
Novel formazan derivatives containing phenylsulfanyl and carbonyl units: synthesis, optical and electrochemical properties†
Gulsen Turkoglu and
Halil Berber*
Department of Chemistry, Faculty of Science, Anadolu University, 26470 Eskisehir, Turkey. E-mail: hlberber@anadolu.edu.tr
First published on 29th September 2016
Abstract
A series of phenylsulfanyl and aryl carbonyl containing formazan derivatives 4a–4h and 5a–5h were synthesized by coupling of substituted phenylhydrazones with diazonium salts of 2-(phenylthio)benzenamine and 5-chloro-2-benzoylbenzenamine in pyridine. The electronic properties of the new formazans were studied by cyclic voltammetry and UV-vis spectroscopy. Fluorescence emission wavelengths were measured in solution, and fluorescence quantum yields and Stokes shifts were calculated. Computational studies were carried out on the tautomer structures formed through intramolecular proton transfer in the formazan backbone, and their HOMO–LUMO energy values were estimated. In addition, the influences emerging from ether, thioether and carbonyl groups on the properties of formazans were elaborated.
Introduction
Recently, formazans have drawn widespread attention in the chemical and biological communities due to their increasing biological, technological and other special applications.1 They are coloured compounds due to the conjugated double bonds in formazan's –NH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NN– backbone. Therefore, a large number of such compounds have recently been prepared.2 Many works have been reported related to the establishment of their syntheses, structural and spectroscopic properties,3 photochemical transformations,4 tautomers5 and redox potential measurements.6 Formazans are also reported to possess a wide spectrum of biological activities such as antiviral,7 antimicrobial,8 anti-inflammatory,9 antifungal,10 anti-tubercular,11 and others.12 They were reported to be used for the synthesis, optical properties and electrochemical behaviours of metal13 and boron complexes as functional materials.14 In addition, a formazan derivative has been used for the modification of a pencil graphite electrode in electrochemical sensor applications.15
C–NN– backbone. Therefore, a large number of such compounds have recently been prepared.2 Many works have been reported related to the establishment of their syntheses, structural and spectroscopic properties,3 photochemical transformations,4 tautomers5 and redox potential measurements.6 Formazans are also reported to possess a wide spectrum of biological activities such as antiviral,7 antimicrobial,8 anti-inflammatory,9 antifungal,10 anti-tubercular,11 and others.12 They were reported to be used for the synthesis, optical properties and electrochemical behaviours of metal13 and boron complexes as functional materials.14 In addition, a formazan derivative has been used for the modification of a pencil graphite electrode in electrochemical sensor applications.15
Formazans form the corresponding tetrazolium salt when oxidized. At the same time given to a living organism these salts are reduced back to formazan depending upon the viability of the organism. These enables the viability of the organism be tested by monitoring formazan formation with spectroscopy. The tetrazolium/formazan couple is a special redox system acting as proton acceptor or oxidant; this system is now widely implemented in different branches of the biological science.16 That is why the spectroscopic investigation of formazans is of importance. Tautomerism in formazans affects their chemical and biological activities, especially their chelating and colour properties. So, it is very important to know the complete scheme of tautomerism and the reaction pathways between different tautomers.17 Therefore, tautomerism, structural and intramolecular hydrogen bonding properties in some formazan derivatives was widely examined by means of DFT.18
Aryl sulphide and their derivatives are valuable intermediates in synthetic organic chemistry as scaffolds for materials chemistry19 for medicinal chemistry due to their biological and pharmacological activities.20 At the same time, diaryl carbonyl compounds are important structural units found in a wide variety of molecules, including natural products,21 compounds of biological and pharmaceutical importance,22 organic materials,23 functional molecules,24 and synthetic intermediates.25 In our previous work, we recently presented detailed structure–property relationship investigations on the new formazans derivatives based on N5-diphenyl ether.26 While the phenyl sulfanyl group is less electron donating than the corresponding aryl ether group, aryl carbonyl group is a strong electron withdrawing. We were therefore interested to investigate the optical and electrochemical performance of the new formazans possessing less electron donating phenyl sulfanyl group and strong electron withdrawing aryl carbonyl unit to compare the diphenyl ether analogues.
As an expanded continuation of our studies related to the synthesis, photophysical and electrochemical properties of new formazan derivatives, in this paper, we present the initial synthesis and characterization of a series of asymmetrically N5-substituted aryl sulphide (4a–4h) and carbonyl derivatives (5a–5h) (Scheme 1). The structures of all synthesized molecules were characterized using elemental analysis and various spectroscopic methods. The UV-vis absorption and fluorescence property of these compounds were also investigated. Electrochemical properties of the new products were explored by cyclic voltammetry. Moreover, the results of the calculations at PBE1PBE/6-311g(2d,2p) level were taken into the consideration to determine the possible tautomer structures and intramolecular hydrogen bonds existing in all synthesized compounds. Through judicious structural variation, we aim to elucidate the effect of substituents on their optical and electrochemical properties.
 | ||
| Scheme 1 The synthesis of the target compounds 4a–4h and 5a–5h. | ||
Results and discussion
Synthesis
Syntheses of substituted phenylhydrazones (1a–1h) were performed according to our previous procedure.26 Synthesis of the studied compounds is depicted in Scheme 1.5-Phenylsulfanyl and 5-arylcarbonyl containing formazan derivatives 4a–4h and 5a–5h were synthesized through the coupling of phenyl diazonium chloride salts with the corresponding substituted phenylhydrazones 1a–1h in a pyridine/NaOH according to published procedures.2a,26 The desired products were obtained intensely colour such as a dark blue, darkish purple, red, brown. The modular nature of this synthetic procedure allows the introduction of a variety of different substitution patterns, including asymmetric derivatives. The yields of products were found to be dependent of the nature of substituent groups in starting material as well as the appropriate reaction temperature and pH medium. Therefore, starting materials were chosen to eliminate the steric effects. Phenylsulfanyl subunit (4a–4h) generally gives rise to higher yields of the end products with respect to diphenylether26 and arylcarbonyl groups (5a–5h). While a coupling reaction of diazonium salt with 1a possessing unsubstituted 1-phenyl group gives a corresponding product 4a in 94% yield, carbonyl group containing compound 5a having electron attractive property results in moderate yield (55%). Compounds 4b, 4e, 5d and 5e were isolated and purified using column chromatography method and the other compounds were recrystallized from methanol with high purity. Their molecular structures were confirmed by FITR, 1H NMR, 13C NMR spectroscopy, LC-MS and elemental analysis. Analytical and spectral data fully support the structure of all target compounds.
Characterizations
The FTIR spectral data of all target formazans (4a–4h and 5a–5h) are given in Experimental section. Observation of N–H weak bands in the range of 3056–3099 cm−1 for 4a–4g and 3058–3074 cm−1 for 5a–5g, excluding 4h (3112 cm−1) and 5h (3271 cm−1), indicates the existing of intramolecular hydrogen bond in formazans.26,27 The sharp characteristic CN stretching bands in formazans are observed between 1591–1606 cm−1 for 4a–4h and 1591–1618 cm−1 for 5a–5h pointing out the existence of intramolecular hydrogen bond (Scheme 2) as excepted.26,28 The observation of intense bands between 1510–1461 cm−1 for 4a–4h and 1473–1498 cm−1 for 5a–5h demonstrates the –NN– group which is well in agreement with the reported FTIR values.3a,26
 | ||
| Scheme 2 Intramolecular hydrogen bonding and tautomerism of the formazans 4a and 5a. | ||
The all of formazans were characterized by 1H NMR spectroscopy (Fig. S3–S18, ESI†), explanatory diagnostic formazan NH shifts in the downfield between 14.85–14.96 ppm for (4a–4e), 4g and 14.38–15.20 ppm for 5a–5g respectively.5b,29 The observations of these values confirm the existence of intramolecular hydrogen bond between Ph1–NH with N–Ph5 in formazan backbone (Scheme 2). NH protons of 4f, 4h and 5h containing electro withdrawing NO2 were obtained in the upfield shift of at 13.30, 13.65 and 11.59 ppm, respectively, compared to other derivatives.8,13b,30 This chemical shifts were indicative of protons deshielded by an electron-withdrawing effect of phenyl rings and double bond resonance in the structure of formazan. The chemical shifts of the aromatic protons are recorded between 8.88–7.03 ppm for 4a–4h and 8.85–6.97 ppm for 5a–5h as expected. While methoxy protons (–OCH3) are observed as singlet between 3.86–3.80 ppm for 4a–4h and 3.82–3.94 ppm for 5a–5h as expected, methyl groups (–CH3) in the compounds 4g and 5g are detected in the upfield (2.30 and 2.40 ppm, respectively).
The characteristic carbon atoms in –N–CN– units of 4a–4h and 5a–5h are recorded in downfield between 164.91–157.58 and 165.98–159.91 ppm, respectively, which are well in agreement with reported literature values in 13C NMR spectrums.26,31 Observation of characteristic peaks in the range of 190.99–196.75 ppm for compounds 5a–5h indicates the existing of carbonyl group. While aliphatic –OCH3 carbon atoms of all compounds are observed in the range of 58.72–50.62 and 49.88–55.67 ppm as expected, methyl groups attached to 4g and 5g appears at 19.98 and 19.99–20.05 ppm, respectively.
Further analysis by mass spectrometry and elemental analysis confirmed the proposed structures of compounds 4a–4h and 5a–5h.
Absorption spectra
The UV-vis absorption properties of all the compounds were carried out in methanol solution and the results are listed in Table 1. All compounds show intense absorption bands in the visible range of the spectrum (between 400 and 600 nm) attributed to characteristic π–π* transitions within the formazan framework.1,26,32 The absorption spectra of as sample compound 4a and 5a within methanol solution are shown in Fig. 1. The synthesized formazans display similar broadband UV-vis absorption characteristic in the visible range of the spectrum that account for their intense colour as excepted.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) and chemical shift values (Δλmax) of compounds 4a–4h and 5a–5h in different solvents
ε) and chemical shift values (Δλmax) of compounds 4a–4h and 5a–5h in different solvents
| Comp. | Substituent | Methanol | 1,4-Dioxane | Toluene | |||
|---|---|---|---|---|---|---|---|
| λmax (nm) (logε) |
Δλmaxa | λmax (nm) (logε) |
Δλmaxa | λmax (nm) (logε) |
Δλmaxa | ||
| a Δλmax = λmax1 (unsubstituted formazan (4a or 5a)) − λmax1 (substituted formazan (4b–4h or 5b–5h)). | |||||||
| 4a | 4-H | 505 (4.75) | — | 512 (4.54) | — | 524 (4.58) | — |
| 4b | 4-OCH3 | 490 (4.36) | 15 | 501 (4.55) | 11 | 515 (4.62) | 9 |
| 4c | 4-Cl | 521 (4.59) | −16 | 525 (4.61) | −13 | 532 (4.61) | −8 |
| 4d | 4-Br | 527 (4.56) | −22 | 527 (4.60) | −15 | 536 (4.60) | −12 |
| 4e | 4-F | 506 (4.58) | −1 | 509 (4.54) | 3 | 522 (4.65) | 2 |
| 4f | 4-NO2 | 524 (4.80) | −19 | 530 (4.77) | −18 | 537 (4.69) | −13 |
| 4g | 3,4-(CH3)2 | 499 (4.76) | 6 | 505 (4.62) | 7 | 515 (4.61) | 9 |
| 4h | 2,4-(NO2)2 | 389 (4.88) | 116 | 483 (4.80) | 29 | 503 (4.81) | 21 |
| 5a | 4-H | 498 (4.88) | — | 501 (4.67) | — | 509 (4.72) | — |
| 5b | 4-OCH3 | 500 (4.78) | −2 | 502 (4.76) | −1 | 512 (4.89) | −3 |
| 5c | 4-Cl | 511 (4.68) | −13 | 512 (4.69) | −11 | 518 (4.71) | −9 |
| 5d | 4-Br | 512 (4.76) | −14 | 517 (4.64) | −16 | 523 (4.72) | −14 |
| 5e | 4-F | 499 (4.75) | −1 | 502 (4.67) | −1 | 511 (4.72) | −2 |
| 5f | 4-NO2 | 530 (4.70) | −32 | 540 (4.74) | −39 | 552 (4.77) | −35 |
| 5g | 3,4-(CH3)2 | 500 (4.79) | 2 | 500 (4.71) | 1 | 503 (4.56) | 6 |
| 5h | 2,4-(NO2)2 | 391 (4.99) | 107 | 386 (4.88) | 115 | 388 (4.88) | 121 |
 | ||
| Fig. 1 Electronic absorption bands of the compounds (a) 4a–4h and (b) 5a–5h in methanol. | ||
The observed wide bands in visible region between λmax1 of 490–527 nm for compounds 4a–4g and λmax1 498–530 nm for 5a–5g in methanol, belong to the characteristic π–π* transitions due to the existence of a delocalized a six-membered in the formazan backbone (–NH–NC–NN–) (Scheme 2).26,27a Conversely, the view of the spectra of ortho-NO2 substituted 4h and 5h are quite different with a strong blue shift between λmax = 385–390 nm in polar protic solvents and λmax = 369–391 nm in all solvents excluding DMSO and DMF, respectively (Tables S1 and S2, see ESI†). This was encountered in a previous study.26,32,33 The replacement of one phenyl sulfanyl substituent for a aryl carbonyl substituent in 4a caused a red shift of maximum absorption (λmax) in methanol, from 505 nm for 4a and 498 nm for 5a. While in 4c, 4d and 4e the observed trend was the same, compounds 4b, 4f, 4g and 4h have a shorter wavelength of maximum absorption to compared 5b, 5f, 5g and 5h.
In accordance to the values in the literature, the second bands observed λmax2 294–310 nm for 4a (4c–4e), 4g and 299–314 nm for 5a (5c–5e), 5g.27b The peaks observed are due to n–π* transitions of –NN– and –CN– groups in formazan main chain. As for the formazans 4b and 4f, 5b and 5f, this band obtained a red shift between 325–378 nm and 361–394 nm respectively, and is believed to be caused from an intramolecular charge transfer within the whole molecule.
Effects of solvent polarity on absorption of these formazans are summarized in Tables S1 and S2, ESI.† Absorption properties of the formazans show slightly dependence on the polarity of the solvents (Fig. 2). Generally, with decreasing polarity of the solvents from in DMSO relative to n-hexane, the position of the absorption maxima of formazans 4a, 4c–4e, 4g and 5a–5e, 5g slightly shifts to red region (Δλmax = 0–17 nm), thus exhibiting a positive solvatochromism. While compounds 4h exhibits usually red shift (Δλmax = 5–25 nm) with increasing polarity of the solvents, excluding DMSO, it exhibits obvious blue shift Δλmax = 88–93 nm in polar protic solvents such as methanol, ethanol. These changes were attributed to hydrogen-bonding interaction between the solute molecule and the solvent molecule.34 In addition, with an increase in solvent polarity, the absorption band of 5h exhibits red shift (Δλmax = 10–37 nm). The absorption maxima of compounds 4b and 4f appear that were red shifted by Δλmax = 0–31 nm and Δλmax = 52–95 nm, respectively, as the polarity of solvent was increased, thus showing clearly a positive solvatochromism. This may be attributed to the stabilization of intramolecular charge transfer within whole molecule. With increasing the polarity of solvents, the absorption compound 5f (Δλmax = 16–72 nm) is shifted to shorter wavelength, thus exhibiting a negative solvatochromism, because the ground state was more stabilized in a solvent cage of already partly oriented solvent molecules with stronger polarity. On the other hand, the spectral shifts in the molecular systems with intramolecular hydrogen bonds are very small. The largest red shift in absorption spectra for compounds 4a–4h has found at 537 nm in toluene for compound 4f and the highest blue shift was obtained at 385 nm in 1-buthanol for compound 4h. While compound 5f, possessing para-NO2 group on 1-phenyl ring, hold the longest absorption wavelength appearing at 568 nm in n-hexane, the shortest absorption wavelength of 369 nm in n-hexane was observed for compound 5h, possessing ortho- and para-NO2 on 1-phenyl ring. This phenomenon can be elaborated by some specific solute–solvent interaction (e.g. H-bonding).26
 | ||
| Fig. 2 UV-vis absorption spectra of compound 4a (left) and compound 5a (right) in various solvents at room temperature. | ||
The absorption spectra of all phenylsulfanyl derivatives 4a–4h were generally observed to be red shifted no more than 7–10 nm and 0–6 nm compared to the corresponding compounds 5a–5h and their diphenyl ether analogue, excluding 1-phenyl-NO2 derivatives, in different solvents. These results were attributed to the hydrogen-bonding interaction between the solute and solvent molecules or the intramolecular charge transfer within the whole formazan structure.
First we will discuss the absorption spectral trends observed upon variation of the 1-phenyl ring substituents in formazans 4a–4h and 5a–5h. As shown in Fig. 1 and Table 1 in methanol solution, while the absorption peak of 4b is red shifted by 15 nm compared to 4a as a result of the electron-donating methoxy moiety, the absorption value (λmax1) of compound 5b is recorded 2 nm longer wavelength than the parent molecule 5a. The λmax1 values for –Cl and –Br substituted derivatives 4c, 4d, and 5c, 5d were noticeably red shifted by 16, 22 and 13, 14 nm, respectively. Cl and Br substituents act as an inductive electron withdrawing and a resonance electron donating effects, which result in two opposite effects. The inductive electron-withdrawing of flour group attached to the 1-phenyl ring brings in 4e and 5e slight red shift as compared to 4a and 5a bearing the unsubstituted-phenyl groups in methanol (Table 1). In the case of para-NO2 substituent, as a strong electron withdrawing unit, the λmax1 values of 4f and 5f were detected towards a red region by 19 nm and 32 nm, respectively. However, the high electron withdrawing effect of the double NO2 substituted 4h and 5h indicate shifts towards shorter wavelength by 116 and 107 nm, respectively, that will decrease the strength of the hydrogen bond between the NH and NN groups due to the formation of a possible hydrogen bond between N–H protons and oxygen of a –NO2 group attached to the o-position of the 1-phenyl ring.20,32 It also antagonizes and therefore weakens the charge transfer. Alkyl substituted 4g and 5g provide blue shift absorption peaks with Δλmax values of 6 nm for 4g and 2 nm for 5g. According to these results, generally electron donating groups at 1-phenyl substituent lead to blue shifts, whereas the electron withdrawing units result in red shifts.
Finally, we studied the absorption properties of 5-phenyl substituted formazans 4a–4h and 5a–5h, which contain aryl sulfide and carbonyl substituents in two different arrangements. The wavelength of maximum absorption of aryl sulfanyl-substituted formazans 4a and 4c–4e were red-shifted from 7 to 15 nm, to compare relative to the aryl carbonyl-substituted analogues 5a and 5c–5e. However, a blue shift in λmax for formazans 4b, 4f, 4g and 4h was observed between 2 to 10 nm relative to 5b, 5f, 5g and 5h (Table 1).
Upon substitution of formazans derivatives containing 5-aryl sulphide and 5-diphenyl ether groups,26 λmax is generally red-shifted to compared formazans derivatives bearing 5-aryl carbonyl groups. This likely may attributed from the strong electron withdrawing properties of the aryl carbonyl rings, which prevents conjugation between the formazan backbone and the 1-phenyl substituent, thereby limiting the length of the π-conjugated system.
Fluorescence spectra
Fluorescence spectra of the target materials under excitation at 470 nm in DMSO solution are depicted in Fig. 3b and 4b. Excitation at the λmax of most formazans showed either no fluorescence or very low intensity, which suggested that fluorescence does not come from the species (intramolecular hydrogen bonding or not hydrogen bonding structures) responsible for λmax absorption. The 470 nm excitation wavelength provided the highest fluorescence level and was a useful wavelength for quantum yield comparison. However all formazan derivatives indicated weakly emissive and modest emission quantum yields. | ||
| Fig. 3 (a) Absorption, (b) normalized fluorescence spectra of 4a–4h in DMSO solution. | ||
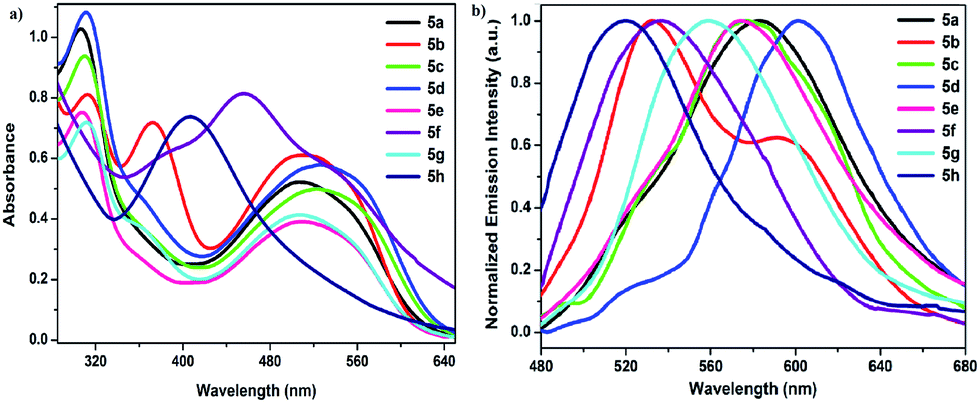 | ||
| Fig. 4 (a) Absorption and (b) normalized fluorescence spectra of 5a–5h in DMSO solution. | ||
All the compounds 4a–4h and 5a–5h displayed green, yellow or orange emission depending on the nature of the whole molecule in DMSO solution (Table 2). As presented in Fig. 3b, compound 4a lacking of substituent on the 1-phenyl ring exhibited yellow fluorescence with a maximum of 577 nm. The emission maximum of compounds 4b, 4e and 4g were all blue-shifted with at 2 nm, 12 nm, and 7 nm, respectively, compared to 4a. Green emission with a blue shift at 58 and 68 nm (519 and 509 nm) was recorded for compounds 4f and 4h, respectively, due to strong electron-withdrawing ability of nitro group on the 1-phenyl ring, compared to 4a.
| Comp. | logεa |
λabsb (nm) | λemb (nm) | Φxc (%) | Sd (nm) | Sd (cm−1) |
|---|---|---|---|---|---|---|
| a Extinction coefficient (1 M−1 cm−1).b Wavelengths of maximum absorbance (λabs) and emission (λem).c Φx was determined by using Rhodamine 6G in ethanol (Φs = 0.95) as the standard.d Stokes shift are calculated as the difference between excitation and emission maxima wavelength. | ||||||
| 4a | 4.65 | 517 | 523, 577 | 10.0 | 107 | 3945 |
| 4b | 4.59 | 521 | 532, 575 | 14.0 | 105 | 3885 |
| 4c | 4.61 | 537 | 519, 588 | 6.0 | 118 | 4269 |
| 4d | 4.59 | 537 | 518, 588 | 9.0 | 118 | 4269 |
| 4e | 4.59 | 521 | 526, 565 | 7.5 | 95 | 3577 |
| 4f | 4.74 | 501 | 519 | 5.5 | 49 | 2008 |
| 4g | 4.65 | 515 | 528, 570 | 8.5 | 100 | 3738 |
| 4h | 4.86 | 409 | 509 | 4.5 | 39 | 1630 |
| 5a | 4.72 | 505 | 582 | 5.4 | 112 | 4094 |
| 5b | 4.78 | 513 | 533, 591 | 2.9 | 121 | 4356 |
| 5c | 4.70 | 520 | 576 | 2.0 | 106 | 3915 |
| 5d | 4.62 | 524 | 600 | 3.7 | 130 | 4610 |
| 5e | 4.59 | 508 | 574 | 2.1 | 104 | 3855 |
| 5f | 4.92 | 456 | 536 | 2.8 | 66 | 2620 |
| 5g | 4.62 | 506 | 559 | 2.6 | 89 | 3387 |
| 5h | 4.87 | 406 | 520 | 1.9 | 50 | 2045 |
Emission maxima of compounds 5a–5h were recorded in the range of 520 and 600 nm in DMSO solution. As presented in Fig. 4b, the fluorescence maximum of 5a (λem = 582 nm) showed a small red shift of 5 nm with respect to 4a. Emission maxima of compounds 5c and 5e are blue shifted about 6 nm and 8 nm relative to 5a, respectively. Moreover, green emission of compounds 5f–5h is clearly blue shifted to by 46, 23 and 62 nm respectively, compared to 5a. The red shifts were observed for 5b and 5d, which showed orange emission with at 591 nm and 600 nm, respectively. The trends observed for the emission spectra mirrored those observed for the absorption spectra.
Comparing emission maxima of compounds 4b, 4d–4f, 4h and 5b, 5d–5f, 5h the replacement of the 5-substituted aryl sulfanyl by the aryl carbonyl group results in slight red shift by 9–18 nm, while 5c, 5g are blue shifted by 11 and 12 nm, respectively. In addition, maximum emission bands of 4a–4h and 5a–5h are generally red shifted by 1 to 35 nm for 4a–4e, 4g, and by 4 to 47 nm for 5a–5f, 5h, compared to diphenyl analogues. These results can explain by the different electron withdrawing-donating capability of the 1,5-substituents and the degree of conjugation depending on the nature of the whole molecule.
The Stokes shifts observed for the compounds 4a–4h were smaller than those of 5a–5h. All formazans with Ar5 = aryl sulfanyl have Stokes shifts ranging from 39 to 118 nm (1630–4269 cm−1) in DMSO, while those with Ar5 = aryl carbonyl have larger Stokes shifts, from 50 to 130 nm (2045–4610 cm−1) in DMSO. The intramolecular CT results in increment of charge density on 1,5-aryl substituents. These values are larger than those observed for formazan analogues based on 5-diphenyl ether (40–100 nm).26 The species created as a result of the resonance structure afforded the distinct emission pattern in 4a–4h and 5a–5h.
The fluorescence quantum yields of compounds 5a–5h have varying values from 1.9% to 5.4%, which are smaller than those of compounds 4a–4h (4.5–14.0%). Compound 4b showed the highest quantum yield of 14.0% in the series of 4a–4h and 5a–5h. The lowest quantum yields were observed for 4h and 5h possessing two electron withdrawing nitro groups (Table 2).26,35 Based on the outcome of fluorescence investigations, quantum yields are slightly dependent on the properties of 1-phenyl electron-donating and -withdrawing substituents. However, the addition of the 5-aryl sulphide substituent clearly increases the emission quantum yield compared to 5-aryl ether (3.8–9.2%)26 and 5-aryl carbonyl substituents. The reason for this is probably due to the intramolecular charge-transfer (ICT) between 5-aryl thioether with 1-aryl substituent in formazan framework.
Electrochemical properties
The redox potentials for compounds 4a–4h and 5a–5h are obtained by cyclic voltammetry (Fig. 5) and summarized in Table 3. The oxidation potentials of formazans are known to be the most characteristic property due to formation of the corresponding tetrazolium salt.6,13b In the anodic region, 1-phenyl-unsubstituted compound 4a (Eox1 = 1.21 and Eox2 = 1.78 V vs. Fc/Fc+) are oxidized via an two irreversible two-electron transfer process corresponding to the production of its radical cation followed by removal of one proton accompanied by ring closure to form the tetrazole cation of the formazan unit (Scheme 3a)13b,26,36 The charge delocalization within systems showing an extended π-conjugation further stabilizes the cation radical intermediate.37 Compound 5a, which provided only one oxidation wave at Eox1 = 1.42 V vs. Fc/Fc+, were oxidized by an irreversible in a single two-electron process. This electrochemical process is coupled with H+ transfer giving directly their tetrazolium cations due to a very fast follow-up chemical reaction (Scheme 3b).6,38 | ||
| Fig. 5 Cyclic voltammograms of 4a–4h and 5a–5h in 0.1 M TBAPF6 in dry dichloromethane; run at a sweep rate of 100 mV s−1 vs. Fc/Fc+, showing oxidation and reduction potentials. | ||
| Comp. | Eox1/Eox2a (V) | Ered1/Ered2a (V) | Eoxonsetb (V) | Eredonsetb (V) | EHOMO/LUMOc (eV) | ECVg (eV) | HOMO/LUMOd (eV) | Ecalgd (eV) |
|---|---|---|---|---|---|---|---|---|
| a Oxidation and reduction potentials from cyclic voltammograms.b Onsets of oxidation and reduction peaks.c HOMO = −(Eoxonset + 4.40) (eV), LUMO = −(Eredonset + 4.40).d HOMO and LUMO energy values predicted at PBE1PBE/6-311g(2d,2p) level and theoretical HOMO–LUMO gaps. | ||||||||
| 4a | 1.21/1.78 | −0.40/−1.10 | 0.94 | −0.15 | −5.34/−4.25 | 1.09 | −4.61/−3.35 | 1.26 |
| 4b | 1.01/1.86 | −0.53/−1.25 | 0.80 | −0.30 | −5.20/−4.10 | 1.10 | −4.44/−3.20 | 1.24 |
| 4c | 1.23/1.84 | −0.34/−1.04 | 1.01 | −0.14 | −5.41/−4.26 | 1.15 | −4.75/−3.56 | 1.19 |
| 4d | 1.24 | −0.34/−1.04 | 0.96 | −0.17 | −5.36/−4.23 | 1.13 | −4.71/−3.48 | 1.23 |
| 4e | 1.27/1.53 | −0.38/−1.06 | 0.99 | −0.18 | −5.39/−4.22 | 1.17 | −4.65/−3.40 | 1.25 |
| 4f | 1.35 | −0.25/−1.18/−1.40 | 1.06 | −0.11 | −5.46/−4.29 | 1.17 | −5.01/−3.84 | 1.17 |
| 4g | 0.88/1.26 | −1.36 | 0.64 | −0.76 | −5.04/−3.64 | 1.40 | −4.52/−3.25 | 1.27 |
| 4h | 1.50 | −0.70/−0.96 | 1.18 | −0.44 | −5.58/−3.96 | 1.62 | −5.26/−3.97 | 1.29 |
| 5a | 1.42 | −0.32/−0.85 | 1.21 | −0.11 | −5.61/−4.29 | 1.32 | −4.65/−3.51 | 1.14 |
| 5b | 0.90/1.29 | −0.67/−1.34 | 0.76 | −0.51 | −5.16/−3.89 | 1.27 | −4.71/−3.32 | 1.39 |
| 5c | 1.08/1.57 | −0.56/−1.10 | 0.89 | −0.42 | −5.29/−3.98 | 1.31 | −4.79/−3.64 | 1.15 |
| 5d | 0.98/1.45 | −0.59/−1.19 | 0.82 | −0.48 | −5.22/−3.92 | 1.30 | −4.76/−3.56 | 1.20 |
| 5e | 1.18/1.62 | −0.45/−1.10 | 0.97 | −0.36 | −5.37/−4.04 | 1.33 | −4.87/−3.57 | 1.30 |
| 5f | 0.76 | −1.37 | 0.57 | −1.20 | −4.97/−3.20 | 1.77 | −5.06/−3.88 | 1.18 |
| 5g | 0.97/1.73 | −0.60/−1.21/−1.59 | 0.82 | −0.49 | −5.22/−3.91 | 1.31 | −4.76/−3.40 | 1.36 |
| 5h | 1.40 | −1.14 | 1.11 | −0.90 | −5.51/−3.50 | 2.01 | −5.14/−3.36 | 1.78 |
 | ||
| Scheme 3 Schematic representation of structure of 4a–4h and 5a–5h for possible oxidation mechanism (a) for 4a–4c, 4e, 4g and 5b–5e, 5g, (b) structure of 5a for possible oxidation mechanism of 4d, 4f, 4h and 5a, 5f, 5h, (c) possible reduction mechanism for 4a–4h and 5a–5h. | ||
We begin by discussing 1-substituted formazans 4a–4h and 5a–5h. The first oxidation peak potentials of 1-phenyl electron-rich 4-methoxy and a weakly electron donating 3,4-dimethyl substituted compounds 4b (Eox1 = 1.01 V vs. Fc/Fc+) and 4g (Eox1 = 0.88 V vs. Fc/Fc+), which show two irreversible oxidation potentials, were more easily oxidized than the parent molecules 4a (Table 3). In comparison to compound 4a, the irreversible first oxidation potential mainly for compounds 4c–4e halogens moiety positively shifted by 0.02 to 0.06 V, which suggest slightly a difficult oxidation requirement. However, compound 4d were oxidized by two electrons process in a single step. Electron withdrawing nitro unit substituted 4f (Eox1 = 1.35 V) and 4h (Eox1 = 1.50 V), which provided only one irreversible oxidation wave, were oxidized in a single two-electron process giving directly their tetrazolium cations. Formazans 4f and 4h were significantly more difficult to oxidized than their 1-phenyl substituted analogues.26
In a series of 5a–5h, the first quasi-reversible oxidation potentials of compounds 5c–5e are negatively shifted by 0.34, 0.44 and 0.24 V, respectively, compared to 5a. All three formazans were oxidized by two electron process corresponding to the formation of the radical cation, then tetrazolium cation of the formazan unit (Scheme 3a). For compounds 5b (Eox1 = 0.90, Eox2 = 1.29 V vs. Fc/Fc+) and 5g (Eox1 = 0.97, Eox2 = 1.73 V vs. Fc/Fc+), two reversible oxidation potential are monitored. In comparison to compound 5a, the first oxidation potentials of compounds 5b and 5g are negatively shifted by 0.52 and 0.45 V, indicating an easier oxidation requirement. Unexpectedly, in a series of 5a–5h, the oxidation potential of nitro substituted 5f (Eox1 = 0.76 V) and 5h (Eox1 = 1.40 V), which have irreversible one oxidation potentials, were recorded lower than that of 5a (Table 3). This state is probably due to the strong electron-withdrawing effects of nitro groups with carbonyl unit in formazans.
In the cathodic region of the first reduction peak potentials of the formazans, observed that reduction of the tetrazolium cation occurs between −0.25 and −0.70 V for 4a–4f, 4h and −0.32 and −0.67 V for 5a–5e, 5g.13b,15,26 Second wave was attributed to the reduction formazan derivatives corresponding to the formation of a anion with a one-electron transfer (Scheme 3c).39 Formazan chain with four nitrogen atoms and one carbon atom conjugated system possessing three aromatic rings displays electronic changes upon both oxidation and reduction as stable anionic and cationic particles.34 There is also agreement between these results and the data given in literature.6,26,38b,40
On the other side, reduction potentials are also affected by substituents. Phenyl substituted compound 4a exhibited two irreversible reduction potentials at Ered1 = −0.40 and Ered2 = −1.10. The p-methoxy substituted 4b at the 1-phenyl ring was reduced at irreversible peak potentials of Ered1 = −0.53 and Ered2 = −1.25 V, while 3,4-methyl-substituted compound 4g was the most difficult to reduce (Ered = −1.36 V). The first reduction potentials of the compounds 4c, 4d and 4e (Ered1 = −0.34, −0.34 and −0.38 V respectively) were relatively shifted more positive by 0.02 to 0.06 V, compared to 4a. This is probably due to the mesomeric effects of halogen groups in 4c–4e. Compound 4f shows three quasi-reversible reduction potentials (−0.25, −1.18 and −1.40 V). In comparison to compound 4a, the first reduction potential mainly for the reduction of the tetrazolium cation positively shifted by 0.15 V, which suggest an easier reduction requirement. For compound 4h, two quasi-reversible reduction potential are monitored, which is substantially more difficult to reduce, with the sequential one electron reductions occurring at −0.70 and −0.96 V vs. Fc/Fc+.
In the series of compounds 5a–5h, 1-phenyl-unsubstituted compound 5a shows two irreversible reduction potential (Ered1 = −0.32 and Ered2 = −0.85 V). Compound 5b and 5g, bearing p-methoxy and 3,4-dimethyl substituted at the 1-phenyl ring, were more difficult to reduce than 5a, with reversible reduction waves observed at Ered1 = −0.67, Ered2 = −1.34 V and Ered1 = −0.60, Ered2 = −1.21, Ered3 = −1.59 V, respectively. The first reversible reduction potential of compounds 5c–5e, bearing Cl, Br and F are negatively shifted by 0.24, 0.27 and 0.13 V respectively, compared to compound 5a. Compound 5f (1-phenyl p-NO2) is the most difficult to reduce, with only one reversible reduction potential occurring at −1.37 V vs. Fc/Fc+. Compound 5h shows one quasi-reversible reduction potential (−1.14 V vs. Fc/Fc+), which is easier to reduce than 5f.
Investigation of Ar5 substituent effects by cyclic voltammetry gave rise to the reversibility of the reduction wave for compounds 5b–5g indicating the fact that formazan anions are stable on the electrochemical timescale of the solvent (DCM) compared to phenylsulfanyl derivatives (Fig. 5). In addition, the first one-electron reduction potentials mainly for the reduction of the tetrazolium cation occurring at −0.32 to −1.37 V of compounds 5a–5f, 5h are slightly more difficult to reduce compared to the first reduction potentials (−0.25 to −0.70 V) of the compounds 4a–4f, 4h. Unexpectedly, 3,4-dimethyl-substituted compounds 5g was also clearly easier to reduce than 4g by ca. 0.76 V.
Formazans 5a–5h generally provided the quasi-reversible oxidation peak, while oxidation peaks of 4a–4h exhibited irreversible ones which are unstable on the electrochemical timescale of the solvent. Furthermore, 5b–5f and 5h, excluding 5a and 5g, are slightly easier to be oxidized with the first oxidation waves occurring from 0.76 to 1.40 V with respect to those of the compounds 4a–4h (0.88 to 1.50 V). The first oxidation potential of 4a–4h and 5a–5h was smaller than that of the dipheny ether analogues, with oxidation waves observed at 1.27–1.56 V.26
The energy gaps as well as the HOMO and LUMO energies were calculated in order to correlate them with the oxidation and reduction potentials of the investigated 4a–4h and 5a–5h. On the basis of the onset potentials (Eoxonset) and (Eredonset), while the HOMO energy levels of 4a–4h were estimated to be between −5.04 and −5.46 eV, the LUMO energy levels were calculated between −3.64 and −4.29 eV, respectively. HOMO energies of compounds 5a–5h vary from −4.97 to −5.61 eV, whereas LUMO energies are between −3.20 and −4.29 eV. The band gaps inclusion of 5-phenyl sulfanyl 4a–4h was smaller at 1.09–1.62 eV in comparison to electrochemical band gaps of electron donating group 5-diphenyl ether analogues (1.17–2.14 eV)26 and electron withdrawing group 5-aryl carbonyl derivatives (1.27–2.0 eV). As shown in Table 3, the anodic onset potential and the HOMO level of 4g and 5f are smaller than those of the other derivatives. Moreover, as expected, reduction of 4g and 5f are more difficult and thus the LUMO levels of them are smaller than those of the other derivatives. The relatively low HOMO and LUMO energies of 4g, 4h, and 5b–5d, 5f–5h can be rationalized by good π–π* transitions in formazan backbone and π-accepting ability of the phenyl sulfanyl, carbonyl, and electron-withdrawing substituents.
Theoretical calculations
To obtain further insight into the effect of tautomer structures and electron distribution on the spectroscopic properties of all synthesized compounds, were performed in gas phase at DFT level using PBE1PBE/6-311g(2d,2p) basis set.18b,26 Thermal free energies of tautomers, important bond distances (Table S3, ESI†) and Frontier Molecular Orbital distributions (Fig. S1 and S2, ESI†) are provided in ESI.†Computational investigations revealed that tautomerisation of aryl formazans with key end atoms, N(1) and N(5), is an intramolecular process occurring between two degenerate forms,1,2a,17,26 which are in tautomeric equilibrium, namely tautomer 1 and tautomer 2. The equilibrium is fully shifted towards the tautomer form 1 or 2 according to the electronic characteristics.1 Possible tautomeric forms of 4a and 5a for target compounds are depicted as a representative example in Fig. 6, respectively. While 4b, 4c and 4d are stable in the form of tautomer 2, compounds 4a, 4e and 4f–4h were observed to be more stable in the tautomer 1. For carbonyl compounds excluding 5a, 5c and 5e tautomer 2 is more stable, while each tautomeric form of compound 5e is available as an equal in gas phase. This phenomenon can be rationalized by substantial substituent effect on the geometry of molecules. The obtained results suggested that both tautomers 1 and 2 can exist in the solution with a certain ratio.
 | ||
| Fig. 6 Molecular structures of the possible tautomeric forms of 4a (above) and 5a (below) with atom numbering. | ||
Each tautomeric form of compounds 4a–4h and 5a–5h comprise two intramolecular hydrogen bonds. In tautomer 1 of molecules 4a–4g have a strong hydrogen bond between H1⋯N2 and relatively weak hydrogen bond between H2⋯N1, whereas in tautomer 2 of compounds 4g and 4h the reverse situation was observed. In the same way, 5a–5d, 5g have relatively stronger hydrogen bond between H2⋯N1 in tautomer 1 according to hydrogen bond between H1⋯N2 in form of tautomer 2. However, compounds 5e, 5f and 5h have stronger intramolecular hydrogen bond in the form of tautomer 2 which leads to the increase of population of its and consequently decrease of the absorption maximum down to 406 nm (5h) owing to the diminishing of conjugation between benzene unit and carbonyl group. These observations support the six-membered intramolecular hydrogen bond structure mentioned in the characterization section and in Scheme 2 of formazans backbone. While hydrogen bonds between H1⋯O1 and H1⋯N2 of compound 4h1 were predicted to be 1.928 and 1.940 Å, respectively, H2⋯O1 and H2⋯N1 hydrogen bonds of compound 4h2 were obtained as 1.789 and 2.135 Å (Table S3, ESI†). These results prove the optical properties and the existence of a hydrogen bond formed between ortho-nitro group with NH proton. All compounds, excluding 4h2, in tautomer 2 have strong hydrogen bonds between H2 and S2 atoms. However, tautomer 1 brings about weak hydrogen bonds H1⋯S1 in all molecules according to calculation. Compound 5a–5h containing carbonyl group, excluding 5f and 5h, has a strong intramolecular hydrogen bond between H1⋯O1.
From the electron density plots in Fig. S1 (ESI†) they can be seen that the HOMO orbitals of 4a–4h are mainly localized on the –NH–NC–NN– framework and phenylsulfanyl group, implying that other phenyl ring moiety has little influence on the electron density distribution. All of the compounds LUMOs is higher that were localized on Ph1–NH–NC–NN–Ph5 backbone. However, compounds 5a–5h the HOMO orbitals are mainly localized on the formazan skeleton, 1-phenyl and 3-phenyl groups, while LOMOs were intensely localized on electron withdrawing carbonyl group and –NH–NC–NN– skeleton (Fig. S2, ESI†). The HOMO orbital of 4a–4g (−4.44 to −5.01 eV) is slightly more delocalized than that of 5a–5g (−4.65 to −5.06 eV) probably due to an increased conjugation length as the distribution over the main formazan framework, electron-rich phenylsulfanyl moieties. The more delocalized HOMOs of 4a–4g explain slightly the red-shifted absorption spectra in comparison to 5a–5g. Also LUMO energy levels for 4a–4g were calculated to be between −3.20 and −3.84 eV. LUMOs of 5a–5g are found between −3.88 and −3.32 eV, which slightly more localized than that of 4a–4g. And compounds 4h and 5h spread the HOMO and LUMO through the 2,4-NO2 substituents in 1-phenyl ring, which could explain the relatively lower energy level by comparison of other substituents. Compound 4h markedly lowers the LUMO level by 0.61 eV (this value for the HOMO level is 0.12 eV), compare to compound 5h (Table 3). This is generally consistent the results that indicate the reduced intensity of the ICT band observed in the absorption spectra. It is generally indicative of a HOMO/LUMO absorption transition to bear a significant charge transfer character. The extent of energy change in HOMO–LUMO levels is almost equal and thus the band gap values are nearly same to that of all compounds. The calculated HOMO–LUMO gaps of formazans 4a–4h and 5a–5h were generally observed to be consistent with the electrochemical band gaps (ECV) (Table 3).
Conclusions
In summary, a series of new formazan derivatives were designed and synthesized in order to investigate the effect of aryl sulfanyl in comparison to aryl carbonyl groups on formazan structure, optic and electrochemical properties. We have shown that the properties of these formazans, including their wavelengths of maximum absorption and emission, quantum yields, and electrochemical properties can be tuned by the introduction of electron-donating (5-phenylsulfanyl) or electron-withdrawing (5-arylcarbonyl) substituents at the formazan backbone. Modest enhancements in emission intensity were observed for the compounds 4a–4h containing phenylsulfanyl groups, which exhibited quantum yields relatively high (14%) compared to other analogues. In addition, this work demonstrated that formazan derivatives have larger Stokes shifts than compounds based diphenyl ether of our previous study. CV studies revealed significantly different behaviour, whereby the reversible reduction potentials of compounds 5a–5h was observed, presumably due to the extremely electron-withdrawing environment created upon the reduction of the formazans backbone. Theoretical calculations on properties of tautomers and HOMO–LUMO energy levels of these compounds were also conducted. The results suggest that a high intramolecular charge-transfer process from the phenylsulfanyl moiety to the formazan backbone occurs in compounds 4a–4g, while weakly intramolecular charge-transfer process from the aryl carbonyl core to the formazan skeleton takes place in compounds 5a–5g. These characterizations of phenylsulfanyl and arylcarbonyl substituted formazans should help in development of new functional material for future applications. Also, compounds 5b–5g may be using as redox-active functional materials.Experimental
Materials and methods
2-(Phenylthio)benzenamine, 5-chloro-2-benzoylbenzenamine, substituted phenylhydrazines, 4-methoxybenzaldehyde and NaOH were purchased from Sigma-Aldrich and used without any further purification. The used solvents, such as ethyl acetate (EtOAc), methanol (MeOH), and ethanol (EtOH) were spectroscopic grade (Sigma-Aldrich). Synthesis of substituted phenylhydrazones (1a–1h) was performed according to literature procedure.26 Thin layer chromatography plates (DC-Alufolien Kieselgel 60 F254) were obtained from Merck, Darmstadt. Isolation of compounds was carried out by using column chromatography over Sigma Silica gel 60 (63–200 μm).Microanalyses were carried out using a Leco CHNS-932 elemental analyser. The melting points were measured on a Gallenkamp apparatus using a capillary tube. 1H and 13C NMR spectra were recorded on a Bruker DPX FT-NMR (500 MHz) spectrometer (SiMe4 as internal Standard) and chemical shifts (δ) are given in ppm. The spectrometer was equipped with a 5 mm PABBO BB-inverse gradient probe. The FTIR spectra were recorded as KBr pellets using a Bruker IFS 66 v/S FTIR Spectrophotometer in a 4000–400 cm−1 range at room temperature. The electronic spectra of the formazans (1 × 10−5 mol L−1) in different solvents were measured using a SHIMADZU UV-3150 UV-VIS-NIR spectrophotometer. The fluorescence spectra of the compounds were recorded on a Cary Eclipse Fluorescence Spectrophotometer (Varian). The fluorescence quantum yields (Φx) for formazan derivatives were determined in the DMSO using the Rhodamine 6G (Φs = 0.95 in ethanol) as a standard by previously described methods.41 The relative fluorescence quantum yields (Φx) were calculated by the comparative method (eqn (1)),42
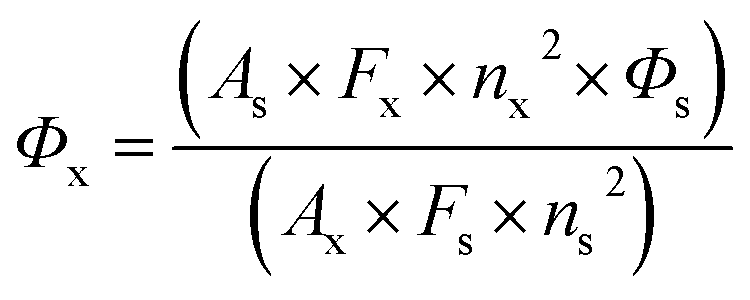 | (1) |
LC-MS studies of the formazans were carried out with an Agilent 1100 Series LC/MSD Trap VL&SL using atmospheric pressure chemical ionization and electrospray with positive and negative ion detection.
Electrochemical measurements were made using a CH-Instruments Model 400A as a potentiostat. A conventional three-electrode configuration consisting of a platinum working electrode, a Pt-wire counter electrode and an Ag-wire reference electrode was employed. All the measurements were run in dry dichloromethane with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte at room temperature under nitrogen atmosphere. Ferrocene was added as a calibrant after each set of measurements, and all potentials reported were quoted with reference to the ferrocene/ferrocenium (Fc/Fc+) couple at a scan rate of 100 mV s−1.
In the calculations, ChemBioDraw Ultra 2013,43 GaussView 5 44 and Gaussian 09 45 package programs were used. Quantum chemical calculations were performed at DFT level of theory using Perdew, Burke and Ernzerhof (PBE1PBE) functionals with 6-311G (2d,2p) basis sets18b,46 without any symmetry restrictions.
Synthesis
1-Phenyl-3-(4-methoxyphenyl)-5-2-(phenylthio)phenylformazans (4a). Compound 4a was obtained from 3a (880 mg, 4.40 mmol) and diazonium salt 2 (1.00 g, 4.40 mmol). Formazan 4a (1.82 g, 94%) was obtained as a light violet powder with a melting point of 126–127 °C. Rf: 0.61 (n-hexane/ethylacetate, 5
:1). FTIR (KBr pellet, cm−1) νmax: 3064, 1352 (N–H w., C–N str.), 1581, 2993 (aromatic ring, CC str., C–H str.), 1606 (CN str. in Schiff base), 1500–1479 (NN of formazans), 1245–1209 (C–O–C), 738 (C–S–C). 1H NMR (500 MHz, DMSO-d6) δ: 14.90 (s, 1H), 8.10 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.5 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H), 7.56 (t, J = 8.0 Hz, 3H), 7.47 (t, J = 7.0 Hz, 2H), 7.32 (d, J = 7.5 Hz, 2H), 7.28 (d, J = 7.5 Hz, 1H), 7.24 (d, J = 7.5 Hz, 3H), 7.05 (d, J = 8.5 Hz, 2H), 3.80 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 160.91, 150.80, 147.64, 136.41, 135.66, 132.02, 131.22, 131.01, 130.93, 130.08, 129.50, 128.60, 128.17, 127.72, 123.34, 121.57, 116.14, 115.22, 56.02. MS, ESI−: m/z (%) 439.1 (M + 1, 9); 438.1 (M, 31); 437.0 (M − 1, 100). MS/MS, ESI−: m/z (%) 330.9 (22); 275.9 (100); 182.9 (92). Anal. calcd for C26H22N4OS: C, 71.21; H, 5.06; N, 12.78; S, 7.31%. Found: C, 70.91; H, 5.29; N, 12.51; S, 7.20%.
1-(4-Methoxyphenyl)-3-(4-methoxyphenyl)-5-(2-(phenylthio)phenyl)formazan (4b). Compound 4b was obtained from 1b (1.50 g, 5.80 mmol) and diazonium salt 2 (1.18 g, 5.80 mmol). The product was purified by column chromatography using a solvent mixture of n-hexane/ethylacetate/CHCl3 (5
:1:2, Rf: 0.72) as eluent resulting in claret solid. Yield: 1.94 g, 71%. mp 258–260 °C; FTIR (KBr pellet, cm−1) νmax: 3060, 1363 (N–H w., C–N str.), 1581, 2997 (aromatic ring, CC str., C–H str.), 1602 (CN str. in Schiff base), 1504–1487 (NN of formazans), 1299–1247 (C–O–C), 744 (C–S–C). 1H NMR (500 MHz, CDCl3) δ: 14.88 (s, 1H), 8.20 (dd, J = 5.7, 3.3 Hz, 2H), 8.11 (d, J = 8.9 Hz, 4H), 7.94 (d, J = 8.9 Hz, 2H), 7.89 (dd, J = 5.7, 3.3 Hz, 2H), 7.50 (dd, J = 5.7, 3.3 Hz, 2H), 7.33 (t, J = 7.2 Hz, 1H), 7.20 (d, J = 7.2 Hz, 2H), 7.00 (d, J = 8.9 Hz, 2H), 3.90 (s, 6H). 13C NMR (126 MHz, CDCl3) δ: 157.58, 154.83, 142.04, 141.58, 137.36, 134.69, 132.09, 1316, 130.81, 126.32, 125.42, 124.48, 124.43, 122.92, 122.59, 122.33, 121.98, 121.24, 119.62, 119.61, 118.35, 118.08, 116.85, 112.56, 50.91, 50.62. MS, ESI+: m/z (%) 469.0 (M + 1, 11); 468.1 (M, 29); 467.1 (M − 1, 100). MS/MS, ESI+: m/z (%) 254.0 (6); 184.9 (100); 135.0 (24). Anal. calcd for C27H24N4O2S: C, 69.21; H, 5.16; N, 11.89; S, 6.84%. Found: C, 69.21; H, 4.97; N, 11.59; S, 6.71%.
1-(4-Chlorophenyl)-3-(4-methoxyphenyl)-5-(2-(phenylthio)phenyl)formazan (4c). Compound 4c was obtained from 1c (1.03 g, 3.90 mmol) and diazonium salt 2 (790 mg, 3.90 mmol) as a claret solid. Yield: 1.71 g, 86%. Rf: 0.83 (n-hexane/ethylacetate, 5
:1). mp 146–147 °C. FTIR (KBr pellet, cm−1) νmax: 3068, 1357 (N–H w., C–N str.), 1585, 3002 (aromatic ring, CC str., C–H str.), 1606 (CN str. in Schiff base), 1508–1483 (NN of formazans), 1249–1168 (C–O–C), 740 (C–S–C). 1H NMR (500 MHz, DMSO-d6) δ: 14.85 (s, 1H), 8.11 (d, J = 8.1 Hz, 1H), 8.02 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 8.6 Hz, 2H), 7.57 (t, J = 7.7 Hz, 1H), 7.46 (d, J = 7.7 Hz, 2H), 7.33 (t, J = 7.7 Hz, 2H), 7.29 (d, J = 7.4 Hz, 2H), 7.24 (d, J = 8.1 Hz, 3H), 7.04 (d, J = 8.7 Hz, 2H), 3.80 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 159.98, 148.26, 146.79, 142.41, 135.27, 134.60, 134.00, 131.05, 130.19, 129.06, 128.94, 127.88, 127.60, 127.52, 122.02, 115.63, 114.50, 55.68. MS, ESI+: m/z (%) 472.1 (M, 23); 473.0 (M + 1, 16); 471.0 (M − 1, 55). MS/MS, ESI+: m/z (%) 332.0 (18); 184.9 (100); 152.0 (5). Anal. calcd for C26H21ClN4OS: C, 66.02; H, 4.48; N, 11.85; S, 6.78%. Found: C, 66.04; H, 4.62; N, 11.70; S, 6.70%.
1-(4-Bromophenyl)-3-(4-methoxyphenyl)-5-(2-(phenylthio)phenyl)formazan (4d). Compound 4d was obtained from 1d (1.0 g, 2.90 mmol) and diazonium salt 2 (590 mg, 2.90 mmol) as a dark violet solid. Yield: 1.39 g, 82%. Rf: 0.66 (n-hexane–ethylacetate, 5
:1). mp 146–147 °C. FTIR (KBr pellet, cm−1) νmax: 3056, 1357 (N–H w., C–N str.), 1585, 2999 (aromatic ring, CC str., C–H str.), 1604 (CN str. in Schiff base), 1506–1481 (NN of formazans), 1247–1168 (C–O–C), 740 (C–S–C). 1H NMR (500 MHz, DMSO-d6) δ: 14.85 (s, 1H), 8.10 (d, J = 8.2 Hz, 1H), 8.02 (d, J = 8.7 Hz, 2H), 7.74 (d, J = 8.7 Hz, 2H), 7.70 (d, J = 8.7 Hz, 2H), 7.57 (t, J = 7.6 Hz, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.33 (t, J = 7.8 Hz, 3H), 7.26 (d, J = 7.6 Hz, 3H), 7.04 (d, J = 8.7 Hz, 2H), 3.85 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 161.98, 160.08, 149.26, 148.76, 143.91, 135.27, 134.60, 133.18, 130.21, 129.91, 129.61, 129.06, 128.93, 127.51, 127.22, 127.12, 122.04, 115.70, 114.50, 55.69. MS, ESI+: m/z (%) 518.1 (M + 2, 11); 517.1 (M + 1, 29); 516.0 (M, 100). MS/MS, ESI+: m/z (%) 332.0 (15); 212.9 (3); 184.9 (100). Anal. calcd for C26H21BrN4OS: C, 60.35; H, 4.09; N, 10.83; S, 6.20%. Found: C, 60.41; H, 4.27; N, 10.59; S, 5.98%.
1-(4-Fluorophenyl)-3-(4-methoxyphenyl)-5-(2-(phenylthio)phenyl)formazan (4e). Compound 4e was obtained from 1e (1.20 g, 4.90 mmol) and diazonium salt 2 (980 mg, 4.90 mmol) as a claret solid. Chromatographic purification using a mixture of n-hexane/ethylacetate (5
:1, Rf: 0.64) as an eluent resulted in 1.52 g yield (63%) with a melting point of 125–127 °C. FTIR (KBr pellet, cm−1) νmax: 3070, 1365 (N–H w., C–N str.), 1585, 3004 (aromatic ring, CC str., C–H str.), 1604 (CN str. in Schiff base), 1490 (NN of formazans), 1255–1168 (C–O–C), 740 (C–S–C). 1H NMR (500 MHz, DMSO-d6) δ: 14.90 (s, 1H), 8.09 (d, J = 8.1 Hz, 2H), 8.02 (d, J = 8.8 Hz, 2H), 7.89 (d, J = 8.8 Hz, 2H), 7.60 (t, J = 7.8 Hz, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.43 (t, J = 8.2 Hz, 1H), 7.28 (t, J = 7.8 Hz, 2H), 7.22 (t, J = 7.5 Hz, 1H), 7.17 (d, J = 7.5 Hz, 3H), 7.03 (d, J = 8.8 Hz, 2H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 164.91, 162.92, 159.96, 147.87, 146.12, 142.10, 136.65, 135.31, 131.82, 130.06, 129.19, 127.82, 127.69, 127.00, 125.78, 124.03, 123.96, 119.50, 117.35, 117.16, 115.27, 114.50, 55.97. MS, ESI−: m/z (%) 457.1 (M + 1, 8); 456.0 (M, 30); 455.1 (M − 1, 100). MS/MS, ESI−: m/z (%) 293.9 (98); 269.0 (100); 225.9 (42); 198.9 (32); 174.9 (42). Anal. calcd for C26H21FN4OS: C, 68.40; H, 4.64; N, 12.27; S, 7.02%. Found: C, 68.57; H, 4.45; N, 11.98; S, 7.07%.
1-(4-Nitrophenyl)-3-(4-methoxyphenyl)-5-(2-(phenylthio)phenyl)formazan (4f). Compound 4f was obtained from 1f (1.00 g, 3.70 mmol) and diazonium salt 2 (890 mg, 3.70 mmol) as a darkish solid. Yield: 0.96 g, 54%, Rf: 0.79 (n-hexane/ethylacetate, 5
:1), mp 187–189 °C. FTIR (KBr pellet, cm−1) νmax: 3099, 1326 (N–H w, C–N str.), 1589, 3008 (aromatic ring, CC str., C–H str.), 1606 (CN str. in Schiff base), 1494 (NN of formazans), 1253–1134 (C–O–C), 767 (C–S–C). 1H NMR (500 MHz, DMSO-d6) δ: 13.30 (s, 1H), 8.27 (d, J = 9.0 Hz, 2H), 8.09 (d, J = 7.9 Hz, 1H), 7.91 (d, J = 8.7 Hz, 2H), 7.63 (d, J = 9.0 Hz, 2H), 7.56 (d, J = 7.2 Hz, 2H), 7.50 (d, J = 7.2 Hz, 3H), 7.43 (t, J = 7.2 Hz, 2H), 7.05 (t, J = 7.9 Hz, 3H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 160.35, 149.85, 149.15, 146.14, 141.88, 139.94, 133.97, 133.81, 131.90, 130.60, 129.54, 129.49, 127.61, 126.82, 126.37, 117.19, 115.12, 114.65, 114.29, 55.72. MS, ESI−: m/z (%) 484.0 (M + 1, 10); 483.1 (M, 27); 482.1 (M − 1, 100). MS/MS, ESI−: m/z (%) 321.0 (100); 296.3 (2); 270.2 (9). Anal. calcd for C26H21N5O3S: C, 64.58; H, 4.38; N, 14.48; S, 6.63%. Found: C, 64.78; H, 4.23; N, 14.16; S, 6.87%.
1-(3,4-Dimethylphenyl)-3-(4-methoxyphenyl)-5-(2-(phenylthio)phenyl)formazan (4g). Compound 4g was obtained from 1g (1.20 g, 4.70 mmol) and diazonium salt 2 (950 mg, 4.70 mmol) as a claret solid. Yield: 1.56 g, 71%. Rf: 0.67 (n-hexane/ethylacetate, 5
:1). mp 126–128 °C. FTIR (KBr pellet, cm−1) νmax: 3070, 1350 (N–H w., C–N str.), 1583, 3012 (aromatic ring, CC str., C–H str.), 1606 (CN str. in Schiff base), 1487 (NN of formazans), 1245–1170 (C–O–C), 738 (C–S–C). 1H NMR (500 MHz, DMSO-d6) δ: 14.96 (s, 1H), 8.09 (d, J = 8.2 Hz, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.60 (t, J = 8.2 Hz, 2H), 7.54 (d, J = 7.7 Hz, 1H), 7.33 (d, J = 7.7 Hz, 1H), 7.28 (t, J = 7.7 Hz, 2H), 7.21 (s, 1H), 7.19 (d, J = 7.1 Hz, 1H), 7.15 (d, J = 8.2 Hz, 3H), 7.04 (d, J = 8.8 Hz, 2H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 159.91, 149.71, 146.41, 141.98, 141.13, 138.28, 136.93, 135.73, 131.96, 131.15, 130.04, 129.44, 127.80, 127.39, 126.84, 125.29, 122.91, 119.48, 118.69, 115.09, 114.49, 55.67, 19.98 (2C). MS, ESI−: m/z (%) 466.1 (M, 37); 465.1 (M − 1, 100). MS/MS, ESI−: m/z (%) 236.9 (7); 182.9 (34). Anal. calcd for C28H26N4OS: C, 72.07; H, 5.62; N, 12.01; S, 6.87%. Found: C, 72.21; H, 5.33; N, 11.77; S, 6.99%.
1-(2,4-Dinitrophenyl)-3-(4-methoxyphenyl)-5-(2-(phenylthio)phenyl)formazan (4h). Compound 4h was obtained from 1h (1.50 g, 4.70 mmol) and diazonium salt 2 (950 g, 4.70 mmol) as a brown solid. Yield: 1.33 g, 53%. Rf: 0.75 (n-hexane/ethylacetate, 5
:1). mp 207–208 °C. FTIR (KBr pellet, cm−1) νmax: 3112, 1367 (N–H w., C–N str.), 1512, 3058 (aromatic ring, CC str., C–H str.), 1591 (CN str. in Schiff base), 1498 (NN of formazans), 1251–1166 (C–O–C), 750 (C–S–C). 1H NMR (500 MHz, DMSO-d6) δ: 13.65 (s, 1H), 8.88 (s, 1H), 8.67 (s, 1H), 8.54 (d, J = 9.6 Hz, 1H), 8.36 (d, J = 12.2 Hz, 1H), 8.10 (d, J = 9.6 Hz, 1H), 8.04 (d, J = 7.4 Hz, 1H), 7.97 (d, J = 8.6 Hz, 1H), 7.78 (d, J = 8.6 Hz, 2H), 7.62 (dd, J = 5.8, 2.1 Hz, 1H), 7.56 (dd, J = 5.8, 2.1 Hz, 2H), 7.50 (d, J = 8.2 Hz, 1H), 7.11 (d, J = 8.8 Hz, 1H), 7.06 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 161.21, 151.02, 148.79, 144.36, 141.60, 138.96, 135.36, 133.54, 131.71, 130.78, 129.97, 129.90, 129.39, 127.96, 125.90, 124.42, 123.45, 122.13, 117.11, 114.54, 113.89, 55.42. MS, ESI+: m/z (%) 529.1 (M + 1, 12); 528.1 (M, 33); 527.0 (M − 1, 100). MS/MS, ESI+: m/z (%) 332.3 (42); 301.1 (49); 185.0 (10). Anal. calcd for C26H20N6O5S: C, 59.08; H, 3.81; N, 15.90; S, 6.07%. Found: C, 58.81; H, 3.65; N, 15.67; S, 5.88%.
5-(2-Benzoyl-5-chlorophenyl)-3-(4-methoxyphenyl)-1-phenylformazans (5a). Compound 5a was obtained from 1a (1.00 g, 4.40 mmol) and diazonium salt 3 (1.02 g, 4.40 mmol). Formazan 5a (1.13 g, 55%) was obtained as a claret red powder with a melting point of 161–162 °C. Rf: 0.64 (n-hexane/ethylacetate, 5
:1). FTIR (KBr pellet, cm−1) νmax: 3064, 1369 (N–H w., C–N str.), 1558, 3006 (aromatic ring, CC str., C–H str.), 1604 (CN str. in Schiff base), 1473 (NN of formazans), 1247–1168 (C–O–C), 1641 (CO). 1H NMR (500 MHz, CDCl3) δ: 15.20 (s, 1H), 8.34 (d, J = 9.4 Hz, 2H), 8.26 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 7.1 Hz, 2H), 7.63 (t, J = 7.1 Hz, 2H), 7.56 (m, 6H), 7.02 (d, J = 9.4 Hz, 2H), 3.94 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 196.51, 159.93, 152.74, 145.29, 143.37, 138.63, 134.01, 132.43, 132.28, 131.55, 129.68, 129.33, 128.46, 128.31, 125.54, 122.85, 122.24, 117.21, 113.84, 55.42. MS, ESI−: m/z (%) 469.1 (M + 1, 33); 468.1 (M, 39); 467.1 (M − 1, 100); MS/MS, ESI−: m/z (%) 312.0 (100); 278.0; (11) 227.9 (12). Anal. calcd for C27H21ClN4O2: C, 69.15; H, 4.51; N, 11.95%. Found: C, 68.90; H, 4.35; N, 11.69%.
5-(2-Benzoyl-5-chlorophenyl)-1,3-bis(4-methoxyphenyl)formazan (5b). Compound 5b was obtained from 1b (1.31 g, 5.16 mmol) and diazonium salt 3 (1.20 g, 5.16 mmol) as a dark blue solid. Yield: 1.60 g, 63%; Rf: 0.54; mp 178–180 °C. FTIR (KBr pellet, cm−1) νmax: 3058, 1371 (N–H w., C–N str.), 1579, 2999 (aromatic ring, C
C str., C–H str.), 1598 (CN str. in Schiff base), 1475 (NN of formazans), 1247–1145 (C–O–C), 1635 (CO). 1H NMR (500 MHz, DMSO-d6) δ: 14.82 (s, 1H), 8.29 (d, J = 9.0 Hz, 2H), 8.21 (d, J = 4.3 Hz, 1H), 7.99 (d, J = 8.9 Hz, 2H), 7.75 (d, J = 7.8 Hz, 2H), 7.69 (t, J = 7.8 Hz, 1H), 7.61 (t, J = 7.8 Hz, 2H), 7.54 (d, J = 4.3 Hz, 1H), 7.43 (d, J = 2.4 Hz, 1H), 7.21 (d, J = 9.0 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 3.84–3.90 (s, 6H). 13C NMR (126 MHz, CDCl3) δ: 196.70, 163.04, 159.91, 147.76, 145.34, 143.87, 138.94, 134.20, 132.69, 132.02, 129.56, 129.25, 128.60, 128.55, 128.40, 125.34, 124.36, 120.93, 116.97, 114.57, 113.76, 55.67, 50.38 (2C). MS, ESI+: m/z (%) 499.1 (M + 1, 54); 498.1 (M, 34); 497.0 (M − 1, 100). MS/MS, ESI+: m/z (%) 256.2 (9); 212.1 (100); 122.0 (31). Anal. calcd for C28H23ClN4O3: C, 67.40; H, 4.65; N, 11.23%. Found: C, 67.67; H, 4.37; N, 11.94%.
5-(2-Benzoyl-5-chlorophenyl)-1-(4-chlorophenyl)-3-(4-methoxyphenyl)formazan (5c). Compound 5c was obtained from 1c (800 mg, 3.07 mmol) and diazonium salt 3 (710 mg, 3.07 mmol) as a violet solid. Yield: 1.28 g, 83%; Rf: 0.82; mp 163–164 °C. FTIR (KBr pellet, cm−1) νmax: 3066, 1369 (N–H w., C–N str.), 1577, 3004 (aromatic ring, C
C str., C–H str.), 1604 (CN str. in Schiff base), 1477 (NN of formazans), 1247–1170 (C–O–C), 1635 (CO). 1H NMR (500 MHz, CDCl3) δ: 15.16 (s, 1H), 8.24 (d, J = 8.5 Hz, 2H), 8.19 (d, J = 8.7 Hz, 1H), 8.01 (d, J = 9.0 Hz, 2H), 7.73 (d, J = 9.0 Hz, 2H), 7.60 (t, J = 8.7 Hz, 1H), 7.52 (d, J = 7.5 Hz, 2H), 7.49 (m, 3H), 7.74 (s, 1H), 6.97 (d, J = 8.5 Hz, 2H), 3.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 196.45, 159.92, 151.13, 145.06, 143.23, 138.52, 137.35, 134.00, 132.43, 132.29, 130.65, 129.60, 129.51, 129.18, 128.44, 128.33, 128.16, 125.78, 124.24, 124.00, 122.16, 117.17, 113.80, 55.32. MS, ESI−: m/z (%) 503.0 (M + 1, 80); 502.0 (M, 35); 501.0 (M − 1, 100); MS/MS, ESI−: m/z (%) 339.9 (100); 312.0 (6); 227.9 (6). Anal. calcd for C27H20Cl2N4O2: C, 64.42; H, 4.00; N, 11.13%. Found: C, 64.68; H, 3.82; N, 11.09%.
5-(2-Benzoyl-5-chlorophenyl)-1-(4-bromophenyl)-3-(4-methoxyphenyl)formazan (5d). Compound 5d was obtained from 1d (1.50 g, 4.90 mmol) and diazonium salt 3 (1.14 g, 4.90 mmol). The product was purified by column chromatography using a solvent mixture of n-hexane/ethylacetate (5
:1) as eluent resulting in as a dark violet solid. Yield: 1.57 g, 58%; Rf: 0.71; mp 163–165 °C. FTIR (KBr pellet, cm−1) νmax: 3062, 1367 (N–H w., C–N str.), 1581, 3002 (aromatic ring, CC str., C–H str.), 1602 (CN str. in Schiff base), 1477 (NN of formazans), 1247–1170 (C–O–C), 1637 (CO). 1H NMR (500 MHz, CDCl3) δ: 15.19 (s, 1H), 8.23 (d, J = 5.0 Hz, 1H), 8.18 (d, J = 7.5 Hz, 2H), 8.03 (dd, J = 9.7, 5.0 Hz, 2H), 7.75 (d, J = 9.7 Hz, 2H), 7.66 (d, J = 10.0 Hz, 2H), 7.61 (t, J = 7.5 Hz, 1H), 7.51 (m, 4H), 6.98 (d, J = 7.5 Hz, 2H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 190.99, 154.45, 145.93, 139.61, 137.75, 133.01, 128.53, 127.04, 126.95, 126.88, 124.17, 123.71, 122.99, 122.67, 120.46, 120.37, 118.68, 116.87, 111.73, 108.34, 49.88. MS, ESI+: m/z (%) 547.0 (M + 1, 27); 546.0 (M, 100); 545.0 (M − 1, 23); MS/MS, ESI+: m/z (%) 334.0 (13); 242.9 (12); 214.9 (100); 180.0 (17). Anal. calcd for C27H20BrClN4O2: C, 59.20; H, 3.68; N, 10.23%. Found: C, 59.31; H, 3.61; N, 9.94%.
5-(2-Benzoyl-5-chlorophenyl)-1-(4-fluorophenyl)-3-(4-methoxyphenyl)formazan (5e). Compound 5e was obtained from 1e (1.25 g, 5.12 mmol) and diazonium salt 3 (1.18 g, 5.12 mmol). The product was purified by column chromatography using a solvent mixture of n-hexane/ethylacetate (5
:1) as eluent resulting in as a violet solid. Yield: 1.68 g, 68%; Rf: 0.68; mp 175–177 °C. FTIR (KBr pellet, cm−1) νmax: 3074, 1371 (N–H w., C–N str.), 1558, 2999 (aromatic ring, CC str., C–H str.), 1591 (CN str. in Schiff base), 1496–1477 (NN of formazans), 1249–1168 (C–O–C), 1627 (CO). 1H NMR (500 MHz, CDCl3) δ: 15.18 (s, 1H), 8.40 (d, J = 8.1 Hz, 2H), 8.25 (d, J = 8.3 Hz, 1H), 8.08 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H), 7.64 (t, J = 7.9 Hz, 1H), 7.56 (d, J = 8.3 Hz, 1H), 7.54 (d, J = 7.9 Hz, 2H), 7.28 (s, 1H), 7.24 (t, J = 7.9 Hz, 2H), 7.02 (d, J = 8.8 Hz, 2H), 3.92 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 196.67, 165.98, 163.96, 159.98, 149.69, 149.66, 145.10, 143.31, 138.70, 134.19, 132.64, 132.24, 129.61, 129.22, 128.47, 128.30, 125.25, 125.23, 125.16, 121.55, 117.13, 116.45116.27113.85, 55.39. MS, ESI+: m/z (%) 487.0 (M + 1, 28); 486.0 (M, 28); 485.0 (M − 1, 100); MS/MS, ESI+: m/z (%) 256.2 (42); 212.1 (100). Anal. calcd for C27H20ClFN4O2: C, 66.60; H, 4.14; N, 11.51%. Found: C, 66.66; H, 4.18; N, 11.30%.
5-(2-Benzoyl-5-chlorophenyl)-1-(4-nitrophenyl)-3-(4-methoxyphenyl)formazan (5f). Compound 5f was obtained from 1f (1.34 g, 4.90 mmol) and diazonium salt 3 (1.14 g, 4.90 mmol) as a darkish solid. Yield: 1.26 g, 50%; Rf: 0.75; mp 191–192 °C. FTIR (KBr pellet, cm−1) νmax: 3068, 1357 (N–H w, C–N str.), 1512, 3004 (aromatic ring, C
C str., C–H str.), 1593 (CN str. in Schiff base), 1498–1475 (NN of formazans), 1251–1168 (C–O–C), 1660 (CO). 1H NMR (500 MHz, CDCl3) δ: 14.38 (s, 1H), 8.29 (d, J = 9.1 Hz, 2H), 8.17 (d, J = 8.8 Hz, 1H), 8.01 (d, J = 8.8 Hz, 2H), 7.90 (d, J = 9.1 Hz, 2H), 7.88 (d, J = 7.2 Hz, 2H), 7.68 (t, J = 7.2 Hz, 1H), 7.64 (dd, J = 8.8, 2.26 Hz, 1H), 7.55 (m, 2H), 7.29 (s, 1H), 7.01 (d, J = 8.8 Hz, 2H) 3.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 196.12, 160.20, 151.51, 147.51, 144.75, 143.55, 137.05, 134.14, 133.89, 133.10, 132.52, 130.31, 130.11, 128.90, 128.70, 127.94, 125.43, 117.95, 117.57, 113.98, 55.40. MS, ESI−: m/z (%) 514.0 (M + 1, 36); 513.0 (M, 28); 512.1 (M − 1, 100); MS/MS, ESI−: m/z (%) 350.9 (100); 296.3 (2); 253.0 (2). Anal. calcd for C27H20ClN5O4: C, 63.10; H, 3.92; N, 13.63%. Found: C, 62.97; H, 3.91; N, 13.51%.
5-(2-Benzoyl-5-chlorophenyl)-1-(3,4-dimethylphenyl)-3-(4-methoxyphenyl)formazan (5g). Compound 5g was obtained from 1g (1.50 g, 5.90 mmol) and diazonium salt 3 (1.36 g, 5.90 mmol) as a dark red solid. Yield: 1.82 g, 62%; Rf: 0.73; mp 164–166 °C. FTIR (KBr pellet, cm−1) νmax: 3074, 1326 (N–H w., C–N str.), 1579, 3018 (aromatic ring, C
C str., C–H str.), 1606 (CN str. in Schiff base), 1481 (NN of formazans), 1245–1170 (C–O–C), 1637 (CO). 1H NMR (500 MHz, DMSO-d6) δ: 14.87 (s, 1H), 8.22 (d, J = 8.9 Hz, 2H), 7.99 (d, J = 8.0 Hz, 1H), 7.75 (s, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.61 (t, J = 7.4 Hz, 2H), 7.53 (t, J = 7.4 Hz, 1H), 7.48 (d, J = 2.3 Hz, 1H), 7.42 (d, J = 7.4 Hz, 2H), 7.41 (d, J = 7.9 Hz, 1H), 7.30 (d, J = 7.9 Hz, 1H), 3.85 (s, 3H), 2.35–2.34 (s, 6H). 13C NMR (126 MHz, CDCl3) δ: 196.42, 159.91, 151.54, 145.24, 143.83, 141.45, 138.72, 137.62, 134.02, 132.42, 132.19, 130.55, 129.69, 129.15, 128.50, 128.42, 124.79, 123.91, 121.53, 121.17, 117.03, 114.73, 113.78, 55.38, 19.99–20.05. MS, ESI+: m/z (%) 497.0 (M + 1, 38); 496.0 (M, 32); 495.1 (M − 1, 100); MS/MS, ESI+: m/z (%) 284.2 (2); 245.0 (10); 212.1 (100). Anal. calcd for C29H25ClN4O2: C, 70.08; H, 5.07; N, 11.27%. Found: C, 70.37; H, 4.87; N, 11.16%.
5-(2-Benzoyl-5-chlorophenyl)-1-(2,4-dinitrophenyl)-3-(4-methoxyphenyl)formazan (5h). Compound 5h was obtained from 1h (1.59 g, 5.80 mmol) and diazonium salt 3 (1.36 g, 5.8 mmol) as a brown solid. Yield: 1.45 g, 52%; Rf: 0.79; mp 228–230 °C. FTIR (KBr pellet, cm−1) νmax: 3271, 1332 (N–H w., C–N str.), 1604, 3060 (aromatic ring, C
C str., C–H str.), 1618 (CN str. in Schiff base), 1448 (NN of formazans), 1249–1132 (C–O–C), 1664 (CO). 1H NMR (500 MHz, DMSO-d6) δ: 11.59 (s, 1H), 8.85 (s, 1H), 8.63 (s, 1H), 8.35 (s, 1H), 8.06 (d, J = 10.0 Hz, 2H), 7.95 (s, 1H), 7.74 (d, J = 7.5 Hz, 2H), 7.50 (m, 5H), 7.04 (d, J = 10.0 Hz, 2H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 196.75, 162.29, 162.26, 159.90, 151.16, 145.13, 142.06, 137.43, 135.17, 134.10, 132.35, 132.20, 130.52, 129.65, 129.51, 129.18, 128.48, 128.33, 128.16, 125.60, 124.24, 124.00, 122.16, 117.23, 55.34. MS, ESI−: m/z (%) 558.0 (M, 25); 557.0 (M − 1, 100); MS/MS, ESI−: m/z (%) 465.1 (7); 315.1 (100); 181.9 (8). Anal. calcd for C27H19ClN6O6: C, 58.02; H, 3.43; N, 15.04%. Found: C, 58.32; H, 3.30; N, 14.76%.
Acknowledgements
We are very grateful to the Anadolu University Research Fund for providing financial support for this project (Projects No: 1004F94 and 1102F027). We are indebted to the Medicinal Plants and the Medicine Research Center of Anadolu University Eskisehir (Turkey) for the NMR measurements and to Anadolu University for the ChemBioDraw Ultra 2013 program.Notes and references
- G. I. Sigeikin, G. N. Lipunova and I. G. Pervova, Russ. Chem. Rev., 2006, 75, 885 CrossRef CAS.
- (a) A. W. Nineham, Chem Rev., 1955, 55, 355 CrossRef CAS; (b) D. E. Berry, R. G. Hicks and J. E. Gilroy, J. Chem. Educ., 2009, 86, 76 CrossRef CAS; (c) P. J. Das and J. Begum, RSC Adv., 2015, 5, 44604 RSC; (d) G. Mariappan, R. Korim, N. M. Joshi, F. Alam, R. Hazarika, D. Kumar and T. Uriah, J. Adv. Pharm. Technol. Res., 2010, 1, 396 CrossRef CAS PubMed; (e) Y. H. Al-Araji, J. K. Shneine and A. A. Ahmed, Int. J. Res. Pharm. Chem., 2015, 5, 41 Search PubMed; (f) A. Bamoniri, B. B. F. Mirjalili and N. Moshtad Arani, J. Mol. Catal. A: Chem., 2014, 393, 272 CrossRef CAS; (g) A. Bamoniri and N. Moshtael-Arani, Bull. Chem. Soc. Jpn., 2015, 88, 662 CrossRef CAS.
- (a) H. Y. Zhang, W. X. Sun, Q. A. Wu, H. Q. Zhang and Y. Y. Chen, Synth. React. Inorg. Met.-Org. Chem., 2000, 30, 571 CrossRef CAS; (b) A. V. Zaidman, I. G. Pervova, A. I. Vilms, G. P. Belov, R. R. Kayumov, P. A. Slepukhin and I. N. Lipunov, Inorg. Chim. Acta, 2011, 367, 29 CrossRef CAS; (c) H. Tezcan, Spectrochim. Acta, Part A, 2008, 69, 971 CrossRef PubMed.
- (a) L. S. Atabekyan, V. A. Barachevskii, S. A. Melkozerov, G. N. Lipunova, I. G. Pervova, I. N. Lipunov and G. I. Sigeikin, High Energy Chem., 2011, 45, 52 CrossRef CAS; (b) I. G. Pervova, V. A. Barachevskii, S. A. Melkozerov, G. N. Lipunova, G. I. Sigeikin and I. N. Lipunov, Photochemistry, 2010, 44, 20 CrossRef CAS.
- (a) A. R. Katritzky, S. A. Belyakov, D. Cheng and H. D. Durst, Synthesis, 1995, 5, 577 CrossRef; (b) V. Zsoldos-Mády, I. Pintér, P. Sándor, M. Peredy-Kajtár and A. Messmer, J. Carbohydr. Chem., 2001, 20, 747 CrossRef.
- G. M. Abou-Elenien, J. Electroanal. Chem., 1994, 375, 301 CrossRef CAS.
- V. K. Pandey and H. S. Negi, Indian Drugs, 1999, 36, 37 CAS.
- (a) A. B. Samel and N. R. Pai, J. Chem. Pharm. Res., 2010, 2, 60 CAS; (b) S. I. Marjadi, J. H. Solanki and A. L. Patel, Eur. J. Chem., 2009, 6, 844 CAS.
- T. Ram, R. Tyagi, B. Goel, K. K. Saxena, V. K. Srivastava and A. Kumar, Indian Drugs, 1998, 35, 216 CAS.
- K. G. Desai and K. R. Desai, J. Heterocycl. Chem., 2006, 43, 1083 CrossRef CAS.
- J. P. Raval, T. N. Akhaja, H. V. Patel, N. H. Patel and S. Patel, Int. J. Pharm. Res., 2009, 1, 62 Search PubMed.
- (a) P. Kumar, C. Nath and K. Shanker, Pharmazie, 1985, 40, 267 CAS; (b) K. Mazaahir, N. Negi and S. D. Gupta, Chem. Pharm. Bull., 1994, 42, 2363 CrossRef; (c) A. S. Shawali and N. A. Samy, J. Adv. Res., 2015, 6, 241 CrossRef CAS PubMed.
- (a) G. N. Lipunova, Z. G. Rezinskikh, T. I. Maslakova, P. A. Slepukhin, I. G. Pervova, I. N. Lipunov and G. I. Sigeikin, Russ. J. Coord. Chem., 2009, 35, 215 CrossRef CAS; (b) N. A. Frolova, S. Z. Vatsadze, A. I. Stasha, R. D. Rakhimov and N. V. Zyk, Chem. Heterocycl. Compd., 2006, 42, 1444 CrossRef CAS; (c) A. Iqbal, M. G. Moloney, H. L. Siddiqui and A. L. Thompson, Tetrahedron Lett., 2009, 50, 4523 CrossRef CAS; (d) I. G. Pervova, P. A. Slepukhin, A. V. Zaidman, G. N. Lipunova and I. N. Lipunov, Russ. J. Coord. Chem., 2010, 36, 213 CrossRef CAS; (e) M. C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., 2014, 126, 1 (Angew. Chem., Int. Ed., 2014, 53, 4118) CrossRef; (f) I. S. Maksakova, I. G. Pervova, G. P. Belov, I. I. Khasbiullin, N. A. Ivanova, E. N. Frolova and I. N. Lipunova, Pet. Chem., 2015, 55, 217 CrossRef CAS.
- (a) M.-C. Chang and E. Otten, Chem. Commun., 2014, 50, 7431 RSC; (b) S. M. Barbon, P. A. Reinkeluers, J. T. Price, V. N. Staroverov and J. B. Gilroy, Chem.–Eur. J., 2014, 20, 11340 CrossRef CAS PubMed; (c) M.-C. Chang, P. Roewen, R. Travieso-Puente, M. Lutz and E. Otten, Inorg. Chem., 2015, 54, 379 CrossRef CAS PubMed; (d) S. M. Barbon, J. T. Price, U. Yogarajah and J. B. Gilroy, RSC Adv., 2015, 5, 56316 RSC; (e) S. M. Barbon, V. N. Staroverov and J. B. Gilroy, J. Org. Chem., 2015, 80, 5226 CrossRef CAS PubMed; (f) M. Hesari, S. M. Barbon, V. N. Staroverov, Z. Ding and J. B. Gilroy, Chem. Commun., 2015, 51, 3766 RSC; (g) S. M. Barbon, J. T. Price, P. A. Reinkeluers and J. B. Gilroy, Inorg. Chem., 2014, 53, 10585 CrossRef CAS PubMed; (h) S. Novoa, J. A. Paquette, S. M. Barbon, R. R. Maar and J. B. Gilroy, J. Mater. Chem. C, 2016, 4, 3987 Search PubMed.
- H. Gorcay, G. Turkoglu, Y. Sahin and H. Berber, IEEE Sens. J., 2014, 14, 2529 CrossRef CAS.
- (a) D. M. Aziz, Anim. Reprod. Sci., 2006, 92, 1 CrossRef CAS PubMed; (b) S. H. Moussa, A. A. Tayel, A. A. Al-Hassan and A. Farouk, Hindawi Publishing Corporation Journal of Mycology, 2013, ID753692 Search PubMed , 1.
- H. Tavakol, Int. J. Quantum Chem., 2012, 112, 1215 CrossRef CAS.
- (a) R. A. King and B. Murrin, J. Phys. Chem. A, 2004, 108, 4961 CrossRef CAS; (b) H. Tezcan and N. Tokay, Spectrochim. Acta, Part A, 2010, 75, 54 CrossRef PubMed; (c) M. Toy, H. Tanak and H. Senoz, Dyes Pigm., 2015, 113, 510 CrossRef CAS.
- (a) T. Okamoto, C. Mitsui, M. Yamagishi, K. Nakahara, J. Soeda, Y. Hirose, K. Miwa, H. Sato, A. Yamano, T. Matsushita, T. Uemura and J. Takeya, Adv. Mater., 2013, 25, 6392 CrossRef CAS PubMed; (b) T. Mori, T. Nishimura, T. Yamamoto, I. Doi, E. Miyazaki, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 13900 CrossRef CAS PubMed.
- (a) G. Liu, J. R. Huth, E. T. Olejniczak, R. Mendoza, P. De-Vries, S. Leitza, E. B. Reilly, G. F. Okasinski, E. Nielsen, S. W. Fesik and T. W. Von Geldern, J. Med. Chem., 2001, 44, 1202 CrossRef CAS PubMed; (b) S. F. Nielsen, E. Nielsen, G. M. Olsen, T. Liljefors and D. Peters, J. Med. Chem., 2000, 43, 2217 CrossRef CAS PubMed; (c) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318 CrossRef CAS PubMed; (d) S. Pasquini, C. Mugnaini, C. Tintori, M. Botta, A. Trejos, R. K. Arvela, M. Larhed, M. Witvrouw, M. Michiels, F. Christ, Z. Debyser and F. Corelli, J. Med. Chem., 2008, 51, 5125 CrossRef CAS PubMed.
- M. Cueto, P. R. Jensen, C. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Nat. Prod., 2001, 64, 1444 CrossRef CAS.
- (a) A. Ghinet, A. Tourteau, B. Rigo, V. Stocker, M. Leman, A. Farce, J. Dubois and P. Gautret, Bioorg. Med. Chem., 2013, 21, 2932 CrossRef CAS PubMed; (b) J. R. Luque-Ortega, P. Reuther, L. Rivas and C. Dardonville, J. Med. Chem., 2010, 53, 1788 CrossRef CAS PubMed; (c) D. R. Guay, Pharmacotherapy, 1999, 19, 6 CrossRef CAS PubMed; (d) G. R. Pettit, B. Toki, D. L. Herald, P. Verdier-Pinard, M. R. Boyd, E. Hamel and R. K. Pettit, J. Med. Chem., 1998, 41, 1688 CrossRef CAS PubMed.
- (a) D. Tanaka, K. Nakagawa, M. Higuchi, S. Horike, Y. Kubota, T. C. Kobayashi, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 3914 CrossRef CAS PubMed; (b) H. Peng, J. Am. Chem. Soc., 2008, 130, 42 CrossRef CAS PubMed.
- (a) J. S. Cisar and B. F. Cravatt, J. Am. Chem. Soc., 2012, 134, 10385 CrossRef CAS PubMed; (b) Q. Qin, J. T. Mague and R. A. Pascal Jr, Org. Lett., 2010, 12, 928 CrossRef CAS PubMed; (c) H. Kim, S. M. So, C. P.-H. Yen, E. Vinhato, A. J. Lough, J.-L. Hong, H.-J. Kim and J. Chin, Angew. Chem., Int. Ed., 2008, 47, 8657 CrossRef CAS PubMed.
- (a) Y. Wei, I. Deb and N. Yoshikai, J. Am. Chem. Soc., 2012, 134, 9098 CrossRef CAS PubMed; (b) J. W. Seo, H. J. Kim, B. S. Lee, J. A. Katzenellenbogen and D. Y. Chi, J. Org. Chem., 2008, 73, 715 CrossRef CAS PubMed.
- G. Turkoglu, H. Berber and I. Kani, New J. Chem., 2015, 39, 2728 RSC.
- (a) V. M. Dziomko, V. M. Ostrovskaya and T. E. Zhukova, Khim. Geterotsikl. Soedin., 1979, 8, 1039 Search PubMed; (b) O. E. Sherif, Monatsh. Chem., 1997, 128, 981 CrossRef CAS.
- (a) G. Arnold and C. Schiele, Spectrochim. Acta, Part A, 1969, 25A, 685 CrossRef; (b) A. P. Zeif, G. N. Lipunova, N. P. Bednyagina, L. N. Shchegoleva and L. I. Chernyavskii, Zh. Org. Khim., 1970, 6, 2590 CAS.
- (a) A. A. Abbas and A. H. M. Elwaby, ARKIVOC, 2009,(x), 65 CAS; (b) Y. A. Ibrahim, A. A. Abbas and A. H. M. Elwaby, J. Heterocycl. Chem., 2004, 41, 135 CrossRef CAS.
- G. Mariappan, R. Korim, N. M. Joshi, F. Alam, R. Hazarika, D. Kumar and T. Uriah, J. Adv. Pharm. Technol. Res., 2010, 1, 396 CrossRef CAS PubMed.
- Y. A. Ibrahim, Tetrahedron, 1997, 53, 8507 CrossRef CAS.
- H. Tezcan and E. Uzluk, Dyes Pigm., 2007, 75, 633 CrossRef CAS.
- H. Tezcan and N. Ozkan, Dyes Pigm., 2003, 56, 159 CrossRef CAS.
- M. Homocianu, A. Airinei and D. O. Dorohoi, J. Adv. Res., 2011, 2, 1 CrossRef.
- M. El-Sedik, N. Almonasy, M. Nepras, F. Bures, M. Dvorák, M. Michl, J. Cermák and R. Hrdina, Dyes Pigm., 2012, 92, 1126 CrossRef CAS.
- M. Lacan, I. Tabakovic and Z. Cekovic, Tetrahedron, 1974, 30, 2911 CrossRef CAS.
- R. Y. Lai, K. Xiangxing, S. A. Jenekhe and A. J. Bard, J. Am. Chem. Soc., 2003, 125, 12631 CrossRef CAS PubMed.
- (a) H. Tezcan, E. Uzluk and M. L. Aksu, J. Electroanal. Chem., 2008, 619–620, 105 CrossRef CAS; (b) H. Tezcan and G. Ekmekci, Acta Chim. Slov., 2010, 57, 189 CAS.
- K. Umemoto, Bull. Chem. Soc. Jpn., 1989, 62, 3783 CrossRef CAS.
- (a) T. Oritani, N. Fukuhara, T. Okajima, F. Kitamura and T. Ohsaka, Inorg. Chim. Acta, 2004, 357, 436 CrossRef CAS; (b) H. Tezcan, H. Senoz and N. Tokay, Monatsh. Chem., 2012, 143, 579 CrossRef CAS.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455 CrossRef.
- S. R. Meech and D. Phillips, J. Photochem., 1983, 23, 193 CrossRef CAS.
- Chemoffice Pro for 13.0.2.3021 Microsoft Windows, Cambridge Scientific Computing: Inc., 875 Massachusetts Avenue. Suite 61. Cambridge MA 2139, USA Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian Inc, Wallingford CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, UV-vis spectra and HOMO and LUMO distributions of target compounds. See DOI: 10.1039/c6ra23008c |
| This journal is © The Royal Society of Chemistry 2016 |