. A. Chabbert, Ann. Inst. Pasteur, 1956, 90, 787–790 CAS.
- L. J. Griffith, W. E. Ostrander, C. G. Mullins and D. E. Beswick, Science, 1965, 147, 746–747 CrossRef CAS PubMed.
- C. J. Lai, J. E. Dahlberg and B. Weisblum, Biochemistry, 1973, 12, 457–460 CrossRef CAS PubMed.
- A. Kumar, A. Kumar and K. S. Prasad, Gene Rep., 2021, 23, 101175 CrossRef CAS.
- E. L. Rieke, T. B. Moorman, E. L. Douglass and M. L. Soupir, Sci. Total Environ., 2018, 610–611, 1173–1179 CrossRef CAS PubMed.
- A. Béjaoui, M. Gharbi, S. Bitri, D. Nasraoui, W. Ben Aziza, K. Ghedira, M. Rfaik, L. Marzougui, A. Ghram and A. Maaroufi, Antibiotics, 2022, 11, 830 CrossRef PubMed.
- H. Krüger, X. Ji, D. Hanke, A. K. Schink, S. Fiedler, H. Kaspar, Y. Wang, S. Schwarz, C. Wu and A. T. Feßler, J. Antimicrob. Chemother., 2022, 77, 2296–2298 CrossRef PubMed.
- A. L. Greninger, A. Addetia, K. Starr, R. J. Cybulski, M. K. Stewart, S. J. Salipante, A. B. Bryan, B. Cookson, C. Gaudreau, S. Bekal and F. C. Fang, Clin. Infect. Dis., 2020, 71, 1896–1904 CrossRef CAS PubMed.
- S. Schwarz, C. Werckenthin and C. Kehrenberg, Antimicrob. Agents Chemother., 2000, 44, 2530–2533 CrossRef CAS PubMed.
- S. M. Toh, L. Xiong, C. A. Arias, M. V. Villegas, K. Lolans, J. Quinn and A. S. Mankin, Mol. Microbiol., 2007, 64, 1506–1514 CrossRef CAS PubMed.
- C. Leão, L. Clemente, M. Cara d'Anjo, T. Albuquerque and A. Amaro, Antibiotics, 2022, 11, 1439 CrossRef PubMed.
- M. Wali, M. S. Shah, T. U. Rehman, H. Wali, M. Hussain, L. Zaman, F. U. Khan and A. H. Mangi, J. Infect. Public Health, 2022, 15, 1142–1146 CrossRef PubMed.
- C. Y. Mei, H. Wu, Y. Wang, Z. Y. Wang, Q. C. Ma, P. C. Shen, Y. Y. Zhou, J. Wang and X. Jiao, Int. J. Antimicrob. Agents, 2021, 57 Search PubMed.
- Y. Liu, Y. Wang, C. Wu, Z. Shen, S. Schwarz, X. D. Du, L. Dai, W. Zhang, Q. Zhang and J. Shen, Antimicrob. Agents Chemother., 2012, 56, 1650–1654 CrossRef CAS PubMed.
- J. Huang, J. Sun, Y. Wu, L. Chen, D. Duan, X. Lv and L. Wang, Int. J. Antimicrob. Agents, 2019, 54, 43–48 CrossRef CAS PubMed.
- L. M. Deshpande, D. S. Ashcraft, H. P. Kahn, G. Pankey, R. N. Jones, D. J. Farrell and R. E. Mendes, Antimicrob. Agents Chemother., 2015, 59, 6256–6261 CrossRef CAS PubMed.
- Y. Tang, L. Dai, O. Sahin, Z. Wu, M. Liu and Q. Zhang, J. Antimicrob. Chemother., 2017, 72, 1581–1588 CrossRef CAS PubMed.
- T. Candela, J. C. Marvaud, T. K. Nguyen and T. Lambert, Int. J. Antimicrob. Agents, 2017, 50, 496–500 CrossRef CAS PubMed.
- M. Sassi, F. Guérin, A. Zouari, R. Beyrouthy, M. Auzou, M. Fines-Guyon, S. Potrel, L. Dejoies, A. Collet, S. Boukthir, G. Auger, R. Bonnet and V. Cattoir, J. Antimicrob. Chemother., 2019, 74, 1469–1472 CrossRef CAS PubMed.
- S. Pang, P. Boan, T. Lee, S. Gangatharan, S. J. Tan, D. Daley, Y. T. Lee and G. W. Coombs, Int. J. Antimicrob. Agents, 2020, 55, 105831 CrossRef CAS PubMed.
- C. Crowe-McAuliffe, V. Murina, K. J. Turnbull, S. Huch, M. Kasari, H. Takada, L. Nersisyan, A. Sundsfjord, K. Hegstad, G. C. Atkinson, V. Pelechano, D. N. Wilson and V. Hauryliuk, Nat. Commun., 2022, 13, 1860 CrossRef CAS PubMed.
- L. M. Almeida, A. Gaca, P. M. Bispo, F. Lebreton, J. T. Saavedra, R. A. Silva, I. D. Basílio-Júnior, F. M. Zorzi, P. H. Filsner, A. M. Moreno and M. S. Gilmore, Front. Public Health., 2020, 8, 518 CrossRef PubMed.
- V. Stojković, M. F. Ulate, F. Hidalgo-Villeda, E. Aguilar, C. Monge-Cascante, M. Pizarro-Guajardo, K. Tsai, E. Tzoc, M. Camorlinga, D. Paredes-Sabja, C. Quesada-Gómez, D. G. Fujimori and C. Rodríguez, Antimicrob. Agents Chemother., 2019, 64, 01074-19 CrossRef PubMed.
- N. M. Andersen and S. Douthwaite, J. Mol. Biol., 2006, 359, 777–786 CrossRef CAS PubMed.
- S. Kimura and T. Suzuki, Nucleic Acids Res., 2009, 38, 1341–1352 CrossRef PubMed.
- G. N. Basturea, K. E. Rudd and M. P. Deutscher, RNA, 2006, 12, 426–434 CrossRef CAS PubMed.
- S. Čubrilo, F. Babić, S. Douthwaite and G. M. Vlahoviček, RNA, 2009, 15, 1492–1497 CrossRef PubMed.
- B. Gutierrez, J. A. Escudero, A. S. Millan, L. Hidalgo, L. Carrilero, C. M. Ovejero, A. Santos-Lopez, D. Thomas-Lopez and B. Gonzalez-Zorn, Antimicrob. Agents Chemother., 2012, 56, 2335–2341 CrossRef CAS PubMed.
- V. S. Lioy, S. Goussard, V. Guerineau, E. J. Yoon, P. Courvalin, M. Galimand and C. Grillot-Courvalin, RNA, 2014, 20, 382–391 CrossRef CAS PubMed.
- Y. Yang, B. Ou, L. Chen, Y. Song, Y. Yang, Q. Zhang, L. Li, W. L. Tham, D. H. Francis and G. Zhu, J. Global Antimicrob. Resist., 2016, 6, 32–38 CrossRef PubMed.
- Y. Ishizaki, Y. Shibuya, C. Hayashi, K. Inoue, T. Kirikae, T. Tada, T. Miyoshi-Akiyama and M. Igarashi, J. Antibiot., 2018, 71, 798–807 CrossRef CAS PubMed.
- J. M. LaMarre, J. B. Locke, K. J. Shaw and A. S. Mankin, Antimicrob. Agents Chemother., 2011, 55, 3714–3719 CrossRef CAS PubMed.
- N. Vázquez-Laslop, H. Ramu and A. Mankin, in Ribosomes, Springer, Vienna, 2011, pp. 377–392 Search PubMed.
- N. Vázquez-Laslop, H. Ramu, D. Klepacki, K. Kannan and A. S. Mankin, EMBO J., 2010, 29, 3108–3117 CrossRef PubMed.
- P. Gupta, S. Sothiselvam, N. Vázquez-Laslop and A. S. Mankin, Nat. Commun., 2013, 4, 1–9 Search PubMed.
- S. Horinouchi and B. Weisblum, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 7079–7083 CrossRef CAS PubMed.
- T. J. Gryczan, G. Grandi, J. Hahn, R. Grandi and D. Dubnau, Nucleic Acids Res., 1980, 8, 6081–6097 CrossRef CAS PubMed.
- B. Beckert, E. C. Leroy, S. Sothiselvam, L. V. Bock, M. S. Svetlov, M. Graf, S. Arenz, M. Abdelshahid, B. Seip, H. Grubmüller, A. S. Mankin, C. A. Innis, N. Vázquez-Laslop and D. N. Wilson, Nat. Commun., 2021, 12, 4466 CrossRef CAS PubMed.
- E. Dzyubak and M. N. F. Yap, Antimicrob. Agents Chemother., 2016, 60, 7178–7188 CrossRef CAS PubMed.
- L. E. de Vries, H. Christensen and Y. Agersø, Mobile Genet. Elem., 2012, 2, 72–80 CrossRef PubMed.
- S. L. Zada, B. Ben Baruch, L. Simhaev, H. Engel and M. Fridman, J. Am. Chem. Soc., 2020, 142, 3077–3087 CrossRef CAS PubMed.
- A. Sonousi, J. C. K. Quirke, P. Waduge, T. Janusic, M. Gysin, K. Haldimann, S. Xu, S. N. Hobbie, S. H. Sha, J. Schacht, C. S. Chow, A. Vasella, E. C. Böttger and D. Crich, ChemMedChem, 2021, 16, 335–339 CrossRef CAS PubMed.
- S. L. Zada, B. Ben Baruch, L. Simhaev, H. Engel and M. Fridman, J. Am. Chem. Soc., 2020, 142, 3077–3087 CrossRef CAS PubMed.
- A. Sonousi, J. C. K. Quirke, P. Waduge, T. Janusic, M. Gysin, K. Haldimann, S. Xu, S. N. Hobbie, S. H. Sha, J. Schacht, C. S. Chow, A. Vasella, E. C. Böttger and D. Crich, ChemMedChem, 2021, 16, 335–339 CrossRef CAS PubMed.
- J. C. K. Quirke, G. C. Sati, A. Sonousi, M. Gysin, K. Haldimann, E. C. Böttger, A. Vasella, S. N. Hobbie and D. Crich, ChemMedChem, 2022, 17, e202200120 CrossRef CAS PubMed.
- M. Juhas, E. Widlake, J. Teo, D. L. Huseby, J. M. Tyrrell, Y. S. Polikanov, O. Ercan, A. Petersson, S. Cao, A. F. Aboklaish, A. Rominski, D. Crich, E. C. Böttger, T. R. Walsh, D. Hughes and S. N. Hobbie, J. Antimicrob. Chemother., 2019, 74, 944–952 CrossRef CAS PubMed.
- T. Matsushita, G. C. Sati, N. Kondasinghe, M. G. Pirrone, T. Kato, P. Waduge, H. S. Kumar, A. C. Sanchon, M. Dobosz-Bartoszek, D. Shcherbakov, M. Juhas, S. N. Hobbie, T. Schrepfer, C. S. Chow, Y. S. Polikanov, J. Schacht, A. Vasella, E. C. Böttger and D. Crich, J. Am. Chem. Soc., 2019, 141, 5051–5061 CrossRef CAS PubMed.
- M. J. Mitcheltree, A. Pisipati, E. A. Syroegin, K. J. Silvestre, D. Klepacki, J. D. Mason, D. W. Terwilliger, G. Testolin, A. R. Pote, K. J. Y. Wu, R. P. Ladley, K. Chatman, A. S. Mankin, Y. S. Polikanov and A. G. Myers, Nature, 2021, 599, 507–512 CrossRef CAS PubMed.
- J. B. Locke, J. Finn, M. Hilgers, G. Morales, S. Rahawi, G. C. Kedar, J. J. Picazo, W. Im, K. J. Shaw and J. L. Stein, Antimicrob. Agents Chemother., 2010, 54, 5337–5343 CrossRef CAS PubMed.
- A. Wright, K. Deane-Alder, E. Marschall, R. Bamert, H. Venugopal, T. Lithgow, D. W. Lupton and M. J. Belousoff, ACS Pharmacol. Transl. Sci., 2020, 3, 425–432 CrossRef CAS PubMed.
- K. Tsai, V. Stojković, D. J. Lee, I. D. Young, T. Szal, D. Klepacki, N. Vázquez-Laslop, A. S. Mankin, J. S. Fraser and D. G. Fujimori, Nat. Struct. Mol. Biol., 2022, 29, 162–171 CrossRef CAS PubMed.
- J. Clancy, B. J. Schmieder, J. W. Petitpas, M. Manousos, J. A. Williams, J. A. Faiella, A. E. Girard and P. R. McGuirk, J. Antibiot., 1995, 48, 1273–1279 CrossRef CAS PubMed.
- P. J. Hajduk, J. Dinges, J. M. Schkeryantz, D. Janowick, M. Kaminski, M. Tufano, D. J. Augeri, A. Petros, V. Nienaber, P. Zhong, R. Hammond, M. Coen, B. Beutel, L. Katz and S. W. Fesik, J. Med. Chem., 1999, 42, 3852–3859 CrossRef CAS PubMed.
- K. Kreander, M. Kurkela, A. Siiskonen, P. Vuorela and P. Tammela, Pharmazie, 2006, 61, 247–248 CAS.
- M. Feder, E. Purta, L. Koscinski, S. Čubrilo, G. M. Vlahovicek and J. M. Bujnicki, ChemMedChem, 2008, 3, 316–322 CrossRef CAS PubMed.
- I. P. Foik, I. Tuszynska, M. Feder, E. Purta, F. Stefaniak and J. M. Bujnicki, Eur. J. Med. Chem., 2018, 146, 60–67 CrossRef CAS PubMed.
- K. Bush and P. A. Bradford, Nat. Rev. Microbiol., 2019, 17, 295–306 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |