
Improving the electrochemical performance of Li1.2Mn0.52Co0.13Ni0.13O2 by surface nitrogen doping via plasma treatment†
Bing Lia,
Chao Lib,
Zulai Caob,
Jing wangb and
Jinbao Zhao*ab
aCollege of Energy, School of Energy Research, Xiamen University, Xiamen, 361005, P. R. China. E-mail: jbzhao@xmu.edu.cn
bState Key Lab of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, P. R. China
First published on 22nd March 2016
Abstract
Nitrogen plasma processing technology is used to introduce nitrogen to the surface of Li1.2Mn0.52Co0.13Ni0.13O2. The material shows a better rate capability and cycle stability than pristine Li1.2Mn0.52Co0.13Ni0.13O2. Compared to the conventional treatment methods, it is a facile and promising way.
The development of the new energy storage field urgently needs high energy density batteries. For the whole battery system, cathode materials have a great impact on the energy density. The Li-rich oxide layer materials, which could be defined as xLi2MnO3·(1 − x)LiMO2 (M = Mn, Co, Ni, etc.), have been regarded as a promising cathode material to meet the demand for high energy lithium batteries due to their high specific capacity of more than 250 mA h g−1.1–4
It is generally considered that the Li2MnO3 component is able to provide additional capacity through the migration of Li+ ion5 and the release of oxygen6 at above 4.5 V. The active process above 4.5 V may also be considered as the release of Li2O.7 This phenomenon could cause a large irreversible capacity loss in the first charge/discharge cycle. The acid treatment has been proved to be an effective way to improve the columbic efficiency.8 However, the electrode material would show a poor cycling stability, mostly owing to the H+/Li+ (ref. 9) exchange during the process of acid treatment. Other methods are surface coated with Li-free insertion materials,10,11 and treated with (NH4)2SO4 solution,12 or Na2S2O8 solution.13 There are also some problems, such as H+/Li+ exchange, tedious steps, low controllability and a mediocre rate performance. It is noteworthy that the removal of a proper amount of Li+ ions from the bulk materials could increase the coulombic efficiency at the first cycle. The surface nitrogen-doping has been proved to effectively improve the electrochemical performance of Li-rich oxide layer material.14 Therefore, we thought that the Li-rich oxide layer cathode materials could be improved in electrochemical performance if they were treated with surface nitrogen-doping and simultaneously extracting an appropriate amount of Li+.
In this work, the nitrogen plasma processing technology is used to modify the Li-rich oxide layer material. It has the following advantages: (1) the degree of surface nitrogen-doping can be easily controlled; (2) a trace amount of Li+ is removed from the bulk material without H+/Li+ exchange, implying no damages to the electrode material; (3) the coulombic efficiency at the first charge/discharge cycle, capacity retention and rate performance have been improved; (4) compared with liquid nitrogen treatment14 or hydrothermal growth in NH3 atmosphere,15 the plasma processing treatment is much more controllable and safe to improve the property of electrode materials.
The Li-rich oxide layer cathode material was prepared through a liquid process reported in our previous report.16 The material obtained is Li1.2Mn0.52Co0.13Ni0.13O2, defined as the pristine material. The pristine material was put into a special porous ceramic crucible, the bottom of which was covered with a thin layer of pristine material. The crucible was placed in a vacuum quartz glass tube, which was evacuated to less than 1 Pa. Then, pure N2 (99.999%) was introduced into this tube at the flow rate of 50 ml min−1. Plasma components were carried out for 2 h. After treatment, the material collected was defined as P1. Plasma was generated by an RF power supply at 1k MHz and 400 W, and the plasma components were located at both ends of the tube.
Scanning electron microscopy (SEM) was carried out on Hitachi 4800 equipment. X-ray diffraction (XRD) patterns were collected through the Rigaku Ultima IV using Cu Kα radiation source operated at 40 kV and 15 mA. X-ray photoelectron spectroscopy (XPS) was measured using the Quantum 2000 (Physical electronics). X-ray fluorescence (XRF) was performed with a Bruker model S8 TIGER. The inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was carried out on the plasma 1000 (NCS Testing Technology, China).
All electrochemical measurements were conducted with the 2016 coin-type cells. The lithium metal sheet (the diameter of 16 mm) was used as the anode of the cells. The active materials mixed with acetylene black and polyvinylidene fluoride in N-methyl-2-pyrrolidone were coated on the Al foil, with the quality ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. The electrolyte solution was 1 M LiPF6 in 1:1:1 (volume ratio) ethylene carbonate, dimethyl carbonate and diethyl carbonate. All the cells were assembled in the argon-filled glove box (Mbraun, Germany). The average loading of active material was adjusted to 3 mg for each coin cell, and the diameter of cathode sheet is 12 mm.
:1:1. The electrolyte solution was 1 M LiPF6 in 1:1:1 (volume ratio) ethylene carbonate, dimethyl carbonate and diethyl carbonate. All the cells were assembled in the argon-filled glove box (Mbraun, Germany). The average loading of active material was adjusted to 3 mg for each coin cell, and the diameter of cathode sheet is 12 mm.
All cells were galvanostatically charged and discharged between 4.8 V and 2.0 V (vs. Li+/Li), using the Land CT2001A (Wuhan, China) battery testing system at room temperature. The rate performance was carried out from 25 mA g−1 (0.1C) to 1250 mA g−1 (5C). All capacities were calculated based on the active material. The electrochemical impedance spectroscopy (EIS) was carried out on an electrochemical workstation (Autolab PGSTA T302N) with frequency range of 0.01 Hz to 10 kHz.
The exhaust gas of the plasma treatment device was introduced into the buffer bottle, filled with the indicator solution. The 0.1% methyl red was used as the indicator. The colour change of indicator was observed, which confirms the generation of nitrogen oxides. Considering the vacuum pre-treatment, the oxygen is more likely from the material Li1.2Mn0.52Co0.13Ni0.13O2 itself. In order to further exclude the environmental interference, a control experiment (without Li1.2Mn0.52Co0.13Ni0.13O2 in the plasma treatment device) was done. The colour of indicator in the control experiment did not change. All the materials were subjected to XRF analysis, and the obtained data are shown in Table 1. After nitrogen plasma treatment, the existence of nitrogen in P1 sample is confirmed, and the mass percentage is about 1.32%.
| Mn/% | Co/% | Ni/% | N/% | |
|---|---|---|---|---|
| Pristine | 66.75 | 16.80 | 16.45 | 0 |
| P1 | 66.67 | 16.75 | 16.58 | 1.32 |
Fig. 1a shows the XRD patterns of pristine and P1 sample. There are no additional diffraction peaks in P1 sample, suggesting that the main structure of pristine material is not damaged during the nitrogen plasma treatment. All the peaks belong to a layer α-NaFeO2 structure, except a small group of diffraction peaks located between 20–25°. The small group of peaks has a relationship with the ordering of Li+, Mn4+ ions in the transition metal layers.17 The intensity ratio of I(003)/I(104) means the degree of cation mixing in the Li-layers,18 and the value of the ratio more than 1.2 is considered to be an acceptable cation mixing.19 The ratio of all samples is 1.49, denoting no structural rearrangement during nitrogen plasma treatment. The lattice parameters of samples analysed by the XRD data through PDXL2 software are listed in Table 2, revealing that the nitrogen plasma treatment does not obviously influence the crystal structure of the Li1.2Mn0.52Co0.13Ni0.13O2, probably owing to the low concentration of nitrogen. The c/a ratio is often regard as a parameter for characterizing hexagonal structure. When the ratio is larger than 4.9, the material can be assumed to possess layered characteristics.20 It can be seen that both the c/a ratio of pristine and P1 sample are larger than 4.9.
 | ||
| Fig. 1 (a) XRD patterns of the pristine and P1 sample; (b) SEM image of P1; (c) SEM image of the pristine sample. | ||
| a = b /Å | c/Å | c/a | |
|---|---|---|---|
| Pristine | 2.8594 | 14.2266 | 4.975 |
| P1 | 2.8616 | 14.2601 | 4.983 |
The SEM images (Fig. 1b and c) show that the micro-sized spherical particles are composed of nano-sized particles. After the nitrogen plasma treatment, the spherical particles are not cracked, but the surface becomes slightly rough. This phenomenon may be caused by the oxygen escaping from the crystal lattices.
Fig. 2 shows the XPS spectra of pristine and P1 samples. Firstly, the existence of nitrogen on the surface is affirmed in the P1 material. The position of Mn 2p3/2 peak in the pristine sample is 642.4 eV, confirming the Mn4+ oxidation states of the pristine material.21,22 However, the Mn 2p3/2 peak shifts slightly to a lower binding energy in the P1 material. The nitrogen-doping on the surface results in weakening the binding energy of Mn–O bonds.14 Due to the same reason, the Co 2p3/2 and Ni 2p3/2 peaks of the P1 sample shifts to a lower binding energy.
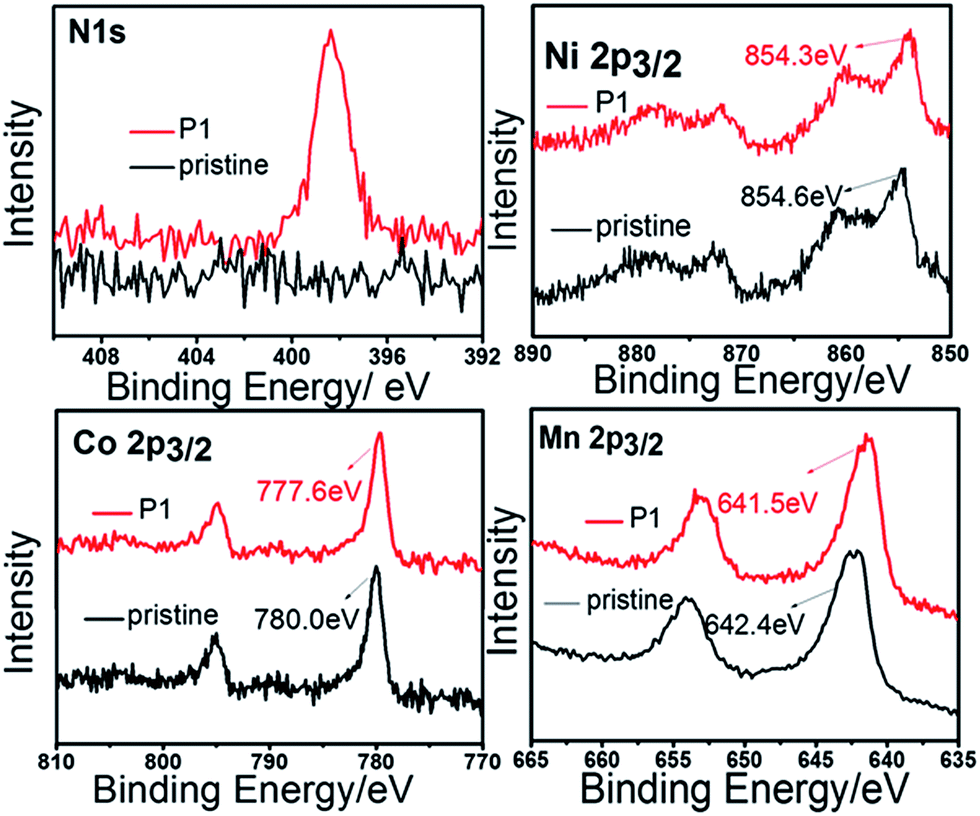 | ||
| Fig. 2 XPS spectra of pristine sample and P1. | ||
The first charge/discharge curves of P1 and the pristine sample under 0.1C rate are shown in Fig. 3a. Both of them could deliver the discharge capacity of 250 mA h g−1. However, the P1 sample has a lower charge capacity than the pristine materials. This may due to the decrease of oxygen, suppressing the release of Li2O, as the previous study.23 The first part below 4.5 V is corresponding to the oxidation of Ni2+ and Co3+, while the other part above 4.5 V is removal of Li2O from Li2MnO3 yielding active MnO2.24 The coulombic efficiency of the first charge/discharge circle is 80.5% for the P1 sample, which is higher than the pristine sample (73.9%). According to the ICP data, the mole ratio of Li/TM (transitional metal) decreases from 1.50 to 1.48, confirming the loss of lithium. It suggests that an appropriate physical extraction of lithium from the initial electrode material may reduce the irreversible capacity loss during the first charge/discharge circle, without reducing the discharge capacity.8,13,25 In order to further discuss the influence of nitrogen on the charge process, the dQ/dV curves are discussed (Fig. 3b). The intensity of oxidation peak at around 4.60 V in P1 is lower than the pristine material, which may be associated with the release of oxygen.7 It is noteworthy that, these two samples have the same discharge dQ/dV curves (Fig. 3b inset), indicating the discharge process is not affected.
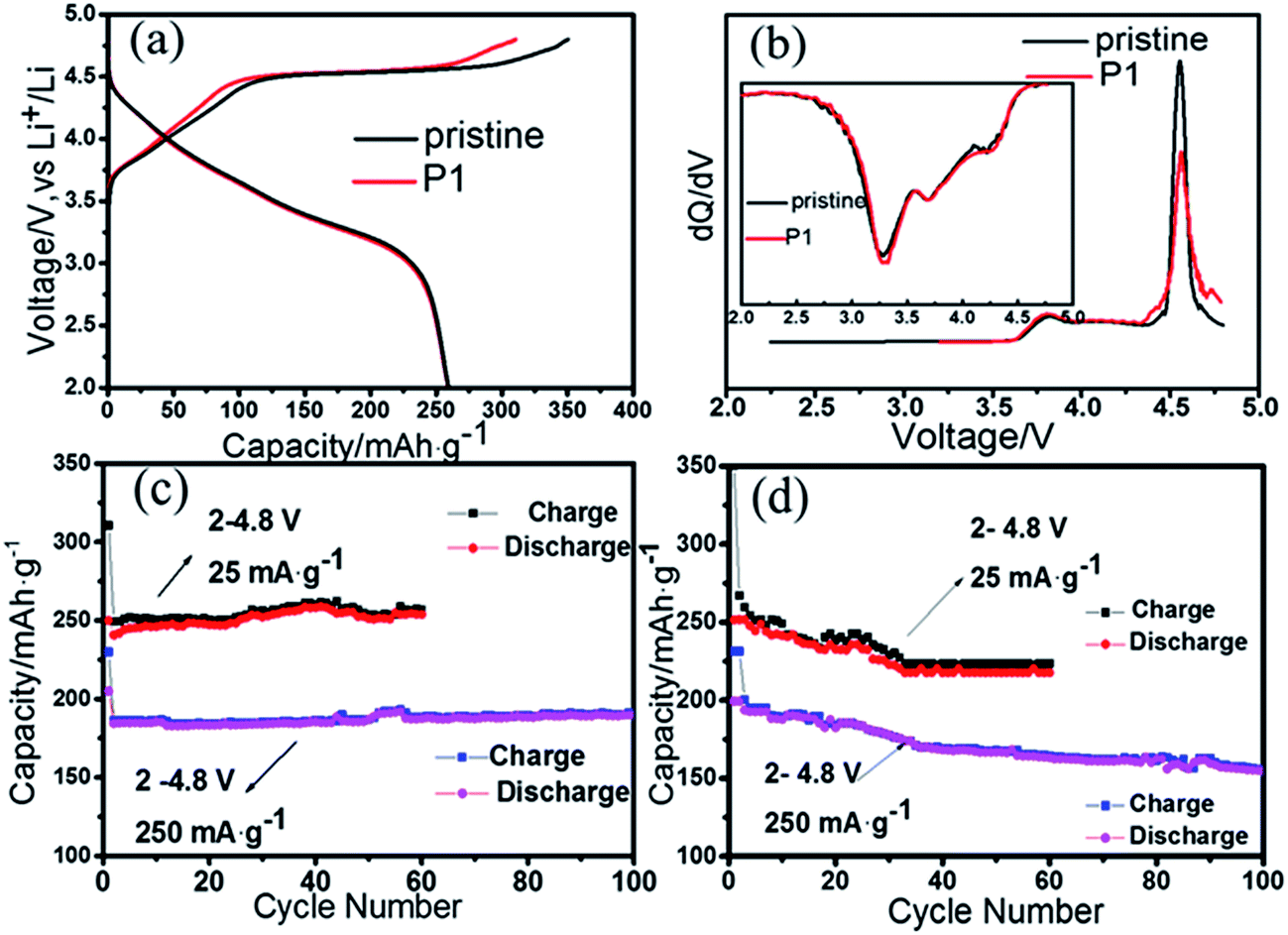 | ||
| Fig. 3 (a) The first charge/discharge curves of all samples; (b) the dQ/dV curves of pristine and P1 samples for the first cycle; (c) cycling performance at 25/250 mA g−1 of P1; (d) cycling performance at 25/250 mA g−1 of pristine sample. | ||
The cycling performance of the cells of the pristine material and P1 are compared in Fig. 3c and d. At a rate of 25 mA g−1 (0.1C), the discharge capacity of P1 is 256.7 mA h g−1 after 60 cycles, while the pristine delivers only 227.8 mA h g−1. The higher discharge capacity of the P1 electrode may benefit from its more stable surface structure of electrode material after nitrogen plasma treatment. At a higher rate, the discharge capacity of the pristine sample decreases to 169.0 mA h g−1 at the 100th cycle, while the P1 sample is 190.9 mA h g−1. Although the improvement is not significant, the data of improved electrochemical performance in this work is as good as others reported.14,26,27 Compared to common method of using liquid nitrogen treatment, the plasma processing method is very convenient.
Fig. 4a shows the rate performance of the cells of the pristine material and P1. The P1 sample can deliver the discharge capacity of 160.0 mA h g−1 at 5C rate. The improved high-rate capability of the P1 sample is probably due to a fast charge transfer kinetics coming from the surface nitridation. Fig. 4b shows the EIS spectra. According to the equivalent circuit model (Fig. 4b inset), the Rs means the resistance of the solution (corresponding to a high frequency), while the Rct represents the charge transfer associated with the Li+ pass through the electrode/electrolyte interface.28 For the P1 sample, the value of Rct is 40.10 Ω, which is far less than the pristine sample (89.90 Ω). The improved rate performance could be attributed to the decrease of Rct. Meanwhile, the diffusion coefficient of Li+ in pristine is 5.16 × 10−15 cm2 s−1, while that of the P1 is 1.49 × 10−14 cm2 s−1 (ESI†). Therefore, it is reasonable to believe that the improvement of the diffusion of Li+ is mainly due to the introduction of nitrogen to the surface of Li1.2Mn0.52Co0.13Ni0.13O2 material.
 | ||
| Fig. 4 (a) Rate performances of P1 and the pristine sample; (b) EIS spectra of P1 and the pristine sample. | ||
The plasma technology was used to improve the electrochemical performance of Li-rich oxide layer cathode material. The material after nitrogen plasma treatment, exhibits an increased coulomb efficiency in the first charge/discharge cycle and an enhanced rate performance. The improved electrochemical performance is attributed to surface nitrogen-doping. Compared to the conventional treatment methods, it is very convenient and could avoid the H+/Li+ exchange, which could damage the structure of materials. Considering usability and safety, it may be a promising way to improve the electrochemical performance of electrode materials for batteries.
Acknowledgements
The authors gratefully acknowledge the financial support from the Key Project of Science and Technology of Fujian Province (2013H6022), the National Natural Science Foundation of China (21321062) and Fostering Talents of Basic Science (J1310024). The authors also appreciate the positive suggestions of Professor Daiwei Liao.Notes and references
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- J. Wang, B. Qiu, H. L. Cao, Y. G. Xia and Z. P. Liu, J. Power Sources, 2012, 218, 128–133 CrossRef CAS.
- F. Amalraj, D. Kovacheva, M. Talianker, L. Zeiri, J. Grinblat, N. Leifer, G. Goobes, B. Markovsky and D. Aurbach, J. Electrochem. Soc., 2010, 157, A1121–A1130 CrossRef CAS.
- F. Li, S. X. Zhao, Y. C. Wang, K. Z. Wang, B. H. Li and C. W. Nan, J. Electrochem. Soc., 2013, 161, A102–A108 CrossRef.
- F. Dogan, B. R. Long, J. R. Croy, K. G. Gallagher, H. Iddir, J. T. Russell, M. Balasubramanian and B. Key, J. Am. Chem. Soc., 2015, 137, 2328–2335 CrossRef CAS PubMed.
- A. R. Armstrong, M. Holzapfe, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- S. Hy, F. Felix, J. Rick, W. N. Su and B. J. Hwang, J. Am. Chem. Soc., 2013, 136, 999–1007 CrossRef PubMed.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, J. Mater. Chem., 2008, 20, 6095–6106 CrossRef CAS.
- Y. Paik, C. P. Grey, C. S. Johnson, J. S. Kim and M. M. Thackeray, Chem. Mater., 2002, 14, 5109–5115 CrossRef CAS.
- F. Wu, N. Li, Y. F. Su, H. Q. Lu, L. J. Zhang, R. An, Z. Wang, L. Y. Bao and S. Chen, J. Mater. Chem., 2012, 22, 1489–1497 RSC.
- Z. Wang, E. Liu, C. He, C. Shi, J. Li and N. Zhao, J. Power Sources, 2013, 236, 25–32 CrossRef CAS.
- D. Y. W. Yu, K. Yanagida and H. Nakamura, J. Electrochem. Soc., 2010, 157, A1177–A1182 CrossRef CAS.
- J. Zheng, S. N. Deng, Z. C. Shi, H. J. Xu, H. Xu, Y. F. Deng, Z. C. Zhang and G. H. Chen, J. Power Sources, 2013, 221, 108–113 CrossRef CAS.
- H. Z. Zhang, Q. Q. Qiao, G. R. Li, S. H. Ye and X. P. Gao, J. Mater. Chem., 2012, 22, 13104–13110 RSC.
- J. Dai, M. Wang, M. Song, P. C. Li, C. Y. Zhang, A. J. Xie and Y. H. Shen, Scr. Mater., 2016, 112, 67–70 CrossRef CAS.
- B. Li, Y. Y. Yu and J. B. Zhao, J. Power Sources, 2015, 275, 64–72 CrossRef CAS.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- L. Liu, N. Q. Zhang, K. N. Sun and T. Y. Yang, J. Phys. Chem. Solids, 2009, 70, 727–731 CrossRef CAS.
- T. Ohzuku, A. Ueda, M. Nagayama, Y. Iwakoshi and H. Komori, Electrochim. Acta, 1993, 38, 1159–1167 CrossRef CAS.
- S. H. Lee, J. S. Moon, M. S. Lee, T. H. Yu, H. Kim and B. M. Park, J. Power Sources, 2015, 281, 77–84 CrossRef CAS.
- Y. H. Wang, X. F. Bie, K. Nikolowski, H. Ehrenberg, F. Du, M. Hinterstein, C. Z. Wang, G. Chen and Y. G. Wei, J. Phys. Chem. C, 2013, 117, 3279–3286 CAS.
- C. Fu, G. Li, D. Luo, Q. Li, J. Fan and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 15822–15831 CAS.
- Y. Wu and A. Manthiram, Electrochem. Solid-State Lett., 2007, 10, A151–A154 CrossRef CAS.
- S. H. Kang, P. Kempgens, S. Greenbaum, A. J. Kropf, K. Amine and M. M. Thackeray, J. Mater. Chem., 2007, 17, 2069–2077 RSC.
- X. Y. Liu, T. Huang and A. S. Yu, Electrochim. Acta, 2015, 163, 82–92 CrossRef CAS.
- G.-Y. Kim, S. B. Yi, Y. J. Park and H. G. Kim, Mater. Res. Bull., 2008, 43, 3543–3552 CrossRef CAS.
- Y. Chen, G. Xu, J. Li, Y. Zhang, Z. Chen and F. Kang, Electrochim. Acta, 2013, 87, 686–692 CrossRef CAS.
- S. J. Shi, J. P. Tu, Y. Y. Tang, X. Y. Liu, X. Y. Zhao, X. L. Wang and C. D. Gu, J. Power Sources, 2013, 241, 186–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation of the Li+ diffusion coefficient. See DOI: 10.1039/c6ra04922b |
| This journal is © The Royal Society of Chemistry 2016 |