
A photoluminescent and electrochemiluminescent probe based on an iridium(III) complex with a boronic acid-functionalised ancillary ligand for the selective detection of mercury(II) ions†
Kyoung-Rok
Kim
,
Jinrok
Oh
and
Jong-In
Hong
 *
*
Department of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-747, Korea. E-mail: jihong@snu.ac.kr
First published on 2nd October 2023
Abstract
Exposure to mercury(II) ions (Hg2+) can cause various diseases such as Minamata disease, acrodynia, Alzheimer's disease, and Hunter–Russell syndrome, and even organ damage. Therefore, real-time and accurate monitoring of Hg2+ in environmental samples is crucial. In this study, we report a photoluminescent (PL) and electrochemiluminescent (ECL) probe based on a cyclometalated Ir(III) complex for the selective detection of Hg2+. The introduction of a reaction site, o-aminomethylphenylboronic acid, on the ancillary ligands allowed a prompt transmetalation reaction to take place between Hg2+ and boronic acid. This reaction resulted in significant decreases of the PL and ECL signals due to the photo-induced electron transfer from the Ir(III) complex to the Hg2+ ions. The probe was applied to the selective detection of Hg2+, and the signal changes revealed a linear correlation with Hg2+ concentrations in the range of 0–10 μM (LOD = 0.72 μM for PL, 8.03 nM for ECL). The designed probe allowed the successful quantification of Hg2+ in tap water samples, which proves its potential for the selective detection of Hg2+ in environmental samples.
Introduction
Rapid industrialisation has resulted in the frequent release of toxic chemicals, which can cause serious environmental problems. Among various released chemicals, the mercury(II) ion (Hg2+) is classified as a persistent, bio-accumulative, and toxic chemical, and is considered a threat to humans and the environment.1 Although Hg2+ is colourless, tasteless, and odourless, it can be easily absorbed into the body through the cell membranes of the skin, respiratory tract, and gastrointestinal tract.2 The assimilation of Hg2+ in the human body can damage the central nervous system, endocrine glands, and organs.3,4 Furthermore, chronic exposure to Hg2+ has been associated with various diseases, such as Minamata disease, acrodynia, Alzheimer's disease, and Hunter–Russell syndrome.5 Consequently, the U.S. Environmental Protection Agency (EPA) has recommended that the maximum level of inorganic mercury in drinking water is 10 nM.6 The common methods employed for the detection of Hg2+ include inductively coupled plasma atomic emission spectroscopy (ICP-AES),7 atomic absorption spectroscopy,8,9 electrochemical analysis,10–12 surface-enhanced Raman spectroscopy (SERS),13,14 piezoelectric detection,15 and colourimetric and fluorometric detection.16 In particular, fluorescent chemosensors for Hg2+ have been developed based on the Hg2+-catalysed reaction and coordination of heteroatoms with Hg2+.17 However, these detection methods are very expensive and time-consuming, and require skilled laboratory professionals. Furthermore, they require bulky and fragile instrumental setup, including an additional optical source.Electrochemiluminescence (ECL) is a luminescence process involving sequential electron transfers between electrogenerated species on an electrode surface.18 Compared with the photoluminescence (PL) process, ECL offers several advantages due to its low background signal and high sensitivity. In addition, ECL rarely requires complicated procedures, complex conditions, or bulky equipment for analysis.18–20 Consequently, ECL has been considered a powerful tool for real-time, on-site sample analysis, and several studies have reported systems that combine the ECL technique with chemosensors.21–41 So far, most ECL molecular probes have been developed using tris(2,2′-bipyridine)ruthenium(II) complexes ([Ru(bpy)32+]) with their characteristically low metal-centred state (MC state), making it challenging to adjust the wavelength with changes in the molecular structure.42 Among various luminophores considered as replacements for Ru(II) complexes, Ir(III) complexes are regarded as promising ECL luminophores due to their high ECL quantum yields and colour tunability through ligand modification by high ligand field stabilization energies.33,43–48 Hong and co-workers reported the first ECL analysis system for the detection of Hg2+ ions based on the reaction of a small molecular probe with these ions.35 Unfortunately, this system could not be applied in field monitoring because large amounts of organic solvents were required. Consequently, the development of new ECL probes with high selectivity and sensitivity toward Hg2+ in aqueous solutions is greatly desired.
Herein, we report a new PL and ECL chemodosimetric probe based on an iridium(III) complex for the determination of Hg2+ (Scheme 1). The design strategy of probe 1 is based on the following: (i) [Ir(piq)2(bpy)][PF6] (piq = 1-phenylisoquinoline, bpy = bipyridine) was selected as a luminophore, and (ii) o-aminomethylphenylboronic acid was introduced into the ancillary ligand as a PL and ECL signalling modulator, as well as a reaction site for the Hg2+ ions.49–53 Probe 1 was expected to exhibit significant PL and ECL changes towards Hg2+ ions in aqueous solutions when compared to other Ir(III)-based probes because this probe has cationic charge and hydrophilic o-aminomethylphenylboronic acid, which imparts high solubility in aqueous solutions. Probe 1 promptly reacted with Hg2+ ions through the transmetalation reaction to generate 1-Hg2+, resulting in decreased PL and ECL signals. Probe 1, as analysed through PL and ECL, exhibited high sensitivity and selectivity in detecting Hg2+ ions within 2 min in aqueous solutions with small amounts of organic solvent (10% acetonitrile). These results imply significant potential for on-site Hg2+ detection, compared with other PL-based chemodosimeters, as illustrated in Table S1.† In addition, probe 1 was successfully applied to ECL analysis of Hg2+ ions in tap water samples.
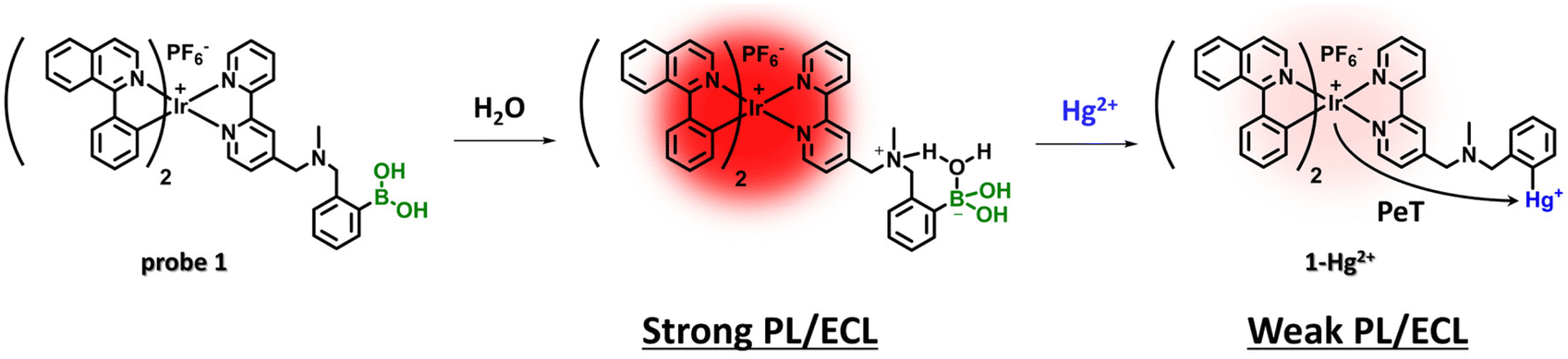 | ||
| Scheme 1 The detection mechanism of probe 1 for Hg2+ ions. | ||
Experimental
Probes 1 and 2 were synthesised as shown in Scheme 2. Detailed synthetic procedures for compounds 1–11 are provided in the ESI.†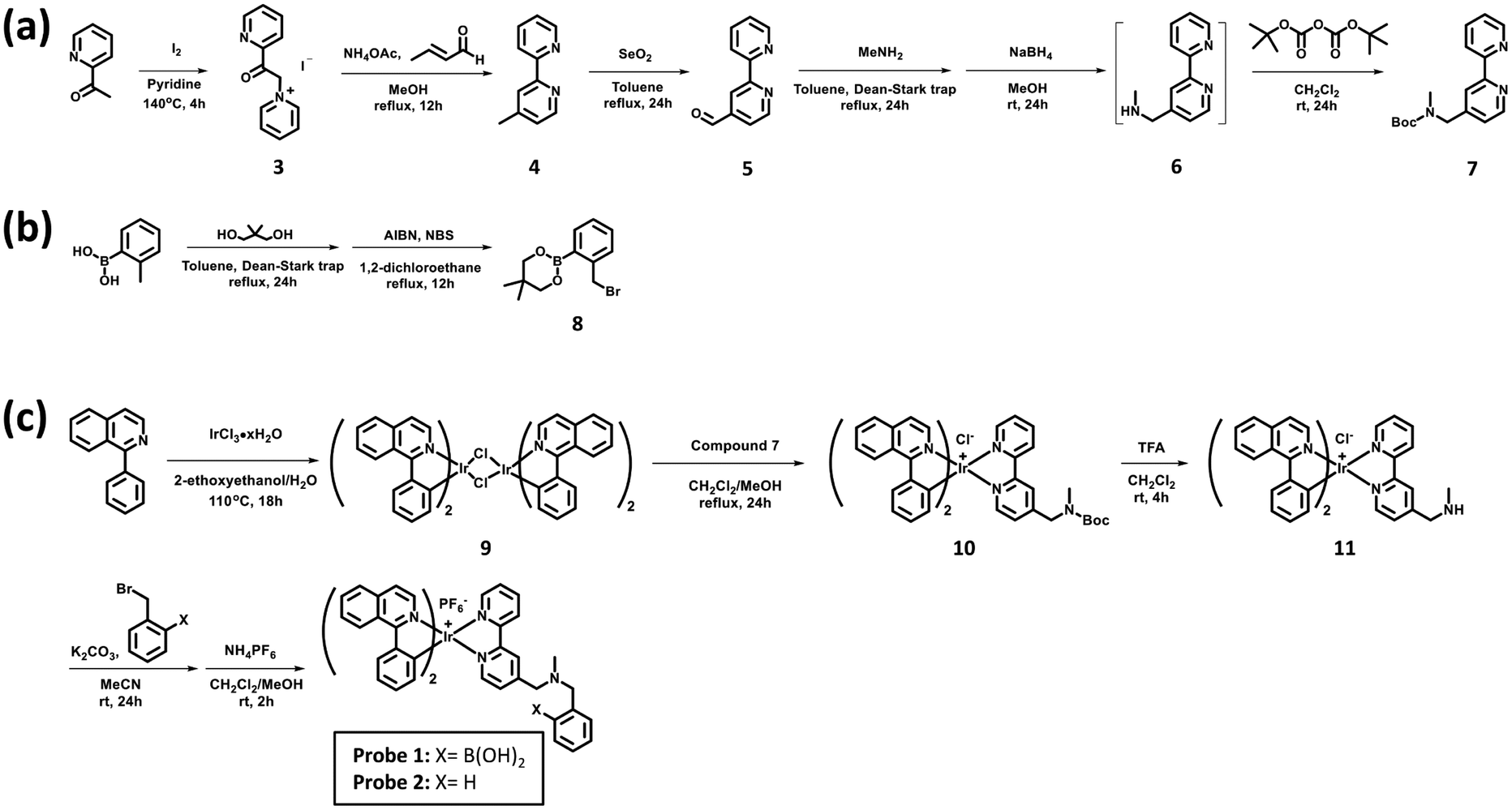 | ||
| Scheme 2 Synthetic routes of (a) compound 7, (b) compound 8, and (c) probes 1 and 2. | ||
Synthesis of 1
A mixture of crude compound 11 (17 mg, 20 μmol), compound 8 (7 mg, 25 μmol), and potassium carbonate (13 mg, 94 μmol) in 0.2 mL of acetonitrile was stirred at room temperature for 12 h. The solvent was evaporated under reduced pressure and extracted with dichloromethane three times. The organic phase was washed with water and brine, dried over anhydrous sodium sulphate, and evaporated. The residue was purified by silica gel column chromatography (acetonitrile/H2O/sat. KNO3(aq) = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1). The product was re-dissolved in dichloromethane/methanol (1:1, 1 mL) and stirred at room temperature for 2 h with potassium hexafluorophosphate (37 mg, 0.2 mmol). The solution was filtered, and the filtrate was evaporated under reduced pressure. The residue was triturated with dichloromethane and diethylether. The resulting precipitate was filtered, washed with diethylether and dried in vacuo to afford 1 (10 mg, 47%). 1H NMR (300 MHz, CDCl3) δ 8.99–8.84 (m, 2H), 8.78 (d, J = 8.2 Hz, 1H), 8.60 (s, 1H), 8.44 (s, 2H), 8.28–8.20 (m, 2H), 8.10 (t, J = 7.3 Hz, 1H), 7.89 (dd, J = 6.2, 3.0 Hz, 2H), 7.80–7.69 (m, 6H), 7.64 (d, J = 5.6 Hz, 1H), 7.40–7.28 (m, 8H), 7.22 (d, J = 7.1 Hz, 1H), 7.10 (t, J = 7.6 Hz, 2H), 6.88 (t, J = 7.0 Hz, 2H), 6.25 (d, J = 7.3 Hz, 2H), 3.82 (d, J = 5.5 Hz, 4H), 2.28 (s, 3H). 13C NMR (126 MHz, CD3CN) δ 168.49, 168.45, 156.01, 155.25, 153.47, 153.38, 150.68, 150.61, 145.57, 145.53, 140.80, 140.68, 139.47, 137.09, 137.07, 135.92, 131.98, 131.87, 130.86, 130.83, 130.75, 130.57, 130.50, 129.14, 129.02, 129.01, 128.64, 128.48, 127.56, 127.54, 127.45, 126.76, 126.75, 126.17, 126.15, 125.63, 124.86, 122.32, 122.30, 121.98, 121.90, 62.75, 58.55, 40.79. LRMS (MALDI-TOF) m/z: [M]+ calc. for C49H40BIrN5O2 934.290, found 934.063. HRMS (FAB) m/z: [M − H2O]+ calc. for C49H38BIrN5O 916.2799, found 916.2793.
:4:1). The product was re-dissolved in dichloromethane/methanol (1:1, 1 mL) and stirred at room temperature for 2 h with potassium hexafluorophosphate (37 mg, 0.2 mmol). The solution was filtered, and the filtrate was evaporated under reduced pressure. The residue was triturated with dichloromethane and diethylether. The resulting precipitate was filtered, washed with diethylether and dried in vacuo to afford 1 (10 mg, 47%). 1H NMR (300 MHz, CDCl3) δ 8.99–8.84 (m, 2H), 8.78 (d, J = 8.2 Hz, 1H), 8.60 (s, 1H), 8.44 (s, 2H), 8.28–8.20 (m, 2H), 8.10 (t, J = 7.3 Hz, 1H), 7.89 (dd, J = 6.2, 3.0 Hz, 2H), 7.80–7.69 (m, 6H), 7.64 (d, J = 5.6 Hz, 1H), 7.40–7.28 (m, 8H), 7.22 (d, J = 7.1 Hz, 1H), 7.10 (t, J = 7.6 Hz, 2H), 6.88 (t, J = 7.0 Hz, 2H), 6.25 (d, J = 7.3 Hz, 2H), 3.82 (d, J = 5.5 Hz, 4H), 2.28 (s, 3H). 13C NMR (126 MHz, CD3CN) δ 168.49, 168.45, 156.01, 155.25, 153.47, 153.38, 150.68, 150.61, 145.57, 145.53, 140.80, 140.68, 139.47, 137.09, 137.07, 135.92, 131.98, 131.87, 130.86, 130.83, 130.75, 130.57, 130.50, 129.14, 129.02, 129.01, 128.64, 128.48, 127.56, 127.54, 127.45, 126.76, 126.75, 126.17, 126.15, 125.63, 124.86, 122.32, 122.30, 121.98, 121.90, 62.75, 58.55, 40.79. LRMS (MALDI-TOF) m/z: [M]+ calc. for C49H40BIrN5O2 934.290, found 934.063. HRMS (FAB) m/z: [M − H2O]+ calc. for C49H38BIrN5O 916.2799, found 916.2793.
Synthesis of 2
A mixture of crude compound 11 (9 mg, 11 μmol), benzyl bromide (4 mg, 23 μmol), potassium iodide (4 mg, 24 μmol), and potassium carbonate (3 mg, 22 μmol) in 0.2 mL of acetonitrile was stirred at room temperature for 12 h. The solvent was evaporated under reduced pressure and extracted with dichloromethane three times. The organic phase was washed with water and brine, dried over anhydrous sodium sulphate, and evaporated. The residue was re-dissolved in dichloromethane/methanol (1:1, 1 mL) and stirred at room temperature for 2 h with ammonium hexafluorophosphate (18 mg, 0.11 mmol). The solution was filtered and evaporated, and the residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 50:1) to afford 2 (6 mg, 53%). 1H NMR (400 MHz, CDCl3) δ 8.92 (dd, J = 5.9, 3.1 Hz, 2H), 8.73 (d, J = 8.2 Hz, 1H), 8.58 (s, 1H), 8.26 (d, J = 8.0 Hz, 2H), 8.15 (t, J = 7.8 Hz, 1H), 7.90 (dd, J = 6.3, 3.1 Hz, 2H), 7.77 (dd, J = 6.4, 3.2 Hz, 4H), 7.72 (d, J = 4.9 Hz, 1H), 7.63 (d, J = 5.4 Hz, 1H), 7.49 (d, J = 20.2 Hz, 1H), 7.36 (m, J = 6.5 Hz, 7H), 7.28 (d, J = 7.3 Hz, 2H), 7.20 (d, J = 6.8 Hz, 1H), 7.11 (t, J = 7.6 Hz, 2H), 6.89 (t, J = 7.4 Hz, 2H), 6.27 (t, J = 6.6 Hz, 2H), 3.70 (d, J = 72.2 Hz, 4H), 2.26 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 169.04, 168.99, 155.65, 155.47, 153.55, 153.41, 150.11, 145.47, 145.41, 140.21, 139.71, 136.99, 132.17, 131.70, 130.87, 130.85, 130.68, 130.66, 128.70, 128.49, 127.99, 127.59, 127.57, 126.93, 126.88, 126.31, 126.26, 125.38, 122.29, 121.98, 121.96, 62.03, 29.68. HRMS (FAB) m/z: [M]+ calc. for C49H39IrN5 890.2835, found 890.2834.
Results and discussion
Spectroscopic properties of 1
We checked the changes in the UV-vis and PL spectra of probe 1 in the presence and absence of Hg2+ in an aqueous medium (pH 7.4, HEPES buffer/CH3CN = 9:1, v/v). Probe 1 displayed an intense absorption peak at 288 nm, which was attributed to the spin-allowed ligand-centred (1LC) transition states (Fig. S1†). Furthermore, the broad absorption peak detected around 330–510 nm was attributed to the mixing of metal-to-ligand charge-transfer (1MLCT and 3MLCT) transitions, ligand-centred (3LC) transitions, and ligand-to-ligand charge-transfer (3LLCT and 1LLCT) transitions.54 When the concentration of Hg2+ ions in solution increased, the absorption bands of probe 1 showed a slight red-shift. Subsequently, the PL response of probe 1 to Hg2+ was evaluated. As the amount of Hg2+ increased, the PL signal (λex = 400 nm) gradually decreased until it reached a plateau after the addition of 10 μM Hg2+ (Fig. 1a). This indicated that probe 1 reacted in 1:1 stoichiometry with Hg2+. The limit of detection (LOD) was 0.73 μM from the linear correlation (R2 = 0.99) between the concentration of Hg2+ ions (in the range of 0–10 μM) and the PL intensity (Fig. 1b).
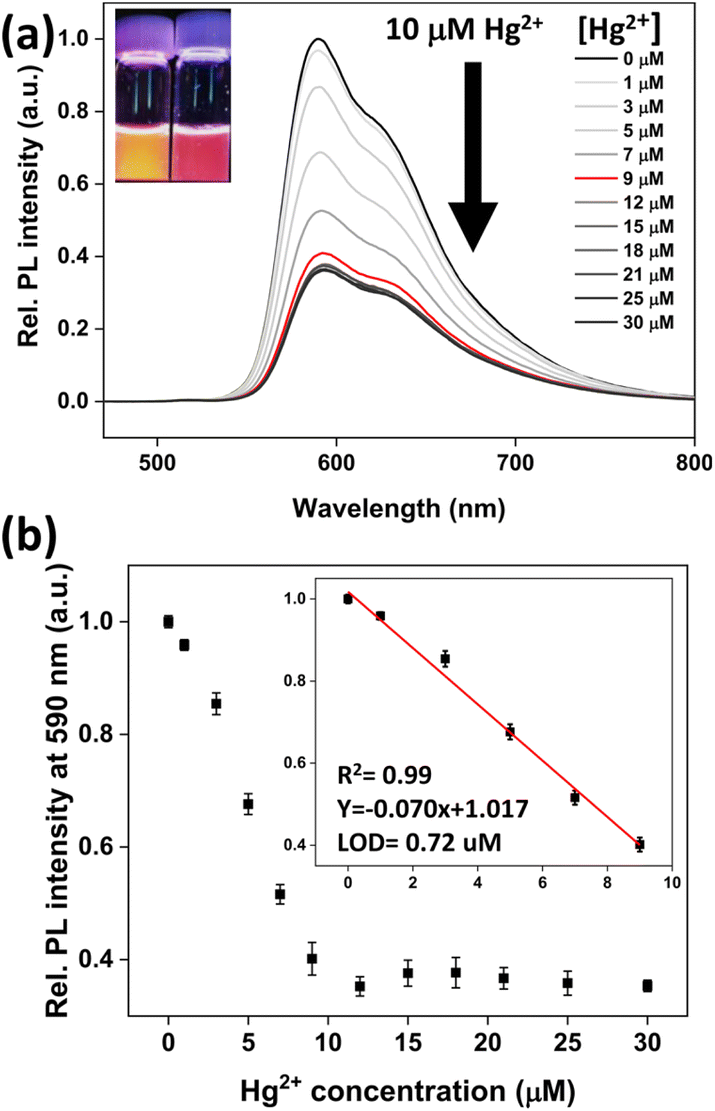 | ||
| Fig. 1 (a) PL emission spectra (λex = 400 nm) of probe 1 (10 μM) with increasing Hg2+ ion concentrations in CH3CN/H2O (1:9 v/v, pH 7.4, 10 mM HEPES). Inset: photographs of the probe solutions without and with Hg2+ under 365 nm UV irradiation. (b) PL intensity changes of probe 1 at 590 nm upon the addition of Hg2+ ions. Inset: a linear correlation between the PL intensity at 590 nm and the concentration of Hg2+. | ||
To further understand the quenching behaviour of probe 1 in the presence of Hg2+, its fluorescence lifetimes before and after the addition of Hg2+ ions were measured by time-correlated single-photon counting, whereby this probe exhibited single-exponential decay with a lifetime of 0.82 μs (Fig. S2a†). However, in the presence of Hg2+ ions, an additional shorter lifetime of ∼0.1 μs was observed, thus revealing double-exponential decay in the PL decay curve. This can be attributed to the generation of the reaction product, 1-Hg2+, via the transmetalation reaction between Hg2+ and boronic acid on the ancillary ligands. Upon the addition of Hg2+, the lifetimes decreased, thus indicating that the quenching mechanism was highly dependent on dynamic quenching (Fig. S2b†).55–57 These changes were observed with the addition of up to 10 μM Hg2+ ions, which were consistent with the signal change trends observed in the PL response experiments.
To determine the selectivity of probe 1 in an aqueous solution, the PL spectra were measured in the presence of various metal ions. As expected, the PL of probe 1 decreased effectively only in the presence of Hg2+, whereas other metal ions exhibited negligible PL signal changes (Fig. 2). This selectivity can be attributed to the transmetalation between phenylboronic acid and Hg2+ ions.49–53 The PL of 1-Hg2+ generated through the reaction between probe 1 and Hg2+ was quenched by the electron transfer from the excited Ir(III) complex to Hg2+.58
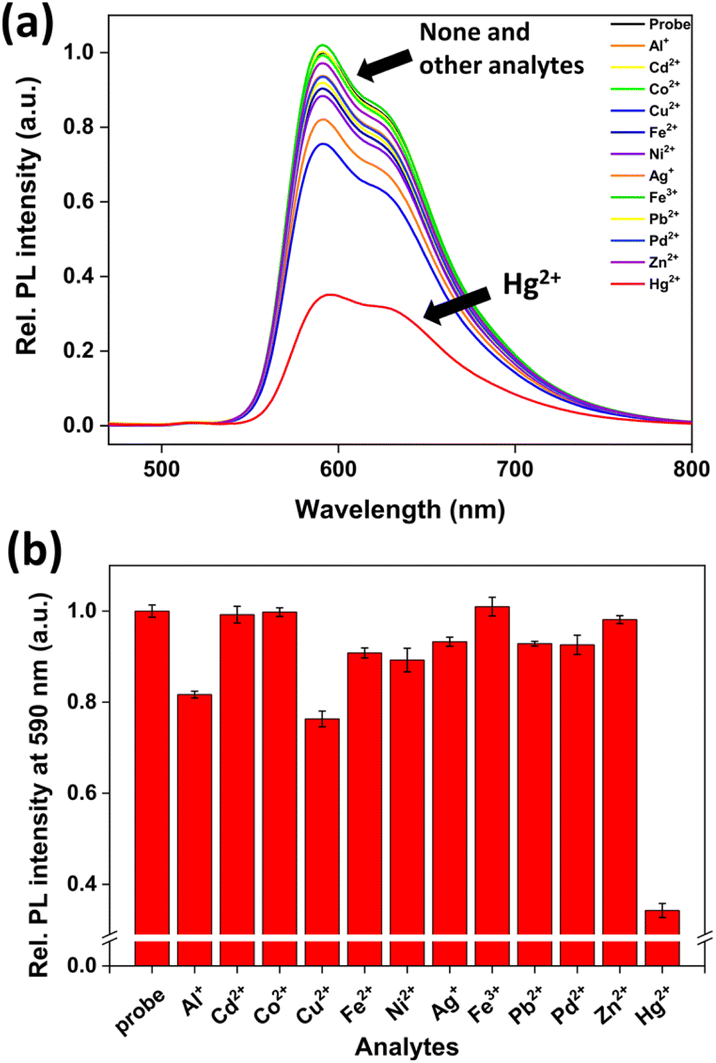 | ||
| Fig. 2 (a) PL emission spectra (λex = 400 nm) of probe 1 (10 μM) in the presence of various analytes (40 μM each; Hg2+, 20 μM) in CH3CN/H2O (1:9 v/v, pH 7.4, 10 mM HEPES). (b) PL emission intensities at 590 nm of probe 1 (10 μM) upon the respective addition of various metal ions (40 μM). | ||
To investigate the probe's pH susceptibility, the PL intensity of probe 1 was measured in the pH range of 4–10 after the addition of Hg2+ (Fig. 3a). The probe showed a significant change in PL in the presence of Hg2+ over a wide pH range of 5–9, thus confirming its high stability in the detection of Hg2+. The relatively weaker Hg2+ responses observed at pH 4 and 10 were presumably due to inefficient formation of boronate and generation of mercury(II) hydroxide, respectively.59,60 Subsequently, the sensing ability toward Hg2+ ions in different ratios of CH3CN–HEPES buffer solution was evaluated (Fig. 3b). Upon increasing the ratio of the HEPES buffer solution in the CH3CN–HEPES buffer solution, a remarkable PL signal change in the presence of Hg2+ was observed since in an aqueous solution o-aminomethylphenylboronic acid undergoes H2O insertion to generate boronate.60 Since the formation of boronate is essential in a transmetalation reaction,61–63 the reaction of probe 1 with Hg2+ ions occurred more efficiently in solutions with high H2O content. Therefore, a significant PL response of probe 1 toward Hg2+ was induced in a 1:9 CH3CN–HEPES buffer solution. The PL intensity of probe 1 was saturated within 2 minutes in the presence of 10 μM Hg2+ ions, indicating that Hg2+ ions could be detected quickly under our experimental conditions (Fig. S3†).
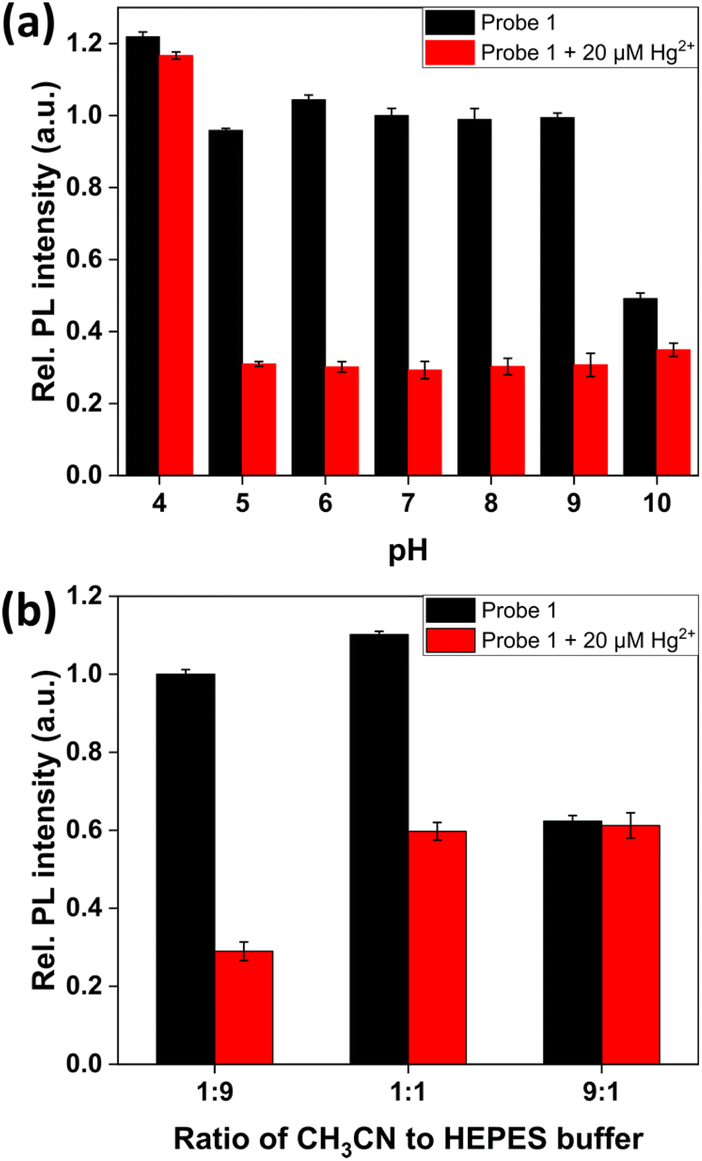 | ||
| Fig. 3 PL intensity changes of probe 1 (10 μM, λem = 590 nm, λex = 400 nm) towards 20 μM Hg2+ ions (a) in CH3CN/H2O (1:9 v/v, pH 7.4, 10 mM HEPES) under varying pH conditions and (b) in different ratios of CH3CN to 10 mM HEPES buffer solution (pH = 7.40). | ||
Proposed sensing mechanism
1H nuclear magnetic resonance (NMR) experiments with probe 1 in CD3CN/D2O (1:1) were then conducted (Fig. S4†). Upon the addition of Hg2+ ions to probe 1, the signals at 3.75 ppm corresponding to the two methylene protons connected to the tertiary nitrogen were slightly downfield-shifted because of the binding of Hg2+ and the tertiary amine. However, after the addition of an excess amount of Hg2+ ions, the proton signals significantly decreased, and two new methylene proton peaks appeared at 3.72 and 3.53 ppm due to the reaction between boronic acid and Hg2+ ions. Similarly, the methyl proton signal at 2.25 ppm moved gradually downfield with the addition of Hg2+ ions with a decreased intensity, while a new peak appeared at 2.21 ppm with an increased intensity. High-performance liquid chromatography coupled with Thermo Scientific linear ion trap-orbitrap mass spectrometry (HPLC-LTQ-orbitrap) analysis was performed to confirm the reaction product generated after the addition of Hg2+ ions (Fig. S5†). Before the addition of Hg2+ ions, an m/z signal appeared at 934.6, which was attributed to probe 1. Upon the addition of 20 μM Hg2+ ions to probe 1, the m/z signal of probe 1 disappeared, and a major peak at 1115.6 appeared, which was attributed to 1-Hg2+. Both NMR and HPLC-LTQ-orbitrap analyses confirmed that probe 1 reacted effectively with Hg2+ through transmetalation to generate 1-Hg2+.
To confirm the role of boronic acid in the PL and ECL signal changes with Hg2+ ions, probe 2 without boronic acid was designed. The PL of probe 1 significantly decreased in the presence of 20 μM Hg2+ ions, while probe 2 hardly showed any PL changes, even after the addition of an excess amount of Hg2+ ions (Fig. 4). These results indicated that the transmetalation of boronic acid with Hg2+ ions is essential in the PL/ECL sensing mechanism.
 | ||
| Fig. 4 PL intensity changes of (a) probe 1 and (b) probe 2 (10 μM) upon the addition of Hg2+ ions in CH3CN/H2O (1:9 v/v, pH 7.4, 10 mM HEPES). | ||
To verify the signal changes in PL, an electrochemical analysis with DPV was performed (Fig. S6 and Table S2†). The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of probe 1 were calculated to be −5.78 and −3.12 eV, respectively. Meanwhile, a mixture of probe 1 and 10 μM Hg2+ ions revealed a new peak at −0.54 V (vs. Fc/Fc+), which was attributed to the LUMO energy level (−4.26 eV) of –Hg+, while the HOMO and LUMO energy levels of the Ir(III) complex were similar to those of probe 1. The LUMO energy level of –Hg+ lies between the HOMO and LUMO levels of the Ir(III) complex (Fig. S7†), which indicated that the PL signals of probe 1 were restrained by the photo-induced electron transfer (PeT) process from the Ir(III) complex to –Hg+. Therefore, probe 1 showed decreased PL signals in the presence of Hg2+. Meanwhile, 1-Hg2+ showed a more stabilised LUMO energy level by 0.13 eV compared to probe 1 due to the mercury cation introduced into probe 1 through the transmetalation reaction. These results are in accordance with the red-shift tendency observed in the UV-vis absorption spectra of probe 1 with Hg2+ ions (Fig. S1†).
ECL properties of 1
Fig. 5a shows the ECL emission profile during cyclic voltammetry in aqueous media (pH 7.4, HEPES buffer/CH3CN = 9:1 v/v, 0.1 M 2-(dibutylamino)ethanol (DBAE), 0.1 M HEPES, 0.1% polidocanol, and 0.1 M tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte). Probe 1 produced a strong ECL signal around 1.05 V. However, the ECL signals gradually decreased as the amounts of Hg2+ increased. Interestingly, the ECL signals showed a linear correlation with Hg2+ concentrations in the range of 0–10 μM (Fig. 5b, R2 = 0.98) with a LOD of 8.03 nM, which was much lower than the LOD determined by the PL method. This value falls below the maximum allowable contaminant level of mercury in drinking water recommended by the U.S. EPA. The high sensitivity of probe 1 demonstrates its ability to effectively detect Hg2+ ions in waste streams, which can have significant adverse effects on the environment.
 | ||
| Fig. 5 (a) ECL intensities of probe 1 (10 μM) upon the addition of Hg2+ in CH3CN/H2O (1:9 v/v, pH 7.4, 100 mM DBAE, 0.1 M HEPES, 0.1% polidocanol and 0.1 M TBAP as the supporting electrolyte) while the potential is swept at a GC disk electrode (diameter 2 mm) in the range of −0.3–1.3 V (scan rate: 0.1 V s−1). (b) ECL intensity changes of probe 1 at 1.05 V upon the addition of Hg2+. Inset: a linear correlation between the ECL intensity at 1.05 V and the concentration of Hg2+. | ||
As shown in Fig. 6, the ECL intensity of probe 1 did not show any significant changes upon the addition of other metal ions; however, a significant decrease in the ECL signal (∼20% that of probe 1) was observed after the addition of 20 μM (2 equiv.) of Hg2+. The designed system showed rapid reaction rate, high sensitivity, and remarkable selectivity toward Hg2+ ions, thus offering a new practical method for field monitoring of Hg2+ ions.
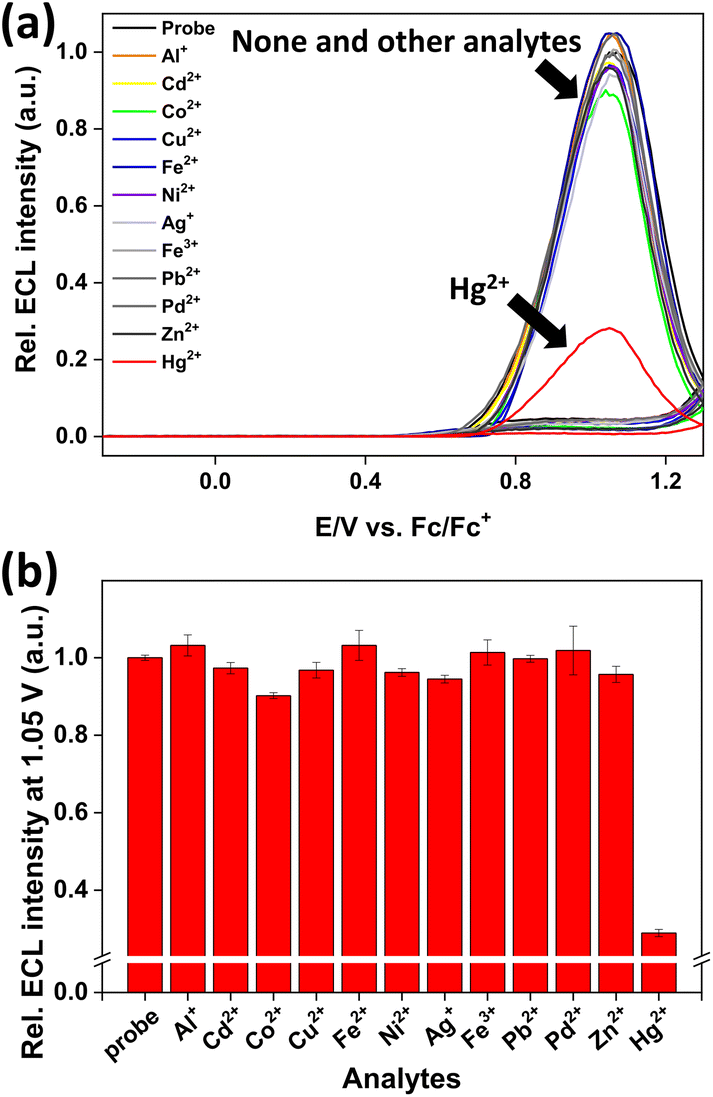 | ||
| Fig. 6 (a) ECL intensities of probe 1 (10 μM) in the presence of various metal ions (40 μM each; Hg2+ 20 μM) in CH3CN/H2O (1:9 v/v, pH 7.4, 100 mM DBAE, 0.1 M HEPES, 0.1% polidocanol, and 0.1 M TBAP as the supporting electrolyte) while the potential is swept at a GC disk electrode (diameter 2 mm) in the range of −0.3–1.3 V (scan rate: 0.1 V s−1). (b) ECL intensities at 1.05 V of probe 1 (10 μM) upon the respective addition of various metal ions (40 μM) and Hg2+ ions (20 μM). | ||
Consequently, probe 1 can be used as a highly selective Hg2+ probe based on ECL analysis (Scheme 3). Probe 1 was directly oxidised on glassy carbon (GC) electrodes to form 1˙+. Subsequently, it was turned into the excited state, 1*, by one-electron transfer from the DBAE radical (DBAE˙), which emitted red ECL. In the presence of Hg2+, probe 1 rapidly reacted with Hg2+via an irreversible transmetalation reaction to generate 1-Hg2+. The excited state of 1-Hg2+ was formed via a mechanism similar to that of probe 1 when oxidation occurs on the GC electrode. However, (1-Hg2+)* was rapidly consumed through an immediate PeT process from (1-Hg2+)* to –Hg+, resulting in a significant decrease in the ECL intensity (Fig. S7†).
 | ||
| Scheme 3 The proposed sensing mechanism of probe 1 for detection of Hg2+ ions through ECL analysis. | ||
Quantification of Hg2+ ions in tap water
To confirm the reliability of the ECL sensing system in real samples, probe 1 was used to quantify Hg2+ ions in tap water using the standard addition method. Analytical solutions were prepared by diluting 1 mL of tap water containing varying concentrations of Hg2+ ions with 1 mL of CH3CN/H2O (1:4 v/v, pH 7.4, 20 μM probe 1, 200 mM DBAE, 0.2 M HEPES, 0.2% polidocanol, and 0.2 M TBAP). Hg2+ ions in the diluted analytical solutions were quantified by comparing ECL values with the calibration curve shown in Fig. 5b. Then, the actual concentrations of Hg2+ ions in tap water samples were accurately determined by multiplying the values obtained from the calibration curve by the dilution factor of 2. First, the ECL emission of probe 1 in tap water was assessed and almost no change in the ECL signal occurred prior to the addition of Hg2+ (Table 1). This observation was made in comparison with the ECL intensity of probe 1 in CH3CN/H2O (as shown in the inset of Fig. 5b), suggesting that the tap water sample contained a negligible amount of Hg2+. As the concentration of Hg2+ ions increased, the ECL signal of probe 1 decreased. The recovery rates for Hg2+ ions ranged from 97.34% to 105.93%, indicating the high reliability and feasibility of this system for the detection of Hg2+ in actual environmental samples.
| [Hg2+]addedb |
Rel. ECL intensity (experiment, n = 3) | [Hg2+]calc'd (μM) | Recovery (%) |
|---|---|---|---|
|
a All ECL intensities were measured along with the cyclic voltammetry (CV) scan in CH3CN/H2O (1:9 v/v, pH 7.4, 100 mM DBAE, 0.1 M HEPES, 0.1% polidocanol, and 0.1 M TBAP) while the potential was swept at a GC working electrode in the range of −0.3–1.3 V vs. Fc/Fc+.
b Concentrations of Hg2+ ions in tap water samples.
|
|||
| 0 μM | 1 ± 0.0167 | — | — |
| 3 μM | 0.7514 ± 0.0028 | 3.1780 ± 0.0347 | 105.93 ± 1.10 |
| 5 μM | 0.6371 ± 0.0071 | 5.0356 ± 0.1145 | 100.71 ± 2.27 |
| 7 μM | 0.5277 ± 0.0040 | 6.8137 ± 0.0655 | 97.34 ± 0.96 |
| 10 μM | 0.3409 ± 0.0068 | 9.8500 ± 0.1107 | 98.50 ± 1.12 |
Conclusions
In summary, we developed a novel photoluminescent (PL) and electrochemiluminescent (ECL) dual signalling probe based on an Ir(III) complex with boronic acid as the reaction site for the selective detection of Hg2+ ions. PL and ECL analyses with the probe showed it had remarkable selectivity for Hg2+ ions with limits of detection of 0.72 μM and 8.03 nM, respectively. Furthermore, the successful quantification of Hg2+ ions in tap water samples using the ECL analysis demonstrated the effectiveness of this probe. Consequently, our system serves as a new proof-of-concept for real-time and on-site sample analysis. We believe that our strategy would be of great help to the advancement of developing ECL-based probes and other fields concerning ECL-based analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NRF grant funded by the Korean Government (MSIT) (No. 2023R1A2C2006049).References
- C. Kumunda, A. S. Adekunle, B. B. Mamba, N. W. Hlongwa and T. T. I. Nkambule, Front. Mater., 2021, 7, 616787 CrossRef.
- B.-J. Ye, B.-G. Kim, M.-J. Jeon, S.-Y. Kim, H.-C. Kim, T.-W. Jang, H.-J. Chae, W.-J. Choi, M.-N. Ha and Y.-S. Hong, Ann. Occup. Environ. Med., 2016, 28, 5–5 CrossRef PubMed.
- J. Mutter, J. Naumann, C. Sadaghiani, R. Schneider and H. Walach, Neuroendocrinol. Lett., 2004, 25, 331–339 CAS.
- M. V. Yigit, A. Mishra, R. Tong, J. Cheng, G. C. L. Wong and Y. Lu, Chem. Biol., 2009, 16, 937–942 CrossRef CAS PubMed.
- A. Anandababu, S. Anandan and M. Ashokkumar, ChemistrySelect, 2018, 3, 4413–4420 CrossRef CAS.
- W.-L. Cui, Z.-H. Zhang, L. Wang, J. Qu and J.-Y. Wang, Spectrochim. Acta, Part A, 2022, 267, 120516 CrossRef CAS PubMed.
- Z. Zhu, G. C. Y. Chan, S. J. Ray, X. Zhang and G. M. Hieftje, Anal. Chem., 2008, 80, 7043–7050 CrossRef CAS PubMed.
- H. Erxleben and J. Ruzicka, Anal. Chem., 2005, 77, 5124–5128 CrossRef CAS PubMed.
- S. Gil, I. Lavilla and C. Bendicho, Anal. Chem., 2006, 78, 6260–6264 CrossRef CAS PubMed.
- S. Gupta, R. Singh, M. D. Anoop, V. Kulshrestha, D. N. Srivastava, K. Ray, S. L. Kothari, K. Awasthi and M. Kumar, Appl. Phys. A, 2018, 124, 737 CrossRef CAS.
- S.-J. Liu, H.-G. Nie, J.-H. Jiang, G.-L. Shen and R.-Q. Yu, Anal. Chem., 2009, 81, 5724–5730 CrossRef CAS PubMed.
- A. Moutcine and A. Chtaini, Sens. Bio-Sens. Res., 2018, 17, 30–35 CrossRef.
- Y. Du, R. Liu, B. Liu, S. Wang, M.-Y. Han and Z. Zhang, Anal. Chem., 2013, 85, 3160–3165 CrossRef CAS PubMed.
- S. J. Lee and M. Moskovits, Nano Lett., 2011, 11, 145–150 CrossRef CAS PubMed.
- D. P. Ruys, J. F. Andrade and O. M. Guimarães, Anal. Chim. Acta, 2000, 404, 95–100 CrossRef CAS.
- G. Chen, Z. Guo, G. Zeng and L. Tang, Analyst, 2015, 140, 5400–5443 RSC.
- H. Shuai, C. Xiang, L. Qian, F. Bin, L. Xiaohui, D. Jipeng, Z. Chang, L. Jiahui and Z. Wenbin, Dyes Pigm., 2021, 187, 109125 CrossRef CAS.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed.
- M. Hesari and Z. Ding, J. Electrochem. Soc., 2015, 163, H3116–H3131 CrossRef.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- M. Schmittel and H.-W. Lin, Angew. Chem., Int. Ed., 2007, 46, 893–896 CrossRef CAS PubMed.
- E. Berni, I. Gosse, D. Badocco, P. Pastore, N. Sojic and S. Pinet, Chem. – Eur. J., 2009, 15, 5145–5152 CrossRef CAS PubMed.
- W. Zhang, D. Zhao, R. Zhang, Z. Ye, G. Wang, J. Yuan and M. Yang, Analyst, 2011, 136, 1867–1872 RSC.
- J.-W. Oh, T. H. Kim, S. W. Yoo, Y. O. Lee, Y. Lee, H. Kim, J. Kim and J. S. Kim, Sens. Actuators, B, 2013, 177, 813–817 CrossRef CAS.
- K. Chen and M. Schmittel, Chem. Commun., 2014, 50, 5756–5759 RSC.
- P. Li, Z. Jin, M. Zhao, Y. Xu, Y. Guo and D. Xiao, Dalton Trans., 2015, 44, 2208–2216 RSC.
- H. Li, A. C. Sedgwick, M. Li, R. A. R. Blackburn, S. D. Bull, S. Arbault, T. D. James and N. Sojic, Chem. Commun., 2016, 52, 12845–12848 RSC.
- Y. Xu, L. Zhang, Y. Liu, Z. Jin, Q. Zhao, F. Yang and D. Xiao, Biosens. Bioelectron., 2016, 77, 182–187 CrossRef CAS PubMed.
- D. Han, M. Qian, H. Gao, B. Wang, H. Qi and C. Zhang, Anal. Chim. Acta, 2019, 1074, 98–107 CrossRef CAS PubMed.
- F. Yuan, K. Hao, S. Sheng, T. H. Fereja, X. Ma, F. Liu, M. N. Zafar, B. Lou, H. Tian and G. Xu, Electrochim. Acta, 2020, 329, 135117 CrossRef CAS.
- Y. Dai, Z. Zhan, L. Chai, L. Zhang, Q. Guo, K. Zhang and Y. Lv, Anal. Chem., 2021, 93, 4628–4634 CrossRef CAS PubMed.
- I.-S. Shin, S. W. Bae, H. Kim and J.-I. Hong, Anal. Chem., 2010, 82, 8259–8265 CrossRef CAS PubMed.
- H. J. Kim, K.-S. Lee, Y.-J. Jeon, I.-S. Shin and J.-I. Hong, Biosens. Bioelectron., 2017, 91, 497–503 CrossRef CAS PubMed.
- S.-Y. Kim, H. J. Kim and J.-I. Hong, RSC Adv., 2017, 7, 10865–10868 RSC.
- H. Rhee, T. Kim and J.-I. Hong, Dalton Trans., 2018, 47, 3803–3810 RSC.
- K.-R. Kim, H. J. Kim and J.-I. Hong, Anal. Chem., 2019, 91, 1353–1359 CrossRef CAS PubMed.
- T. Kim and J.-I. Hong, ACS Omega, 2019, 4, 12616–12625 CrossRef CAS PubMed.
- J. Park, T. Kim, H. J. Kim and J.-I. Hong, Dalton Trans., 2019, 48, 4565–4573 RSC.
- H. J. Kim, T. Kim and J.-I. Hong, Sens. Actuators, B, 2020, 307, 127656 CrossRef.
- T. Kim, H. J. Kim, I.-S. Shin and J.-I. Hong, Anal. Chem., 2020, 92, 6019–6025 CrossRef CAS PubMed.
- Y. Namkoong, J. Oh and J.-I. Hong, Chem. Commun., 2020, 56, 7577–7580 RSC.
- H. Wei and E. Wang, Luminescence, 2011, 26, 77–85 CrossRef CAS PubMed.
- J. M. Fernandez-Hernandez, E. Longhi, R. Cysewski, F. Polo, H.-P. Josel and L. De Cola, Anal. Chem., 2016, 88, 4174–4178 CrossRef CAS PubMed.
- E. Kerr, E. H. Doeven, D. J. D. Wilson, C. F. Hogan and P. S. Francis, Analyst, 2016, 141, 62–69 RSC.
- J. I. Kim, I.-S. Shin, H. Kim and J.-K. Lee, J. Am. Chem. Soc., 2005, 127, 1614–1615 CrossRef CAS PubMed.
- E. Longhi, J. M. Fernandez-Hernandez, A. Iordache, R. Fröhlich, H.-P. Josel and L. De Cola, Inorg. Chem., 2020, 59, 7435–7443 CrossRef CAS PubMed.
- Z. M. Smith, E. Kerr, E. H. Doeven, T. U. Connell, N. W. Barnett, P. S. Donnelly, S. J. Haswell and P. S. Francis, Analyst, 2016, 141, 2140–2144 RSC.
- Y. Zhou, K. Xie, R. Leng, L. Kong, C. Liu, Q. Zhang and X. Wang, Dalton Trans., 2017, 46, 355–363 RSC.
- A. Chatterjee, M. Banerjee, D. G. Khandare, R. U. Gawas, S. C. Mascarenhas, A. Ganguly, R. Gupta and H. Joshi, Anal. Chem., 2017, 89, 12698–12704 CrossRef CAS PubMed.
- X. Guo, J. Huang, Y. Wei, Q. Zeng and L. Wang, J. Hazard. Mater., 2020, 381, 120969 CrossRef CAS PubMed.
- S. W. Lee, S. Y. Lee and S. H. Lee, Tetrahedron Lett., 2019, 60, 151048 CrossRef CAS.
- S. Subedi, L. N. Neupane, P. K. Mehta and K.-H. Lee, Dyes Pigm., 2021, 191, 109374 CrossRef CAS.
- S. Subedi, L. N. Neupane, H. Yu and K.-H. Lee, Sens. Actuators, B, 2021, 338, 129814 CrossRef CAS.
- M. Lepeltier, B. Graff, J. Lalevée, G. Wantz, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Org. Electron., 2016, 37, 24–34 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 2006 Search PubMed.
- A. S. Tanwar, L. R. Adil, M. A. Afroz and P. K. Iyer, ACS Sens., 2018, 3, 1451–1461 CrossRef CAS PubMed.
- A. S. Tanwar, R. Parui, R. Garai, M. A. Chanu and P. K. Iyer, ACS Meas. Sci. Au, 2022, 2, 23–30 CrossRef CAS PubMed.
- J. H. He, Y. Y. Cheng, T. Yang, H. Y. Zou and C. Z. Huang, Anal. Chim. Acta, 2018, 1035, 203–210 CrossRef CAS PubMed.
- M. Tian, C. Wang, Q. Ma, Y. Bai, J. Sun and C. Ding, ACS Omega, 2020, 5, 18176–18184 CrossRef CAS PubMed.
- X. Sun, B. M. Chapin, P. Metola, B. Collins, B. Wang, T. D. James and E. V. Anslyn, Nat. Chem., 2019, 11, 768–778 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- C. F. R. A. C. Lima, A. S. M. C. Rodrigues, V. L. M. Silva, A. M. S. Silva and L. M. N. B. F. Santos, ChemCatChem, 2014, 6, 1291–1302 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01266b |
| This journal is © The Royal Society of Chemistry 2023 |