
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-specific X-ray induced dynamics in liquid methanol†
Clara-Magdalena
Saak
 a,
Isaak
Unger
a,
Barbara
Brena
a,
Carl
Caleman
ab and
Olle
Björneholm
*a
a,
Isaak
Unger
a,
Barbara
Brena
a,
Carl
Caleman
ab and
Olle
Björneholm
*a
aDepartment of Physics and Astronomy, Box 516, 751 20 Uppsala, Sweden. E-mail: olle.bjorneholm@physics.uu.se
bCenter for Free-Electron Laser Science, Deutsches Elektronen-Synchrotron, DE-22607 Hamburg, Germany
First published on 26th June 2019
Abstract
Complex chemical and biochemical systems are susceptible to damage from ionising radiation. However, questions remain over the extent to which such damage is influenced by the nature of the surrounding chemical environment, which can consist of both hydrophobic and hydrophilic domains. To gain fundamental insight into the first crucial mechanistic steps of radiation damage in such systems, we need to understand the initial radiation response, i.e. dynamics occurring on the same timescale as electronic relaxation, which occur in these different environments. Amphiphilic molecules contain both hydrophobic and hydrophilic domains, but the propensity for charge delocalisation and proton dynamics to occur in these different domains has been largely unexplored so far. Here, we present carbon and oxygen 1s Auger spectra for liquid methanol, one of the simplest amphiphilic molecules, as well as its fully deuterated equivalent d4-methanol, in order to explore X-ray induced charge delocalisation and proton dynamics occurring on the few femtosecond timescale. Unexpectedly, we find a similar propensity for proton dynamics to occur at both the carbon and oxygen site within the lifetime of the core hole. Our results could serve as a model for decay processes that are likely to occur in other more complex amphiphilic systems.
1 Introduction
In the study of radiation damage, a central question is how to determine the extent of the damage caused by a single, localised ionisation event. In other words, how fast and far does the perturbation spread to the surrounding molecular environment due to the sequence of events occurring upon ionisation? The initial steps of charge migration induced by the creation of a core electron vacancy can already occur within the first few femtoseconds following ionisation via two pathways, electronic decay cascade1,2 and ultrafast bond dissociation,3 with the latter being especially important if one of the bonding partners is hydrogen. This charge migration leads to the subsequent ionisation or protonation of neighbouring molecules creating highly reactive short-lived molecular intermediates, which may then undergo further dynamics and/or reactions. Therefore, it is important to know the likelihood that core holes decay without involvement of other molecules versus excitation being passed on to a neighboring molecule.The probability for a given molecule to relax via one of these non-local decay channels is determined by the intermolecular structure of the environment the ionised site is embedded within, and therefore on the nature of the interactions of the site with its environment. In complex chemical systems, particularly the biochemical systems in which radiation damage is most relevant, such a wide variety of different modes and strengths of molecular interactions are available that disentangling the influence of different domains is unfeasible at present. We therefore need to look for a simpler system which can capture some of the environmental variety found in nature. One class of molecules displaying properties covering a range of modes and strengths of intermolecular interaction are amphiphilic molecules. Such molecules are composed of functional groups with distinctly different properties: Hydrophilic groups, which are usually characterised by charge polarisation, the formation of hydrogen bonds and strong interaction with charges and, in contrast, hydrophobic groups that are usually apolar, forming only weak dispersion interactions and weak interactions with charges. These differences between the functional groups lead to the formation of hydrophilic and hydrophobic domains in a liquid, and the pronounced differences between these molecular domains are a major driver in the formation of structure and interfaces in a liquid environment (e.g. membranes and micelles).
On the macroscopic scale, these hydrophilic/hydrophobic interactions help give rise to important bulk-level properties in the context of surfaces and interfaces, for example at the solid–liquid interface they are crucial in determining the wettability of the surface,4,5i.e. the ability of a liquid to maintain contact with the surface. Such properties are a collective manifestation of the individual molecular-level hydrophobic/hydrophilic interactions described above.
To investigate how different chemical environments influence the extent and effects of ionisation, here we have chosen to study methanol (CH3OH) as a simple model system for amphiphilic systems in general. The hydrophobic methyl group (–CH3) contains electronic states very similar to those of methane, while the hydrophilic hydroxyl group (–OH) is similar to water,6 thereby capturing the essential properties of the different types and strengths of interaction we are interested in. In addition, we want to study if and how domain formation around the different functional groups of a single amphiphilic molecule affects its post-ionisation decay. The intermolecular structure of liquid methanol has been discussed in numerous publications.7–11 The hydroxyl groups form “short, highly non-linear chains” connected by hydrogen bonds,7 which are 2.7 monomer units long on average with very few chains containing more than five molecules,7 resulting in different local environments surrounding the two functional groups; relatively denser packing of neighbouring molecules around the hydroxyl groups and looser packing around the methyl groups.
The experimental technique we employ in this work is Auger electron spectroscopy, which is well suited for studying the local environment of a molecule and its interactions with other molecules, since it combines chemical selectivity (via 1s ionisation) and sensitivity to the valence states that participate in any inter- and intramolecular interaction and bonding. In addition, Auger spectroscopy provides temporal information about processes occurring on the timescale of the core-hole lifetime, i.e. a few femtoseconds. The possible post-ionisation decay channels discussed in this work are shown in Fig. 1. Initial core ionisation at the carbon or oxygen site leaves the molecule in a highly excited 1s−1 state. For isolated molecules in the gas phase the dominant relaxation pathway for the intermediate state is Auger decay (>99%).12 In this mechanism, sometimes referred to as KVV Auger, a valence electron fills the 1s−1 vacancy and the excess energy is dissipated by the emission of a secondary electron from a valence state, leaving the initially neutral molecule or atom in a double valence-vacancy (v−2) final state of charge (2+). This decay mechanism is sketched in the centre of Fig. 1 and referred to as ‘local decay’ throughout the text. When there are neighbouring molecules sufficiently close to the ionised site, such as in molecular clusters and the condensed phase, the decay process can also involve the electronic states of these neighbouring molecules. Depending on the exact mechanism, these ‘non-local decay’ channels have been termed ‘interatomic coulombic decay’ (ICD)13–16 or ‘electron transfer mediated decay’ (ETMD).17,18 In Fig. 1 these pathways are labeled as O1 and C1. In the case of ICD, emission of the secondary electron occurs on a neighbouring molecule, and in the case of ETMD an electron from an adjacent molecule fills the core hole. Both of these decay channels lead to a final state in which the total charge (2+) is distributed over two molecules, which leads to a lower energy of the final states and hence higher kinetic energy (KE) of the secondary electron. These non-local process do not require any nuclear dynamics.
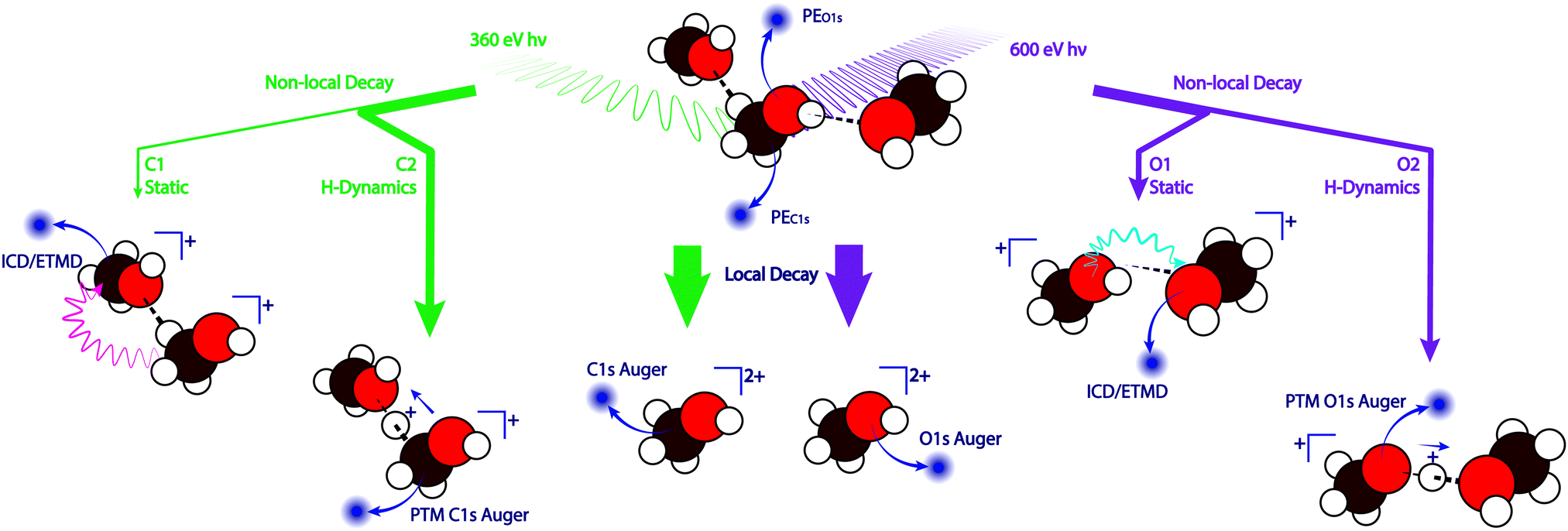 | ||
| Fig. 1 Possible decay channels discussed in this work. Decay paths after carbon ionisation are indicated in green, and the corresponding channels at the oxygen site are shown in purple. The ejected photoelectrons (PE) and secondary electrons are shown in dark blue. The majority of excited states decay via a local pathway (centre). The static non local decay mechanism (C1 and O1) shown here is ‘interatomic coulombic decay’ (ICD) but this pathway also includes ‘electron transfer mediated decay’ (ETMD). The pink and blue wavy arrows (C1 and O1) indicate the energy transfer of the ICD process. Any decay that occurs in conjunction with proton dynamics is denoted as C2 and O2 here. This figure shows the example of local Auger decay in a proton-transferred structure (PTM-Auger), but proton dynamics can also precede the other non-local decay channels mentioned above. | ||
If the lifetime of the intermediate 1s−1 state is long enough, some nuclear relaxation can occur before the electronic decay into the final state. In the case of the C 1s−1 and O 1s−1 the lifetime of the core-hole is on the order of a few femtoseconds.19 Nuclear dynamics occurring on this timescale have been described in previous publications.3,20,21 In the case of water a proton transfer during the lifetime of the O 1s−1 intermediate state has been studied in detail by Thürmer et al.22 What is unusual about this system is that the water O 1s−1 state is not dissociative in the gas phase, but dissociation becomes allowed in the hydrogen bonded condensed phase.
Any decay process involving such proton dynamics have been termed ‘proton transfer mediated charge separation’ (PTM-CS),22,23 since the loss of the proton decreases the total charge on the ionised molecule. This charge separation lowers the energy of the final state, which can be observed spectroscopically as an increase in kinetic energy of the secondary electron and therefore an increase of the relative intensity on the high kinetic energy flank of the Auger spectrum. The proton-transfer decay pathways are denoted as non-local channels O2 and C2 in Fig. 1.
In order to separate the PTM-CS feature from other non-local decay channels, deuterium is substituted for hydrogen in the sample. The higher mass of the isotope leads to decreased dispersion of the wave packet in the excited state, i.e. a higher percentage of states decays close to the ground state geometry.22 All other non-local processes are unaffected by the isotopic substitution, as the geometric and electronic structure of the system is not significantly altered by the isotopic substitution. The PTM-CS contribution can therefore be identified by comparing the high kinetic energy flank of the Auger region of a light/normal system and its fully deuterated equivalent.
Here, we investigate how the propensity for different non-local core hole decay pathways shown in Fig. 1 differ between the carbon and oxygen sites in liquid methanol, due to the different intermolecular bonding patterns of the hydrophilic hydroxyl (–OH) and hydrophobic methyl (–CH3) groups. We find that, in liquid methanol, local Auger decay remains the dominant channel for both the carbon and oxygen sites. The non-local decay of the –OH group largely mirrors the behaviour of liquid water,22 with a significant fraction of ionised sites decaying via static non-local decay and via channels involving hydrogen dynamics. The carbon vacancy shows a lesser contribution from static non-local decay channels but a surprisingly large isotope effect, which implies that significant proton dynamics occur during the lifetime of the C 1s−1 vacancy.
2 Experimental
To study the effect of proton dynamics and charge separation on the post-ionisation processes in liquid methanol, a series of Auger (O 1s and C 1s) spectra were recorded for liquid methanol alongside its fully deuterated equivalent d4-methanol. The gas phase contribution to each spectrum was subtracted from the Auger traces, yielding the ‘liquid-only’ traces, which were then normalised to their total area.To approximate the relationship between local/molecular Auger decay channels and non-local decay channels in the liquid phase spectra the gas phase spectrum is convoluted with a Gaussian function, which effectively broadens it, and shifting it towards higher kinetic energy, as outlined for water clusters by Öhrwall et al.14 The comparison between the liquid-only traces and the broadened gas phase then yields the spectral regions that contain signal intensity that cannot be assigned to local decay. The width of the broadening Gaussian function is determined by an additional broadening factor that is determined from the relative gas and liquid phase photoionisation spectrum. A detailed outline of the procedure that was used to determine the additional broadening factor can be found in the Supplementary methods section of the ESI.†
The liquid-only traces of methanol and d4-methanol are then subtracted from one another (methanol – d4-methanol), in order to produce a difference spectrum (total area = 0) showing the relative redistribution of signal intensity from low to high kinetic energy due to proton dynamics.
Experiments were performed at the SOL3 endstation24 located at the U49-2/PGM1 beamline25 of the BESSY II storage ring at Helmholtz-Zentrum Berlin. The target was a 20 μm diameter liquid jet, which was perpendicularly intersected with linearly polarised soft X-ray radiation. The hemispherical electron analyser was mounted at magic angle (∼55°) relative to the horizontal polarization of the synchrotron radiation. Gas phase spectra were recorded by disaligning the liquid jet and measuring on the evaporating solvent. In order to avoid charging of the sample, 50 mM NaCl (Sigma Aldrich, >99.8% purity) was added to both methanol and d4-methanol, to ensure sufficient conductivity of the liquid. The liquid jet was operated at a sample flow-rate of 1.15 ml min−1. C 1s data was obtained with a photon energy of 360 eV and the corresponding O 1s data at 600 eV. At these photon energies the beamline provides a flux of ∼1013 photons s−1. During operation of the liquid jet the pressure in the interaction chamber is maintained in the range of 10−4 to 10−3 mbar by a combination of turbopumps and liquid nitrogen cool-traps; details of the differential pumping system are provided in a publication by Seidel et al.24 which describes the experimental endstation in detail. When the liquid jet is not mounted on the experiment the base pressure of the interaction chamber is ∼10−7 mbar. The kinetic/binding energy scale was calibrated on the lowest binding energy valence peak of the liquid. The binding energy of this peak was previously reported to be 9.99 eV.26 This work does not fully consider effects of the streaming potential,27 which has been observed to significantly alter the apparent binding energy in the case of liquid water.28–30 However, to the best of our knowledge no genuine binding energy has been reported for liquid methanol. Therefore, the absolute binding/kinetic energy values may be different from those reported here, but this does not affect the overall results, discussion or conclusions of this work.
When interpreting the results from these measurements there are two effects to take into consideration: Firstly, the lifetimes of the O 1s−1 and C 1s−1 excited states are not identical, but 4 fs and 6 fs,19 respectively. The longer lifetime of the C 1s core-hole allows for greater dispersion of the wavepacket in the excited state, making the C 1s Auger more sensitive to any changes in the final state energy due to structural changes. Secondly, since the Auger electrons carry relatively low kinetic energy, ∼250 eV and 500 eV for the C 1s and O 1s respectively, the mean free path of the electrons in the liquid is ∼1–2 nm.31 This means that the Auger spectra contain contributions not only from the bulk but also from the surface layer, where the intermolecular distances may be somewhat different.32 To disentangle these bulk and surface contributions is currently not feasible, and remains a subject for future research.
To complement the experimental data with computational support, we performed Density Functional Theory (DFT) calculations of the methanol molecule and dimer using the Gaussian16 program,33 using the hybrid B3LYP functional and the triple zeta aug-cc-pVTZ (augmented valence triple Z plus polarization)34 basis set. First we computed the relaxed ground state geometries of the single methanol molecule and its dimer. In the dimer, a hydrogen bond forms between the hydrogen bonded to the oxygen atom of one molecule and the oxygen atom of the other molecule. We then simulate a core hole in the hydrogen bond donor oxygen atom via the equivalent core approximation35 and performed a geometry optimisation of the molecule. In the equivalent core approximation the oxygen atom is substituted by a fluorine atom and a positive charge is added. The increased nuclear charge of the fluorine atom approximates the increased coulombic potential experienced by the valence electrons due to the core hole. The additional positive charge is necessary to maintain the same number of valence electrons as in the case of the un-substituted system.
3 Results and discussion
Auger spectra for the isolated gas phase methanol molecule are shown in Fig. 2. These spectra consist of transitions from the C 1s−1 and O 1s−1 intermediate state to a multitude of final states with two valence holes, both localized on the molecule, as schematically shown as local decay in Fig. 1. The gas phase Auger spectrum of methanol has previously been described in detail by Rye et al.6 As discussed therein, the highest energy Auger electrons result from transitions involving the outer-valence states of the molecule, such as the non-bonding lone pairs of the oxygen site. Transitions involving the inner-valence states then appear at progressively lower kinetic energy. Rye et al. also point out that the C 1s and O 1s Auger spectra closely resemble those of methane and water, respectively, with only a few transitions shifting in energy. This indicates that the OH group is still expected to resemble the behaviour previously observed in water (see above). | ||
| Fig. 2 Liquid-only spectra of methanol (red) and d4-methanol (blue) are compared to the shifted gas-phase (grey) and its broadened equivalent (green). A difference trace (orange) of the methanol spectrum and the broadened gas-phase is shown to illustrate the contributions of non-local decay (see main text). The C 1s spectra are shown in the top and the O 1s in the bottom panel. The C 1s gas phase and broadened gas phase traces are shifted by +3.7 eV and the corresponding O1s traces by +4.6 eV. | ||
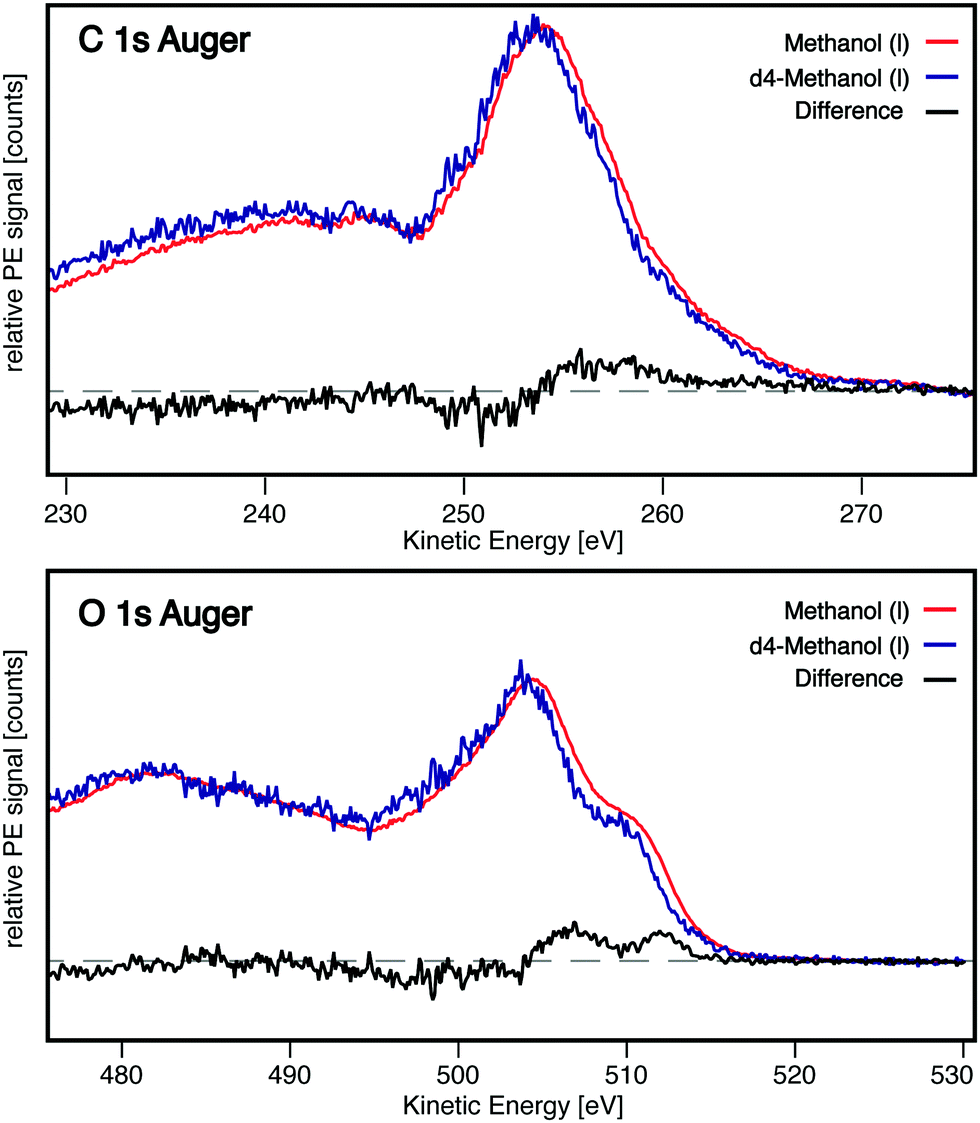
The C 1s and O 1s Auger spectra of the gas phase are shown in Fig. 2, these traces are shifted by +3.7 eV in the case of C 1s ionisation and +4.6 eV for the corresponding O 1s trace. The reason for this shift is outlined later in the text. The gas phase C 1s Auger spectrum shows a broad main peak at ∼250 eV kinetic energy and a progression of smaller features at lower kinetic energy. Additionally, two small features can be identified between 255–265 eV. The gas phase O 1s Auger spectrum has a prominent main feature at ∼500 eV, with a pronounced shoulder at ∼505 eV. At low kinetic energy (<500 eV) a progression of smaller peaks is found, similar to the C 1s Auger. The C 1s and O 1s Auger spectra of liquid methanol and d4-methanol all contain significant contributions from the gas phase of evaporating molecules surrounding the liquid jet. The gas phase subtracted liquid-only traces are displayed in Fig. 2. The liquid-only C 1s spectrum shows one broad main peak at 253 eV, and similar to the gas phase spectrum, a series of smaller peaks at lower kinetic energy. However, these features are broadened in the liquid phase, such that they merge and are no longer fully resolved in our data. The liquid-only O 1s Auger spectrum is comprised of two overlapping peaks at 504 and 509.5 eV kinetic energy. The lower kinetic energy region of the spectrum (<495 eV), similar to the C 1s region, features a series of overlapping broad peaks. Both the C 1s and O 1s Auger liquid-only traces feature some background contribution from inelastic scattering in the lower kinetic energy region (below the main peak).
To describe the local/molecular Auger decay channels in the liquid phase spectra, which are equivalent to the gas phase transitions, one needs to account for additional polarisation screening of the charges in the excited and final state by the condensed phase. This can be approximated by convoluting the gas phase spectrum with a Gaussian function, which effectively broadens it, and shifting it towards higher kinetic energy, as described for water clusters by Öhrwall et al.14 A detailed outline of the procedure that was used to determine the additional broadening factor can be found in the Supplementary methods section of the ESI.† The full-width-at-half-maximum of the broadening function that was used to obtain the broadened gas phase spectra shown in this work was determined to be 2.46 eV for C Auger and 3.27 eV for O Auger. The comparison between the liquid-only traces (methanol and d4-methanol) and the broadened gas phase is shown in Fig. 2.
Any signal intensity that cannot be described by the broadened gas phase trace is most likely due to non-local decay channels involving the neighbouring molecules of the ionisation site, e.g. static non-local decay channels C1 and O1 of Fig. 1. Both ICD14–16 and ETMD17,18 have been observed to occur for solutes in the aqueous medium.36 These non-local decay channels do not require any nuclear dynamics to occur and lead to a final state in which the double vacancy is shared between two molecules. From the comparison shown in Fig. 2 it becomes apparent that these channels significantly contribute to the decay processes at the oxygen site and less so to the carbon site. In the case of the C 1s Auger the broadened gas phase and the liquid-only trace are very similar in their shape, the only difference is on the high kinetic energy flank, which contains a small broad, featureless contribution that cannot be described by the broadened gas phase spectrum. In the O 1s region, however, there is a significant difference between the broadened gas phase and the liquid-only traces, specifically the high kinetic energy shoulder of the main peak which can clearly be seen in the bottom panel of Fig. 2.
Due to the lifetime of the intermediate core hole state (4 and 6 fs19) both local and non-local decay channels can occur in parallel to nuclear dynamics in the 1s−1 state.3,20,23,31 In order to separate decay events that occur in the ground state geometry from those involving proton dynamics, we compare the liquid-only traces of methanol and d4-methanol, as these systems have different timescales for the nuclear dynamics. Fig. 3 shows the comparison between the liquid-only spectra of methanol and its deuterated equivalent and indicates an appreciable isotope effect in the O 1s Auger region, similar to the effect previously reported for water,22i.e. a relative increase of intensity on the high kinetic energy flank of the methanol spectra compared with d4-methanol. A similar effect is observed for the C 1s Auger. This redistribution of relative signal intensity indicates that proton transfer dynamics are taking place on the oxygen and carbon site of methanol during the lifetime of the intermediate 1s−1 state. The mechanism of the proton transfer is sketched in Fig. 1.
 | ||
| Fig. 3 The normalised liquid-only traces of methanol (red) and d4-methanol (blue) are subtracted from one another to yield a difference trace (black). The positive and negative intensity of the difference trace illustrated the change in final state energy due to proton dynamics during the lifetime of the core hole. The C 1s Auger case is shown in the top and the O 1s Auger in the bottom panel. | ||
To confirm whether the excited state shows the same or similar dissociative behaviour in methanol as it does in water we performed a set of DFT simulations. The computed ground state structure of methanol is shown in Fig. 4 structure (a) and the corresponding geometry optimised structure of the excited state is shown in Fig. 4 structure (b). The core hole ionised excited state is simulated by means of the equivalent core approximation35 where the oxygen is substituted by a F+ atom, which we denote as O(*). This approach is also often referred to as the (Z + 1) approximation.37 The computed OH bond length in the isolated methanol molecule is 0.961 Å and 0.951 Å in the excited state. Since all bonds stay intact when the O(*) core hole is introduced it can be concluded from these structures that the excited state is expected to be non-dissociative in the isolated molecule.
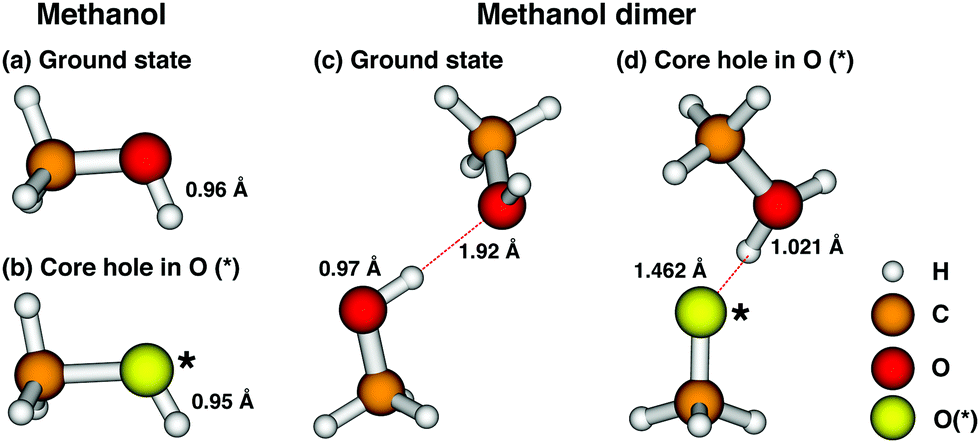 | ||
| Fig. 4 Computed geometries of methanol and methanol dimer in the ground state and the core hole excited state. Structure (a) shows the methanol molecule in its ground state, with the O–H bond length indicated. Structure (b) shows the equivalent core approximation of the 1s core excited oxygen in methanol: the excited oxygen site is substituted by a F+ atom, and denoted as O(*). The resulting slightly reduced O–H bond length is indicated. Structure (c) shows the ground state methanol dimer. The O–H bond length and the distance between the proton and the oxygen of the opposite molecule are given. Structure (d) shows the equivalent core approximation in the case of the methanol dimer. In the approximated core excited state the proton migrates to the neighboring molecule along the axis of the hydrogen bond. The distance between the O(*) atom and the proton, as well as the O–H bond length are given. | ||
In the case of the methanol dimer (Fig. 4 structure (c)) a (O–H⋯O) hydrogen bond is formed, as expected, leading to an elongation of the O–H bond to 0.969 Å in the ground state, compared with the isolated molecule. The intermolecular distance between the proton and the oxygen of the hydrogen bond acceptor molecule is 1.922 Å. The excited state structure of the dimer was computed in the same manner as for the isolated molecule and is shown in Fig. 4 structure (d). After geometry optimisation, the proton previously bound to the core excited site has migrated toward the oxygen atom of the other molecule. In this relaxed structure the newly formed O–H bond is 1.021 Å long, while the O(*) atom lies at 1.462 Å from the proton. This migration of the proton means that the excited state becomes dissociative once the O–H group is engaged in a hydrogen bond, as opposed to the non-dissociative exited state of the methanol monomer.
In the experiment the molecules are not expected to fully relax into the excited state structure as shown in Fig. 4(d) due to the limited lifetime of the state, but it is reasonable to assume that the system will move towards this structure until it decays into the final state via an electronic decay channel. These results therefore show that the O 1s−1 core excited state is expected to be dissociative when the methanol molecule is embedded into a hydrogen bond network but non-dissociative in the gas phase, mirroring the behaviour that was previously reported for water.22
In summary, our measurements are consistent with the expectation that the O 1s−1 excited state of the methanol hydroxyl group largely decays like water, showing a significant contribution from non-local decay channels and an isotope effect in the high kinetic energy region. In the case of C 1s ionisation our measurements indicate a weaker non-local decay contribution, compared with the hydroxyl group, but we observe proton dynamics that are of a similar propensity to those observed after O 1s ionisation, within the accuracy of our measurements. This is unexpected because it has been previously argued that for PTM-CS to occur there needs to be a hydrogen bond in the ground state geometry.38 In the case of methanol only few, weak C–H⋯O hydrogen bonds are expected to be formed.8,11 These results suggest either there are more carbon-based hydrogen bonds than we previously thought, or that proton transfer can occur in the absence of strong hydrogen bonding, which we discuss later in the text.
As shown above, there are noticeable differences in non-local decay propensity between the carbon and oxygen sites. Since these non-local decay channels (labelled C1 and O1 in Fig. 1) strongly modulate with intermolecular distance to the next neighbour, any differences in the immediate environments of these sites could explain the differences observed in our experiment. The partial radial distribution functions (RDF) of liquid methanol have been described in detail previously.7 Here we focus our discussion on the three nearest non-hydrogen neighbour distances as reported by Yamaguchi et al.:7 O–O (2.7 Å), C–O (3.5 Å) and C–C (3.8 to 4 Å). The O–O distance is significantly shorter than any nearest neighbor distance involving carbon, while the larger distance to the nearest (non-hydrogen) neighbour around carbon reflects the absence of strong hydrogen bonds formed with the methyl group. Our observation of a lower non-local decay propensity at the carbon site can therefore be attributed to the difference in the distance to the nearest neighbor at these sites. Structural differences and environments are not only important for understanding non-local effects in methanol, but they also play a critical role in determining the local proton transfer dynamics, as we discuss next. The decay channels (labelled C2 and O2) involving proton dynamics are sketched in Fig. 1.
The equivalent core (Z + 1) approximation, which was employed in the methanol dimer simulations of the O 1s−1 excited state, can be a helpful tool to qualitatively understand how core-ionised species behave in a molecule.37 The equivalent substitution to CH3FH+ in the case of CH3O(1s−1)H+ ionisation for the C(1s−1)H3OH+ state would be the NH3OH+ molecule. Both of these model molecules will most likely dissociate a proton, as was shown for O 1s−1 in Fig. 4. The doubly bonded fluoride-like center is expected to be much more unstable than the ammonium-like group, which would correspond to a steeper potential in the O 1s−1 state. During the short lifetimes of the oxygen and carbon core-hole, the proton is most likely not fully transferred to its nearest neighbour. It is more likely the O–H bond elongates and the proton moves towards a Zundel-like39 structure in which the proton is shared between two molecules. The electronic decay then occurs in this proton-transferred geometry. This mechanism is outlined in the equation below, where we use O 1s ionisation in a methanol dimer as an example. Non-covalent interactions, such as hydrogen bonds are denoted by ‘⋯’, and elongated covalent bonds by ‘—’. The second column shows the equivalent core approximation, where applicable:

We may consider the initial elongation of the C–H or O–H bond in the intermediate state to initiate a proton transfer that completes after relaxation into the electronic final state. In the case of oxygen ionisation, specifically, the ejected proton may then diffuse along the hydrogen bond network of the hydrophilic domain via a Grotthuss-like mechanism, further separating the two final-state charges.
Since the proton initially moves through a Zundel-like structure, there needs to be sufficient electron density (δ−) close to the proton to stabilise the positive charge site created by the dissociation event. Formation of a hydrogen bond in the ground state immobilises the molecules in exactly the geometry which is necessary for a proton to be transferred in the excited state. To estimate the population of hydrogen bonds formed at the C and O sites, we again look at the intermolecular radial distribution functions,7 since the formation of a hydrogen bond reduces the average distance to the nearest non-hydrogen neighbour. In the case of methanol, the only site which can act as a hydrogen-bond acceptor and receive a proton is the non-bonding lone pair of oxygen. The distance to the next oxygen site is therefore a good indicator for the probability of proton transfer. Since the intermolecular C–O distance is ∼1 Å longer than O–O (see above) we may assume stronger interaction of the proton with the oxygen lone pair on an adjacent OH group in the ground state, than with the –CH3 group. It is therefore more likely for a proton to be transferred in the O 1s−1 excited state. The population of lone pairs around O–H is also higher, due to the strong O–H⋯O hydrogen bonds formed in methanol.40 C–H⋯O hydrogen bonds have been observed for methanol in water,41 where the –CH3 group can only interact with an oxygen lone pair. In pure methanol this is not the case, since the hydroxyl groups preferentially interact with each other, thereby creating hydrophobic domains. Only a small fraction of molecules are expected to form these bonds at any given moment.
All considerations outlined so far would suggest we should expect significantly less proton dynamics at the carbon site, due to the lower population of C–H⋯O hydrogen bonds. Unexpectedly, however, our results show the isotope effect in the carbon region is of similar magnitude to the oxygen site. Next, we discuss two possible explanations of this unusually large isotope effect in the carbon Auger, beyond the difference in core hole lifetime of the two sites.
Firstly, it might simply be that there are more C–H⋯O hydrogen bonds formed in the liquid than we previously thought. In contrast, it could be the case that the proton transfer in the excited state is facilitated by proximity to an oxygen site, but without a hydrogen bond forming. In this case the polarisation screening of the charges in the condensed phase would have to be sufficiently strong to lower the proton transfer barrier in the excited state.
The second effect to consider is that non-dissociative proton dynamics in the excited state can also lead to an isotope effect. The umbrella vibration of ammonia has previously been observed to lead to similar effects on the Auger spectrum as the dissociation of a C–H or O–H bond.42 To judge if such effects influence the decay at a specific group, we should consider if there is additional rearrangement at the ionisation site in conjunction with the proton transfer. For the –OH group, there is no need for significant rearrangement since the oxygen site is left with a single bond (O−–CH3) upon loss of a proton. For the –CH3 group, however, loss of the proton most likely also leads to rearrangement of the remaining two C–H bonds from a tetrahedral sp3 to a planar sp2 geometry. This rearrangement happens in all likelihood in synchronicity with the dissociation of the C–H bond and may lead to an additional isotope effect. With our data it is not possible to pin down which of these effects drives the unexpectedly high rate of proton dynamics at the carbon site, since we cannot differentiate between dissociation and rearrangement dynamics. Simulations, as were performed for aqueous ammonia solutions,42 might be helpful to address this question in the future.
Now we turn our attention to what happens after autoionisation has taken place, and we are left with the final state charges. Liquid methanol displays only weak autoprotolysis, as well as weak acidity. However, the dissociation we observe upon core level ionisation leads to the creation of free protons that are not present in the ground state. This means we observe X-ray induced acidity, similar to the photoacidity of X–H bonds reported in connection with HOMO–LUMO excitation.43
The difference in non-local decay propensity between carbon and oxygen observed here leads, for oxygen ionisation, to a distribution of the total charge in the final state over two molecules, whereas core-level ionisation of the methyl group most likely leads to a higher local charge and less charge separation. The strongly interacting hydroxyl group can distribute the total final state charge to its neighbours. This sharing of charge can occur both via non-local decay or through migration of a proton. The increased contribution of these channels to relaxation of the system leads to many single unit charges that are shared between hydrophilic sites. In contrast, the carbon group can delocalise its final state charge to a lesser degree via non-local decay, as the neighboring molecules are further away leading to a small propensity for non-local decay, as discussed above. The only pathway to reduce the total charge in the final state is therefore the dissociation of a C–H bond.
Another difference between the hydrophilic and hydrophobic regions is the way the charges of the final states are screened. Small charges will be more strongly screened in a hydrophilic environment, making the effective charge ‘per unit real charge’ larger in a hydrophobic medium. Similarly, the valence vacancies created by electronic decay can be more strongly stabilised in the hydrophilic domain, namely by the oxygen lone pairs, than by the non-polar alkyl groups.
On the longer post-Auger timescale, these differences in charge delocalisation dynamics between the hydrophilic and hydrophobic regions will affect further fragmentation and radical formation, i.e. the first steps in radiation damage in a more complex network of hydrophilic/hydrophobic domains.
4 Conclusions
The carbon and oxygen site of methanol show different propensities for post-ionisation decay channels. The C 1s−1 and O 1s−1 vacancies mainly decay via the local Auger pathway, however a larger subset of the O 1s−1 states decay non-locally through a mechanism that involves the electronic states of its immediate neighbours. This different propensity for local/non-local decay means that the final states following C 1s or O 1s ionisation show either more localised charge density (C) or shared charge states (O). This is because the local structure around the ionisation site determines decay channel propensity. Carbon mainly displays local decay because of its weak intermolecular interaction with its neighbours, and the resulting larger distances to the next atom. Conversely the oxygen site decays non-locally to a higher degree, because of its strong, directional intermolecular interactions which decrease the distance to the next atom.Proton transfer during the 1s−1 excited state appears to be a general pathway as long as strong intermolecular interactions such as hydrogen bonding lower the barrier for a shared proton Zundel-like state to be formed between both participating molecules. Both in the case of –CH3 and –OH a significant isotope effect, and therefore propensity for C–H or O–H bond dissociation, has been observed. This dissociation allows for a delocalisation of the (2+) final state charge without involving the electron density of the neighboring molecules in the decay process.
While water and methanol show different dissociation constants in the ground state, their behaviour becomes more similar in the core excited state which we have studied here. The additional driving force for dissociation in the excited state is the same for both methanol and water. Excited state C–H or O–H dissociation is therefore not dependent on ground state dissociation, but they both need the same geometry, namely a hydrogen-bond-like arrangement in which an oxygen lone-pair is positioned on axis with the C–H or O–H bond.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Robert Seidel for his support with the SOL3 experimental endstation. We thank the Helmholtz-Zentrum Berlin (HZB) for the allocation of synchrotron radiation beamtime and the BESSY II staff for support during the beamtime. Funding from the Swedish Research Council (VR) for the projects VR 2017-04162, VR 2018-00740, and VR 2013-03940 is acknowledged. CC acknowledges program-oriented funds of the Helmholtz Association through the Center for Free-Electron Laser Science at Deutsches Elektronen-Synchrotron (DESY). CMS acknowledges funding from the Swedish Foundation for International Cooperation in Research and Higher Education (STINT).References
- V. Stumpf, K. Gokhberg and L. S. Cederbaum, Nat. Chem., 2016, 8, 237–241 CrossRef CAS PubMed.
- D. Céolin, N. V. Kryzhevoi, C. Nicolas, W. Pokapanich, S. Choksakulporn, P. Songsiriritthigul, T. Saisopa, Y. Rattanachai, Y. Utsumi, J. Palaudoux, G. Öhrwall and J.-P. Rueff, Phys. Rev. Lett., 2017, 119, 263003 CrossRef PubMed.
- P. Morin and C. Miron, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 259–266 CrossRef CAS.
- S. Maier and M. Salmeron, Acc. Chem. Res., 2015, 48(10), 2783–2790 CrossRef CAS PubMed.
- E. A. Vogler, Adv. Colloid Interface Sci., 1998, 74, 69–117 CrossRef CAS PubMed.
- R. R. Rye, T. E. Madey, J. E. Houston and P. H. Holloway, J. Chem. Phys., 1978, 69, 1504–1512 CrossRef CAS.
- T. Yamaguchi, K. Hidaka and A. K. Soper, Mol. Phys., 1999, 96, 1159–1168 CrossRef CAS.
- G. Yukhnevich and E. Tarakanova, J. Mol. Struct., 1998, 447, 257–261 CrossRef CAS.
- S. Kashtanov, A. Augustson, J.-E. Rubensson, J. Nordgren, H. Ågren, J.-H. Guo and Y. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 104205 CrossRef.
- K. Lin, X. Zhou, Y. Luo and S. Liu, J. Phys. Chem. B, 2010, 114, 3567–3573 CrossRef CAS PubMed.
- Y. Tamenori, K. Okada, O. Takahashi, S. Arakawa, K. Tabayashi, A. Hiraya, T. Gejo and K. Honma, J. Chem. Phys., 2008, 128, 124321 CrossRef CAS PubMed.
- M. O. Krause, J. Phys. Chem. Ref. Data, 1979, 8, 307–327 CrossRef CAS.
- L. S. Cederbaum, J. Zobeley and F. Tarantelli, Phys. Rev. Lett., 1997, 79, 4778–4781 CrossRef CAS.
- G. Öhrwall, R. F. Fink, M. Tchaplyguine, L. Ojamäe, M. Lundwall, R. R. T. Marinho, A. Naves de Brito, S. L. Sorensen, M. Gisselbrecht, R. Feifel, T. Rander, A. Lindblad, J. Schulz, L. J. Sæthre, N. Mårtensson, S. Svensson and O. Björneholm, J. Chem. Phys., 2005, 123, 054310 CrossRef PubMed.
- T. Jahnke, H. Sann, T. Havermeier, K. Kreidi, C. Stuck, M. Meckel, M. Schöffler, N. Neumann, R. Wallauer, S. Voss, A. Czasch, O. Jagutzki, A. Malakzadeh, F. Afaneh, T. Weber, H. Schmidt-Böcking and R. Dörner, Nat. Phys., 2010, 6, 139–142 Search PubMed.
- M. Mucke, M. Braune, S. Barth, M. Förstel, T. Lischke, V. Ulrich, T. Arion, U. Becker, A. Bradshaw and U. Hergenhahn, Nat. Phys., 2010, 6, 143–146 Search PubMed.
- J. Zobeley, R. Santra and L. S. Cederbaum, J. Chem. Phys., 2001, 115, 5076–5088 CrossRef CAS.
- R. Santra, J. Zobeley and L. Cederbaum, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 245104 CrossRef.
- C. Nicolas and C. Miron, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 267–272 CrossRef CAS.
- I. Hjelte, M. Piancastelli, R. Fink, O. Björneholm, M. Bässler, R. Feifel, A. Giertz, H. Wang, K. Wiesner, A. Ausmees, C. Miron, S. Sorensen and S. Svensson, Chem. Phys. Lett., 2001, 334, 151–158 CrossRef CAS.
- C. Richter, D. Hollas, C.-M. Saak, M. Förstel, T. Miteva, M. Mucke, O. Björneholm, N. Sisourat, P. Slavíček and U. Hergenhahn, Nat. Commun., 2018, 9, 4988 CrossRef PubMed.
- S. Thürmer, M. Ončák, N. Ottosson, R. Seidel, U. Hergenhahn, S. E. Bradforth, P. Slavíček and B. Winter, Nat. Chem., 2013, 5, 590–596 CrossRef PubMed.
- P. Slavíček, N. V. Kryzhevoi, E. F. Aziz and B. Winter, J. Phys. Chem. Lett., 2016, 7, 234–243 CrossRef PubMed.
- R. Seidel, M. N. Pohl, H. Ali, B. Winter and E. F. Aziz, Rev. Sci. Instrum., 2017, 88, 073107 CrossRef PubMed.
- T. Kachel, Journal of large-scale research facilities, 2016, 2, A72 CrossRef.
- M. Faubel, B. Steiner and J. P. Toennies, J. Chem. Phys., 1997, 106, 9013–9031 CrossRef CAS.
- N. Preissler, F. Buchner, T. Schultz and A. Lübcke, J. Phys. Chem. B, 2013, 117, 2422–2428 CrossRef CAS PubMed.
- B. Winter, R. Weber, W. Widdra, M. Dittmar, M. Faubel and I. V. Hertel, J. Phys. Chem. A, 2004, 108, 2625–2632 CrossRef CAS.
- N. Kurahashi, S. Karashima, Y. Tang, T. Horio, B. Abulimiti, Y.-I. Suzuki, Y. Ogi, M. Oura and T. Suzuki, J. Chem. Phys., 2014, 140(17), 174506 CrossRef PubMed.
- G. Olivieri, A. Goel, A. Kleibert, D. Cvetko and M. A. Brown, Phys. Chem. Chem. Phys., 2016, 18, 29506–29515 RSC.
- S. Thürmer, R. Seidel, M. Faubel, W. Eberhardt, J. C. Hemminger, S. E. Bradforth and B. Winter, Phys. Rev. Lett., 2013, 111, 173005 CrossRef PubMed.
- K. R. Wilson, R. D. Schaller, D. T. Co, R. J. Saykally, B. S. Rude, T. Catalano and J. D. Bozek, J. Chem. Phys., 2002, 117, 7738–7744 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision B.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- B. Brena, Y. Luo, M. Nyberg, S. Carniato, K. Nilson, Y. Alfredsson, J. Åhlund, N. Mårtensson, H. Siegbahn and C. Puglia, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 195214 CrossRef.
- I. Unger, R. Seidel, S. Thürmer, M. N. Pohl, E. F. Aziz, L. S. Cederbaum, E. Muchová, P. Slavíček, B. Winter and N. V. Kryzhevoi, Nat. Chem., 2017, 9, 708–714 CrossRef CAS PubMed.
- S. Hüfner, Photoelectron Spectroscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 3rd edn, 2003 Search PubMed.
- I. Unger, S. Thürmer, D. Hollas, E. F. Aziz, B. Winter and P. Slavíček, J. Phys. Chem. C, 2014, 118, 29142–29150 CrossRef CAS.
- G. Zundel and H. Metzger, Z. Phys. Chem., 1968, 58, 225–245 CrossRef CAS.
- B. M. Pettitt and P. J. Rossky, J. Chem. Phys., 1983, 78, 7296–7299 CrossRef CAS.
- Y. Yu, W. Fan, Y. Wang, X. Zhou, J. Sun and S. Liu, J. Phys. Chem. B, 2017, 121, 8179–8187 CrossRef CAS PubMed.
- I. Unger, D. Hollas, R. Seidel, S. Thürmer, E. F. Aziz, P. Slavíček and B. Winter, J. Phys. Chem. B, 2015, 119, 10750–10759 CrossRef CAS PubMed.
- W. Domcke and A. L. Sobolewski, Science, 2003, 302, 1693–1694 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp02063b |
| This journal is © the Owner Societies 2019 |