
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Following carbon condensation by in situ TEM: towards a rational understanding of the processes in the synthesis of nitrogen-doped carbonaceous materials†
Diana
Piankova
 *,
Janina
Kossmann
,
Hannes
Zschiesche
,
Markus
Antonietti
,
Nieves
López-Salas
and
Nadezda V.
Tarakina
*
*,
Janina
Kossmann
,
Hannes
Zschiesche
,
Markus
Antonietti
,
Nieves
López-Salas
and
Nadezda V.
Tarakina
*
Department of Colloid Chemistry, Max-Planck Institute of Colloids and Interfaces, Science Park Golm, 14476 Potsdam, Germany. E-mail: nadja.tarakina@mpikg.mpg.de
First published on 9th November 2022
Abstract
Porous carbonaceous materials obtained from biomass have been an important class of CO2 sorbents since ancient times. Recent progress in carbon-based adsorbent technology is based on the implication of the concept of heteroatom doping. In this respect, the synthesis of carbonaceous materials through one-step condensation of cheap nitrogen-containing molecular precursors is an attractive strategy for obtaining such N-doped carbons. The design of the adsorbents obtained by this route relies on the careful adjustment of synthesis parameters, such as the temperature, the heating rate, and the atmosphere. However, in most cases, the latter choice remains rather empirical due to the lack of a fundamental understanding of the condensation mechanism of molecular precursors. In this work, we followed the structural, morphological, and chemical evolution of a molecular precursor (uric acid) at the nanoscale using a combination of in situ condensation inside a scanning transmission electron microscope with ex situ analysis of the products of condensation at different temperatures, atmospheres, and heating rates, and correlate our findings with the CO2 sorption properties of the obtained materials. We showed that varying pressures and reaction rates result in particles with different porosity. The porosity of the surface of the particles during the early stages of condensation governs the subsequent release of volatiles and the development of a hierarchical pore structure. We found that synthesis in vacuum enables effective condensation at considerably low temperatures (500 °C). Using a higher heating rate (10 °C min−1) suppresses structural ripening and preserves the optimal size of micropores, thus giving a CO2 uptake twice as high compared to samples synthesized in a nitrogen atmosphere, which is commonly used, preserving the same selectivity.
Introduction
CO2 cycling for climate remediation starts with the efficient sorption and capture of CO2.1 The materials exploited for this purpose have to fulfill several criteria: high uptake and selectivity towards CO2, low price, reusability, sustainability.2 MOFs3 and zeolites4,5 demonstrate very high CO2 uptake and selectivity due to the presence of tuned structural pores with partly open-metal sites.6 However, complicated ligand structures and the presence of metals, which are in short supply, make MOFs and zeolites expensive and unsustainable. The use of carbonaceous materials can overcome these drawbacks: they are metal-free and can be obtained from biomass.7 Even though bare carbon materials are reported to lack affinity to CO2 (thus, lower uptake and selectivity), this issue was recently overcome by introducing nitrogen into their structure, e.g., using nitrogen-containing organic precursors.8,9 Nitrogen changes the electron density distribution within the carbonaceous structure and creates basic sites for CO2 capture.10A straightforward way to obtain nitrogen-containing carbonaceous materials is the condensation of a molecular precursor which already contains a high amount of nitrogen and is “noble” (hard to oxidize).11,12 The direction of the condensation at the molecular scale can be estimated if the initial molecule has functional groups which can polymerize13 or condense.14 However, cross-linking occurs randomly and sometimes poorly for most molecular precursors, leading to low carbon yields and disordered residues with low functionality. Therefore, the design of the porous structure and pore size distribution favorable for CO2 adsorption – if no porogen15 or template16 is applied – is more challenging than for crystalline materials. The porous structure and the properties of condensates depend greatly on synthetic conditions (temperature, pressure, heating rate), the choice of which often remains empirical due to the lack of a fundamental understanding of the condensation process. Usually, the influence of synthetic conditions on morphology and the porous structure is studied after the condensation is completed. In contrast, in situ observations of the condensation process would allow direct monitoring of porosity formation, and consequently, it would help set the base for a more controlled synthesis of carbonaceous materials. The condensation of molecular precursors occurs at the nano- and microscale, the scale at which transmission electron microscopy (TEM) is effective.
In situ TEM studies related to this topic are purely fundamental. They are mainly focused on the temperature-induced transformations (mainly graphitization) of polymeric precursors,17,18 while to the best knowledge of the authors, nothing is reported about the condensation of molecular precursors. Sharma et al.17 reported the formation of glassy carbon through in situ heating of phenol–formaldehyde fiber. The study revealed the structural dynamics in the formation of glassy carbon: rearranging of fullerenes, strongly curved graphene sheets, and small 2D graphene shapes, which all co-exist in glassy carbon. Schierholz et al.18 investigated not only the graphitization in PAN fibers but also morphological changes preceding this process.
Our study aims to broaden a fundamental understanding of the evolution of porosity during the condensation process of a molecular precursor and to apply this knowledge to design porous carbonaceous materials with enhanced adsorption properties. To realize this goal, we followed the structural and morphological evolution of uric acid at the nanoscale while in situ heating inside a scanning transmission electron microscope. Moreover, we correlated the results with ex situ observations after condensation in a nitrogen atmosphere and under vacuum at different temperatures. Uric acid, bound into the crystal structure by multiple H-bonds and thereby not volatile, is used as a model molecular precursor for condensation to a highly nitrogen-containing carbonaceous material. Using condensation of uric acid as a model system, we identified several specific steps in the condensation process, which allowed us to adjust synthetic conditions to obtain new carbonaceous materials in a controllable way at considerably low temperatures.
Experimental
Materials
Uric acid (≥99%) was purchased from Roth and used as received.Synthesis
Uric acid condensates were synthesized by heat treatment of uric acid precursor. For the synthesis in nitrogen atmosphere 1 g of the precursor was placed into a ceramic crucible and covered with a ceramic lid. The oven with the sample was purged for 20 minutes with nitrogen and then heated until the desired temperature (500 °C, 700 °C and 800 °C) with a heating rate of 1 °C min−1 with constant nitrogen flow. The final temperature was held for 2 h. After cooling down to ambient temperatures, the crucible was removed and the product was obtained as a black powder. For the synthesis in vacuum laboratory a Schlenk line was built and connected to a vacuum pump. For the samples synthesized in vacuum, heating rates of 1 °C min−1 and 10 °C min−1 and final temperatures of 250 °C, 325 °C, 375 °C, 400 °C, and 500 °C were used.The pressure of nitrogen atmosphere was 1 bar, in vacuum ∼1.5 × 10−4 bar (Schlenk line), the high vacuum of the TEM during the in situ heating experiment 3 × 10−10 bar. Vacuum and TEM vacuum will be referred to as “low pressure” conditions throughout the text.
Characterization
Thermogravimetric analysis (TGA) was performed in a NETZSCHTG 209 F1 device using nitrogen as a carrier gas and a heating rate of 10 °C min−1 in a Pt crucible from 25 to 1000 °C. TGA coupled with a mass spectrometer (TGA–MS) has been performed using a thermos microbalance TG2019 F1 Libra (Netzsch, Selb Germany). Elemental chemical analysis (ECA) was carried out using a vario MICRO cube CHNOS Elemental Analyzer (Elementar Analysensysteme GmbH, Langenselbold) in the CHNS mode and a 2mgChem80s Method. X-ray powder diffraction (XRD) patterns were recorded using a Bruker D8 Advance diffractometer with Cu-Kα radiation (λ = 1.5418 Å), 2θ from 5° to 70°. 1625 steps and 1 step per second. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo Fisher Scientific Escalab 250 Xi.Nitrogen adsorption and desorption isotherms at 77 K and 298 K, and CO2 adsorption and desorption isotherms at 273 K and 298 K were measured using a Quantachrome Quadrasorb SI apparatus. Samples were degassed at 150 °C under vacuum for 20 h before each measurement. The specific surface area of each material was obtained from the nitrogen adsorption data (P/P0 < 0.3) using the Brunauer–Emmett–Teller (BET) method. The value was obtained after applying the method in the linear region with the best correlation. Total pore volume (VT) and pore size distribution was obtained by Quenched Solid Density Functional Theory (QSDFT) model with slit/cylindrical pore shape using nitrogen adsorption data at 77 K.
The Ideal Adsorbed Solution Theory (IAST) values were calculated by eqn (1) applied to isotherms at 298 K:
 | (1) |
The Isosteric Heat of Adsorption (Qst) was calculated using the Clausius–Clapeyron equation (eqn (2)):
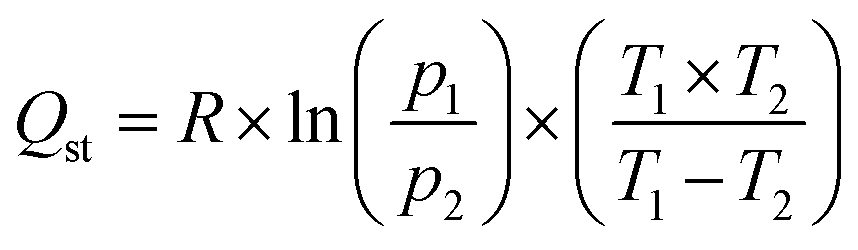 | (2) |
Transmission electron microcopy (ex situ studies)
For high-resolution transmission electron microscopy (HRTEM), energy-filtered electron reduced density function (EF-eRDF) analysis and scanning transmission electron microscopy (STEM) observations, samples were sonicated in ethanol for 15 minutes; the obtained suspensions were drop-casted to a Cu grid with lacey carbon support (purchased from Ted Pella) and dried for 15 minutes. The STEM and HRTEM studies were performed using a double Cs corrected JEOL JEM-ARM200F (S)TEM operated at 80 kV and equipped with a cold-field emission gun, Jeol secondary electron (SE), bright field (BF), annular dark-field (ADF) detectors, and a Gatan Quantum GIF spectroscopy system. Selected-area electron diffraction (SAED) patterns for EF-eRDF analysis were recorded using a Gatan US1000FTXP 2 CCD camera in GIF mode using a camera length of 1.5 cm (signal in reciprocal space up to 20 Å−1). Before the acquisition, SAED patterns were zero-loss energy-filtered; a slit of 15 eV was used to allow mainly elastically scattered electrons to contribute to the signal. Recorded SAED patterns were converted into azimuthally averaged scattered intensity profiles and further processed (fitted and Fourier transformed) to obtain electron reduced density functions (eRDFs) using the eRDF software19 with the same fitting parameters for the whole set of samples. The eRDF describes the probability of finding a pair of atoms at a particular distance, thus allowing to probe short-range order in the structure, and is especially useful for describing amorphous, poorly crystalline, and nano-materials. The cut-off distance of the eRDF provides information about the size of coherently scattering units. The calibration of the camera length was performed using an Au nanoparticles standard (ca. 10 nm size Au particles sputtered onto a carbon grid) following the procedure described in.20 The pore wall thickness was measured using secondary electron STEM images. In particular we measure areas where shells of the particles are broken, giving a cross-sectional view on the shell. For each sample, 60 measurements have been performed, and the average size and standard deviation were calculated.In situ scanning transmission microscopy heating experiment
For the in situ heating experiment, an in situ heating/biasing holder fusion select from Protochips and heating E-chips produced by Protochips were used. In the central part of the heating chip, there is a conductive membrane with 9 round windows, which of them has carbon film support with smaller holes (Fig. S1a and b†). Similar to ex situ studies, the suspension of uric acid in ethanol after 15 minutes of sonication was drop-casted onto the heating chip and dried for 15 minutes. For the in situ experiment, a heating rate of 10 °C min−1 was used. To monitor the process but minimize the exposure of the sample to the electron beam (to avoid beam damage) the beam was turned on only each 50 °C (in the 25–250 °C temperature range) or each 25 °C (in the temperature range 250–550 °C) for quick imaging. Until 250 °C no changes in the morphology of the samples can be observed.Particles of the precursor were found in each window on the carbon support and in the holes. A heating rate of 10 °C min−1 was used and a 50 °C step increase in the temperature. While heating from one temperature to another and while moving to new windows/holes for imaging, the beam was turned off to avoid long exposure under the electron beam. The longest flat region in the heating ramp (Fig. S1c†) at 250 °C corresponds to the process of main change in the sample. STEM enables more precise control over the exposed area in comparison to TEM since STEM allows imaging of a region of interest, not exposing to the radiation the areas outside of the chosen region; moreover, the electron dose can be controlled through a different time of scanning. To distinguish the processes occurring on the surface and in the bulk of the particles, we used Jeol secondary electron (SE) and bright-field (BF) STEM detectors, as in the case of the ex situ studies.
Results and discussion
Characterization of the precursor
Uric acid (UA), the metabolic degradation product of purine nucleobases, is a suitable precursor because of its high stability against oxidation, low volatility in combination with a very high nitrogen content of 33 wt% (C/N ratio of 1.4).21 First, we performed thermogravimetrical analysis-mass spectrometry (TGA–MS) in nitrogen atmosphere of U.A. to analyze its thermal stability and the fractions of volatile components formed upon heating (Fig. S2†). The curve was also used to define reference temperatures for the synthesis and the in situ heating experiment inside the electron microscope. According to these results, the main mass loss begins at 345 °C, with the most significant rate of change at 425 °C. TGA–MS results show that it is accompanied by an abrupt release of H2O, CO2, HCN, CH4, and NH3 at this temperature. At 650 °C, a second, less abrupt release of less amount of volatiles occurs. Therefore, the thermal analysis implies that the condensation of uric acid is expected to occur mainly between 350 °C and 450 °C under nitrogen atmosphere, while at later stages, the carbonaceous framework sinters and rearranges under the elimination of other entities.Synthesis in nitrogen atmosphere
To follow the condensation in nitrogen atmosphere, we heat-treated U.A. at 500, 700, and 800 °C with a heating rate of 1 °C min−1. Hereafter, samples will be named according to their condensation temperature, atmosphere, and heating rate, like U.A. “temperature”-“gas”-“rate” (e.g., UA500-N2-1 stands for uric acid heat-treated at 500 °C under nitrogen atmosphere with a heating rate of 1 °C min−1 – list of all the samples can be found in Table S1†). According to elemental chemical analysis (Table 1), the carbon/nitrogen ratio of the condensation product obtained at 500 °C is 1.4, the same as for untreated uric acid. With increasing the condensation temperature, the nitrogen content drops slightly to a C/N ratio of 1.8 for UA700-N2-1 and 2.7 for UA800-N2-1.| Sample | ECA (at%) | S BET (m2 g−1) | V T (cm3 g−1) | Max. CO2c (mmol g−1) | ||||
|---|---|---|---|---|---|---|---|---|
| C | N | O | H | C/N | ||||
| a Specific surface area determined using the Brunauer–Emmett–Teller (BET) method. b Total pore volume determined by QSDFT method applied to the adsorption branch of nitrogen isotherms at 77 K. c Obtained by CO2 physisorption at 273 K. | ||||||||
| UA500-N2-1 | 41.0 | 28.6 | 7.3 | 23.1 | 1.4 | 41 | 0.14 | 1.1 |
| UA700-N2-1 | 50.8 | 27.6 | 7.0 | 14.6 | 1.8 | 139 | 0.29 | 2.0 |
| UA800-N2-1 | 59.0 | 22.0 | 6.9 | 12.1 | 2.7 | 140 | 0.37 | 1.4 |
To study the structure of the condensates, we performed X-ray powder diffraction (XRD) measurements, as well as high-resolution TEM (HRTEM) and energy-filtered electron reduced density function (EF-eRDF) analysis. XRD patterns (Fig. S3†) of the samples demonstrate only one broad peak of graphitic stacking at 27° indicating the disordered character of the materials. The latter was further verified by HRTEM imaging (Fig. S4a†), which corroborated the lack of long-range order in the materials (amorphous halo in the FFT). From EF-eRDF analysis, the short-range order is found to extend only to ∼7 Å (Fig. S4b and c†) (a detailed description of the EF-eRDF analysis can be found in the ESI†).
The pore structure and CO2 adsorption capability of the obtained carbonaceous materials were analyzed using physisorption experiments. Nitrogen adsorption and desorption isotherms measured at 77 K are shown in Fig. 1a (SBET and total pore volume are given in Table 1). A low nitrogen uptake for UA500-N2-1 at low relative pressures indicates the presence of a very small number of micropores. The slight increase in nitrogen uptake at high relative pressures indicates the presence of a small number of mesopores. Isotherms of UA700-N2-1 and UA800-N2-1 show a mix of Type IV and Type I shapes according to the IUPAC. They have a more considerable uptake at lower relative pressures compared to UA500-N2-1, pointing to the presence of micropores, and a strong hysteresis loop at higher relative pressures, indicating the development of some mesopores.
 | ||
| Fig. 1 (a) N2 adsorption/desorption isotherms at 77 K and (b) CO2 adsorption isotherms at 273 K of the samples synthesized in nitrogen atmosphere. Secondary electron (SE) and bright-field (BF) images of: (c) UA500-N2-1 and (d) UA700-N2-1. SE detector is more sensitive to the signals from the surface of the particles while bright-field acquires transmitted signal which allows seeing the inner structure (bulk) of the particles. Schematic morphology representation of the samples (e) UA500-N2-1 and (f) UA700-N2-1. Dark grey – the core of the particle, white spots are the pores located in the core, light grey color is a shell around the particles. | ||
The hysteresis loop closes at 0.42 P/P0, which can be rationalized with the sample containing mesopores of different sizes and also mesopores having ink-bottle neck shapes. CO2 adsorption isotherms of these three samples are shown in Fig. 1b. The difference in CO2 uptake of UA500-N2-1 and UA800-N2-1 is unexpectedly low, considering the increase of SBET from UA500-N2-1 to UA800-N2-1. This low uptake of nitrogen and, at the same time, high affinity to CO2 of UA500-N2-1 results in a high CO2 selectivity (IAST CO2/N2 of 68). UA700-N2-1 attains a maximum CO2 uptake of 2.00 mmol g−1, which is 1.4 times higher than the CO2 uptake of UA800-N2-1. Since nitrogen sorption isotherms of these two materials are similar, the differences in CO2 uptake arise from the higher nitrogen content of UA700-N2-1. Here it is important to point out that, despite the smaller kinetic diameter of CO2 at 273 K with respect to N2 at 77 K (d(N2) = 3.64 Å, d(CO2) = 3.30 Å), no unequivocal conclusions can be drawn regarding the size of the micropores of the samples because of the large number of nitrogen heteroatoms that they contain and the insensitivity that the NLDFT kernels show when using CO2 as an adsorbate.22
Ex situ scanning transmission electron microscopy (STEM) study using a secondary electron (SE) detector and a bright-field (BF) detector enables simultaneous analysis of external morphology and the internal structure of the samples at the nanoscale. These two signals revealed the formation of the core–shell particles with a significant difference in the shell structure between UA500-N2-1 and UA700-N2-1 (see Fig. 1c and d). While UA500-N2-1 exhibits a shell tightly attached to the core, the shell of UA700-N2-1 detaches from the core and ruptures on some particles. These findings explain the results from nitrogen and CO2 sorption. At 500 °C, the core is not accessible for N2 because of the tightly attached shell (i.e., low N2 uptake of U500-N2-1). Although the shell appears smooth on the surface, the CO2 uptake of 1.1 mmol g−1 from the sorption study suggests that the shell is highly microporous (micropores are not spatially resolvable with the SE detector). Such high CO2 uptake can be explained by a sort of sieving effect of highly selective fine micropores (<1 nm)23 in the shell: these micropores adsorb CO2 but hinder the adsorption of N2 since the kinetic diameter of the latter is larger compared to CO2 (Fig. 1e). It is worth noting that a similar adsorption behavior was also observed for C1N1 materials obtained from guanine.24 When increasing the condensation temperature to 700 °C, the shell partly ruptures and detaches from the particle, which herein facilitates the access of N2 to the core. The increase of CO2 uptake with increasing the temperature to 700 °C could be caused by micropores of the core that becomes more accessible. Alternatively, not taking into account the micropores of the shell, CO2 can adsorb on both sides of the shell, as illustrated in Fig. 1f. When increasing the temperature to 800 °C, no considerable morphology changes were observed (Fig. S5†). Thus, a microporous shell formed around the particles in nitrogen atmosphere obscures the development of interconnectivity of the pores at low temperatures (500 °C) by the formation of the core–shell structure. The origin of the core–shell structure may come from the surface melting, which occurs before the melting temperature in crystalline materials. Uric acid is crystalline up to 380 °C,25 and for most organic compounds, decomposition (accompanied by condensation into a carbonaceous material) starts before or shortly after the melting stage. Presumably, the melted surface led to the formation of a thin layer, which during the decomposition, retained the morphology and transformed into the shell, forming core–shell particles. We performed an in situ STEM condensation experiment to understand the mechanism of porosity development and identify at which stage of the condensation process the shell is formed.
In situ scanning transmission electron microscopy
Since any process connected to the repolymerization from the gas phase or the elimination depends on pressure, our in situ experiment aims not only to visualize the condensation process itself but also to understand how ultra-high vacuum conditions influence it.All crystalline precursor particles of uric acid were imaged at 25 °C before the in situ heating. They all vary in size (200 nm to 4 μm) and shape (Fig. S6a†), and their surface looks smooth (Fig. 2a, SE detector). Once the temperature reached 250 °C, we observed the first changes based on the signal from SE detectors; in particular, particles started to appear porous at the surface (Fig. 2b, SE detector, mesopores of size 18 ± 5 nm). Continuing imaging at 250 °C at higher magnifications (Fig. 2f and Video 1†), we noticed slight shrinking and gradual release of matter (mass transfer) from the near-surface part of the particle while the overall shape of the particle was kept unchanged (denoted with violet arrows in Fig. 2f). After 4 minutes, the particle also started to change inside – becoming more “emptied” (Fig. 2c, SE, slightly larger mesopores of 21 ± 6 nm on the surface) while no further mass loss from the near-surface region was detected. Another particle, which was not continuously exposed to the electron beam, exhibited the same trend (Fig. S7†), proving that the electron beam does not change the mass release but only accelerates the process (more information on the taken precautions on exposure of the specimen to the electron beam are also described in the method section and in the ESI†). Notably, mass release in bigger particles occurs slower (Fig. S6b†). After 64 minutes at 250 °C (Fig. 2d, BF), the whole particle looked empty. We believe that the abovementioned changes at 250 °C can be assigned to the main mass release observed at 425 °C in the TGA–MS curve in nitrogen atmosphere. The shift of the mass release to 250 °C can be explained by the ultra-high vacuum in TEM.18 The intense elimination of the matter at such a low temperature can indicate an overall shift of equilibrium toward the sublimation rather than condensation, confirmed by significant volume loss. Thus, the condensation process accompanied by the mass release resulted in the following morphological changes in the particles: first, the surface of the particles became mesoporous (contrary to the samples synthesized in nitrogen atmosphere); second, the subsequent mass loss took place from the bulk of the particle through the porous shell leading to the formation of hollow particles.
 | ||
| Fig. 2 Secondary electron (SE) and bright-field (BF) STEM images of the particle (a) at 25 °C before the heating, (b) immediately after reaching 250 °C, (c) after 4 minutes at 250 °C, (d) after 64 minutes at 250 °C, (e) immediately after reaching 475 °C, (f) sequentially recorded BF images recorded from the area marked with violet rectangular on (b). The images demonstrate external mass loss during the heating at 250 °C. Arrows are used as guidance to the eye showing the external mass loss (Video 1† – BF and SE detector). The schemes above the SE images display the particle changes in porosity and morphology. | ||
In the temperature range of 250–350 °C, we observed significant shrinkage of different particles of the specimen. Larger particles (∼several micrometers) demonstrated considerably more noticeable shrinkage than smaller particles (less than 1 micrometer). However, larger particles lay on the carbon support, which could have modified the heat transfer (Fig. S8†). In parallel, sintering of the particles occurs; the particles become dense due to the redistribution of the pore volume (Fig. S8†). At 425 °C, we detected the second mass loss, occurring through bubble formation (Fig. 3 and Video 2†). Small bubbles are formed in the entire particle volume. They coalesce with each other forming larger bubbles, which finally escape from the inside of the particle, forming a porous network. The movement of the bubbles inside the particles indicates a high viscosity liquid-like behavior. A similar bubble formation process was predicted for softening coals; however, it was never observed experimentally.26,27 One has to notice that the release of the volatiles occurred slightly different for the particles primarily exposed (Fig. 3b) and unexposed to the electron beam (Fig. 3a). In the particle primarily exposed to the electron beam (Fig. 3b), the surface remains unperturbed (Video 2,† SE detector), while in the particles unexposed to the electron beam (Fig. 3a), escaping off the bubbles considerably modifies the morphology of the surface (Video 3,† SE detector). Shrinking, sintering, and softening are most probably related to the second mass release observed on the TGA–MS curve at 650 °C (Fig. S2†).
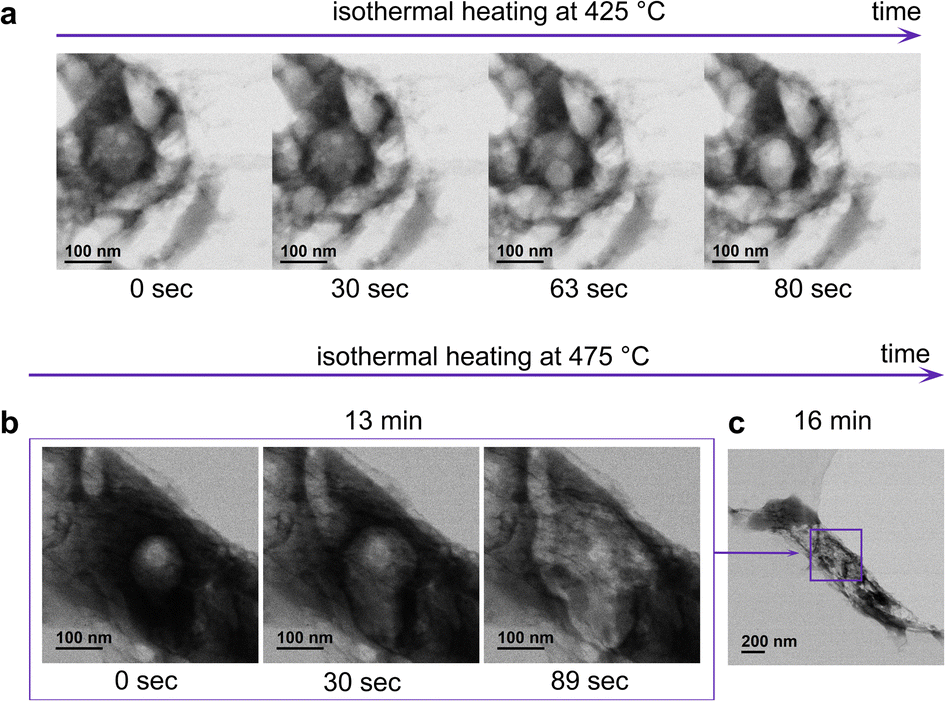 | ||
| Fig. 3 (a) BF images demonstrating “bubbling” at 425 °C, observed in some particles which had not been exposed to the electron beam before (Video 2† – SE and BF detector); (b) sequentially recorded BF images at 475 °C demonstrating mass loss from the bulk of the same particle as in Fig. 2 (Video 3† SE and BF detector); (c) BF image of the particle at lower magnification. | ||
Thus, the crucial insight from the in situ condensation in TEM is that in vacuum, condensation occurs at lower temperatures (250 °C) compared to nitrogen atmosphere, and leads to the formation of the hollow particles with a mesoporous surface (∼21 nm).
The significant difference in the morphology of the particles synthesized in nitrogen and vacuum arises from the difference in the gas pressures used during the synthesis (nitrogen 1 bar, ultra-high vacuum of TEM is 3 × 10−10 bar). This emphasizes that the gas pressure used during synthesis plays a pivotal role in the porosity development (this will be discussed more in detail in the last section). Since by using low gas pressure, we could avoid the formation of a microporous shell (which hinders further porosity development) and considerably reduce the temperature of synthesis, we decided to perform the condensation also in a vacuum oven (∼1.5 × 10−4 bar) and characterize the adsorption behavior of the obtained materials.
Optimization of the synthesis using vacuum
When changing the atmosphere from nitrogen to vacuum (1.5 × 10−4 bar), condensation of uric acid begins at lower temperatures of 325/375 °C, at which no XRD peaks corresponding to uric acid are detected anymore after reaction, and the peak of graphitic stacking at 27° appears (Table S2, Fig. S9 and S10†). To track the evolution of sorption properties and the morphology transformation in vacuum, we heat treated uric acid at 400 and 500 °C. To understand the influence of the heating rate, we also used a higher heating rate of 10 °C min−1 (the same as used for the in situ STEM experiment). With heating up to 400 °C and 500 °C in vacuum the high nitrogen content is kept, resulting in a C/N ratio of 1.4 Table 2.| Sample | ECA (at%) | S BET (m2 g−1) | V T (cm3 g−1) | Max. CO2c (mmol g−1) | ||||
|---|---|---|---|---|---|---|---|---|
| C | N | O | H | C/N | ||||
| a Specific surface area determined using the Brunauer–Emmett–Teller (BET) method. b Total pore volume determined by QSDFT method applied to the adsorption branch of nitrogen isotherms at 77 K. c Obtained by CO2 physisorption at 273 K. | ||||||||
| UA400-vac-1 | 39.2 | 28.6 | 9.1 | 23.1 | 1.4 | 73 | 0.22 | 0.9 |
| UA500-vac-1 | 41.5 | 29.8 | 7.8 | 20.9 | 1.4 | 118 | 0.29 | 2.3 |
| UA500-vac-10 | 44.4 | 30.3 | 7.1 | 18.2 | 1.5 | 94 | 0.25 | 2.0 |
Ex situ STEM analysis revealed the expected morphological difference between vacuum and nitrogen samples: the absence of the core–shell structure of the vacuum samples (Fig. S11† and 4a). The surface of the vacuum samples is mesoporous: 9 ± 5 nm for UA400-vac-1 and 14 ± 5 nm for UA500-vac-1. The particles of the sample UA500-vac-10 look like poked balloons, which surface has even larger mesopores with a broad distribution of their size – 21 ± 11 nm. Thus, we achieved the formation of the particles with a morphology similar to the one observed during in situ measurements, with the only difference that the particles are not hollow. This can arise from the less intense mass release (due to rebalancing sublimation and condensation processes) as well as the absence of the period of isothermal heating at the particular condensation temperature, as has been done during the in situ experiment.
 | ||
| Fig. 4 (a) Secondary electron (SE) and bright-field (BF) STEM images of UA500-vac-1, (b) N2 adsorption/desorption isotherms at 77 K of the samples synthesized in vacuum, (c) their CO2 adsorption isotherms at 273 K, (d) selectivity values for nitrogen and vacuum samples calculated from the ideal adsorbed solution theory (IAST), (e) Qst-CO2 uptake dependence. | ||
The nitrogen sorption isotherms of the samples synthesized in vacuum at different temperatures show an adsorption knee at low relative pressures typically ascribed to Type I isotherms and the presence of micropores. The sample with a larger knee at low relative pressures was UA500-vac-1. We analyzed the sample using Ar at 87 K to understand further whether a high volume of large micropores developed on its pore structure (Fig. S12†). The analysis shows that the sample comprises only a small volume of micropores of ca. 1 nm. As observed on the direct carbonized samples, the presence of hysteresis loops at medium and high relative pressures (Type IV feature according to the IUPAC) indicates the presence of mesopores of different sizes (Fig. 4b). The pore size distribution obtained by applying the NLDFT method to the adsorption branch of the isotherms further corroborates this observation (Fig. S13†). The specific surface of the samples was calculated using the BET method (Table 2). The samples show similar pore volumes and specific surface areas. Interestingly, the specific surface area of UA400-vac-1 is almost two times higher, and the pore volume is 1.5 times higher than that of UA500-N2-1 (Tables 1 and 2), which goes in line with the observations made by TEM.
Despite its more developed porous structure, UA400-vac-1 demonstrates lower CO2 uptake than UA500-N2-1 (Fig. 4c). The less concave shape of UA400-vac-1 isotherms at low relative pressures further indicates a lower affinity to CO2, arising from the presence of nitrogen functionalities with low strength of binding to CO2.28 With increasing the temperature from 400 °C to 500 °C (with the heating rate of 1 or 10 °C min−1), the CO2 uptake doubles, and the CO2 isotherms become concave at low relative pressure, indicating a high affinity of CO2 to the materials.
Not only high CO2 uptake but also high selectivity of CO2 capture and strength of binding (affinity of CO2 to the materials) are crucial to obtain a superior CO2 adsorbent. The ideal adsorbed solution theory (IAST) was used to estimate CO2/N2 selectivity of the materials (Fig. S14 and S15†) and their isosteric heat of adsorption (Qst) was calculated from isotherms recorded at different temperatures. As shown in Fig. 4d, when using the heating rate of 1 °C min−1, independent of the atmosphere used, the selectivity of the samples decreases with increasing temperature. Even though the specific surface area of the material obtained at low temperatures (400–500 °C) is low, it comprises special selective CO2 pores formed at the early stages of the condensation. A similar trend was also reported by Oschatz and Antonietti.29 Indeed, for materials obtained in nitrogen atmosphere, the shell comprises these selective micropores, and at higher temperatures, less selective pores from the core of the particles contribute to the overall uptake, herein leading to the decrease of CO2 selectivity. In vacuum, at 400 °C, small micropores adsorb selectively due to the size exclusion effect; however, with further heating to 500 °C, the selectivity decreases with the increase of the size of the micropores. The enlargement of the micropores might occur similarly to mesopores, which expanded from 18 to 21 nm during 4 minutes of the isothermal heating in situ. However, as reported in the literature, when increasing the heating rate, the small micropores formed at earlier stages can be preserved,30 and a sort of “freezing” allows improving the selectivity. For example, increasing the heating rate from 1 °C min−1 to 10 °C min−1 in vacuum (UA500-vac-1 and UA500-vac-10) leads to a drastic increase in the IAST value from 35 to 61.
Fig. 4e shows the isosteric heat of adsorption (Qst) at different loadings. A high Qst value indicates strong interaction of CO2 with the material. As mentioned before, the strength of interaction of CO2 with a material strongly depends on the chemical structure of the material (i.e., nitrogen content and functionalities). Remarkably, the change in the nitrogen bonding upon increasing the synthesis temperature from 400 to 500 °C yielded an increase of Qst. X-ray photoelectron spectroscopy (XPS) investigations on the materials revealed that the N 1s scan of UA500-vac-1 and UA500-N2-1 displays a higher amount of pyridinic-electron-rich nitrogen with high affinity to CO2 (ref. 25) and a lower amount of pyrrolic-electron-poor nitrogen; for UA400-vac-1 it is the other way around (Fig. S16†). The increase in pyridinic nitrogen content with increasing the carbonization temperature was already reported for other carbonaceous materials.31 This highlights the significance of the temperature factor in the design of synthetic conditions for obtaining carbonaceous materials with a high affinity to CO2. Although for vacuum samples, porous structure develops already at 400 °C, nitrogen functionalities with high affinity to CO2 start to prevail only at 500 °C. This establishes 500 °C as a threshold temperature, at which both high nitrogen content is preserved, and electron-rich nitrogen functionalities start to dominate. The heat of adsorption of UA500-vac-10 is above 50 kJ mol−1, indicating an almost chemical sorption mechanism. The high nitrogen content with a high amount of electron-rich nitrogen and particular distribution of the micropores result in special structural sites with high affinity to CO2 that could be responsible for such strong binding of CO2.
Elaboration on the condensation stages and implication in the final porosity of materials
Based on the above-reported results and the existing experimental and theoretical reports on condensation in literature,32–34 we propose three possible stages of the molecular precursor transformations.Condensation is the stage that mainly determines the obtained materials' porosity. The pore development during this stage is defined by the interplay of heat transfer and mass transfer (release of by-products from the surface and the bulk) at particular temperatures and pressure. The surface of the particles presumably has a slightly higher temperature than the bulk of the particle; thus, the release of gases and cross-linking occurs on the surface of the particles earlier than in bulk. If we use low pressures (e.g., in vacuum), the cross-linking of the molecules is accompanied by the easy release of gases leading to the formation of the mesoporous surface (∼21 nm). The presence of mesopores on the surface makes the subsequent release of the different volatiles from the bulk of particles easier. As a result, formed condensates develop a hierarchical pore structure on both surface and bulk of particles, making the entire particle volume accessible for gases during sorption. When using high gas pressure (N2 flow), the release of volatiles from the surface is partially suppressed, which, combined with cross-linking, leads to the formation of the microporous shell. Thus, mass and heat transfer at different pressure conditions of the synthesis enables controlling porosity development. Using a higher heating rate at the same gas pressures causes more abrupt changes during mass and heat transfer (e.g., more rapid release of gases and the formation of ‘poked-balloon’ morphologies), suppresses structural ripening, and preserves optimal size of micropores.
Condensation in vacuum allows for lower condensation temperatures yielding materials with high nitrogen content and a well-developed porous structure. The selectivity and strength of binding of CO2 can be tailored by adjusting the micropore fraction (by changing the heating rate) and nitrogen bonding (by changing the temperature of the synthesis).
Conclusions
To our knowledge, this is the first study reporting on in situ TEM condensation of a molecular precursor. Combining in situ condensation inside a scanning transmission electron microscope with ex situ analysis of the products of condensation at different temperatures and atmospheres, we were able to follow the structural, morphological, and chemical evolution of the uric acid precursor at the nanoscale upon heating and correlate it with sorption properties of the obtained materials. We showed how one could control and tune the formation of pores in nitrogen-containing carbonaceous materials by varying pressures and reaction rates. These parameters change how the porosity of the surface develops, forming particles with mesopores (in vacuum) or microporous (in nitrogen) surfaces. Since this process co-occurs with cross-linking, the porous structure of the surface governs the subsequent release of volatiles and the development of the hierarchical pore structure. Using these findings, we demonstrated that synthesis in vacuum at 500 °C allows doubling the CO2 uptake compared to synthesis in nitrogen, while using a higher heating rate (10 °C min−1) allows keeping the same selectivity of the material. We believe that the findings from this study provide fundamental insights into the condensation process, which can be applied to a broad range of molecular precursors giving a new perspective on the design of novel nitrogen-doped carbonaceous materials for various applications.Author contributions
DP performed ex situ and in situ TEM analysis and wrote the draft. JK synthesized and characterized the materials, HZ performed in situ TEM analysis and developed the data treatment procedure. The work was supervised and conceptualized by MA, NL-S and NT. The manuscript was revised by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge The Max Planck Society for financial support. We also thank Antje Voelkel for the help with TGA and TGA–MS analysis.Notes and references
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 Search PubMed.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. Albahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 Search PubMed.
- Z. Zhang, Y. Zhao, Q. Gong, Z. Lib and J. Li, Chem. Commun., 2013, 49, 653–661 Search PubMed.
- R. B. Lin, D. Chen, Y. Y. Lin, J. P. Zhang and X. M. Chen, Inorg. Chem., 2012, 51, 9950–9955 Search PubMed.
- M. R. Hudson, W. L. Queen, J. A. Mason, D. W. Fickel, R. F. Lobo and C. M. Brown, J. Am. Chem. Soc., 2012, 134, 1970–1973 Search PubMed.
- R. Poloni, K. Lee, R. F. Berger, B. Smit and J. B. Neaton, J. Phys. Chem. Lett., 2014, 5, 861–865 Search PubMed.
- A. Sharma, J. Jindal, A. Mittal, K. Kumari, S. Maken and N. Kumar, Carbon Materials as CO2 Adsorbents: a Review, Springer International Publishing, 2021, vol. 19 Search PubMed.
- M. Perovic, Q. Qin and M. Oschatz, Adv. Funct. Mater., 2020, 30, 1908371 Search PubMed.
- Y. Xia, R. Mokaya, G. S. Walker and Y. Zhu, Adv. Energy Mater., 2011, 1, 678–683 Search PubMed.
- Á. Sánchez-Sánchez, F. Suárez-García, A. Martínez-Alonso and J. M. D. Tascón, ACS Appl. Mater. Interfaces, 2014, 6, 21237–21247 Search PubMed.
- N. López-Salas and M. Antonietti, Bull. Chem. Soc. Jpn., 2021, 94, 2822–2828 Search PubMed.
- M. Antonietti and M. Oschatz, Adv. Mater., 2018, 30, 1706836 Search PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 Search PubMed.
- R. Walczak, B. Kurpil, A. Savateev, T. Heil, J. Schmidt, Q. Qin, M. Antonietti and M. Oschatz, Angew. Chem., Int. Ed., 2018, 57, 10765–10770 Search PubMed.
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237–8265 Search PubMed.
- S. Zeng, S. Liu, Y. Qi, L. Cui, Q. Dai and C. Bai, RSC Adv., 2018, 8, 11462–11468 Search PubMed.
- S. Sharma, C. N. Shyam Kumar, J. G. Korvink and C. Kübel, Sci. Rep., 2018, 8, 1–12 Search PubMed.
- R. Schierholz, D. Kröger, H. Weinrich, M. Gehring, H. Tempel, H. Kungl, J. Mayer and R. A. Eichel, RSC Adv., 2019, 9, 6267–6277 Search PubMed.
- J. Shanmugam, K. B. Borisenko, Y. J. Chou and A. I. Kirkland, SoftwareX, 2017, 6, 185–192 Search PubMed.
- A. M. M. Abeykoon, H. Hu, L. Wu, Y. Zhu and S. J. L. Billinge, J. Appl. Crystallogr., 2015, 48, 244–251 Search PubMed.
- J. Kossmann, R. Rothe, T. Heil, M. Antonietti and N. López-Salas, J. Colloid Interface Sci., 2021, 602, 880–888 Search PubMed.
- A. V. Cornette, J. Villarroel-Rocha, K. Sapag, R. Delgado Mons, J. P. Toso and R. H. López, Carbon, 2020, 168, 508–514 Search PubMed.
- X. Hu, M. Radosz, K. A. Cychosz and M. Thommes, Environ. Sci. Technol., 2011, 45, 7068–7074 Search PubMed.
- J. Kossmann, T. Heil, M. Antonietti and N. López-Salas, ChemSusChem, 2020, 13, 6643–6650 Search PubMed.
- A. R. Izatulina, V. v. Gurzhiy, M. G. Krzhizhanovskaya, N. v. Chukanov and T. L. Panikorovskii, Minerals, 2019, 9, 373 Search PubMed.
- P. R. Solomon, M. A. Serio and E. M. Suuberg, Prog. Energy Combust. Sci., 1992, 18, 133–220 Search PubMed.
- M. S. Oh, W. A. Peters and J. B. Howard, AIChE J., 1989, 35, 775–792 Search PubMed.
- G. Lim, K. B. Lee and H. C. Ham, J. Phys. Chem. C, 2016, 120, 8087–8095 Search PubMed.
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 Search PubMed.
- Z. Yang and R. Mokaya, Microporous Mesoporous Mater., 2008, 113, 378–384 Search PubMed.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 Search PubMed.
- M. Inagaki, F. Kang, Fundamental Science of Carbon Materials, 2014 Search PubMed.
- S. Kumar, V. R. Battula and K. Kailasam, Carbon, 2021, 183, 332–354 Search PubMed.
- L. Borchardt, Q. L. Zhu, M. E. Casco, R. Berger, X. Zhuang, S. Kaskel, X. Feng and Q. Xu, Mater. Today, 2017, 20, 592–610 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta05247d |
| This journal is © The Royal Society of Chemistry 2022 |