
Faraday Discussions meeting Catalysis for Fuels
Nico
Fischer
*a,
Simon A.
Kondrat
*b and
Mzamo
Shozi
*c
aCatalysis Institute and DST-NRF Centre of Excellence in Catalysis c*change, Department of Chemical Engineering, University of Cape Town, 7701, Cape Town, South Africa. E-mail: nico.fischer@uct.ac.za
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: KondratSA@cardiff.ac.uk
cSchool of Chemistry and Physics, University of KwaZulu-Natal, Durban, 4000, South Africa. E-mail: Shozim2@ukzn.ac.za
First published on 24th April 2017
Abstract
Welcome to Africa was the motto when after more than 100 years the flag ship conference series of the Royal Society of Chemistry, the Faraday Discussions was hosted for the first time on the African Continent. Under the fitting topic ‘Catalysis for Fuels’ over 120 delegates followed the invitation by the conference chair Prof. Graham Hutchings FRS (Cardiff Catalysis Institute), his organizing committee and the co-organizing DST-NRF Centre of Excellence in Catalysis c*change (http://www.cchange.ac.za). In the presentations of 21 invited speakers and 59 posters, cutting edge research in the field of catalysis for fuels, designing new catalysts for synthetic fuels, hydrocarbon conversion in the production of synthetic fuels and novel photocatalysis was presented over the two-day meeting. The scene was set by the opening lecture of Prof. Enrique Iglesias (UC Berkeley) and wrapped-up with the concluding remarks by Philip Gibson (SASOL).
Introduction
From the 24th to the 26th of January, the first time ever Faraday Discussion on the African Continent was held in Cape Town, South Africa. The Faraday Discussion conference series has been organized by the Royal Society of Chemistry for over 100 years and promotes all fields of physical chemistry. In the recent past about 11–12 Faraday Discussion events take place per year and while traditionally held in the United Kingdom, meetings have been organised in the Americas, mainland Europe as well as Asia, and now also Africa. Under the fitting topic ‘Catalysis for Fuels’ the organizing committee chaired by Prof. Graham Hutchings FRS comprised researchers from the Netherlands and the UK as well as members of the South African DST-NRF Centre of Excellence in Catalysis c*change, a collaborative network of 10 South African Universities working on various aspects of catalysis and acting as co-host for the event.A number of highly regarded researchers, of international standing, accepted the invitation by the committee to present their current work at the meeting. The remaining two thirds of the oral contributions were selected via a rigorous reviewing process from abstract submissions. In the end novel cutting edge research was presented in 21 oral and 59 poster presentations over three days. Of the approximately 120 delegates about 50% were from South African institutions, with an additional 15% from other African countries and the balance international delegates.
Due to the nature of the Faraday Discussions researchers who missed the actual event can still engage with the presented research through the published papers in the respective volume of the Faraday Discussion journal and even follow the discussion emanating from this work recorded in the same publication. The present conference report is therefore meant to be a short sneak preview to raise the readers interest in diving deeper into the science presented at this unique event.
Opening lecture
Prof. Enrique Iglesia (UC Berkeley) began his lecture with an overview of the required properties of a fuel (specifically high energy density and control of volatility) and then a range of pathways to take various platform molecules through to these fuels. The first example provided by Prof. Iglesia was that of understanding the challenging process of methane oxidative coupling to form higher hydrocarbons. Through interpretation of the reaction kinetics, combined with isotopic labelling studies, the role of water in the formation of OH radicals and their involvement in gas phase H-abstraction to enhance C2 yields was discussed. The carbonylation reaction of dimethyl ether (DME) to methyl acetate and the catalytic homologation of DME both provided examples for the importance of understanding acid strength and also confinement effects in dictating catalytic activity and reaction selectivity. Specifically, the requirement of 8 membered ring zeolite channels for DME carbonylation and large pores for the solvation of specific transition states in selective DME homologation highlighted the significance of confinement effects.Prof. Iglesia then turned to the theme of Fischer–Tropsch catalysts and in particular addressed two highly debated topics: the first being CO dissociation and the other chain growth mechanisms. Crucial to the context of this discussion was that attempts to address mechanistic questions, associated with Fischer–Tropsch chemistry, should consider that reaction surfaces have very high CO surface coverage. Density functional theory (DFT) and infrared studies from Neurock, Iglesia and co-workers were cited, which stated that supramolecular carbon monoxide coverage was possible on small Ru201 clusters via the formation of geminal adsorbed species. In regard to carbon monoxide dissociation, the prevalence of an H-assisted pathway was advocated on Co and Ru surfaces at high coverage. The second concept discussed by Iglesia was how to rationalise the formation of relatively long chain hydrocarbons from CHx, produced from CO activation, on surfaces with dense CO monolayer coverage, which would impede CHx diffusion. An explanation offered was that there are relatively few chain growth sites, but that the growth rate is very high. Evidence for this was provided from further DFT studies on Ru218, with high CO coverage, which stated that CnH2n−1 species perturb the local CO coverage and enhance H-assisted CO activation near this chain growth site.
Finally, the lecture focused on oxygen removal from biomass as a potential fuel source and provided detailed background on the potential for condensation and esterification reactions in this field (the theory of which was discussed in detail in Neurock and co-workers’ paper in session 1). In summary, the opening lecture by Prof. Iglesia brought together the discussed range of catalytic reactions for fuel production and emphasised the importance of the systematic study, via theory and kinetic analysis, of the elementary steps within these reactions, in order to acquire fundamental knowledge of carbon chain building for fuel synthesis.
Session 1: Catalysis for fuels insights from theory
Following the opening lecture the first session of the Faraday Discussion meeting, chaired by Dr Paul Collier (Johnson Matthey, UK), explored how modern theoretical methods aid our understanding of catalysis for energy provision. The opening paper, presented by Prof. Nora de Leeuw (University College London and Cardiff University, UK), reported on micro-kinetic simulations of hydrazine decomposition on a Cu(111) surface (DOI: 10.1039/c6fd00186f). Its high hydrogen content and low production/transport cost makes hydrazine a viable hydrogen fuel. However, due to the high expense of the conventional Ir/Al2O3 catalyst, alternative more affordable metal catalysts are require to scale up the process. The paper focuses on two different micro-kinetic models, which make use of the calculated reaction and activation barrier energies, along with reaction rate constants for 52 elementary steps of hydrazine decomposition. The first model considered pre-adsorbed hydrazine on the Cu(111) in a temperature programmed desorption simulation, while the second simulated a batch reactor with all gas reactants and products being allowed to adsorb and desorb freely, until equilibrium is reached. It was found for both models that the dominant product of the reaction was ammonia, demonstrating that Cu(111) is not suitable for hydrogen production. However, as the authors stated, it can be concluded that the technique can be utilised in screening for hydrazine decomposition on alternative Cu surfaces with lower metal co-ordination to ascertain the feasibility of hydrogen production. A general consensus in the discussion of the paper was that the micro-kinetic model was an effective tool to predict yield and selectivities for many different systems at a wide range of reaction conditions (Fig. 1).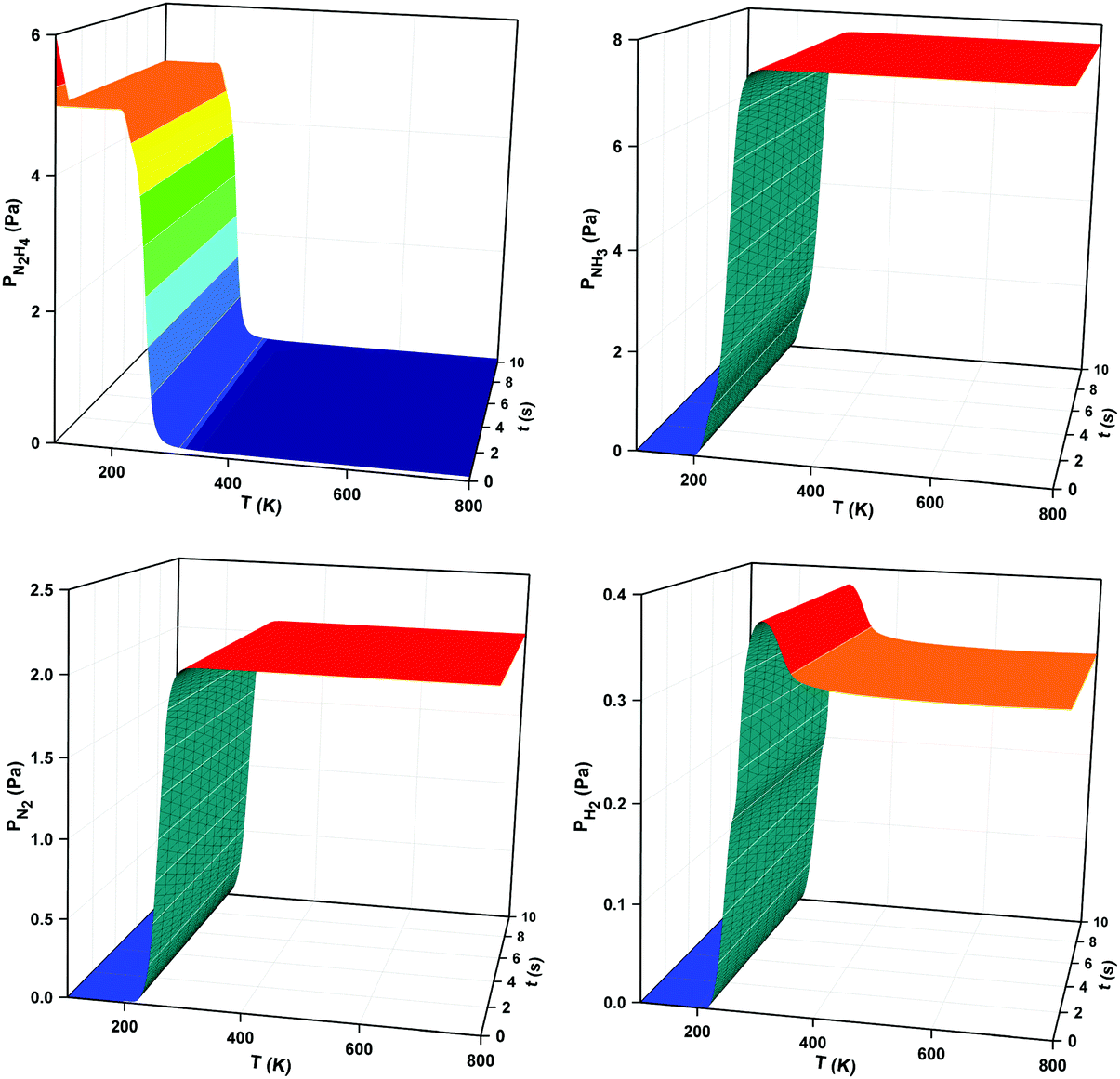 | ||
| Fig. 1 N2H4, NH3, N2 and H2 evolution from Cu(111) surface as a function of temperature and time for an initial N2H4 pressure of 6 Pa with a 1 K min−1 heating rate in the batch reactor simulation. Reproduced from DOI: 10.1039/c6fd00186f | ||
The second paper (DOI: 10.1039/c6fd00226a) of the session from Prof. Matthew Neurock (University of Minnesota, USA) continued the application of DFT calculations to investigate the elementary steps of aldol condensation and esterification reactions from aldehyde, alcohol and hydrogen mixtures over Cu/SiO2, alluded to in Prof. Iglesias opening lecture on biomass conversion. This attractive route for coupling and oxygen removal, which conventionally requires basic catalysts, was demonstrated to proceed on the non-basic Cu/SiO2. The first principles study on a Cu(111) surface showed that surface alkoxides readily form from hydrogen addition to a carbon centre of an aldehyde or proton transfer between adsorbed alcohols. The surface alkoxide was shown to withdraw electron density from the Cu surface and subsequently have significant anionic character. This alkoxide anion was reported to then behave as a base, which catalysed α-H abstraction from the aldehyde during aldol condensation or nucleophilic attack of the carbonyl of the aldehyde to form C–O bonds during esterification reactions. The interesting observations from this theoretical study stimulated debate on the possibility of tuning reaction selectivity and options for catalyst design through maximising the proportion of low index Cu faces to enhance activity.
The next paper discussed also presented a theoretical study to provide context to previous experimental work. In this case, Tracey van Heerden (University of Cape Town, South Africa) presented a DFT study of inverse Co-alumina based Fischer–Tropsch catalysts (DOI: 10.1039/c6fd00201c). The premise of the study is that the metal support interaction can be represented as a metal–ligand interaction with the support acting as a supra-molecular ligand. A range of single aluminium oxide ligands on Co(111) were tested for feasibility, with four ligand configurations being considered to have stable positions. Further analysis of ligand stability in H2/H2O atmospheres suggested that the OAl(OH)2 ligand (labelled AM2) was the most relevant under reaction conditions. After refinement of the ligand attachment to Co(111) and Co(100) surfaces using the surface energies together with Wulff constructs it was found that calculated Co crystal morphology was affected by the presence of the ligand, with a higher Co(100)/Co(111) ratio reported when the ligand was present. A key finding reported in the paper was that, in addition to the effect on Co particle morphology, the OAl(OH)2 ligand strongly interacted with adsorbed CO, which was proposed to demonstrate that the OAl(OH)2 acts as a promotor in CO dissociation during Fischer–Tropsch reactions. The paper clearly demonstrated that relatively simple calculations in a well thought-out experiment can provide insight into such highly complicated reactions, such as Fischer–Tropsch chemistry (Fig. 2).
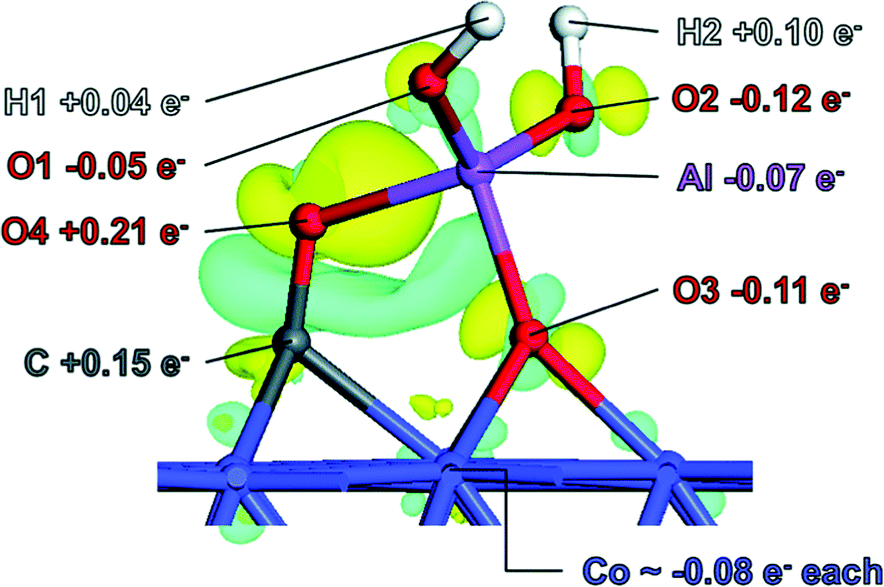 | ||
| Fig. 2 Charge density difference plot for CO-adsorbed system with AM2 ligand. Blue denotes loss of charge and yellow gain. Reproduced from DOI: 10.1039/c6fd00201c | ||
After tea the session resumed with the paper from Dr Kees-Jan Weststrate (SynCat, the Netherlands) titled Understanding FTS selectivity: the crucial role of surface hydrogen (DOI: 10.1039/c6fd00191b). The paper reported on surface science studies of CH3, CO and H adsorbed on the surface of Co(0001) at a range of coverages. At low coverage of CH3ads dehydrogenation to CH followed by coupling to C2H2 was observed, while higher coverage of CH3ads or pre adsorbed H resulted in hydrogenation to methane. It was asserted that under Fischer–Tropsch reaction conditions the ratio of occupied hydrogen sites over free sites (θH/θH*) dictated reaction selectivity. Experiments also showed that COads suppressed dissociative adsorption of hydrogen. Pre adsorption of Hads was found to limit CO adsorption, although it was stipulated that under reaction conditions θCO is not affected by adsorbed hydrogen, as the CO would bind more strongly. Alternatively the author suggested that θCO defines the quantity of Hads next to it. It was concluded that CO pressure indirectly effects θH/θH* and therefore reaction selectivity.
The paper from Dr Pieter van Helden (Sasol Technology Group, South Africa) continued the discussion of cobalt based Fischer–Tropsch catalysis, with a computational micro-kinetic study of the initial steps of the reaction (DOI: 10.1039/c6fd00197a). While micro-kinetic studies have previously been reported, the manuscript describes the first multi-site study. The model presented utilised a multi-site FCC cobalt model, consisting of the dominant Co(111) and Co(100) surfaces along with a step site represented by the Co(211) surface. Using a low coverage DFT data set provided reaction rates that where comparable to experiment; however the underlying mechanism was not correct as the pressure-rate relationship and reaction selectivity didn’t correlate with experimental data. Echoing comments from Prof. Iglesias opening lecture, it was concluded that high CO coverage would have a significant effect and that such parameters should be considered in future calculations. It was hypothesised that high CO coverage would destabilise CHx species and influence the stability of adsorbed H. By implementing a number of justifiable modifications to the model more appropriate pressure-rate relationships were found. It was concluded that the Fischer–Tropsch mechanism comprises processes proceeding on different sites, steps responsible for C1 production and terraces for chain growth (Fig. 3).
 | ||
| Fig. 3 Summary of proposed C2 reaction network for FTS starting from C1. Site A/Co(111) and site B/Co(100). Reproduced from DOI: 10.1039/c6fd00197a | ||
Prof. Emiel Hensen (Eindhoven University of Technology) presented the final talk of the first session, which also discussed micro-kinetic studies of the Fischer–Tropsch reaction (DOI: 10.1039/c6fd00205f). In this case the paper investigated the influence of the elementary reaction steps on chain-growth probability, with a stepped Ru(11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 1) surface. It was reported that the chain-growth mechanism is dependent on the CH dehydrogenation and C hydrogenation steps. The author formulated two kinetic concepts for these steps, the degree of chain-growth probability control and the thermodynamic degree of chain-growth probability control. Steps that control chain-growth are different from those that control the overall Fischer–Tropsch reaction rate. Below the optimum for obtaining long hydrocarbon chains the dehydrogenation reaction is limited by high surface coverage, while above the optimum the hydrogenation reaction limits chain-growth. In agreement with previous talks the critical role of adsorbed H and free site coverage has been shown to fundamentally influence product selectivity and distribution.
1) surface. It was reported that the chain-growth mechanism is dependent on the CH dehydrogenation and C hydrogenation steps. The author formulated two kinetic concepts for these steps, the degree of chain-growth probability control and the thermodynamic degree of chain-growth probability control. Steps that control chain-growth are different from those that control the overall Fischer–Tropsch reaction rate. Below the optimum for obtaining long hydrocarbon chains the dehydrogenation reaction is limited by high surface coverage, while above the optimum the hydrogenation reaction limits chain-growth. In agreement with previous talks the critical role of adsorbed H and free site coverage has been shown to fundamentally influence product selectivity and distribution.
Session 2: Designing new catalysts for synthetic fuels
The second day of the Faraday Discussion was dominated by a session focusing on New Catalysts for Synthetic Fuels chaired by Prof. Neil Coville (University of the Witwatersrand, South Africa). In the opening paper Prof. Ding Ma from Peking University reported on ethylene pre-treated cobalt based Fischer–Tropsch catalyst and the effect of the pre-treatment on selectivity (DOI: 10.1039/c6fd00194g). Alumina supported Co catalysts were either exposed to an ethylene/hydrogen mixture after reduction or indeed instead of reduction in hydrogen. Co particles directly activated in ethylene at 450 °C showed a larger average crystallite size with graphitic carbon species on the surface. Exposure of a hydrogen activated catalyst to ethylene at 250 °C yielded smaller metal crystallites with atomic and polymeric carbon depositions. The carbon depositions showed enhancement of the methane and low hydrocarbon selectivity at the expense of the C12+ fraction. The olefin to paraffin ratio in the C2–C4 product fraction was significantly reduced in the presence of graphitic carbon on the catalyst's surface. The authors argue that no crystallite size or allotrope dependency can explain the observed selectivity trends but argue, based on DFT calculations, that carbon on the cobalt surface increases the CO dissociation barrier while enhancing hydrogenation over C–C coupling.Carbon also plays a significant role in the work presented by Prof. Freek Kapteijn (Delft University of Technology, The Netherlands), who reported on the newest developments in utilizing iron bearing metal organic frameworks (MOF) as catalyst precursors for the iron based high temperature Fischer–Tropsch synthesis (DOI: 10.1039/c6fd00198j). Commercial MOFs were pyrolysed in nitrogen at 500 °C followed by passivation. After this treatment iron is present as nano-crystalline Fe2O3 in a carbon matrix except for the sample obtained from a MOF with specifically high decomposition temperature and N functionalities in the linker groups which showed θ-Fe3C cementite. Depending on the precursor geometry average pore diameters and to a lesser extent BET surface area of the pyrolysis products vary. The samples were tested in the Fischer–Tropsch synthesis both as is and after promotion with potassium. The authors observed a linear increase of mass specific activity with decreasing iron oxide and carbide precursor size. This was not reflected in the surface specific activity and was more pronounced in the presence of the promoter. For two catalysts no influence of the promoter could be observed which was attributed to either a lack of contact of the promoter and the active material or overpromotion due to a relatively low iron surface area. The catalytically best performing catalyst contained traces of S, Mn and Cu residues from the MOF synthesis which were argued to act as additional promoters (Fig. 4).
 | ||
| Fig. 4 Different MOF morphologies applied as catalyst pre-cursors in the discussed study (a) MIL-68, (b) MIL-88A, (c) MIL-100, (d) MIL-101, (e) MIL-127 and (f) the configuration of Fe trimers and super tetrahedra. Reproduced from DOI: 10.1039/c6fd00198j | ||
The last paper discussed before tea was presented by Prof. Michael Claeys (University of Cape Town, South Africa). Using a novel in situ magnetometer the potential re-oxidation of cobalt crystallites under simulated high conversion Fischer–Tropsch conditions was studied (DOI: 10.1039/c6fd00200e). Monodisperse Co3O4 nanoparticles were prepared via a surfactant free synthesis route and deposited on SiO2 Stöber spheres. These catalysts were reduced mildly in order to avoid sintering and subsequently exposed to steam and hydrogen mixtures of different ratios. It could be shown conclusively that smaller Co crystallites undergo more re-oxidation than larger crystallites. The nature of this oxidation event was also reported to be pre-dominantly the formation of cobalt silicates and only to a small extent the formation of CoO. In the presence of CO in the gas atmosphere, the formation of cobalt silicates still dominates but more CoO is formed, suggesting that to achieve the formation of cobalt oxide O species from the dissociation of CO have to be present on the metal surface. Higher water partial pressures in turn hinder their removal – a process termed indirect oxidation to CoO by water. The direct oxidation leads to the formation of silicates (Fig. 5).
 | ||
| Fig. 5 Loss of magnetisation, i.e. oxidation of Co to oxides or silicates at different water to hydrogen partial pressures in the presence (open circles) and the absence (filled circles) of CO. Reproduced from DOI: 10.1039/c6fd00200e | ||
In a second paper by researchers from the University of Cape Town, presented by Thulani Nyathi (DOI: 10.1039/c6fd00217j), the effect of Co3O4 crystallite size on the activity and stability during CO preferential oxidation in hydrogen rich atmospheres was discussed. Cobalt oxide nanoparticles of well-defined sizes (between 4 and 15 nm) were synthesized using a reverse micelle approach and subsequently deposited onto a commercial alumina carrier without introducing significant metal support interactions. Using in situ techniques, namely a magnetometer and a capillary based XRD cell, the activity towards CO oxidation but also towards H2 oxidation and methanation was directly correlated to the bulk phase composition at various reaction temperatures. A maximum CO2 yield was measured for all catalysts at the temperature just below full O2 conversion. A further increase resulted in a loss in CO2 yield, a decrease in conversion as well as in a reduction of the catalyst to CoO. At even higher temperatures CoO reduces to metallic Co which acts under the present conditions as an excellent methanation catalyst. Interestingly, upon cooling the reactor, all catalysts underwent a re-oxidation which was paralleled by the recovery of Co oxidation activity although in bulk only CoO was detected. Comparing the fully oxidized catalysts, i.e. at temperatures below the reduction threshold, a volcano type behaviour was reported for the surface specific CO oxidation activity as a function of crystallite size, peaking at approximately 8.5 nm.
Methanol synthesis was introduced by Dr Simon Kondrat (Cardiff Catalysis Institute, UK) reporting on the effect of trace amounts of sodium on a Cu–ZnO methanol synthesis and water gas shift catalyst (DOI: 10.1039/c6fd00202a). Making use of a supercritical CO2 antisolvent technique, a sodium free zincian georgeite precursor was synthesized in contrast to the conventional zincian malachite which usually contains sodium residue originating from the precipitation agent. Via the use of high purity grade metal precursors as well as the deliberate addition of sodium acetate to the reaction mixture, different levels of sodium in the catalyst precursor were achieved. Interestingly the sample with the highest Na+ levels (2.12%) formed a malachite type structure during uncontrolled aging over 24 hours with a low degree of Zn incorporation. After calcination this sample also exhibited the largest CuO crystallites (6 nm) while all other samples showed a similar CuO crystallite size (approx. 2 nm). If the high sodium sample is not allowed to age but calcined directly, no difference in CuO crystallite size can be detected as function of Na+ concentration. However, after reduction the copper surface area of the not aged high Na+ sample was significantly lower than the other catalyst. The authors hypothesise that the Na+ present reduced the Cu–Zn interactions by oxidizing reduced Zn to ZnOx at the periphery of Cu in turn reducing the apparent Cu surface area. The activity of the obtained catalysts in the low temperature water gas shift reaction could be directly correlated with the Na+ content, with the sodium free sample displaying the highest activity. Although more complicated in its interpretation due to the combination of CO2 activation through the reverse water gas shift reaction and CO hydrogenation, the methanol yield was also observed to increase with decreasing Na+ content.
The direct activation of CO2 to methanol over PdZn catalyst was presented by Prof. Michael Bowker (Cardiff Catalysis Institute, UK) (DOI: 10.1039/c6fd00189k). The catalysts were prepared by physically mixing Pd and Zn acetylacetonate in ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:10 with ZnO, TiO2 or Al2O3. As a reference, pure Pd was also deposited on the support materials. The mixture was evacuated under temperature and subsequently calcined at 500 °C in an air flow. After reduction in a diluted hydrogen stream all catalysts exhibited the characteristic diffraction pattern of the PdZn alloy and, in case of a 1:10 ratio of Pd to Zn, ZnO. While pure Pd does show activity for the direct formation of MeOH from CO2, which can even be enhanced when treating the sample at high temperatures on a ZnO support, the nano-crystalline alloys formed via the described decomposition technique were significantly more active. The authors postulate that the PdZn alloy approaches the electronic properties of Cu explaining the observed activity.
:1 or 1:10 with ZnO, TiO2 or Al2O3. As a reference, pure Pd was also deposited on the support materials. The mixture was evacuated under temperature and subsequently calcined at 500 °C in an air flow. After reduction in a diluted hydrogen stream all catalysts exhibited the characteristic diffraction pattern of the PdZn alloy and, in case of a 1:10 ratio of Pd to Zn, ZnO. While pure Pd does show activity for the direct formation of MeOH from CO2, which can even be enhanced when treating the sample at high temperatures on a ZnO support, the nano-crystalline alloys formed via the described decomposition technique were significantly more active. The authors postulate that the PdZn alloy approaches the electronic properties of Cu explaining the observed activity.
Another alternative MeOH synthesis catalyst was presented by Dr Alberto Roldan (Cardiff University, UK) with CO hydrogenation over greigite (Fe2S4), an isomorph to Fe3O4. With DFT based methods the two Fe absorption sites of the {111} facet were investigated regarding the competitive reactions of CO hydrogenation and CO dissociation. Two intermediates, namely HCOH and H2CO were identified. Both studied Fe sites preferentially yield MeOH under mild reaction conditions, once only via HCOH, and once in a competitive mechanism between the two intermediates. The authors conclude that the sulphide surface represents a selective reaction site for direct CO hydrogenation without CO dissociation.
In the last paper of the second session Dr Marien Bremmer (Leiden University, The Netherlands) reported on in situ TEM studies of the Boudouard reaction on ε-Co nanoparticles. The particles were synthesised using the hot injection method, decomposing dicobalt carbonyl in the presence of oleic acid. After washing, a 2-propanol suspension of the nanoparticles was injected into the nano-reactor followed by drying at 80 °C for two days. In the reactor the sample was heated under N2 to 300 °C in order to remove residual surfactant and subsequently to 500 °C in diluted hydrogen to reduce any possible oxides – although not directly observed. At 500 °C a synthesis gas mixture (H2/CO = 2) was introduced for 100 minutes before removing the hydrogen and exposing the particles to a diluted CO atmosphere. From this point on, the formation of carbon layers was observed. The layers formed at the interface of the cobalt particle and previous C layers and exerted strain on the particle. Twinning was observed as a consequence. Detection of CO2 in the reactor off-gas confirmed the Boudouard reaction as the source of the carbon (Fig. 6).
 | ||
| Fig. 6 Growth of a new carbon layer on the surface of one Co nanoparticle interlayered between the Co surface and previous carbon layers. Reproduced from DOI: 10.1039/c6fd00185h | ||
Session 3: Hydrocarbon conversion in the production of synthetic fuels
The opening talk for the third session was given by Prof. Avelino Corma (Instituto de Tecnología Química, Spain) on opportunities in upgrading biomass crudes obtained from lignocellulose (DOI: 10.1039/c6fd00208k). The upgrading of biocrudes, in this case pine wood chips, into fuel occurs via a multistep process as shown in Fig. 7. The biocrude obtained from hydrothermal liquefaction undergoes hydrotreating using a commercial NiMo catalyst supported on alumina or is first mixed with 70–90 wt% Vacuum Gas Oil (VGO) and the mixture is cracked in fluid catalytic cracking (FCC) unit. The hydrocarbon stream from this process can be fully processed in conventional refineries and is also close to the specifications of commercial fuels.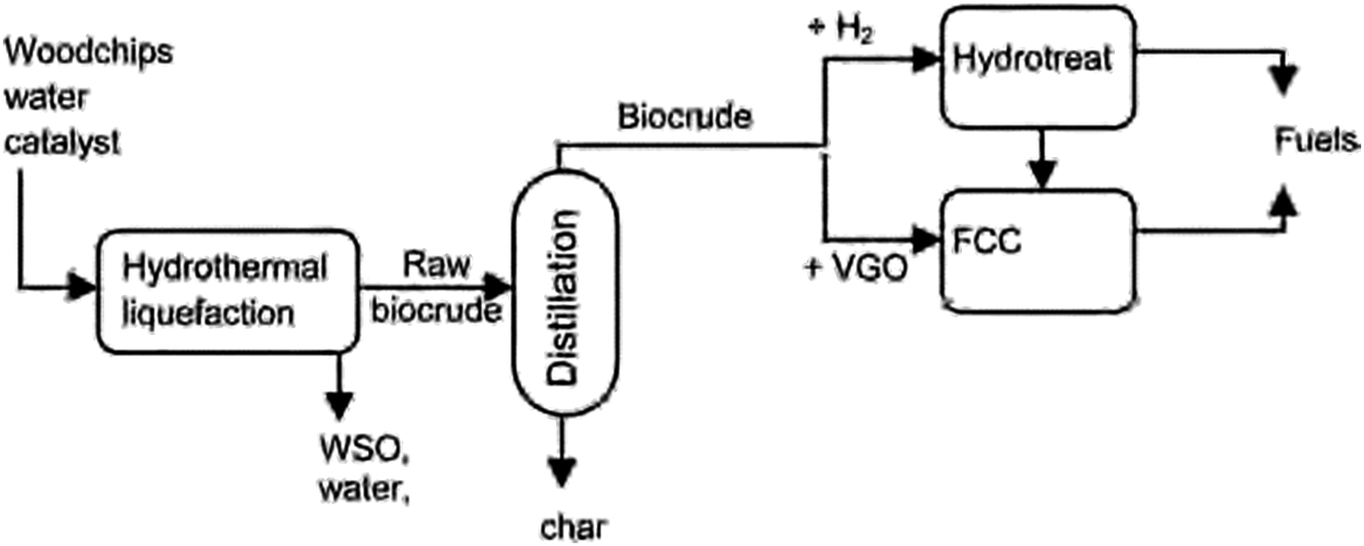 | ||
| Fig. 7 Two step process for the preparation of biocrude and biocrude upgrading options. Reproduced from DOI: 10.1039/c6fd00208k | ||
The second talk of the session by Prof. Burtron Davis (University of Kentucky, USA) was entitled “Fischer–Tropsch synthesis. Evaluation of an aluminium small channel reactor” (DOI: 10.1039/c6fd00179c). The Fischer–Tropsch synthesis was compared between a small channel compact heat exchange reactor constructed from aluminium and a continuous stirred tank reactor. The aluminium reactor was found to be advantageous in terms of superior heat conductivity and lower manufacturing cost, although the stainless steel reactor can operate at higher temperature–pressure regions. Selectivity and activity of the catalyst were reported to be similar. It was also reported that the small channel reactor can be restarted after an intended and unintended shutdown, with the activity and selectivity remaining unchanged.
The influence of zeolite topology on axial deactivation patterns in the methanol to hydrocarbons (MTH) reaction (DOI: 10.1039/c6fd00187d) was presented by Prof. Unni Olsbye (University of Oslo, Norway). Fig. 8 shows the representative conversion versus time on stream plots for the various zeolite topologies which were subjected to spatio-temporal studies. Each catalyst was tested at three different contact times, all giving 100% initial conversion. The applied contact time, τ0, required to obtain full initial conversion is different for each catalyst and the data in Fig. 8 was therefore obtained at different applied contact times. It was found that the critical contact time increased with a decrease in pore size. The conversion capacity of the larger 12-ring topologies, mordenite and beta, decreased with an increase in contact time; in contrast to the 10-ring zeolites with pore sizes smaller than ZSM-5, for which the conversion capacity increased with an increase in contact time. Deactivation studies revealed that zeolite topology affects not only catalyst lifetime and product distribution, but also modifies the axial mode of catalyst deactivation. The study overall improved the understanding of deactivation in the MTH reaction and also revealed that the axial deactivation pattern and critical contact time change with zeolite topology, which had not been previously addressed.
 | ||
| Fig. 8 Methanol conversion versus time-on-stream at 400 °C over the various catalysts tested. Experimentally measured data are represented by symbols while simulated deactivation curves are represented by dashed lines. τ0 = 4 g h−1 mol−1 was applied over ZSM-22-h and ZSM-23-h whereas for the rest of the catalysts τ0 = 2 g h−1 mol−1 was used. Reproduced from DOI: 10.1039/c6fd00187d | ||
The last talk in the session was given by Prof. David Lennon (University of Glasgow, UK) entitled “An assessment of hydrocarbon species in the methanol-to-hydrocarbon reaction over a ZSM-5 catalyst” (DOI: 10.1039/c6fd00195e). Multiple techniques, namely GC (gas chromatography), TPO (temperature programmed oxidation), EPR (electron paramagnetic resonance), IR (infrared) and most notably INS (inelastic neutron scattering) were used to evaluate the MTH reaction using commercial ZSM-5 over a range of conditions. The reaction profile at 350 °C showed that dosing with methanol initially leads to the formation of dimethyl ether through the dimerization of chemisorbed methoxy species. The formed dimethyl ether is subsequently a precursor for the production of olefinic and aromatic products. TPO revealed that an extended time on stream at 350 °C results in the formation of retained carbonaceous material while EPR showed formation of undefined aromatic radical cations of low spin density. IR revealed the co-existence of sp2 and sp3 hybridised C–H entities due to the presence of catalyst hydroxyl groups and retained hydrocarbonaceous species and INS confirmed the information from IR although the assignment for the retained hydrocarbonaceous species was not yet possible due to the complexity of the composition of the hydrocarbon pool.
Session 4: Novel photocatalysts
The last day of the Faraday Discussion focussed on Novel Photocatalysts with the first talk of the session by Prof. Kazunari Domen (University of Tokyo, Japan) entitled “Particulate photocatalyst sheets for Z-scheme water splitting: advantages over powder suspension and photoelectrochemical systems and future challenges” (DOI: 10.1039/c6fd00184j). Z-scheme water splitting via photocatalysis is based on the two-step excitation of an oxygen evolution photocatalyst (OEP) and a hydrogen evolution photocatalyst (HEP) (Fig. 9). In this paper, SrTiO3:La,Rh was used as the HEP and BiVO4:Mo as the OEP using a photocatalyst sheet fabricated in the form SrTiO3:La,Rh/Au/BiVO4:Mo. The sheet split pure water more efficiently than a powder suspension and photoelectrochemical systems. The effects of H+/OH− concentration overpotentials are also reduced in the case of the sheet compared to photoelectrochemical systems. This makes the photocatalyst sheet suitable for efficient large-scale applications. | ||
| Fig. 9 Schematic energy diagrams of photocatalytic water splitting via (a) one-step excitation and (b) two-step excitation (Z-scheme). CB and VB stand for conduction band and valence band, respectively. Reproduced from DOI: 10.1039/c6fd00184j | ||
The next paper was presented by Prof. Detlef Bahnemann (Leibniz University Hannover, Germany) on whether laser-flash-photolysis-spectroscopy was a non-destructive technique (DOI: 10.1039/c6fd00193a). Diffuse-reflectance laserflash-photolysis measurements were made on TiO2 powders to study their morphological and optical properties. In anatase the formation of electron–hole pairs, followed by energy and charge transfer to the TiO2 lattice upon illumination with intense laser pulses is observed. The TiO2 surface with regions of high defect density was found to undergo irreversible changes, such as removal of the lattice oxygen resulting in the formation of oxygen vacancies which enhances the surface reactivity of the TiO2 particles. The phase transition to rutile TiO2 occurs via an oxygen release from the TiO2 surface induced by the electron/energy transfer and was confirmed by the presence of Ti3+ centres in the illuminated powder. It was concluded that upon illumination with intense pulsed laser light, structural changes of the TiO2 photocatalyst may occur which is essential for the evaluation of underlying photocatalytic reactions.
The final talk for this session by Dr Katherine Holt (University College London, UK) reported on the “In situ spectroscopic monitoring of CO2 reduction at copper oxide electrode”. The reduction of CO2 was studied via cyclic voltammetry using a CuO-modified electrode and KHCO3 as the electrolyte solution (Fig. 10). The stability of CuO under potential cycling in 0.5 M KHCO3 in the absence and presence of CO2 was studied and the CuO electrodes were analysed via Raman and XPS after application of different potentials. Raman and XPS revealed that the reduction of the Cu content is inhibited in the presence of CO2 which indicates that some of the charge passed may contribute to reduction of adsorbed CO2 rather than reduction of the catalyst. In support of other studies, it was also shown that the CuO surface allows for CO2 activation at potentials positive of the reduction of CuO.
 | ||
| Fig. 10 Schematic of adsorbed solution species at −0.4 V in (a) argon deoxygenated 0.5 M KHCO3 and (b) CO2 saturated 0.5 M KHCO3. Reproduced from DOI: 10.1039/c6fd00183a | ||
Concluding remarks
It was Philip Gibson's (Sasol Technology group, South Africa) task to find the right words for the concluding remarks. Focusing on the key messages and findings, a review of the four sessions was presented highlighting the scientific quality of the discussed work, both during the oral as well as poster sessions. The special nature of the format of the Faraday Discussions was mentioned and the value of the resulting discussions stressed. Overall the organizing committee as well as the Royal Society of Chemistry were complemented on having the vision to bring this prestigious event to South Africa and making it an astounding success.A very interesting general comment by Philip Gibson was his observation that implementation of novel research in the field of heterogeneous catalysis is becoming increasingly difficult. Even in case of research with a close link to the industrial conditions and application, the transfer from the university laboratories to the private sector represents a significant hurdle which has become increasingly difficult to overcome. A large number of factors, only some of which are economy based, influence the decision making process and have overall reduced the willingness to take risks regarding novel processes. Although more difficult, researchers were encouraged to continue striking a balance between fundamental and applied research approaches and to strive for implementation of new research in tackling global challenges.
| This journal is © The Royal Society of Chemistry 2017 |