
Real-time femtomolar detection of cancer biomarkers from photoconjugated antibody–phage constructs†
M.
Brasino
a and
J. N.
Cha
*ab
aMaterials Science and Engineering Program, University of Colorado, Boulder, USA. E-mail: jennifer.cha@colorado.edu
bDepartment of Chemical and Biological Engineering, University of Colorado, Boulder, USA
First published on 4th October 2016
Abstract
Here we describe novel covalent conjugates of antibody–phage for the detection of multiple cancer biomarkers using real time immuno-polymerase chain reaction (immuno-PCR). While the conventional process of immuno-PCR utilizes DNA-conjugated antibodies, chemical modification of antibodies not only reduces antibody affinity but also creates a heterogeneous population of products. However, phage naturally encapsulate genomic DNA, which can be used as a PCR template. To produce covalently conjugated antibody–phage constructs without recombinant antibody expression or chemical modification of antibodies, we incorporated a photocrosslinkable non-canonical amino acid within an antibody-binding domain displayed on one of the phage coat proteins. To correlate antigen presence to a specific DNA sequence, the phage genomes were modified with domains that recognized specific sets of primers. The crosslinked antibody–phage conjugates were then tested in a sandwich-type immunoassay using real-time PCR where low pg ml−1 concentrations of antigen could be detected and identified from a single solution containing a mixture of three different types of cancer biomarkers.
Introduction
The detection of specific biomarkers with high sensitivity is critical for early diagnosis and treatment. Because many key protein biomarkers are found at concentrations below the detection limits of standard enzyme linked immunoassays (ELISAs), new diagnostic tools are continuously being sought.1–3 One strategy has been to use immuno-PCR, which first requires labeling detection antibodies with synthetic oligonucleotides and utilizing polymerase chain reaction (PCR) to generate amplified signals in response to an antigen binding event.4–6 Because each antibody can be labeled with a distinct nucleic acid sequence, immuno-PCR can potentially be used for multiplexed analysis.3 However, a significant drawback of immuno-PCR is that covalent conjugation of DNA to antibodies can not only hamper the affinity and specificity of the antibody to its antigen but also lead to highly heterogeneous populations of antibody–DNA conjugates.7 This can lead to varying, erroneous, or large background signals.8 While antibodies produced through recombinant expression can potentially be site-specifically modified, many sequences of clinically relevant antibodies are often unknown, and recombinant expression frequently leads to lower antigen affinities.8–10 As such, these methods are often difficult to apply for immuno-PCR.As a means of overcoming these issues, we demonstrate here the use of genetically engineered bacteriophage encasing a multi-kilobase circular genome that can be amplified to detect and identify specific antigens in solution. Because the amplifiable phage DNA is well-protected within the phage capsid, it neither interferes with nor impedes the antigen binding event (Scheme 1). Furthermore, the phage are easily amplified in Escherichia Coli (E. coli), negating the need for costly chemically functionalized synthetic oligonucleotides. To fabricate the antibody–phage constructs, we show here methods to covalently attach the constant fragment (Fc) region of monoclonal detection antibodies to phage by expressing an antibody binding peptide on the p3 capsid proteins that also incorporates the photocrosslinkable non-canonical amino acid p-benzoyl-L-phenylalanine (pBPA) (Scheme 1). By introducing covalent crosslinks between the antibody and the phage, binding of the phage to other antibodies in solution or on an assay surface was effectively eliminated. To use the phage genome to type and quantify the antigen in solution, the DNA of each antibody–phage construct was modified to be recognized by specific primer sets used to amplify portions of the viral DNA. Since each primer set will only amplify one specific phage sequence, multiplexed detection of several biomarkers in solution could be performed in real time by employing a universal Taqman probe (Scheme 1). Immunoassays run from the antibody–phage conjugates showed low pg ml−1 detection limits of three different cancer biomarkers in a single solution in fetal bovine serum.
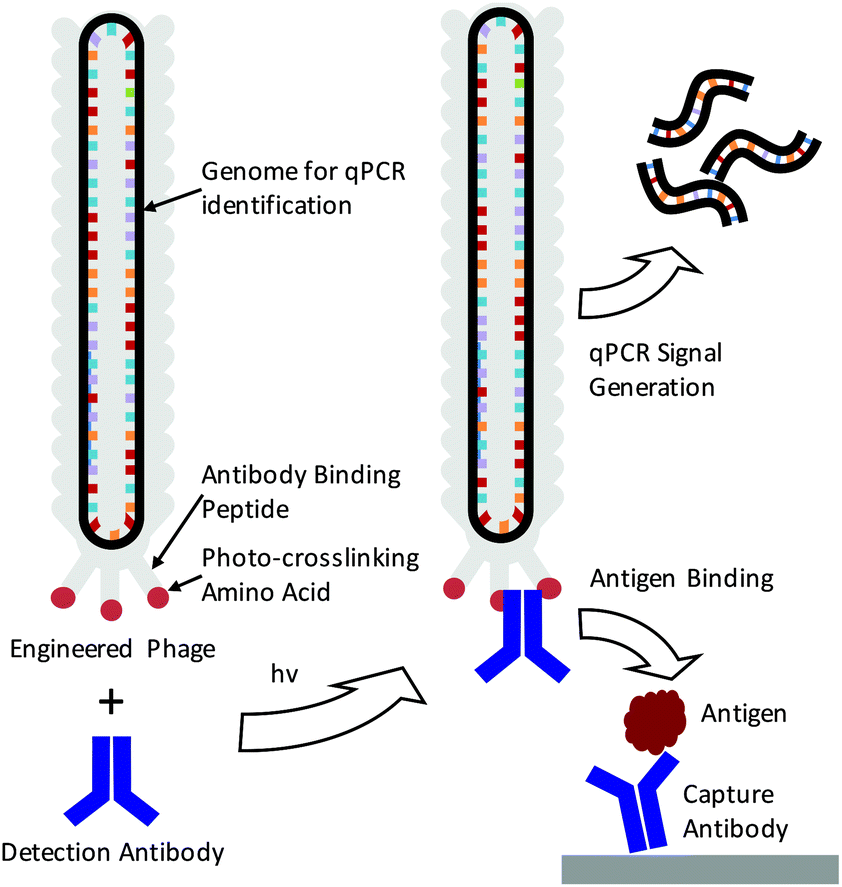 | ||
| Scheme 1 Depiction of immuno-PCR enabled by photo-crosslinked antibody–phage conjugates. Phage are genetically engineered to express an antibody binding peptide containing a photocrosslinking non-canonical amino acid (pBPA). Phage are mixed with detection antibody and irradiated to create antibody–phage conjugates. These conjugates bind to antigens in an immunoassay while being detected and identified through real time PCR amplification of the phage genome. | ||
Results and discussion
In order to fabricate the antibody–phage constructs, Fd derived filamentous phage were utilized due to their tolerance for genetic modification, ability to grow to high titers in E. coli and the relatively small size as compared to other strains.11,12 The viruses grow from a single stranded circular DNA genome that becomes packaged into the viral capsid which contains distinct capping proteins at either end. One end of the phage expresses three to five copies of the p3 protein which is often used to express peptides, or single chain variable fragments that bind particular targets.13–15 In order to attach antibodies to the phage, we have recently developed methods to express a protein A domain (Z domain) from each of the p3 proteins which can non-covalently associate with the Fc portions of detection antibodies.16 While this does allow production of antibody-conjugated phage, the non-covalent and relatively low affinity interactions between the Z domain and the antibody can lead to antibody dissociation from the phage leaving an unoccupied Z domain.17,18 This event then allows the phage to bind other antibodies in solution or on a surface, making multiplexed detection difficult or leading to high background signals. In order to introduce covalent bridges between the Z domain on the phage and the Fc portion of the detection antibodies, a non-canonical amino acid p-benzoyl-L-phenylalanine (pBPA) was inserted at position glutamine 32 (Q32) on the Z domain through amber codon suppression. This amino acid site was chosen based on previous studies by Hui et al. who showed that this led to optimal crosslinking between the Z domain and mouse IgG1 antibodies.19 In addition, since each antibody is capable of binding to two Z domains that are in close proximity,20 we explored the use of conjugating antibodies to phage that express two repeated Z domains per p3 protein. This was done to see if having additional Z domains on the p3 proteins would increase the avidity of the antibody for the p3 proteins at the phage tip. For this, four distinct phage types were created by modifying the phage p3 coding sequence and these were labeled PhA–PhD. Phage PhA expressed one pBPA substituted Z domain per p3 while phage PhB expressed two pBPA substituted Z domains per p3. Phage PhC was designed to produce two repeated Z domains per p3 protein but only one contained a pBPA substitution at Q32. Finally, phage PhD expressed two repeated Z domain, neither of which contained a pBPA substitution and was used as a control for photo-crosslinking. The sequences for each phage construct were inserted between the natural p3 signal peptide and mature protein coding sequence within each phage genome.After producing the phage in E. coli and purifying via PEG–NaCl precipitation, all of the different types of pBPA-Z domain expressing phage were irradiated for 3 h under a 365 nm UV lamp in the presence of an anti-tumor necrosis factor alpha (anti-TNFα) antibody at a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of antibody:phage (42 nM phage). As an additional control, phage were also irradiated in the absence of antibody. The resulting conjugates were first characterized through denaturing, non-reducing SDS PAGE (Fig. 1A). The expected molecular weight of p3 fused to one or two Z domains is 49 kDa and 56 kDa respectively and bands corresponding to this protein within each phage are visible at the bottom of the gel in Fig. 1A. The primary antibody band is at approximately 160 kDa as to be expected for a non-reduced mouse IgG, and is accompanied by two less intense bands at lower molecular weights corresponding to partially reduced antibody fragments. After photoirradiating the pBPA containing phage with antibody, higher molecular weight bands at approximately 220 kDa and 270 kDa were observed which correspond to an antibody crosslinked to either one or two p3 fusion proteins (Fig. 1A). These bands were only present when antibody was added to the phage and irradiated and were not present with control phage that expressed Z domains with no pBPA substitution (Fig. 1A, PhD). As the Fc portion of each antibody can bind to two Z domains, this may explain the presence of a band at approximately 270 kDa.20 However, because no significant difference in band intensities were seen between phage sets PhA–PhC, it can be deduced that in the current design, increasing the number of Z domains per p3 did little to improve antibody binding and covalent conjugation.
:1 molar ratio of antibody:phage (42 nM phage). As an additional control, phage were also irradiated in the absence of antibody. The resulting conjugates were first characterized through denaturing, non-reducing SDS PAGE (Fig. 1A). The expected molecular weight of p3 fused to one or two Z domains is 49 kDa and 56 kDa respectively and bands corresponding to this protein within each phage are visible at the bottom of the gel in Fig. 1A. The primary antibody band is at approximately 160 kDa as to be expected for a non-reduced mouse IgG, and is accompanied by two less intense bands at lower molecular weights corresponding to partially reduced antibody fragments. After photoirradiating the pBPA containing phage with antibody, higher molecular weight bands at approximately 220 kDa and 270 kDa were observed which correspond to an antibody crosslinked to either one or two p3 fusion proteins (Fig. 1A). These bands were only present when antibody was added to the phage and irradiated and were not present with control phage that expressed Z domains with no pBPA substitution (Fig. 1A, PhD). As the Fc portion of each antibody can bind to two Z domains, this may explain the presence of a band at approximately 270 kDa.20 However, because no significant difference in band intensities were seen between phage sets PhA–PhC, it can be deduced that in the current design, increasing the number of Z domains per p3 did little to improve antibody binding and covalent conjugation.
 | ||
| Fig. 1 Phage PhA expressed one pBPA substituted Z domain per p3 while phage PhB expressed two pBPA substituted Z domains per p3. Phage PhC was designed to produce two repeated Z domains per p3 protein but only one contained a pBPA substitution at Q32. Phage PhD expressed two repeated Z domains, neither of which contained a pBPA substitution and was used as a control for photo-crosslinking. (A) Phage were irradiated with or without 10:1 molar excess of anti-TNFα antibody, denatured and separated on a non-reducing SDS poly acrylamide gel shown, stained with Coomae blue. (B) Antibody–phage conjugates prepared as in (A) were tested for binding affinity to TNFα or additional anti-TNFα antibody via ELISA using an anti-phage antibody conjugated with HRP which creates a color change to indicate phage binding. The resulting solutions and their respected absorbance (415 nm) are shown. | ||
In order to utilize the antibody–phage conjugates in immunoassays, it is critical that the constructs not only possess high affinity for the antigen but also that binding to other antibodies in solution or on a surface be completely eradicated. In order to test the photo-crosslinked anti-TNFα-phage for binding to either TNFα or any capture anti-TNFα antibodies on a surface, phage ELISAs were run. For this, the photocrosslinked antibody–phage conjugates were diluted to 1.7 nM in 0.1% PBST and reacted against magnetic beads bound with either 133 nM anti-TNFα antibodies alone or with the capture anti-TNFα antibodies reacted with 32 nM TNFα and blocked with BSA. This was followed by stringent washing with 0.1% PBST to remove unbound conjugates. Next, horseradish peroxidase (HRP) conjugated anti-M13 antibodies were added to detect any anti-TNFα-phage bound to the surface. As shown in Fig. 1B, both the photo-crosslinked (PhA–PhC) and non-crosslinked (PhD) antibody–phage conjugates show comparable affinities to TNFα (conjugates showed relatively little binding to beads without TNFα). However, out of the four different antibody–phage constructs made, only the phage that expressed a single pBPA substituted Z domain (PhA) showed almost no affinity to the capture antibodies on the surface. While the gel electrophoresis analysis (Fig. 1A) did show that all of the antibody–phage constructs tested have some Z-modified p3 proteins that are not covalently attached to an antibody, it is clear from the ELISA studies that having a single pBPA substituted Z domain per p3 is optimal for preventing the phage from binding to additional antibodies.
The antibody–phage construct PhA (anti-TNFα-phage) was next photoirradiated for longer times to see if this would more effectively prevent binding to capture anti-TNFα antibodies on a surface. As shown in Fig. 2A, SDS PAGE analysis gave a similar band pattern as seen in Fig. 1B, with bands corresponding to one or two p3 proteins crosslinked to an antibody increasing in intensity as irradiation time is extended to 3 hours, and then increasing very little when irradiation is increased to 14 h. The decrease in the band intensity corresponding to free p3 proteins indicates that roughly half of the three to five proteins attached per phage particle are bound to an antibody. These conjugates were also tested for binding to anti-TNFα and TNFα by running phage ELISAs. First, photoirradiating the phage–antibody constructs for 0.5 h was found to lead to an increase in TNFα binding as compared to zero-time, presumably due to photocrosslinking preventing antibody–phage dissociation. Upon longer photoirradiation, the anti-TNFα-phage were found to significantly lose their affinity for the capture antibodies on the surface but still maintain binding to the antigen. Finally, photoirradiating for 14 h was found to further lower the antibody–phage binding to the surface bound antibodies.
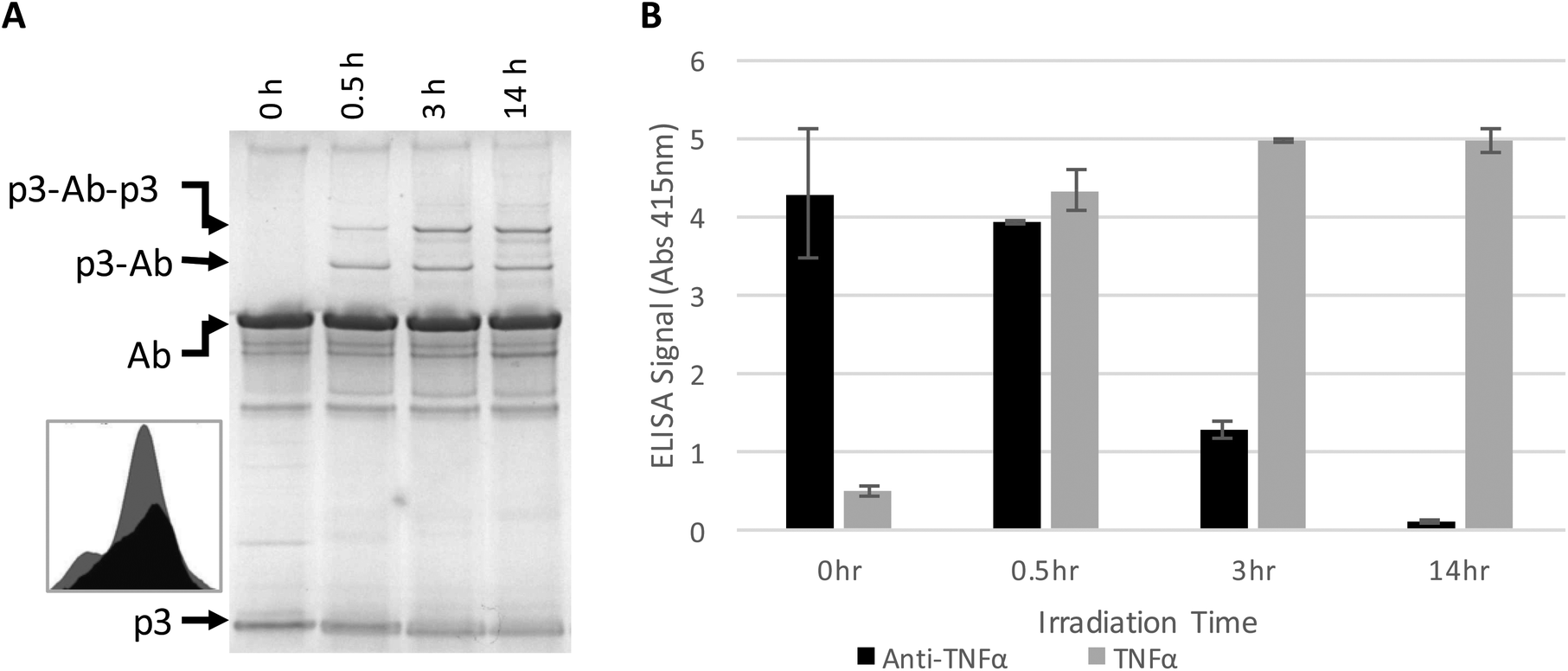 | ||
| Fig. 2 Phage expressing only a single Z domain containing a single pBPA was mixed with anti-TNFα and irradiated for the number of hours listed. (A) The resulting conjugates were denatured and separated on a non-reducing SDS poly acrylamide gel shown, stained with Coomaise blue. Inset figure shows band intensity profiles corresponding to unconjugated p3 with 0 h (grey) and 14 h (black) of irradiation. (B) Phage conjugates irradiated for various amounts of time were tested for binding affinity to TNFα or additional anti-TNFα via ELISA as in Fig. 1. Results show the average and standard deviation of two independent measurements. | ||
Next, the anti-TNFα-phage constructs were tested as scaffolds for immuno-PCR assays. For this, different concentrations of TNFα ranging from 100 ng ml−1 to 0.1 pg ml−1 were reacted in fetal bovine serum (FBS) with streptavidin coated magnetic beads that had been pre-reacted with biotinylated anti-TNFα capture antibodies. After removing free antigen, 1.6 nM solutions of the anti-TNFα-phage were reacted with the antigen coated beads Next, PCR reactions were run by resuspending the beads in a PCR reaction mix that contained primers (Table S1†), polymerase, dNTPs and reaction buffer and submitted to 25 cycles of amplification. The resulting PCR products were then detected by agarose gel electrophoresis (Fig. S1†). While a small amplification product was observed for zero antigen, which was likely due to nonspecific binding of phage conjugates to the bead substrates, a clear trend in amplification intensity could be observed even at low TNFα concentrations.
Although sensitive, gel electrophoresis is slow to run, laborious, less quantitative and cannot be performed in real-time, so we next chose to test the antibody–phage conjugates in real-time PCR assays. For this, although a DNA staining dye such as SYBR Green could be used, because these dyes can also cause high background signals due to binding to primer dimers for example, a universal Taqman probe was utilized instead. Since Taqman probes are designed to produce signal only upon binding to the PCR template during polymerization, high sensitivities can be obtained and in real-time. For this, we designed a 29mer Taqman probe that was complementary to a sequence of the amplified phage product (but separate from the sequences recognized by the primer set) and also contained a 5′ FAM dye, a 3′ IBFQ quencher and an internal quencher 9 bases in from the 5′ end (Table S1†). First, this Taqman probe was used to measure amplification in real time starting from various concentrations of phage in solution and in the presence of magnetic beads which would later be used as an immunoassay substrate. As shown in Fig. S2A and S2B,† real-time PCR analysis showed high sensitivity for femtomolar concentrations of phage. Next, we ran a real-time PCR immunoassay against varying concentrations of TNFα diluted in FBS and immobilized on capture antibody functionalized magnetic beads using photocrosslinked anti-TNFα-phage. After washing to remove unbound antibody–phage, the magnetic beads were transferred directly to the Taqman qPCR reaction mix for real-time PCR. As shown in Fig. 3, real-time analysis showed that the assay was capable of detecting down to ∼10 pg ml−1 TNFα in solution. The delta threshold cycle (ΔCt) values are the difference in threshold cycle for that concentration of antigen versus zero antigen. Therefore, the more sensitive the assay or the lower KD between antibody and antigen, larger ΔCt values would be obtained for a given amount of antigen. As a control, non-photo-crosslinked anti-TNFα-phage were also run against varying concentrations of TNFα bound to the capture antibody coated beads. As shown in Fig. S3,† no crosslinking led to the anti-TNFα antibody dissociating from the phage which generated similar ΔCt values irrespective of antigen concentration.
 | ||
| Fig. 3 Phage are conjugated with antibodies for the three targets listed and used to detect dilutions of said target in a sandwich type immunoassay using Taqman probe based qPCR for signal quantification. ΔCt is the difference in threshold cycle for the listed concentration of target from a no-target control. Data points for each target represent the average and standard error of two independent assays. | ||
In order to develop the antibody–phage conjugates for multiplexed detection of different cancer biomarkers, two additional antibody–phage conjugates that recognize IL-1β and IL-6 were produced using the photoirradiation techniques used to produce the anti-TNFα-phage. For multiplexed detection, since each antibody–phage needs to encode a DNA sequence that is recognized by a specific primer set, we inserted a unique forward primer sequence into the non-coding region of the phage genome for each of the different types of antibody–phage. In order to make sure that there was no erroneous cross-talk between the different primer sets and the different types of antibody–phage, PCR reactions were run from each antibody–phage in the presence of the three different primer sets. As shown in Fig. S4,† only in the presence of the correct primers with the corresponding antibody–phage conjugates (correct phage genome) was any DNA amplification observed.
As had been done with the anti-TNFα-phage, the anti-IL-6-phage and anti-IL-1β-phage were tested in real-time PCR assays against different concentrations of IL-6 and IL-1β phage. Assays for each antigen were performed twice and the average ΔCt values as a function of antigen concentration are shown in Fig. 3 for each antigen, along with errors bars corresponding to standard deviation. Although all of the phage conjugates could detect down to 10 pg ml−1 concentrations, the relatively high affinity of anti-IL6 antibodies for IL-6 gave this assay the highest sensitivities, which was also corroborated by ELISAs (Fig. S5†). By building such calibration curves for each antibody–phage against each antigen, we could then use the ΔCt values to determine in real time both the type and amount of each antigen in solution. These studies also showed the limit of detection of each antigen to be below most clinically relevant concentrations.21,22
Finally, in order to test the antibody–phage conjugates in multiplexed assays, different mixtures of the three antigens (TNFα, IL-6, IL-1β) were reacted in FBS at concentrations of 10 pg ml−1 with a mixture of beads conjugated with the respective capture antibodies. Next, after removing unbound antigen, the 3 different antibody–phage conjugates were added to the beads. Unbound antibody–phage conjugates were removed by washing as before, and then aliquoted into 3 separate wells to which different phage specific primers were added. Real-time PCR assays were then run as before. The ΔCt values were correlated with the calibration curves obtained for each target (Fig. S6†) and used to determine the concentrations of antigens within each mixture. Fig. 4 shows the average concentrations indicated by five independent assays, along with error bars corresponding to standard deviations. While there was some variability in signal such that some of the solutions yielded ΔCt values that correlated to slightly higher than 10 pg ml−1 antigen, overall the results were consistent and more importantly showed minimal signal for antigens not in the mixture. These results therefore show minimal off target effects and demonstrated clear distinctions between all mixtures tested and the potential applicability of these antibody–phage constructs for real time multiplexed immuno-PCR assays.
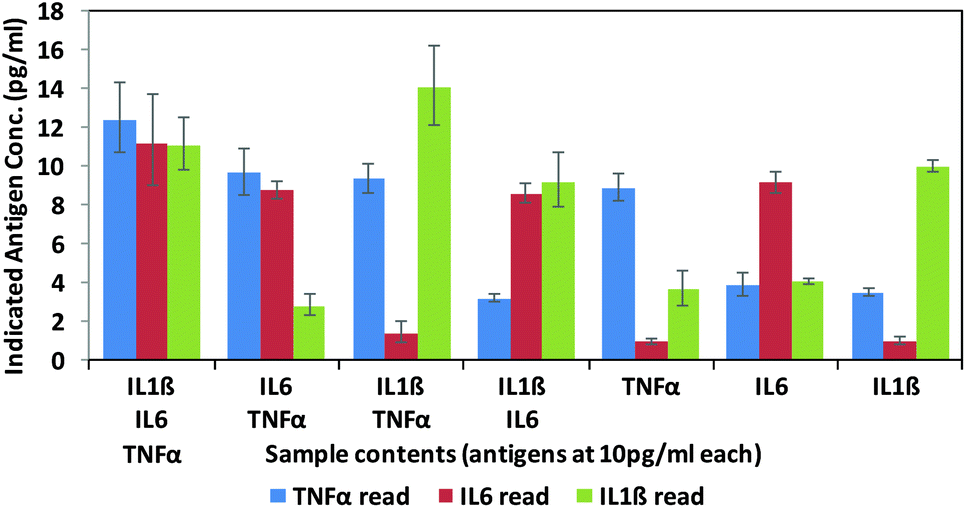 | ||
| Fig. 4 Multiplex assay results of various combinations of targets as calculated by the standard curves created for each target individually (ESI†). Targets listed for each sample are at 10 pg ml−1. Results are the average and standard error of five independent assays. | ||
Materials and methods
Phage design and production
The genome of phage fUSE5 (derived from phage Fd, but containing a tetracycline resistance marker and additional restriction sites) was modified using standard molecular cloning techniques to insert two concatenated copies of the antibody binding peptide (Z domain) or a single copy with the stop codon (TAG) substituted for Q32 as fusions to the N-terminus of capsid protein p3.23 A PCR mutagenesis reaction was then used on the genome bearing two copies of Z domain to generate versions bearing the Q32 mutation within one or both copies. Phage bearing pBPA at these mutations were produced by transforming Escherichia Coli (E. coli) strain K91BlueKan with both the phage genomes and the pEVOL-pBpF plasmid. The pEVOL-BpF plasmid was procured from Addgene and contained both the orthogonal tRNA and amino acyl RNA synthetase required for translation of p-benzoyl-L-phenylalanine (pBPA) at amber stop codons.24,25 Transformants were selected on tetracycline and chloramphenicol containing plates before being pricked and amplified in 40 ml of LB containing ∼300 mM pBPA (Chem-Inpex, Cat: 05110), 20 μg ml−1 tetracycline (Sigma), 50 μg ml−1 chloramphenicol (Sigma) and 0.1% L-arabinose (Spetrum Chem Corp) for the induction of the pEVOL-pBpF. Phage were amplified from the transformants for approximately 20 h, after which cultures were spun at 10000g to remove E. coli from the phage laden supernatant. Phage were precipitated from the supernatant through the addition of a solution of 20% weight per volume 8000 MW PEG (Sigma) in 2.5 M NaCl at a 1/5th volumetric ratio followed by incubation at 4 °C overnight and centrifugation at 10000g for 15 min. The resulting phage pellet was re-suspended in 1 ml of PBS (pH 7.6) and spun at 22000g for 5 min to remove remaining cellular debris. Phage were precipitated again for 1 h followed by centrifugation at 22000g for 15 min. The phage pellet was then re-suspended in 200 ml of phosphate buffered saline at pH 7.6 and stored at 4 °C until use. Phage concentration was determined by UV spectrophotometry following previously reported methods.26 Incorporation of pBPA within p3 appeared to be sufficiently efficient as the phage yield from pBPA containing phage versions was similar to those without.
Photo-conjugation and characterization of antibody–phage via ELISA and SDS PAGE gel
Phage were diluted to a final concentration of 2.5e13 particles per ml (42 nM) in PBS and mixed with a 1:10 molar excess of mouse IgG1 clone Mab1 raised against recombinant TNFα (eBioscience). Antibody–phage mixtures were then transferred to a 0.6 ml polypropylene tube and placed ∼1.5 inches under a 365 nm UV lamp such that the power received was measured to be 810 μW cm−2. Antibody–phage conjugates were irradiated for 3 h. The extent of conjugation was determined by denaturing irradiated antibody–phage mixtures and separating them on a non-reducing SDS PAGE gel (Novex 4–12% BisTris gel, Life Technologies). Protein bands were visualized via coomaisse staining (Thermo Fisher Scientific). ELISA was used to measure antibody–phage conjugate affinity by first suspending 25 μg of streptavidin functionalized magnetic beads (Dynabeads myONE streptavidin T1, Life Technologies) in a 25 μl solution of 133 nM biotinylated antiTNFα (Mab11, eBioscience) or 32 nM biotinylated recombinant TNFα (eBioscience) in 0.05% PBST. These coated beads were then mixed with the antibody–phage conjugate solutions diluted in 0.1% PBST such that the final phage concentration was 1.6 nM. Beads were washed twice with 0.1% PBST and bound antibody–phage conjugates were detected by the addition of an antiphage antibody conjugated to HRP (horse radish peroxidase) (GE healthcare) followed by two more washes and the HRP substrate ABTS (2,2′-azinobis [3-ethylbenzothiazoline-6-sulfonic acid], Sigma). In preparation for use in PCR based assays, irradiated antibody–phage conjugates were first tested as templates for PCR using phage specific primers (Integrated DNA Technology) and Phusion polymerase (New England Biolabs) and found to yield similar concentrations of PCR products to non-irradiated phage, indicating minimal UV induced damage to the phage genome.
Immunoassays using photo-conjugated antibody–phage constructs
Immunoassays were done with antibody–phage conjugates that were irradiated as above for 14 h in the presence of antibody at 20:1 antibody:phage molar ratios. Unbound antibody was then removed by PEG–NaCl precipitating the phage from solution and re-suspending to 1e12 particles per ml (1.6 nM). In the immunoassays for only a single antigen, 25 μg of streptavidin functionalized magnetic beads (Dynabeads myONE streptavidin T1, Life Technologies) were suspended in a 25 μl solution of 133 nM biotinylated capture antibody (purchased from Invitrogen for anti-IL6 or ILβ, eBioscience for anti-TNFα) for 1 h followed by blocking with fetal bovine serum (FBS) (Sigma Aldrich) for an additional hour. Targets were then diluted to the indicated concentration in FBS and incubated with the coated and blocked beads for 1 h. Beads were removed from the target solution and re-suspended in the antibody–phage conjugate solution for 1 h followed by four washes with 0.1% PBST to remove un-bound conjugates. One fifth of the beads were then used for PCR analysis by diluting them in Taqman universal mastermix II (Life Technologies) along with phage specific primers (600 nM, Integrated DNA Technologies) and Taqman probe (250 nM, Integrated DNA Technologies). Quantitative PCR was then performed in an Applied Biosystems QuantStudio6 Flex real time PCR system. Threshold change in normalized fluorescence was set to 0.5 for all results shown. The multiplex protocol was performed identically except that each assay was done with a mixture of beads bound to three different capture antibodies under the same ratios as above (still using 25 μg of beads total per assay), and a mixture of antibody–phage conjugates (each at 1.6 nM) was used to detect the targets rather than just one.
Conclusions
We have demonstrated here the novel engineering of covalently-linked antibody–phage constructs in which antibodies can be incorporated without recombinant expression and without affecting their antigen-binding properties. The crosslinked antibody–phage were prevented from binding to any other antibodies in solution or on a surface but can still recognize the specific antigen target. The naturally encased phage genome was utilized as a template for PCR amplification and could be recognized by a distinct primer set for identification. Furthermore, by utilizing a universal Taqman probe, real-time PCR assays were run with the antibody–phage to build quantitative correlation between a measurable ΔCt value and a specific antigen concentration in solution. Using such calibration curves, a multiplexed assay was developed to detect combinations of TNFα, IL-6, and IL-1β dispersed in a single solution of FBS at concentrations of 10 pg ml−1 each, which is a low and clinically relevant concentration. Results of the multiplexed assay showed that the antibody–phage both detected and quantified antigens with high sensitivity. In addition to immuno-PCR, the antibody–phage constructs developed here can also be applied to nanomedicine applications such as targeted drug delivery or in vivo imaging contrast agents.9,27–29 Future work will expand the breadth of different antibody–phage conjugates to detect a wider variety of different biomarkers. In addition, we will implement these phage antibodies for biomarker detection in clinical patient samples.Acknowledgements
We thank the Biofrontiers next generation sequencing facility and the director Dr Jaime Prior Kirshner for assistance with real time PCR assays. We thank Prof. Itai Benhar for providing us with the phage containing the fUSE5-ZZ genome. We also thank Prof. George P. Smith for providing us with K91BlueKan cells and instructions for their use. We thank Prof. Joel Kaar and Joseph Plaks for advising on the incorporation of pBPA within the Z domain. We thank Prof. Andrew Goodwin for help in editing the manuscript. This work was supported by a NSF Career Award (DMR-1056808) and a NIH award (1R21EB020911-01).Notes and references
- J. F. Rusling, C. V. Kumar, J. S. Gutkind and V. Patel, Analyst, 2010, 135, 2496–2511 RSC.
- S. M. Hanash, Genome Med., 2011, 3, 66 CrossRef PubMed.
- N. Rifai, M. A. Gillette and S. A. Carr, Nat. Biotechnol., 2006, 24, 971–983 CrossRef PubMed.
- T. Sano, C. L. Smith and C. R. Cantor, Science, 1992, 258, 120–122 Search PubMed.
- C. M. Niemeyer, M. Adler and R. Wacker, Trends Biotechnol., 2005, 23, 208–216 CrossRef PubMed.
- I. Burbulis, K. Yamaguchi, A. Gordon, R. Carlson and R. Brent, Nat. Methods, 2005, 2, 31–37 CrossRef PubMed.
- E. R. Hendrickson, T. M. Truby, R. D. Joerger, W. R. Majarian and R. C. Ebersole, Nucleic Acids Res., 1995, 23, 522–529 CrossRef PubMed.
- S. A. Kazane, D. Sok, E. H. Cho, M. L. Uson, P. Kuhn, P. G. Schultz and V. V. Smider, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3731–3736 CrossRef PubMed.
- Y.-C. Guo, Y.-F. Zhou, X.-E. Zhang, Z.-P. Zhang, Y.-M. Qiao, L.-J. Bi, J.-K. Wen, M.-F. Liang and J.-B. Zhang, Nucleic Acids Res., 2006, 34, e62 CrossRef PubMed.
- A. Frenzel, M. Hust and T. Schirrmann, Front. Immunol., 2013, 4, 217 Search PubMed.
- G. P. Smith, Science, 1985, 228, 1315–1317 Search PubMed.
- M. Brasino, J. H. Lee and J. N. Cha, Anal. Biochem., 2015, 7–13 CrossRef PubMed.
- J. McCafferty, A. D. Griffiths, G. Winter and D. J. Chiswell, Nature, 1990, 348, 552–554 CrossRef PubMed.
- D. W. Domaille, J. H. Lee and J. N. Cha, Chem. Commun., 2013, 49, 1759 RSC.
- J. H. Lee, D. W. Domaille and J. N. Cha, ACS Nano, 2012, 6, 5621–5626 CrossRef PubMed.
- M. D. Brasino and J. N. Cha, Analyst, 2015, 140, 5138–5144 RSC.
- H. Bar, I. Yacoby and I. Benhar, BMC Biotechnol., 2008, 8, 37 CrossRef PubMed.
- I. Yacoby, M. Shamis, H. Bar, D. Shabat and I. Benhar, Antimicrob. Agents Chemother., 2006, 50, 2087–2097 CrossRef PubMed.
- J. Z. Hui and A. Tsourkas, Bioconjugate Chem., 2014, 25, 1709–1719 CrossRef PubMed.
- J. Deisenhofer, Biochemistry, 1981, 20, 2361–2370 CrossRef PubMed.
- G. Kleiner, A. Marcuzzi, V. Zanin, L. Monasta, G. Zauli, G. Kleiner, A. Marcuzzi, V. Zanin, L. Monasta and G. Zauli, Mediators Inflammation, 2013, 2013, e434010 Search PubMed.
- Z. R. Yurkovetsky, J. M. Kirkwood, H. D. Edington, A. M. Marrangoni, L. Velikokhatnaya, M. T. Winans, E. Gorelik and A. E. Lokshin, Clin. Cancer Res., 2007, 13, 2422–2428 CrossRef PubMed.
- J. K. Scott and G. P. Smith, Science, 1990, 249, 386–390 Search PubMed.
- J. W. Chin, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11020–11024 CrossRef PubMed.
- T. S. Young, I. Ahmad, J. A. Yin and P. G. Schultz, J. Mol. Biol., 2010, 395, 361–374 CrossRef PubMed.
- C. F. Barbas, D. R. Burton and G. J. Silverman, Phage Display: A Laboratory Manual, CSHL Press, 2004 Search PubMed.
- J. H. Lee and J. N. Cha, Anal. Chem., 2011, 83, 3516–3519 CrossRef PubMed.
- D. P. Lobo, A. M. Wemyss, D. J. Smith, A. Straube, K. B. Betteridge, A. H. Salmon, R. R. Foster, H. E. Elhegni, S. C. Satchell and H. A. Little, et al. , Nano Res., 2015, 8, 3307–3315 CrossRef PubMed.
- H. Zhang, Y. Xu, Q. Huang, C. Yi, T. Xiao and Q. Li, Chem. Commun., 2013, 49, 3778–3780 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an01904h |
| This journal is © The Royal Society of Chemistry 2017 |