
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic hydrotreatment of Alcell lignin fractions using a Ru/C catalyst†
Arjan
Kloekhorst
and
Hero Jan
Heeres
*
University of Groningen, Chemical Engineering, ENTEG, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: H.J.Heeres@rug.nl; Tel: +31 503634174
First published on 17th June 2016
Abstract
We here report the catalytic hydrotreatment of three different Alcell lignin fractions using a Ru/C catalyst in a batch reactor set-up (400 °C, 4 h, 100 bar H2 intake, 5 wt% catalyst on lignin). The fractions, obtained by a solvent fractionation scheme from Alcell lignin, differ in composition and molecular weight. The resulting product oils were characterized by various techniques, such as GC-MS-FID, GC × GC-FID, GPC, and 13C-NMR, to gain insight into the relationship between the feed and product yield/composition on a molecular level. The lowest molecular weight fraction (Mw = 660 g mol−1) gave the highest product oil yield after catalytic hydrotreatment (>70 wt% on lignin fraction). The main differences in molecular composition for the product oils were observed and are related to the chemical structure of the different feed fractions and less on the molecular weight. The highest amounts of valuable alkylphenolics (8.4 wt% on intake) and aromatic compounds (4.2 wt% on intake) in the product oils were obtained with the lowest molecular weight fraction. This fraction also contained the highest amounts of aliphatic hydrocarbons after the hydrotreatment reaction (14.0 wt% on intake), which were primarily linked to the presence of extractives in the Alcell lignin feed, that accumulate in this low molecular weight fraction during solvent fractionation.
1 Introduction
Research and development activities on the conversion of lignocellulosic biomass for renewable energy generation, transportation fuels and biobased chemicals have intensified during the past decade.1–4 Lignocellulosic biomass consists mainly of carbohydrates and lignin, with variable amounts of proteins, triglycerides, minerals and other minor components.5 A large number of technologies have been developed for cellulose and hemi-cellulose valorisation. Examples are aerobic and anaerobic fermentation (e.g. bioethanol, lactic acid, succinic acid) and chemo-catalytic processes (e.g. sorbitol, xylitol).6Lignin is the third most abundant bio-polymer in lignocellulosic biomass. It is an amorphous polyphenolic thermoset with different types of linkages between the aromatic nuclei (see Fig. 1 for details). The main linkages (about 50–60% depending on wood type) in lignin are C–O–C ether bonds (β-O-4 and a small amount of α-O-4). The remaining linkages are primarily C–C double bonds (5-5 and β-5). The high amounts of aromatic fragments in lignin make it an excellent feedstock for the production of aromatic biobased chemicals, such as benzene, toluene, xylene (BTX) and alkylphenolics. This is widely recognized now, and the topic has attracted considerable interest from academia and industry.7–13 A number of technologies have been proposed with different levels of process severity, including (catalytic) solvolysis, hydrothermal liquefaction, catalytic hydrotreatment, pyrolysis, and catalytic cracking (Fig. 2).
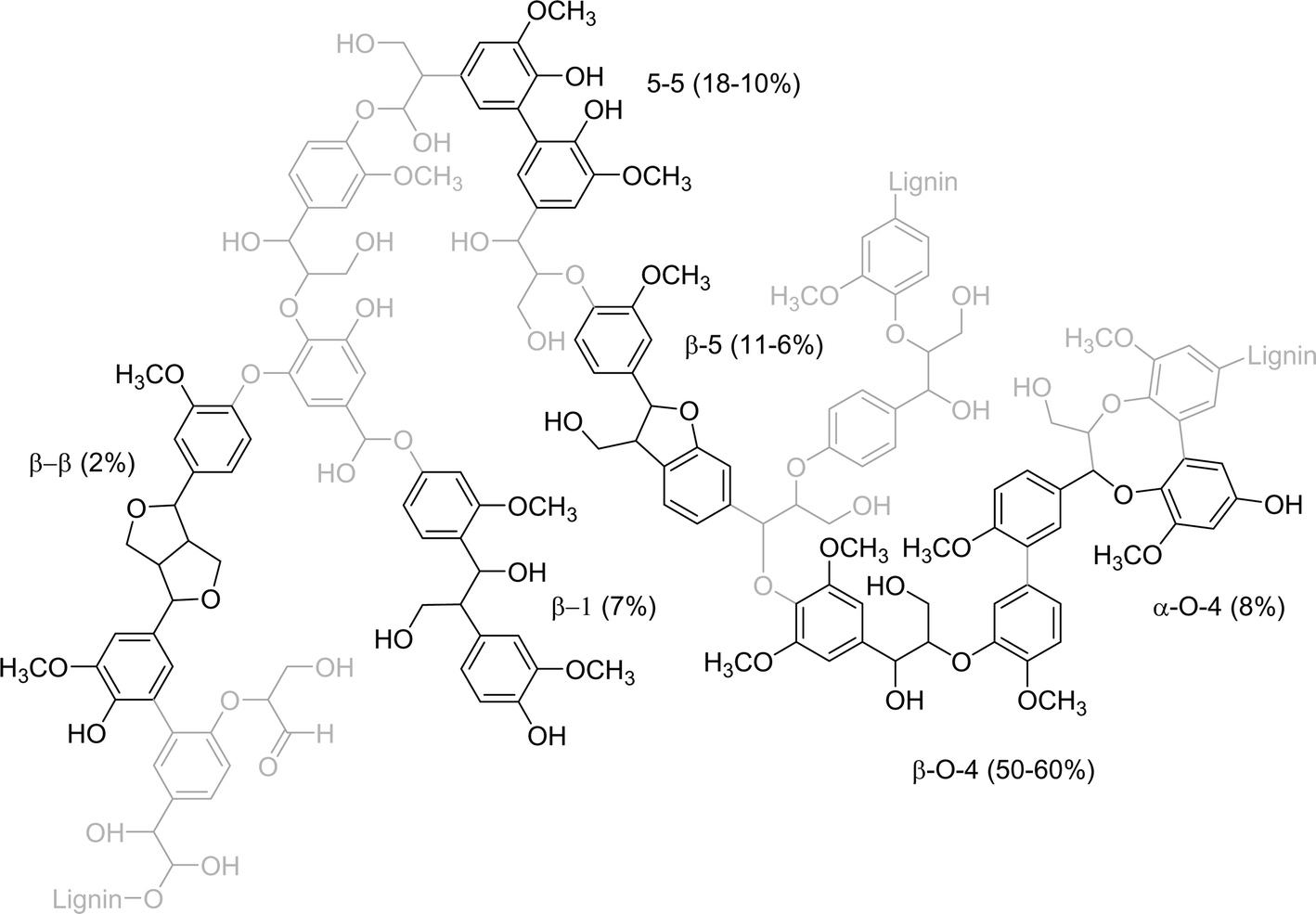 | ||
| Fig. 1 Structural model of spruce lignin (softwood) based on the works of Adler et al.14 and Karhunen et al.,15 with the amounts of different linkages.16 | ||
 | ||
| Fig. 2 Various thermochemical pathways and process parameters (P, T) for the depolymerisation of lignin, adapted from Joffres et al.17 Reproduced with permission. | ||
Lignin valorisation to low molecular weight aromatics and phenolics requires depolymerisation of the lignin structure. However, this is not sufficient as the aromatic nuclei in lignin are heavily substituted, e.g. with at least a 3-carbon side chain with multiple substituents and additional methoxy and hydroxyl groups. For aromatics synthesis, full deoxygenation is required, whereas methoxy group removal is essential for alkylphenolics.
Catalytic hydrotreatment is considered a very promising upgrading technology for lignin and combines depolymerisation and deoxygenation in a single process. It involves contacting the lignin source with hydrogen and a heterogeneous catalyst under elevated conditions.18–24 Typically, harsh conditions are employed, viz. temperatures between 170–450 °C and pressures between 34 and 360 bar. The product oil yields range from 13 to 100%, depending on the catalyst and reaction conditions. The exact chemical composition of the oil is often not given, and the process conditions differ considerably; this hampers the identification of the best catalyst systems to obtain high aromatics and alkylphenolics yields, the most valuable components in the mixture (800–1200 euros per ton). In addition, various lignin sources are used which makes the development of lignin structure–lignin oil composition relations cumbersome.
We here report a systematic study on the catalytic hydrotreatment of Alcell lignin and Alcell lignin fractions, obtained by solvent fractionation, using Ru/C as the catalyst in the absence of an external solvent. As such, the molten lignin, later diluted with products, acts as the solvent. This simplifies the characterisation of products considerably as incorporation of solvent or solvent fragments in the product portfolio (as often observed) can be avoided. The lignin fractions and the resulting product oils were characterized by different techniques, such as GC-MS-FID, GC × GC-FID, GPC, and 13C-NMR, to gain insight into the relationship between the lignin feed and the hydrotreated product oil compositions. A global reaction network is proposed to explain the experimental data.
2 Materials and methods
2.1 Chemicals
Ruthenium on carbon (5 wt% Ru) in the form of a powder was obtained from Sigma Aldrich. Hydrogen (>99.99%) and nitrogen (>99.8%) gases were obtained from Hoekloos. Tetrahydrofuran (THF), methanol, diethylether, and di-n-butylether (DBE) were obtained from Sigma Aldrich (99.99%) and used as such. Alcell® lignin, produced by Repap, Canada from mixed hardwood, was kindly supplied by the Wageningen University and Research Center (WUR), the Netherlands. The sugar content is less than 0.3 wt% on dry basis (see Gosselink for details).252.2 Fractionation of the Alcell lignin
The fractionation of Alcell lignin was performed according to a published procedure by Thring et al.,26 and a representative scheme is shown in Fig. 3. In the first step, the Alcell lignin was continuously extracted in a standard Soxhlet set-up for 72 h with diethylether as the solvent. The diethylether soluble fraction was collected, and the solvent was removed under vacuum (40 mbar, 40 °C) to give a brown solid in 20 wt% yield. This first fraction is abbreviated as F1. The remaining insoluble lignin fraction was dried under vacuum (40 mbar, 50 °C). It was subsequently suspended in methanol (500 ml) and agitated in an ultrasonic bath for 20 minutes and left to settle for 10 min. After settling, the suspension was filtered, and the remaining solid was re-suspended in methanol, agitated and allowed to settle until the methanol phase was essentially colourless and transparent. The combined methanol soluble fractions were dried under vacuum (40 mbar, 50 °C) for 24 h giving 55 wt% of a brown solid. This second fraction is abbreviated as F2. The remaining solids after the methanol extraction were dried under vacuum (40 mbar, 50 °C) for 24 h and isolated as a brown solid in 25 wt% yield. This last fraction is abbreviated as F3. All fractions were milled into a fine powder and stored in a freezer (−17 °C). | ||
| Fig. 3 Fractionation scheme for Alcell lignin using solvent extraction. | ||
2.3 Catalytic hydrotreatment reactions
The catalytic hydrotreatment experiments were performed in a 100 ml batch autoclave (Buchi). The autoclave has a maximum operating temperature of 400 °C and a maximum operating pressure of 350 bar. The reactor is surrounded by a metal block containing electrical heating elements and channels allowing the flow of cooling water. The reactor content was stirred mechanically using a Rushton type turbine with a gas induced impeller. Temperature and pressure were monitored online and logged on a PC.The reactor was filled with 15 g of Alcell lignin or an Alcell lignin fraction (F1–F3) and a catalyst (0.75 g of Ru/C, 5 wt% on lignin) and subsequently flushed with hydrogen and pressurized to 120 bar at room temperature for leak testing. Subsequently, the pressure was reduced to 100 bar. Stirring was started (1200 rpm), and the reactor content was heated to the intended reaction temperature with a rate of about 8 °C min−1. The reaction time was set at t = 0 h when the pre-determined temperature (400 °C) was reached. The reactions were performed in batch mode regarding both the gas phase and the liquid phase, and as such, consumed hydrogen was not replenished. The pressure and temperature were recorded during the reactions. After the pre-determined reaction time (4 h), the reactor was cooled to room temperature with a rate of about 40 °C min−1. The pressure at room temperature was recorded for determination of the amount of gas phase components formed during the reaction. Subsequently, the pressure was released to atmospheric pressure, and the gas phase was collected in a 3 L Tedlar gas bag for determination of the composition (GC). The liquid product was collected with a syringe and weighed. The liquid phase after the reaction consists of two layers, namely, an organic layer and an aqueous layer. The layers were separated by decanting. The water content of both phases was analysed by Karl Fischer titration. The reactor was rinsed several times with acetone, and the combined acetone fractions were collected. After evaporation of the acetone overnight at room temperature, the weight of the remaining liquid phase was determined and added to the organic product phase. The H2 uptake for each experiment was calculated based on pressure and temperature recordings and gas phase compositions before and after the reaction and is expressed as NL per kg lignin. The details of the calculations are given by Ardiyanti.27
2.4 Analytical protocols
GC-MS-FID analyses were performed using a Hewlett Packard 5890 series II plus with a Quadrupole Hewlett Packard 5972 MSD and a FID. The GC is equipped with a Restek RTX-1701 capillary column (60 × 0.25 mm i.d. and 0.25 μm film thickness), and the product exiting the column is split in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and directed to the MSD and FID. The injector temperature was set at 250 °C. The oven temperature was kept at 40 °C for 4 minutes and then heated up to 250 °C at a rate of 4 °C min−1. Before analyses, the organic samples were diluted with tetrahydrofuran (THF) and 1000 ppm di-n-butyl ether (DBE) was added as an internal standard.
:1 ratio and directed to the MSD and FID. The injector temperature was set at 250 °C. The oven temperature was kept at 40 °C for 4 minutes and then heated up to 250 °C at a rate of 4 °C min−1. Before analyses, the organic samples were diluted with tetrahydrofuran (THF) and 1000 ppm di-n-butyl ether (DBE) was added as an internal standard.
GC × GC-FID analysis was performed on organic samples using a trace GC × GC from Interscience equipped with a cryogenic trap system and two columns: an RTX-1701 capillary column (30 m × 0.25 mm i.d. and 0.25 μm film thickness) connected by a meltfit to an Rxi-5Sil MS column (120 cm × 0.15 mm i.d. and 0.15 μm film thickness). An FID was used. A dual jet modulator was applied using carbon dioxide to trap the samples. Helium was used as the carrier gas (continuous flow 0.6 ml min−1). The injector temperature and FID temperature were set at 250 °C. The oven temperature was kept at 40 °C for 5 minutes and then heated up to 250 °C at a rate of 3 °C min−1. The pressure was set at 70 kPa at 40 °C. The modulation time was 6 seconds. Before analyses, the organic samples were diluted with tetrahydrofuran (THF) and 1000 ppm di-n-butyl ether (DBE) was added as an internal standard.
For GC × GC compound identification, a standard lignin oil sample (Alcell lignin, 5 wt% of Ru/TiO2 on lignin intake, 4 h, with 100 bar of initial H2 pressure) was analyzed using a GC × GC-HRTOFMS from JEOL equipped with a cryogenic trap system and two columns: an RTX-1701 capillary column (30 m × 0.25 mm i.d. and 0.25 μm film thickness) connected by a meltfit to an Rxi-5Sil MS column (200 cm × 0.15 mm i.d. and 0.15 μm film thickness). The products were analysed using a TOFMS JMS-T100GCV 4G detector with a scanning speed of 50 Hz and a mass range of 35–600 m/z. Helium was used as the carrier gas (0.8 ml min−1). The injector temperature and TOFMS temperature were set at 250 °C. The oven temperature was kept at 40 °C for 5 minutes and then heated up to 250 °C at a rate of 3 °C min−1. The pressure was set at 77.5 kPa at 40 °C. The modulation time was 6 seconds. The MS spectra plots were analyzed using GC Image® software.
GPC analyses of the lignin oils and feeds (Alcell lignin and fractions F1–F3) were performed using an HP1100 from Hewlett Packard equipped with three MIXED-E columns (300 × 7.5 mm, PLgel 3 μm) in series and a GBC LC 1240 RI detector. The average molecular weights were determined using the PSS WinGPC Unity software from Polymer Standards Service. The following operating conditions were used: THF as an eluent at a flow rate of 1 ml min−1, a pressure of 140 bar, a column temperature of 42 °C, and an injection volume of 20 μl with sample concentrations of 10 mg ml−1. Toluene was used as a flow marker, and polystyrene samples with different molecular weights were used as the calibration standard.
The elemental composition (C, H and N) of the lignin oils and feeds (Alcell lignin, fractions F1–F3) was determined using a Euro Vector 3400 CHN-S elemental analyzer. The oxygen content was determined by difference. All experiments were carried out in duplicate.
The water content of the organic samples was determined by Karl Fischer titration using a Metrohm Titrino 758 titration device. A small amount of sample (ca. 0.03–0.05 g) was added into an isolated glass chamber containing Hydranal® (Karl Fischer Solvent, Riedel de Haen). The titrations were carried out using the Karl Fischer titrant Composite 5K (Riedel de Haen). All measurements were performed in duplicate.
The 13C-NMR spectra were recorded on a Varian 500 at 125 MHz using a 90° pulse and an inverse-gated decoupling sequence with a relaxation time of 4 seconds. The number of scans was at least 10000. The samples were dissolved in DMSO-d6 and measured at 60 °C.
The gradient 1H,13C-HSQC spectra were acquired using a Varian 400 spectrometer equipped with a One NMR Probe. 100 mg of sample was dissolved in 0.6 ml of DMSO-d6, and the spectra were recorded at RT. The conditions of the HSQC were as follows: 2048 data points were acquired over a 16 ppm (14 to −2 ppm) spectral width with an acquisition time of 320 ms with a 1.8 s relaxation delay for the 1H dimension. 64 time increments with a spectral width of 40 ppm (50–90 ppm) were taken for the 13C dimension. The spectra were processed using MestReNova software. The amounts of linkages were determined by integration of the correlation peaks of relevant linkages (A (β-O-4), B (β-β), C (β-5), D (β-1, α-O-4)) and dividing them by the area of the total aromatic region (S, G).28
Thermogravimetric analysis (TGA) data of the original Alcell lignin and the fractionated Alcell lignins were determined using a Mettler-Toledo analyser (TGA/SDTA851e). About 5–7 mg of sample was placed into fused α-Al2O3 crucibles. The samples were heated under an argon atmosphere with a heating rate of 10 °C min−1 in a temperature range between 30–900 °C.
2.5 Gas phase analysis
The gas phases were collected after reaction and stored in a gas bag (SKC Tedlar 3 litre sample bag (9.5′′ × 10′′)) with a polypropylene septum fitting. GC-TCD analyses were performed using a Hewlett Packard 5890 Series II GC equipped with a Porablot Q Al2O3/Na2SO4 column and a molecular sieve (5A) column. The injector temperature was set at 150 °C and the detector temperature at 90 °C. The oven temperature was kept at 40 °C for 2 minutes and then heated up to 90 °C at 20 °C min−1 and kept at this temperature for 2 minutes. The columns were flushed for 30 seconds with the gas sample before starting the measurement. A reference gas was used to quantify the results (55.19% H2, 19.70% CH4, 3.00% CO, 18.10% CO2, 0.51% ethylene, 1.49% ethane, 0.51% propylene and 1.50% propane).3 Results and discussion
3.1 Characterization of the Alcell lignin fractions
The Alcell lignin feed was divided into three fractions by using a solvent extraction protocol developed by Thring et al.26 (Fig. 3). The yield of the individual fractions (Table 1) was close to those reported by Thring. The elemental composition and molecular weight distributions were determined and compared to the original Alcell lignin (see Table 1 for details). The F2 and F3 fractions have a slightly lower C content than the Alcell lignin feed. However, the F1 fraction has a considerably higher C content, which, as will be shown later, is not the result of a major difference in lignin structure but due to the presence of significant amounts of extractives such as long chain fatty acids/esters.| Alcell lignin | F1 | F2 | F3 | |
|---|---|---|---|---|
| a Isolated yield. b Determined by GPC using polystyrene standards. | ||||
| Yield (wt%)a | 20 | 55 | 25 | |
| M w (g mol−1)b | 1720 | 660 | 1640 | 3680 |
| Elemental composition, dry base (wt%) | ||||
| Carbon | 65.2 | 67.1 | 64.5 | 64.6 |
| Hydrogen | 5.8 | 7.1 | 5.7 | 5.4 |
| Oxygen | 29.0 | 25.8 | 29.7 | 29.7 |
| H/C ratio | 1.07 | 1.26 | 1.06 | 1.00 |
| O/C ratio | 0.33 | 0.29 | 0.35 | 0.34 |
Thermogravimetric analyses of the original Alcell lignin and the fractions thereof were performed under Ar to determine the thermal properties of the different fractions (Fig. 4). As is evident from the figure, particularly the TGA of F1 differs considerably from the others. The initial mass loss for F1 occurs at a lower temperature and also the amount of residue is by far lower than those for the other fractions. This is likely due to the fact that F1 is of lower molecular weight and less condensed, although the presence of residual fatty acids/esters may also play a role (vide infra).
 | ||
| Fig. 4 Thermogravimetric analyses of the original Alcell lignin and the fractions derived thereof (F1–F3). | ||
The molecular weight distributions were determined by GPC, and the results are given in Table 1 and Fig. 5. The ether soluble fraction (F1) has a considerably lower molecular weight than the Alcell lignin feed. The methanol soluble fraction (F2) has an almost similar molecular weight (1640 g mol−1) to that of the starting Alcell lignin (1720 g mol−1), whereas the residue (F3) shows a much higher molecular weight of 3680 g mol−1. Thus, the solvent extraction procedure is very suitable to obtain fractions with a range of molecular weight distributions.
 | ||
| Fig. 5 Molecular weight distributions for Alcell lignin and the fractions derived thereof (F1–F3). | ||
The molecular compositions of the Alcell lignin and the fractions derived thereof were investigated by 13C-NMR (DMSO-d6, 60 °C), and the results are shown in Fig. 6 and Table 2. Clear differences are observed for fraction F1 and the two other fractions. F1 contains significant amounts of aliphatic carbons between δ = 10–35 ppm, whereas the peak intensity in this region is considerably lower for F2 and F3. In addition, peaks are present in the δ = 170–210 ppm region arising from the presence of carboxylic acid and ester groups. Again, these are essentially absent in fractions F2 and F3. It is highly likely that the Alcell lignin used in this study contains significant amounts of extractives in the form of long chain fatty acids and esters, likely due to incomplete separation in the isolation procedure of the Alcell lignin from the lignocellulosic biomass, which is in line with the observations reported by Thring.26 These extractives accumulate in the diethylether fraction F1 and apparently have a reasonable solubility in diethylether.
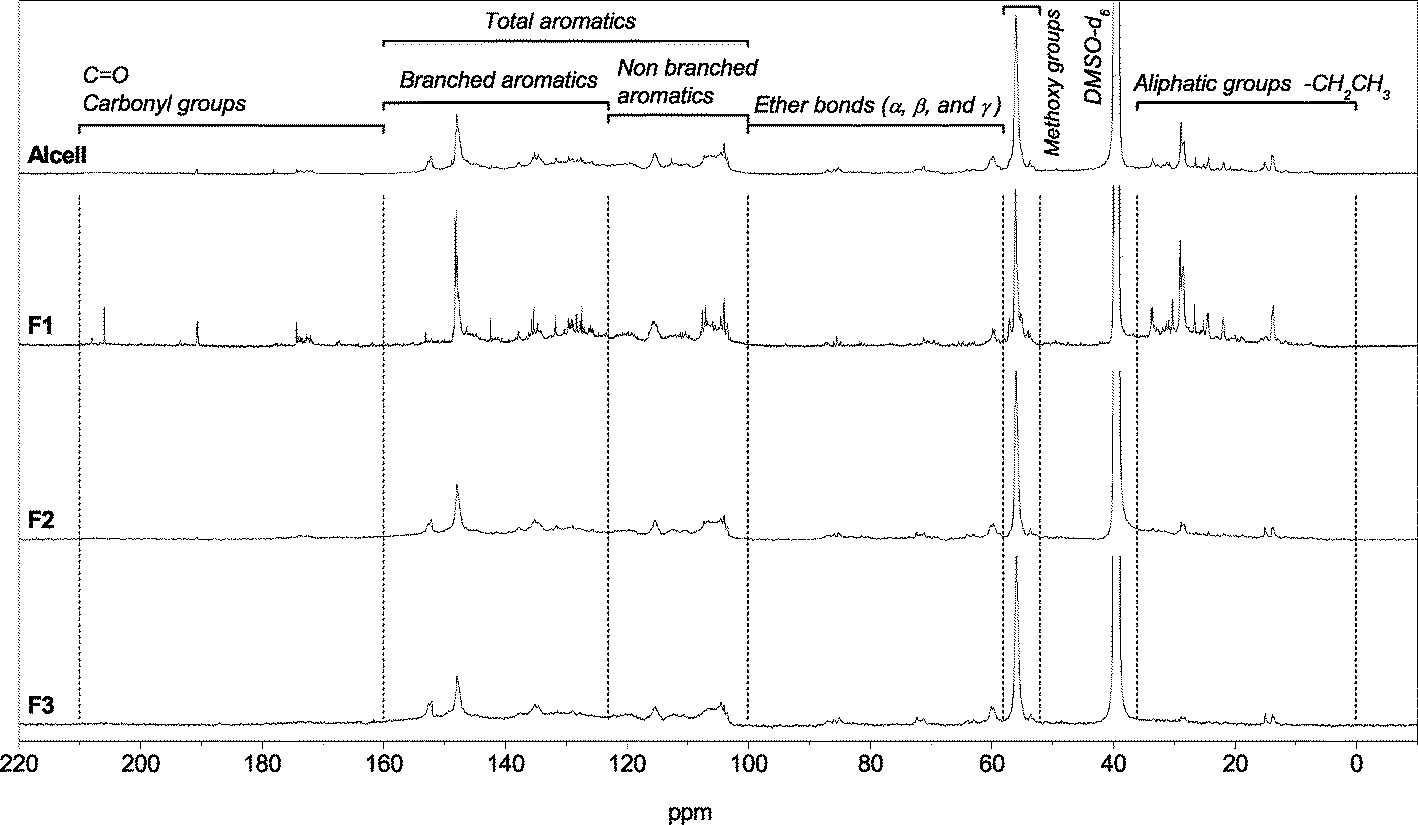 | ||
| Fig. 6 13C-NMR spectra of the original Alcell lignin and the three fractions derived thereof (F1–F3). | ||
| Chemical shift region (ppm) | Type of carbon | Carbon content (% of all carbon) | |||
|---|---|---|---|---|---|
| Alcell | F1 | F2 | F3 | ||
| a Ratio of the area of aliphatic carbons (δ = 36–0 ppm) and aromatic carbons (δ = 160–100 ppm). | |||||
| 0–36 | Aliphatic groups | 17.1 | 32.8 | 12.5 | 7.6 |
| 52–58 | Methoxy groups | 16.6 | 12.1 | 17.6 | 19.3 |
| 58–100 | Ether bonds (α, β and γ) | 6.3 | 2.5 | 7.7 | 5.0 |
| 100–160 | Total aromatics | 59.5 | 50.2 | 61.3 | 67.5 |
| 100–123 | Non-branched aromatics | 25.1 | 20.3 | 23.5 | 26.3 |
| 123–160 | Branched aromatics | 34.4 | 29.9 | 37.8 | 41.2 |
| 160–210 | Carbonyl groups | 0.5 | 2.3 | 0.7 | 0.6 |
| Aliphatic/aromatic ratioa | 0.29 | 0.65 | 0.20 | 0.11 | |
An attempt was made to determine the amount of extractives (as stearic acid equivalents) by comparing the integrals of the characteristic methyl end groups of long chain fatty acids at around δ = 14 ppm with that of the resonance of the methoxy peak of lignin at about δ = 56 ppm. The integration ratio was found to be 1 to 6.5, revealing that the amount of extractives in the Alcell F1 fraction is about 15 mol%. Assuming an average molecular weight of 166 g mol−1 for an Alcell lignin monomer (e.g. 4-propylguaiacol) and a molecular weight of 284.5 g mol−1 for a representative fatty acid (stearic acid), F1 contains about 21 wt% of extractives as stearic acid equivalents. Given the fact that the F1 yield is about 20 wt% on lignin intake, the Alcell lignin feed contains about 4 wt% of extractives as stearic acid equivalents. The presence of the extractives in both F1 and the Alcell lignin has a significant effect on the subsequent catalytic hydrotreatment reactions and particularly the composition of the product oils, as will be shown later on. In addition, they also have an effect on the molecular weight distributions, particularly those for F1, and suggest that the molecular weight of the lignin fraction is actually higher than those reported in Table 1.
The composition of the lignins in terms of characteristic chemical groups was semi-quantified by dividing the 13C-NMR into distinct regions and by subsequent peak integration of the various regions using methods developed by Hu et al.,29 Ingram et al.,30 Xia et al.31 and Robert.32 The normalized integration results for Alcell lignin and the lignin fractions are given in Table 2. F1 contains by far the largest proportion of aliphatic groups, which are at least partly associated with the presence of the extractives. It is of interest to compare the chemical compositions of fractions F2 and F3, for which negligible amounts of extractives are present. The amount of OMe groups is higher for fraction F3 (19.3%) than that for F1 (12.1%). Both the trend and the absolute value of the OMe groups are in line with the data reported by Thring using the TAPPI method (the amount of OMe groups in the Alcell lignin sample by using our method is 16.6% versus 16.5% by Thring).26 This suggests that 13C-NMR is a suitable technique to quantify the functional groups in the lignin samples. In addition, the higher amount of OMe groups for F3 reveals that this fraction contains more syringol groups and, as such, shows that the hardwood character of the Alcell lignin is best preserved in this high molecular weight fraction.
The amount of aliphatic carbons for F2 is slightly higher than that for F3, indicating that F2 is slightly more aliphatic and less aromatic in nature than F3. This statement is also supported by the elemental composition data (Table 1) and the gradient HSQC measurements (Fig. 7 and 8).
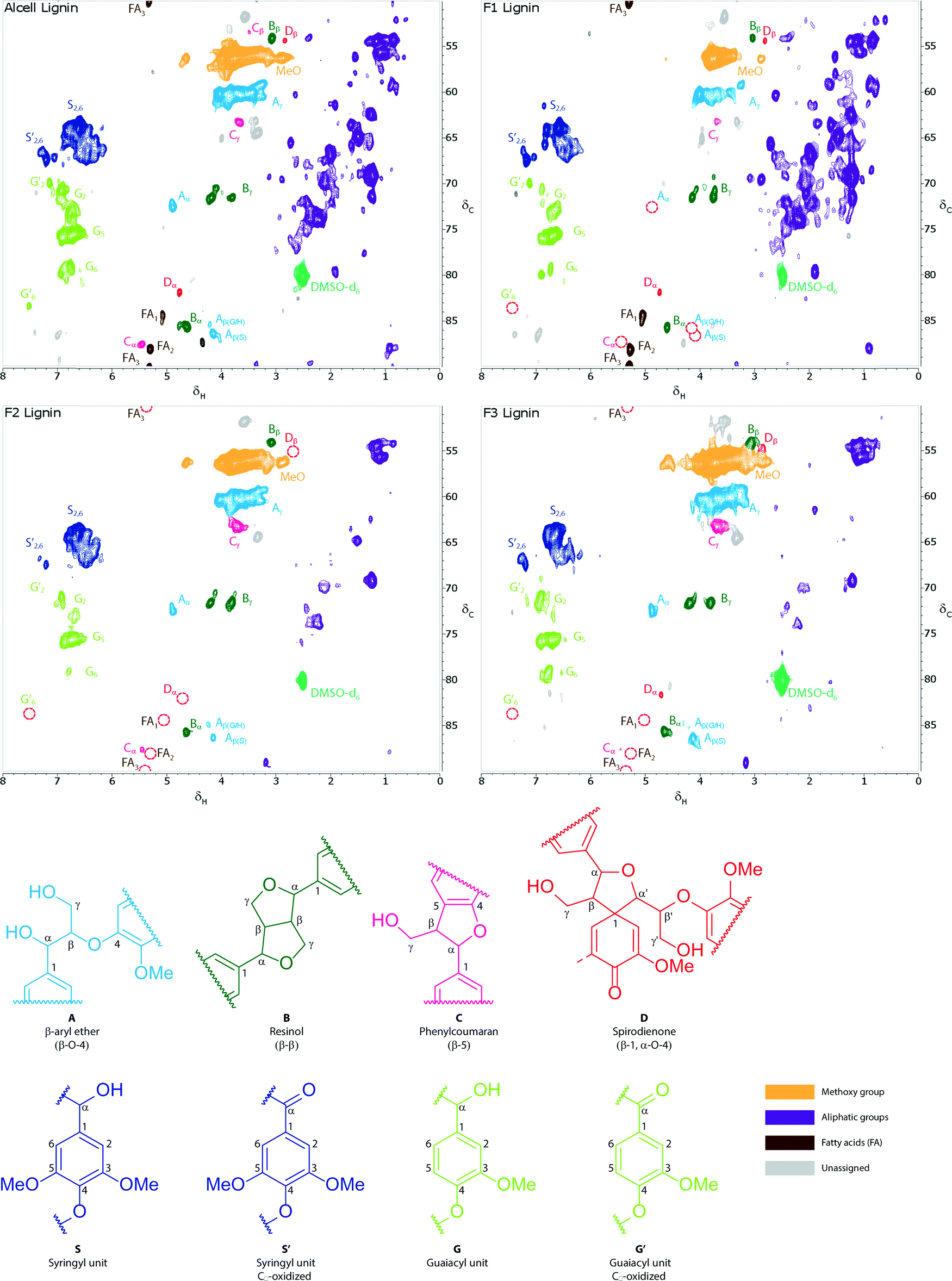 | ||
| Fig. 7 Gradient (folded) HSQC spectra (δC = 50–90, δH = 0–8 ppm) of the Alcell lignin and the fractionated lignins. The contours are coloured according to the type of linkage. The light grey contours are unknown regions. The red circles indicate contour peaks which are not present in all fractions (e.g. peaks from fatty acids, which are only present in F1). | ||
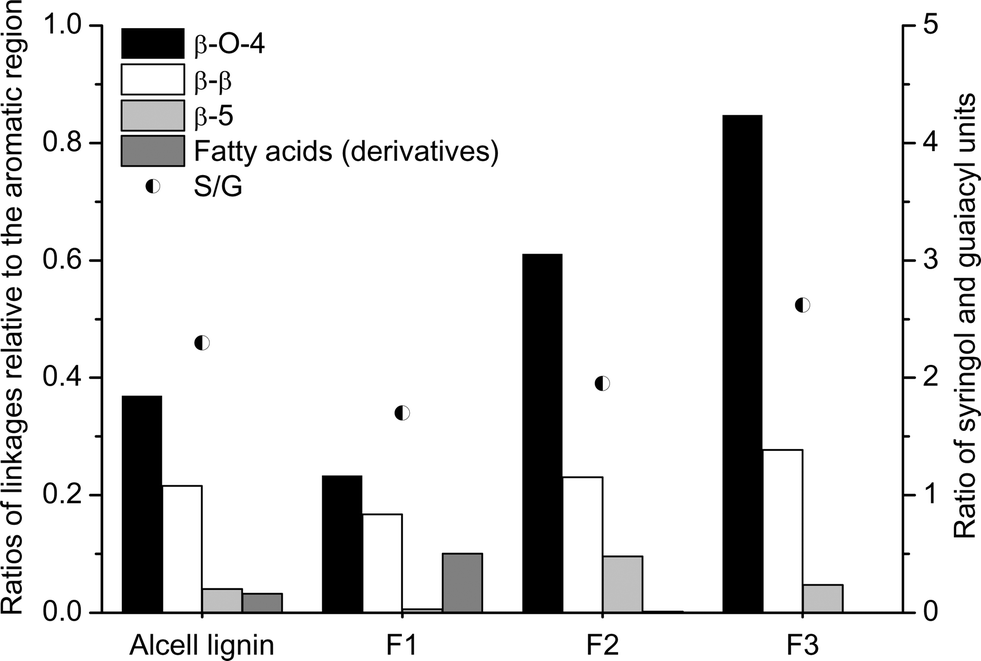 | ||
| Fig. 8 Ratio of linkages relative to the aromatic region and the S/G ratio in Alcell lignin and the lignin fractions. | ||
A more detailed analysis of the lignin structures was performed by using gradient HSQC. The spectra for the original Alcell lignin and the fractionated lignins are given in Fig. 7 and 8. Identification of the different linkages is based on the work of Tran et al.,28 Willker et al.,33 Wen et al.,34,35 del Rio et al.,36 Yuan et al.,37 and Ralph et al.38 The spectra for the different fractions show large differences regarding the amounts of linkages (particularly β-O-4 and β-5) and the amount of fatty acid (derivatives).
A clear increase in the amount of β-O-4 linkages is observed when going from fraction F1 to F3. Thus, it appears that F3 is relatively enriched in these linkages. A clear trend for the β-5 linkages is not present; it is highest for F2 and lowest for F1. Surprisingly, the amount of β-β linkages is not affected by the fractionation of the Alcell lignin and is present in similar amounts in all the fractions. The higher amount of β-O-4 linkages in F2 and F3 indicates that these fractions are likely easier to depolymerise. The syringol to guaiacol ratio (S/G ratio) was also determined, and the ratio is a strong function of the fraction considered, with a higher ratio for F3 (Fig. 8). These findings are in agreement with the results of alkaline nitrobenzene oxidations reported by Thring et al.26 and the 13C-NMR measurements as provided in Table 2.
The HSQC measurements also confirm the presence of substantial amounts of fatty acid (derivatives) in the F1 fraction.
3.2 Catalytic hydrotreatment experiments
The catalytic hydrotreatment experiments with the three different lignin fractions and the original Alcell lignin were performed in a batch set-up at 400 °C, 100 bar H2 initial pressure, and 4 h reaction time using 5 wt% Ru/C on lignin as the catalyst. These harsh conditions were selected on the basis of earlier studies by our group for a catalyst screening study on the hydrotreatment of Alcell lignin with supported noble metal catalysts.23The results for the experiments are summarised in Table 3. After reaction, two main product phases were identified, a liquid phase (about 71–73 wt% on lignin intake) and a gas phase (about 10 wt% on lignin intake, excluding hydrogen gas). Solid formation by e.g. repolymerisation of reactive lignin fragments was very low and quantification proved very difficult. The liquid phase consisted of two immiscible layers, a dark/light brown organic phase (lignin oil, 61–71% on lignin intake) and a clear aqueous layer (3–12 wt% on lignin intake). For the original Alcell lignin, F2, and F3, the organic phase was below the aqueous phase. For the F1 fraction, the organic phase resided on top of the aqueous phase, indicating that the reaction product of this fraction has a lower density than those of F2 and F3 and is in line with the presence of apolar aliphatic structures arising from the extractives in F1 (vide infra). The original Alcell lignin, F1, and F2 gave a product oil with a relatively low viscosity, while the product oil for fraction F3 was more viscous. The lignin oils contain large amounts of carbon (>82.6 wt% on dry basis) and a limited amount of water (1.4–8.5 wt%) indicating the presence of large amounts of relative apolar organics, which was confirmed by GC × GC-FID analysis and other techniques (vide infra). The formation of an aqueous phase indicates the formation of substantial amounts of water by hydrodeoxygenation reactions.
| Lignin type | Alcell lignin | F1 | F2 | F3 |
|---|---|---|---|---|
| a 400 °C, 4 h, 100 bar H2 of initial pressure. b Excluding the carbon content of the aqueous phase. c Ethylene and propylene were not detected; the remainder is hydrogen. | ||||
| Organic phase (wt% on lignin intake) | 64.7 | 71.1 | 62.0 | 61.2 |
| Aqueous phase (wt% on lignin intake) | 9.5 | 2.9 | 8.7 | 11.9 |
| Gas phase (wt% on lignin intake)c | 11.5 | 9.3 | 10.9 | 12.5 |
| Carbon dioxide (mol%) | 14.5 | 7.1 | 13.8 | 20.9 |
| Carbon monoxide (mol%) | 2.2 | 8.3 | 3.0 | 2.6 |
| Ethane (mol%) | 2.1 | 2.4 | 1.9 | 2.9 |
| Propane (mol%) | 0.9 | 1.9 | 0.7 | 1.3 |
| Methane (mol%) | 23.1 | 23.3 | 22.4 | 32.9 |
| Total balance (wt% on lignin intake) | 86 | 83 | 82 | 86 |
| Total carbon balance (wt%) | 90 | 97b | 90b | 89b |
| Hydrogen uptake (Nl kg−1 lignin) | 345 | 340 | 320 | 250 |
| Elemental composition of the lignin oil (wt% dry basis) | ||||
| Carbon | 82.5 | 82.6 | 84.0 | 84.7 |
| Hydrogen | 9.5 | 10.9 | 9.5 | 7.7 |
| Oxygen | 7.7 | 6.5 | 6.3 | 7.3 |
Mass balance closures were reasonable and all above 82 wt% on lignin intake. The losses are likely due to i) an underestimation of the amount of gas phase components formed as the procedure to quantify the amount of gas phase component is elaborate and prone to experimental errors and ii) difficulties in quantitative removal of the liquid reaction products from the reactor due to accumulation in linings and valves. The carbon balance closure was above 90%. Hydrogen uptake was shown to be in the range of 250–350 Nl kg−1 lignin, indicating the substantial hydro(deoxy)genation activity of the catalyst, which is in line with the elemental composition of the lignin oil products and the formation of substantial amounts of water.
4 Composition of the gas and liquid phases
4.1 Gas phase composition
The gas phase composition for all product gases was determined, and the results are given in Table 3. Besides the remaining hydrogen, indicating that the reactions were not carried out under hydrogen starvation conditions, the main gas phase products were CH4 (23–33 mol%), CO2 (7–21 mol%) and CO < 8 mol%). The gas phase composition depends on the hydrotreatment feed. The amount of CO2 increases in the order F1 < F2 < F3, whereas the opposite trend is observed for CO. The CO2 and CO are likely formed by gasification reactions of various reactive lignin fragments. For instance, it is well known that Ru catalysts are active for the gasification of Alcell lignin in supercritical water at 400 °C (3.3 wt% Alcell in water).39 Although in our case the water content is much lower, such gasification reactions may also occur to a significant extent. In addition, the CO/CO2 ratio, particularly for fraction F1, may also be affected by the presence of extractives (partly in the form of long chain fatty acid/esters) which are known to be prone to hydrodeoxygenation reactions (see eqn (1) for details).40 GC-MS-FID measurements on the liquid phase (vide infra) indeed reveal the large formation of heptadecane, a strong indication that deoxygenation of the extractives by CO/CO2 formation occurs to a significant extent. So far, we do not have a sound explanation for the CO/CO2 ratio as a function of the feed used for the experiments, although this ratio may also be affected by the water gas shift reaction. | (1) |
 | (2) |
| CO + H2O ↔ CO2 + H2 CO2 + 4H2 ↔ CH4 + 2H2O | (3) |
4.2 Compositions of the organic and aqueous phases
The elemental compositions of the feeds and lignin oils were determined, and the results are shown in Fig. 9 in the form of a van Krevelen plot. | ||
| Fig. 9 Van Krevelen plot for the feeds and products of the catalytic hydrotreatment reaction (400 °C, 100 bar H2 initial pressure, 4 h, 5 wt% of Ru/C). | ||
Large differences in the H/C ratio of the product oils of the three fractions are observed (1.09–1.58), although the O/C ratios (0.06–0.07) are essentially similar. Apparently, the product oil from F1 is more aliphatic in nature than those of F2 and F3, indicating a high level of hydro(deoxy)genation for F1. However, direct comparison is cumbersome as also the feed composition (F1–F3) differs considerably. As such, it is better to compare the relative change in H/C ratio, as shown by the dotted lines from the feed to the product in Fig. 9. The slope of these lines is a measure of the hydrogenation/hydrodeoxygenation level of the feeds. Clearly, hydrotreatment of F1 is more facile, while it is more sluggish for F3. This is also in line with the experimentally determined hydrogen uptakes (Table 3), which was highest for F1 (340 Nl kg−1) and lowest for F3 (255 Nl kg−1).
All organic product oils were analysed using GC × GC-FID and GC-MS-FID to determine the product composition. A representative example of a GC-MS-FID chromatogram is given in Fig. 10 (F1 and F3 lignin oils), showing a large number of volatile components (>300). The main products are (alkyl substituted) phenolics, aromatics and alkanes (cyclic and linear). Remarkable differences in composition are evident for both fractions and particularly at longer retention times. F1 contains significant amounts of long chain hydrocarbons, which are most likely arising from the hydro(deoxy)genation of the extractives (fatty acid derivatives). In line with this statement is the absence of such compounds in the product of fraction F3.
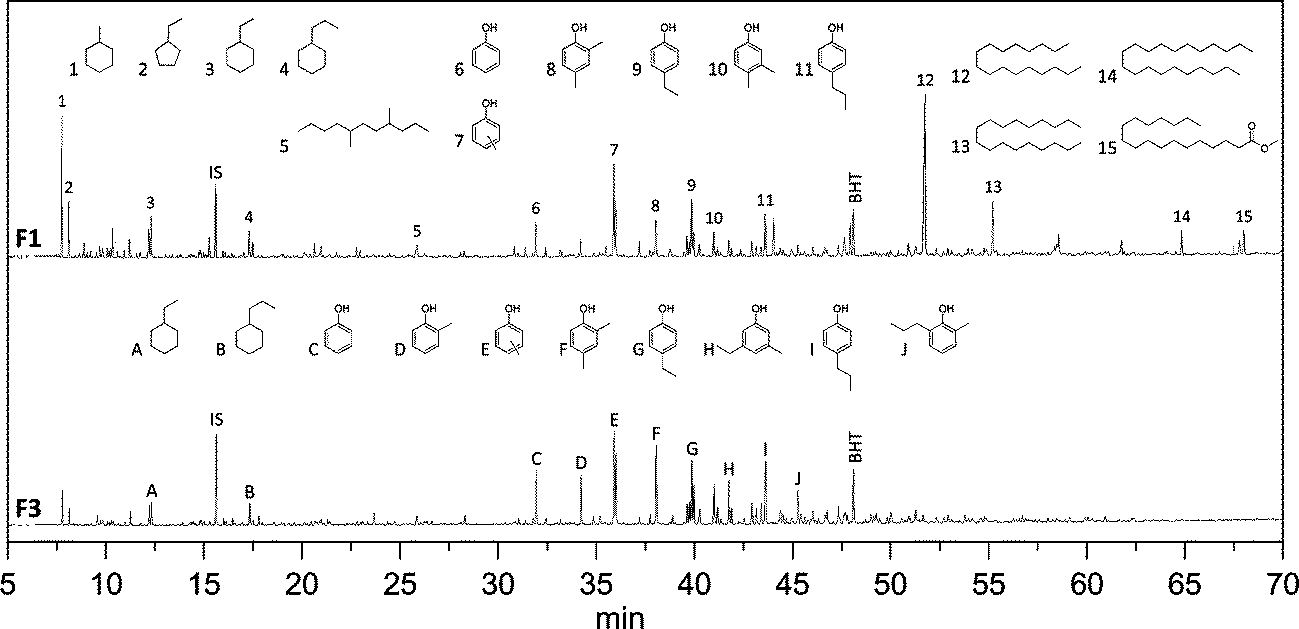 | ||
| Fig. 10 GC-MS-FID chromatogram for the product oils of F1 and F3 after catalytic hydrotreatment (IS: internal standard, BHT: stabiliser in THF). | ||
The product samples were also analysed by GC × GC-FID to gain more insight into the various organic product classes (e.g. aromatics, alkylphenolics, aliphatics) present. GC × GC has been shown to be very suitable for the rapid assessment of major organic compound classes in an organic bioliquid.46,47 Representative chromatograms of the products are given in Fig. 11, including the selected product classes. Clearly, a good separation between the various organic product classes is possible and discrete regions are visible.
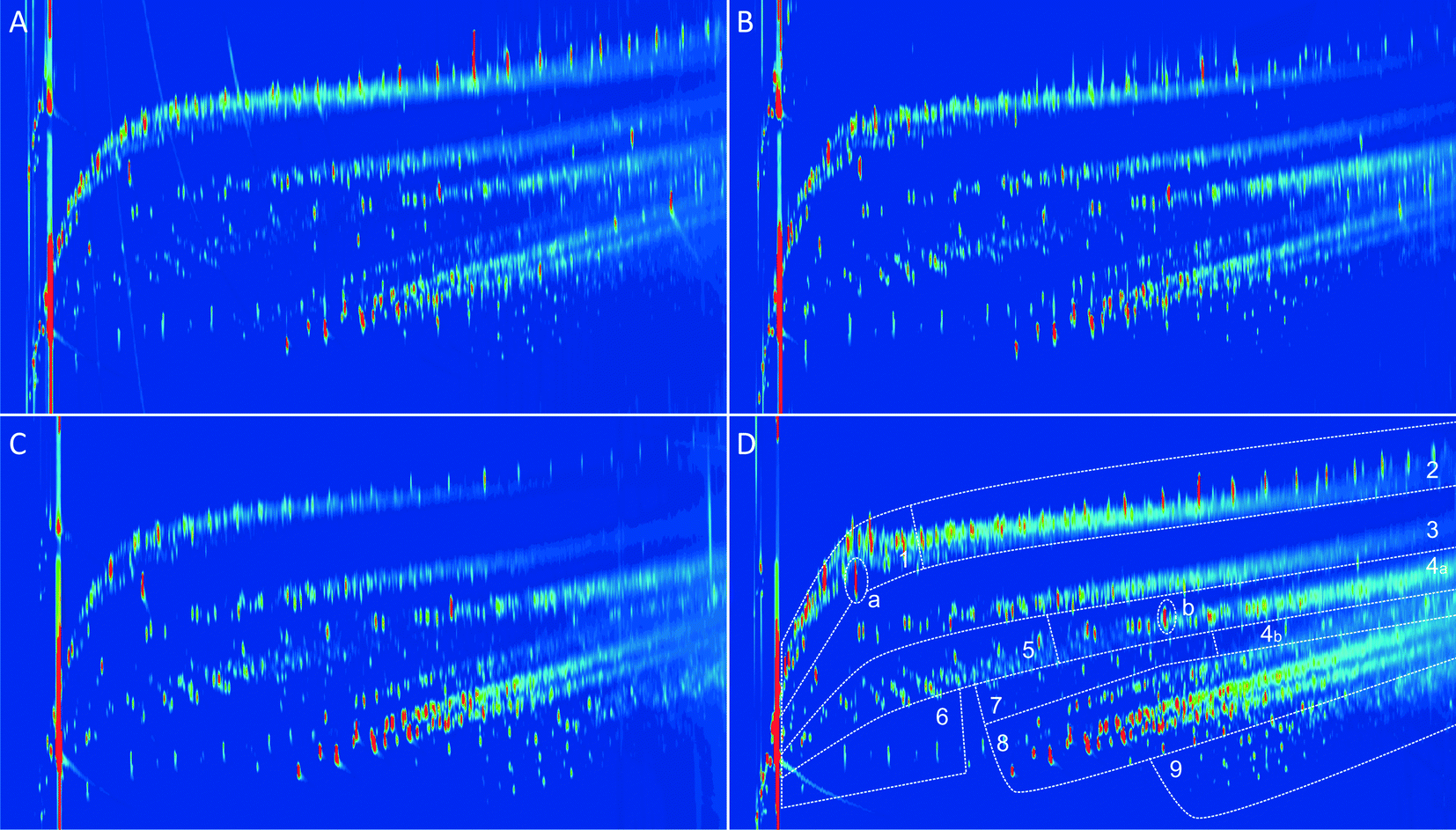 | ||
| Fig. 11 GC × GC-FID chromatograms of the organic phase from A) F1, B) F2, C) F3, and D) unfractionated Alcell. 1 = mainly cyclic alkanes, 2 = mainly linear/branched alkanes, 3 + 4 = aromatics, 4a = naphthalenes, 4b = polycyclic aromatic hydrocarbons, 5 = ketones/alcohols, 6 = acids, 7 = guaiacols, 8 = alkylphenolics, 9 = catechols. In addition, a = internal standard and b = BHT (stabilizer in THF). | ||
Quantification of the various product classes was performed, and the results are given in Table 4. The highest amount of total GC detectable components was found in the F1 fraction (27.6 wt% on lignin intake) and the lowest for F3 (13.4 wt% on lignin). A clear correlation is present between the total GC × GC-FID detectable components and the molecular weight of the product oils (vide infra), with lower molecular weight products leading to a higher level of GC detectable components. The main GC × GC-FID detectable product classes in the different samples are alkylphenolics, aromatics, linear/branched/cyclic alkanes, and some ketones/alcohols. An overview of the main individual components in the various product classes for the F1 oil is given in Table 5.
| Compound classes | Lignin oils (wt% on lignin intake) | |||
|---|---|---|---|---|
| Alcellb | F1 | F2 | F3 | |
| a 400 °C, 100 bar H2 initial intake, 4 h, with 5 wt% Ru/C. b Average of two experiments. | ||||
| Alkylphenolics | 9.0 | 8.4 | 5.3 | 6.8 |
| Guaiacols | 0.1 | 0.1 | 0.1 | 0.0 |
| Catechols | 1.1 | 0.4 | 0.3 | 1.6 |
| Aromatics | 3.2 | 4.2 | 3.4 | 1.8 |
| Linear/branched alkanes | 3.2 | 8.6 | 3.3 | 0.8 |
| Cyclic alkanes | 3.7 | 5.4 | 3.3 | 1.1 |
| Ketones/alcohols | 1.4 | 0.5 | 1.5 | 1.3 |
| Total GC × GC | 21.4 | 27.6 | 17.2 | 13.4 |
| Compounds | Structure |
|---|---|
| a 400 °C, 100 bar H2 initial intake, 4 h, with 5 wt% Ru/C. | |
| Alkylphenolics | |
| a. Phenol |
|
| b. m/p-Cresol | |
| c. 4-Ethylphenol | |
| d. 4-Propylphenol | |
| Aromatics | |
| e. Toluene |

|
| f. Ethylbenzene | |
| g. Naphthalene | |
| h. Indene | |
| Cyclic/linear/branched alkanes | |
| i. Methylcyclohexane |

|
| j. Ethylcyclohexane | |
| k. Heptadecane | |
| l. Nonadecane | |
The main component classes for the F1 fraction oil are the linear/branched and cyclic alkanes which are formed to about 14 wt% on lignin. In contrast, the F3 fraction oil contains by far less alkanes (1.9 wt% on lignin). Similar trends can also be made for the aromatics, with the highest amounts for F1 and by far less for F3, with an intermediate value for F2. Thus, not only the amount of the GC × GC-FID detectable components but also the composition of the oils is a strong function of the Alcell lignin fraction used in the hydrotreatment reaction.
As mentioned earlier, the maximum total amount of GC × GC-FID detectable components is only 27.6 wt% (for fraction F1), indicating the presence of large amounts of non-GC detectable compounds, which are presumed to be of a higher molecular weight. Therefore, GPC analysis was performed on the product oils (see Fig. 12 for details).
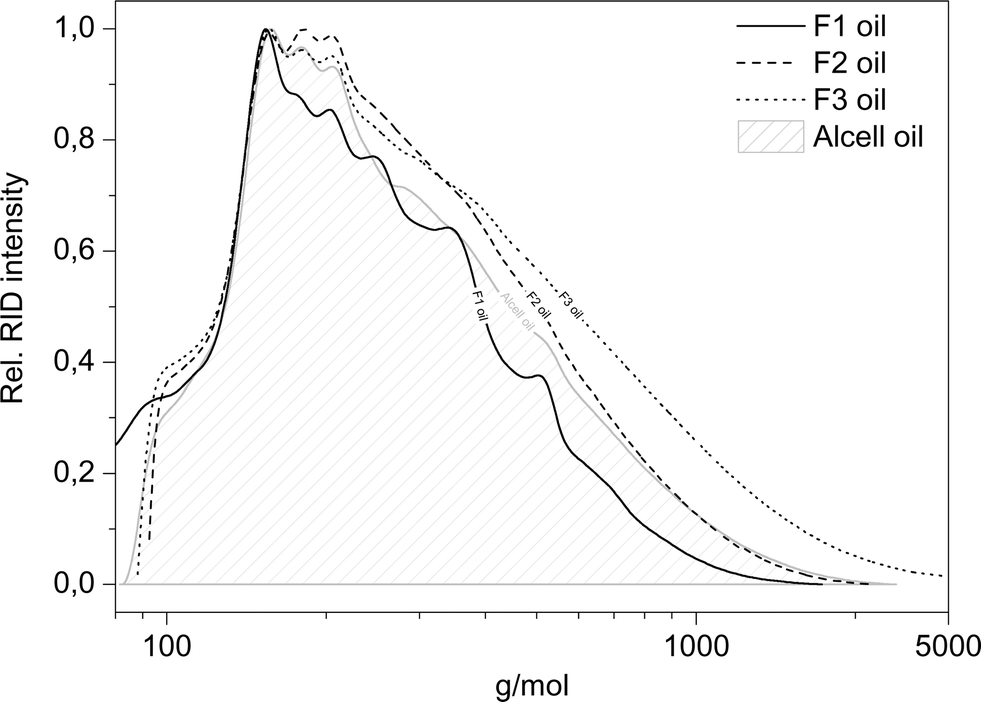 | ||
| Fig. 12 Molecular weight distribution of the product oils (400 °C, 4 h, 100 bar initial H2, 5 wt% Ru/C). | ||
Clearly, the molecular weights of the lignin oils are significantly lower than those of the corresponding lignin feeds (Fig. 5), indicating that all four lignin feeds are significantly depolymerised during the catalytic hydrotreatment process. The F1 product oil shows the lowest value for the molecular weight after catalytic hydrotreatment, whereas those for F2 and F3 fractions are substantially higher. A clear correlation is present between the molecular weights of the product oils and the total GC detectable components, where the lowest molecular weight fraction also shows the highest amount of GC detectable components.
Of interest is a comparison of the relative molecular weight reduction during the catalytic hydrotreatment for the four lignin sources (Alcell and fractions F1–F3). The results are given in Table 6 and show that the relative molecular weight reduction is by far the highest for F3 and lowest for F1. Apparently, a reaction time of 4 h is sufficient to break the largest proportion of the easily cleavable ether linkages for all fractions, supported by 13C-NMR studies (vide infra). However, the C–C linkages are apparently more difficult to cleave as the product oils still contain considerably amounts of higher molecular weight lignin fragments (GPC).
| Feed Mw (g mol−1) | Oil Mw (g mol−1) | Reduction (%) | |
|---|---|---|---|
| Alcell | 1720 | 316 | 82 |
| F1 | 660 | 269 | 60 |
| F2 | 1640 | 324 | 80 |
| F3 | 3680 | 410 | 89 |
The molecular composition of the lignin oils was also determined by 13C-NMR, the advantage of the above GC techniques being that the composition of the complete sample is measured and not solely the GC detectable fraction. The molecular composition was semi-quantified using the same method used for the determination of the lignin feed samples. The normalized integration results for both the starting materials and the resulting lignin oils are given in Table 7. In addition, the 13C-NMR spectra of Alcell lignin and all hydrotreated product oils are shown in Fig. 13.
| Chemical shift region (ppm) | Type of carbon | Carbon content (% of all carbon) | |||
|---|---|---|---|---|---|
| Alcell oil | F1 oil | F2 oil | F3 oil | ||
| a Ratio of the area of aliphatic carbons (δ = 36–0 ppm) and aromatic carbons (δ = 160–100 ppm). | |||||
| 0–36 | Aliphatic groups | 47.0 | 54.5 | 45.1 | 37.4 |
| 52–58 | Methoxy groups | 0.7 | 0.9 | 0.5 | 0.4 |
| 58–100 | Ether bonds (α, β and γ) | 0.0 | 0.1 | 0.6 | 0.5 |
| 100–160 | Total aromatics | 51.9 | 44.5 | 53.4 | 61.0 |
| 100–123 | Non-branched aromatics | 17.9 | 18.4 | 15.0 | 21.3 |
| 123–160 | Branched aromatics | 34.0 | 26.1 | 38.4 | 39.7 |
| 160–210 | Carbonyl groups | 0.3 | 0.2 | 0.4 | 0.8 |
| Aliphatic/aromatica | 0.91 | 1.22 | 0.84 | 0.61 | |
 | ||
| Fig. 13 13C-NMR of the Alcell lignin, the lignin oil from the catalytic hydrotreatment of the Alcell lignin oil, the hydrotreated ether fraction (F1 oil), the hydrotreated methanol fraction (F2 oil) and the hydrotreated residue fraction (F3 oil). | ||
Clearly, major differences are present between the Alcell lignin and the product oils. The most prominent features are the absence of remaining OMe groups (δ = 52–58 ppm) in the NMR spectra of the 4 product oils after catalytic hydrotreatment and a considerable increase in the aliphatic (δ = 0–36 ppm) and aromatic resonances (Table 7), in line with the observations by GC × GC-FID. Furthermore, characteristic resonances of the ether bonds (δ = 58–100 ppm) in the lignin structure, clearly visible in the Alcell lignin feed (6.3%), are nearly absent (<0.6%) in the product oils, indicating that these are easily cleaved during the catalytic hydrotreatment process. The aliphatic carbon content is the highest for the oil from the F1 fraction, which correlates with the GC × GC-FID and elemental analysis data.
5 Discussion
On the basis of the chemical composition (GC, NMR), the molecular weight data, gas phase composition, and elemental composition of the hydrotreated lignin oils, a reaction network is proposed for the hydrotreatment reaction of Alcell lignin and fractions thereof using Ru/C (Fig. 14).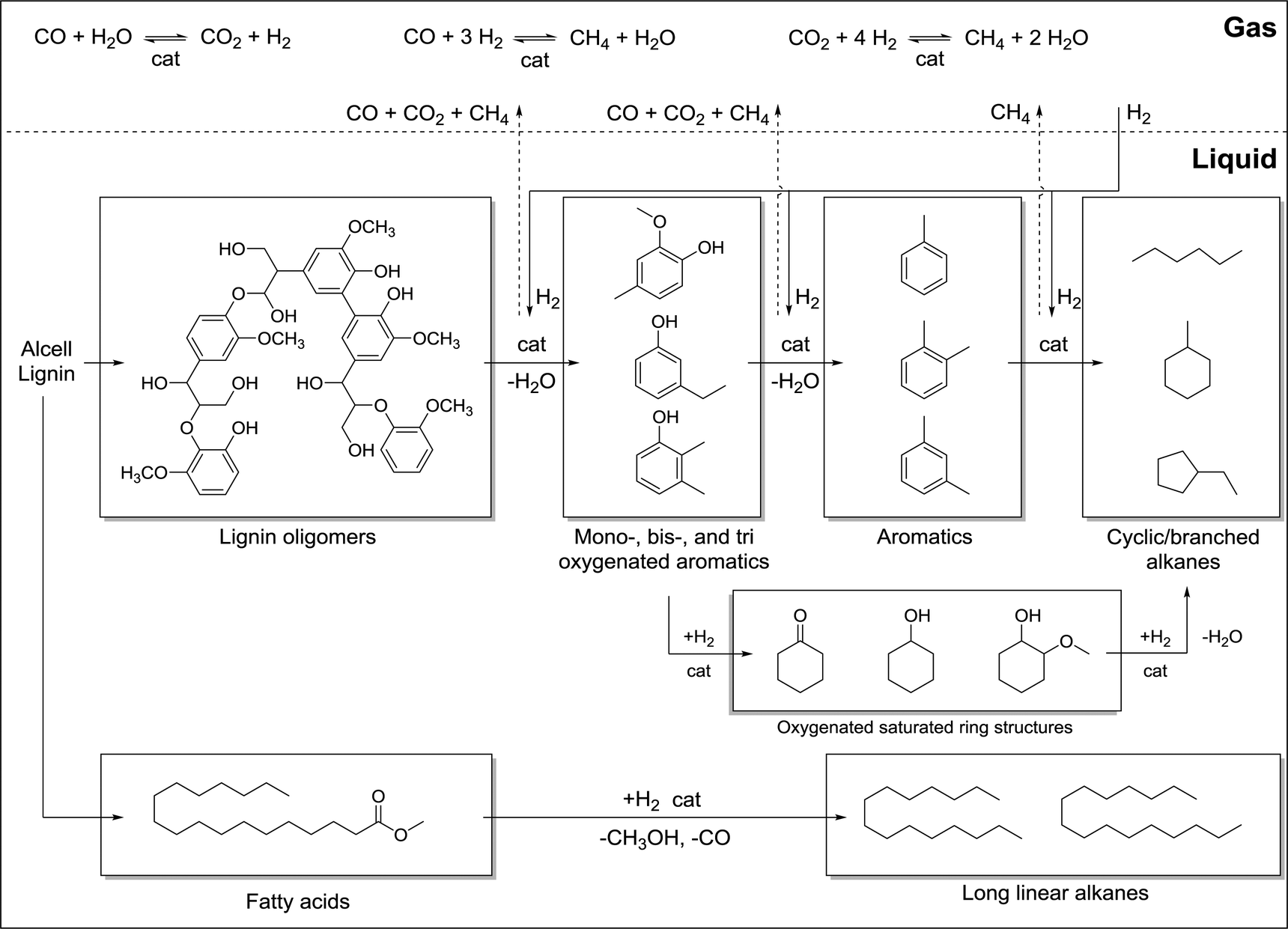 | ||
| Fig. 14 Proposed pathway for the catalytic hydrotreatment of Alcell lignin with Ru/C. | ||
Within the window of experimental conditions (high H2 pressures, 400 °C, reaction times up to 4 h), lignin is converted by a number of parallel and consecutive pathways. In the initial stage of the reaction, the lignin depolymerise thermally and/or catalytically to lower molecular weight lignin oligomers at temperatures as low as 230–260 °C.48 This depolymerisation reaction is relatively slow on the timescale of the reaction time (4 h) and full depolymerisation to solely monomeric compounds was not obtained for all fractions after reaction, although the molecular weights of all product oils are in a narrow range (Table 6). The use of a catalyst is very important, which is confirmed by the observations that a reaction without a catalyst only leads to solid products.23 Thermal degradation of Alcell lignin at 400 °C to a (partly) liquid product is possible as was shown by de Wild et al. However, liquid yields are low and the reactions were performed in a pyrolysis setup under conditions differing substantially from this study (450–500 °C with very fast heating times (1 s) and very short reaction times).49
The lignin oligomers react further to lower molecular weight oxygenated aromatics with the concomitant formation of water and methane. The ether linkages are most probably cleaved in this stage of the reaction, which is supported by model compound studies.7,8 In addition, these bonds have the lowest bond dissociation energy, as shown by thermodynamic calculations. Methoxy group hydrogenolysis also occurs to a significant extent, as is evident from the disappearance of the characteristic 13C-NMR resonances of the methyl group in the lignin oils (Table 7) and the formation of substantial amounts of methane. As such, methoxy removal in oligomeric lignin units, besides that in monomeric compounds, cannot be excluded a priori.
The oxygenated low molecular weight aromatics likely react in two parallel pathways to alkanes, viz. i) hydrogenation of the aromatic C–C double bonds to form saturated cyclic ketones and alcohols followed by subsequent hydrogenations and ii) the hydrodeoxygenation of the oxygenated aromatics to aromatics, followed by overhydrogenation to alkanes. The desired alkylphenolics and aromatic compounds are intermediates in this sequence and can only be formed in substantial amounts when the rate of (over)hydrogenation of aromatic C–C double bonds is retarded, e.g. by optimisation of process conditions and by the identification of catalysts that are less prone to this reaction. In this respect, the performance of the Ru/C catalyst used in this study is far from optimal and substantial amounts of aliphatic hydrocarbons are formed. The largest proportion of the long chain linear alkanes present in the product oils is likely formed from the hydrotreatment of the extractives and particularly from the fatty acids and esters present in the Alcell lignin feed. These extractives are present in an estimated amount of 4 wt% on the Alcell lignin feed and tend to accumulate in the low molecular weight fraction of the solvent extraction process.
An overview of the relevant changes in composition and molecular properties upon the hydrotreatment of the various fractions is given in Fig. 15.
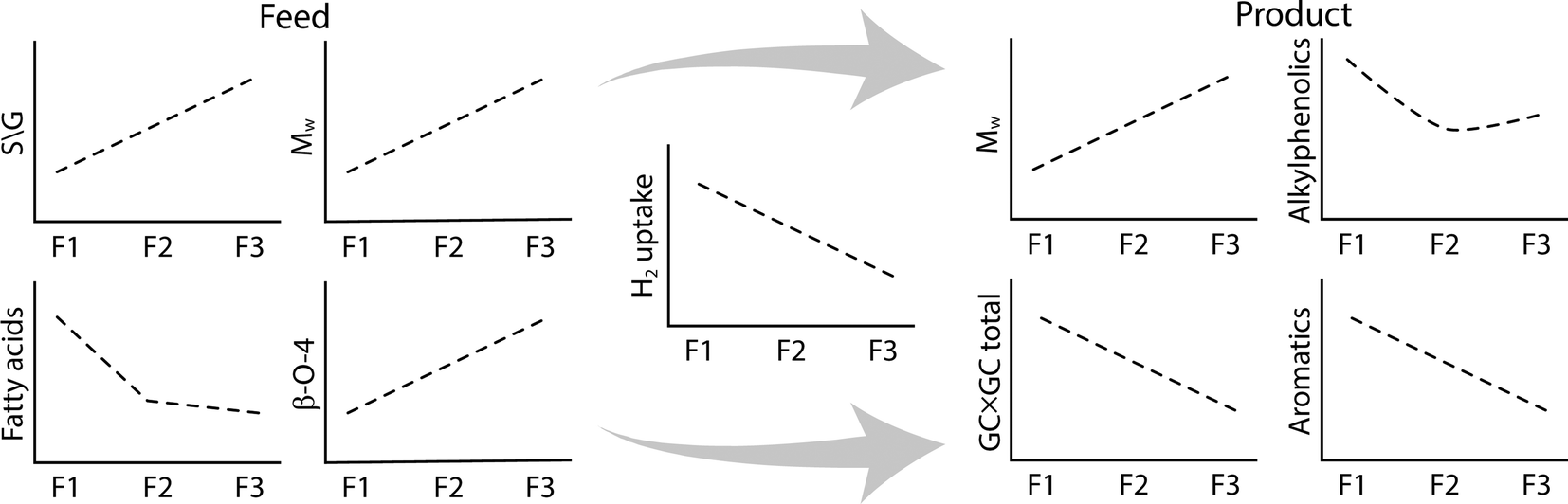 | ||
| Fig. 15 Overview of the differences between the feed and the products of the fractionated lignin. | ||
The most reactive fraction is the low molecular weight fraction F1. This fraction shows the highest hydrogen uptake, as well as the highest increase in H/C ratio although this is overestimated due to the presence of extractives which also react with hydrogen in the process. The observation that fraction F2 is by far more reactive than F3 in the catalytic hydrotreatment reaction may be related to differences in molecular structure and/or molecular weight. Thring et al. reported distinct differences in the level of condensation of Alcell lignin fractions by performing oxidations with alkaline nitrobenzene.26 F3 was shown to comprise the highest amount of condensed structures, rendering it the least amenable to oxidative degradation. This was rationalised by the incorporation of some non-lignin materials during the process such as carbohydrate derived products50 as well as from the reaction of furfural with lignin to form new carbon–carbon linkages.51
However, the molecular weight also plays a role. The molecular weight of the original feed fractions increases in the order F1 to F3 and this is also the case for the hydrotreated product oil fractions. However, the relative decrease is the highest for the F3 fraction. The F3 feed fraction contains the highest amounts of ether linkages and these are known to be cleaved more facilely than C–C linkages.
The product compositions of the lignin oils differ considerably (Table 4). When aiming for the highest amounts of alkylphenolics and aromatics, the use of the F1 fraction seems to be preferred: 8.4 wt% alkylphenolics and 4.2 wt% aromatic compounds on lignin intake. These amounts are slightly underestimated as F1 also contains significant amounts of extractives which do not contribute to the formation of these compound classes.
6 Conclusions
The catalytic hydrotreatment of three Alcell lignin fractions, obtained by a solvent extraction procedure, using Ru/C as the catalyst and in the absence of a solvent was investigated, and the results were compared with the parent Alcell lignin. Distinct differences in the molecular composition and molecular weight distributions of the product oils were observed. These are likely due to differences in molecular weights and molecular structures of the fractions.Full breakdown of the oligomeric structure of all lignin feeds to monomers proved to be impossible under the prevailing reaction conditions as was evident from the molecular weight distributions of the product oils. This is attributed to a relatively slow rate of cleavage of C–C bond linkages in the lignin structure. The rate of the hydro(deoxy)genation reactions is a function of the lignin fraction used and was highest for the low molecular weight fraction F1 and lowest for the highest molecular weight fraction F3, as was evident from the hydrogen uptake during the experiments, the elemental composition of the product oil and the molecular composition of the product oil.
Highest yields of valuable alkylphenolics (8.4 wt% on intake) and aromatic compounds (4.2 wt% on intake) were obtained with the lowest molecular weight fraction F1. This fraction also contained the highest amounts of aliphatic hydrocarbons (14.0 wt% on intake). However, the bulk of these hydrocarbons is formed by hydrodeoxygenation reactions of extractives such as fatty acids/esters present in the Alcell lignin feed (around 4 wt%) that accumulated in fraction F1 during solvent fractionation.
These findings indicate that improvements in the yields of low molecular weight aromatics and alkylphenolics from lignin are possible by proper selection of the lignin feed and particularly by using feeds that have a low molecular weight as well as a low content of condensed structures. As such, a two-step lignin valorisation approach involving an efficient depolymerisation step (for example a base catalyzed depolymerisation, a hydrothermal liquefaction or fast pyrolysis) followed by a catalytic hydrotreatment step may be very advantageous. These studies are in progress and will reported in due course.
Acknowledgements
Hans van der Velde (University of Groningen, Department of Organic Chemistry) is acknowledged for performing the elemental analysis and Wim Kruizinga for help with the 13C-NMR measurements. We also thank Jan Henk Marsman, Leon Rohrbach, Erwin Wilbers, Marcel de Vries and Anne Appeldoorn for technical support. This work was conducted in the framework of the Dutch national project “LIGNOVALUE”, contract no. EOSLT-05011. Financial support from Agentschap NL is gratefully acknowledged.References
- M. Balat, H. Balat and C. Oz, Prog. Energy Combust. Sci., 2008, 34, 551–573, DOI:10.1016/j.pecs.2007.11.001.
- A. E. Atabani, A. S. Silitonga, I. A. Badruddin, T. M. I. Mahlia, H. H. Masjuki and S. Mekhilef, Renewable Sustainable Energy Rev., 2012, 16, 2070–2093, DOI:10.1016/j.rser.2012.01.003.
- J. J. Bozell, Clean, 2008, 36, 641–647, DOI:10.1002/clen.200800100.
- T. Dickerson and J. Soria, Energies, 2013, 6, 514–538, DOI:10.3390/en6010514.
- A. Boisen, T. B. Christensen, W. Fu, Y. Y. Gorbanev, T. S. Hansen, J. S. Jensen, S. K. Klitgaard, S. Pedersen, A. Riisager, T. Stahlberg and J. M. Woodley, Chem. Eng. Res. Des., 2009, 87, 1318–1327, DOI:10.1016/j.cherd.2009.06.010.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, 2004, Technical Report NREL/TP-510-35523 Search PubMed.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103, 10.1039/C3EE43081B.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Amen-Chen, H. Pakdel and C. Roy, Bioresour. Technol., 2001, 79, 277–299, DOI:10.1016/S0960-8524(00)00180-2.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869–883, 10.1039/c2cy00500j.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41, DOI:10.1002/ceat.201000270.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624, DOI:10.1021/acs.chemrev.5b00155.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201510351.
- E. Adler, Wood Sci. Technol., 1977, 11, 169–218 CrossRef CAS.
- P. Karhunen, P. Rummakko, J. Sipilä, G. Brunow and I. Kilpeläinen, Tetrahedron Lett., 1995, 36, 169–170, DOI:10.1016/0040-4039(94)02203-N.
- D. Dimmel, in Lignin and Lignans: Advances in Chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press, 2010, pp. 1–10 Search PubMed.
- B. Joffres, D. Laurenti, N. Charon, A. Daudin, A. Quignard and C. Geantet, Oil Gas Sci. Technol., 2013, 68, 753–763, DOI:10.2516/ogst/2013132.
- D. Meier, R. Ante and O. Faix, Bioresour. Technol., 1992, 40, 171–177 CrossRef CAS.
- D. Meier, J. Berns, O. Faix, U. Balfanz and W. Baldauf, Biomass Bioenergy, 1994, 7, 99–105 CrossRef CAS.
- A. Oasmaa, R. Alen and D. Meier, Bioresour. Technol., 1993, 45, 189–194 CrossRef CAS.
- J. Pepper and Y. Lee, Can. J. Chem., 1969, 47, 723–727, DOI:10.1139/v69-118.
- A. Kloekhorst, Y. Shen, Y. Yie, M. Fang and H. J. Heeres, Biomass Bioenergy, 2015, 80, 147–161, DOI:10.1016/j.biombioe.2015.04.039.
- A. Kloekhorst and H. J. Heeres, ACS Sustainable Chem. Eng., 2015, 3, 1905–1914, DOI:10.1021/acssuschemeng.5b00041.
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Green Chem., 2015, 17, 4921–4930, 10.1039/c5gc01641j.
- R. J. A. Gosselink, Lignin as a renewable aromatic resource for the chemical industry, Wageningen University, Wageningen, 2011 Search PubMed.
- R. W. Thring, M. N. Vanderlaan and S. L. Griffin, J. Wood Chem. Technol., 1996, 16, 139–154 CrossRef CAS.
- A. R. Ardiyanti, Hydrotreatment of Fast Pyrolysis Oil, Catalyst Development and Process-Product Relations, PhD thesis, University of Groningen, Groningen, 2013 Search PubMed.
- F. Tran, C. S. Lancefield, P. C. J. Kamer, T. Lebl and N. J. Westwood, Green Chem., 2015, 17, 244–249, 10.1039/C4GC01012D.
- J. Hu, R. Xiao, D. Shen and H. Zhang, Bioresour. Technol., 2013, 128, 633–639, DOI:10.1016/j.biortech.2012.10.148.
- L. Ingram, D. Mohan, M. Bricka, P. Steele, D. Strobel, D. Crocker, B. Mitchell, J. Mohammad, K. Cantrell and C. U. Pittman, Energy Fuels, 2008, 22, 614–625 CrossRef CAS.
- Z. Xia, L. Akim and D. Argyropoulos, J. Agric. Food Chem., 2001, 49, 3573–3578, DOI:10.1021/jf010333v.
- D. Robert, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer Berlin Heidelberg, 1992, pp. 250–273 Search PubMed.
- W. Willker and D. Leibfritz, Magn. Reson. Chem., 1998, 36, S79–S84, DOI:10.1002/(SICI)1097-458X(199806)36:133.0.CO;2-Z.
- J. Wen, B. Xue, F. Xu, R. Sun and A. Pinkert, Ind. Crops Prod., 2013, 42, 332–343, DOI:10.1016/j.indcrop.2012.05.041.
- J. Wen, S. Sun, B. Xue and R. Sun, Materials, 2013, 6, 359–391, DOI:10.3390/ma6010359.
- J. C. del Rio, J. Rencoret, G. Marques, J. Li, G. Gellerstedt, J. Jimenez-Barbero, A. T. Martinez and A. Gutierrez, J. Agric. Food Chem., 2009, 57, 10271–10281, DOI:10.1021/jf900815x.
- T. Yuan, S. Sun, F. Xu and R. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614, DOI:10.1021/jf2031549.
- J. Ralph and L. Landucci, in Lignin and Lignans: Advances in Chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press, 2010, pp. 137–243 Search PubMed.
- M. Osada, O. Sato, K. Arai and M. Shirai, Energy Fuels, 2006, 20, 2337–2343, DOI:10.1021/ef060356h.
- M. Snare, I. Kubickova, P. Maki-Arvela, K. Eranen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715, DOI:10.1021/ie060334i.
- D. C. Elliott and T. R. Hart, Energy Fuels, 2009, 23, 631–637, DOI:10.1021/ef8007773.
- J. Wildschut, I. Melian-Cabrera and H. J. Heeres, Appl. Catal., B, 2010, 99, 298–306, DOI:10.1016/j.apcatb.2010.06.036.
- D. C. Elliott, L. J. Sealock and E. G. Baker, Ind. Eng. Chem. Res., 1993, 32, 1542–1548, DOI:10.1021/ie00020a002.
- R. H. Venderbosch, A. R. Ardiyanti, J. Wildschut, A. Oasmaa and H. J. Heeres, J. Chem. Technol. Biotechnol., 2010, 85, 674–686, DOI:10.1002/jctb.2354.
- J. Ekerdt and A. Bell, J. Catal., 1979, 58, 170–187, DOI:10.1016/0021-9517(79)90255-0.
- J. H. Marsman, A. Kloekhorst, L. Rohrbach and H. J. Heeres, Manuscript in preparation, 2015 Search PubMed.
- J. H. Marsman, J. Wildschut, F. Mahfud and H. J. Heeres, J. Chromatogr. A, 2007, 1150, 21–27 CrossRef CAS PubMed.
- M. Brebu and C. Vasile, Cellul. Chem. Technol., 2010, 44, 353–363 CAS.
- P. de Wild, R. Van der Laan, A. Kloekhorst and E. Heeres, Environ. Prog. Sustainable Energy, 2009, 28, 461–469, DOI:10.1002/ep.10391.
- A. Klemola and G. Nyman, Pap. Puu-Pap. Och. Tra., 1966, 48, 595–603 CAS.
- M. Wayman and M. Chua, Can. J. Chem., 1979, 57, 2599–2602, DOI:10.1139/v79-420.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy00523c |
| This journal is © The Royal Society of Chemistry 2016 |