
Competition between molecular and dissociative adsorption of hydrogen on palladium clusters deposited on defective graphene
Alejandra Granjaa,
Julio A. Alonsoab,
Iván Cabriaa and
María J. López*a
aDepartamento de Física Teórica, Atómica y Óptica, Universidad de Valladolid, 47011, Valladolid, Spain. E-mail: maria.lopez@fta.uva.es; Fax: +34-983-423013
bDonostia International Physics Center, 20080 San Sebastián, Spain
First published on 8th May 2015
Abstract
The contribution of Pd doping to enhance the hydrogen storage capacity of porous carbon materials is investigated. Using the Density Functional Formalism, we studied the competition between the molecular adsorption and the dissociative chemisorption of H2 on Pd clusters anchored on graphene vacancies. The molecular adsorption of H2 takes place with energies in the range of 0.7–0.3 eV for adsorption of one to six hydrogen molecules. Six molecules saturate the cluster, and additional hydrogen could only be adsorbed, with much smaller adsorption energies, at farther distances from the cluster. Dissociative chemisorption is the preferred adsorption channel from one to three hydrogen molecules, with adsorption energies in the range of 1.2–0.6 eV. After the first three molecules are dissociatively chemisorbed, three additional hydrogen molecules can be adsorbed non-dissociatively onto the Pd cluster with adsorption energies of 0.5 eV. The desorption of Pd–H complexes is prevented in all cases because the Pd clusters are firmly anchored to graphene vacancies. Our results are very promising and show that Pd clusters anchored on graphene vacancies retain their capacity to adsorb hydrogen and completely prevent the desorption of Pd–H complexes that would spoil the hydrogen releasing step of the cycle.
1 Introduction
The successful storage of hydrogen is a technological requirement to boost its use in electric cars powered by hydrogen fuel cells. However, the technological problem of storing 5.5 wt% hydrogen1 at room temperature and moderate pressures remains elusive and efforts are being invested in different directions.2 The most promising technologies focus on the storage of hydrogen adsorbed in light solid materials, such as porous carbons. These materials exhibit a reasonably high storage capacity3,4 of about 6 wt% hydrogen at low temperatures (77 K) but their capacity drops dramatically to about 1 wt% hydrogen at room temperatures and moderate pressures, which is far from the technological requirement. Optimization of the size and shape of the pores yields some improvement5,6 but the hydrogen storage capacity of these materials remains too low for practical application. The main difficulty arises from the small adsorption energy,7–10 below 100 meV, of hydrogen to the pore walls.A recent experimental work by Contescu et al.11 indicated an enhancement in the hydrogen storage capacity of porous carbon materials doped with palladium. It is, therefore, of great interest to understand and explain the mechanisms through which the Pd dopant contributes to the enhancement of the storage capacity of these materials. Recent computer simulations of the structure of nanoporous carbons performed by some of the authors12 indicate that the walls of the pores are one atom thick planar or curved graphene-like layers containing defects. These results suggest that the pore walls of these materials can be modeled through a combination of pristine and defective graphene layers. Previous work has focused on the study of hydrogen adsorption on palladium atoms13,14 and clusters15 deposited on pristine graphene. We have shown15 that hydrogen adsorption takes place with enhanced adsorption energies, which seems to indicate a beneficial effect of palladium doping on the hydrogen storage capacity of porous carbons. However, since the bonding of the palladium clusters with the pristine graphene layer is relatively weak, about 1 eV, desorption of Pd–H complexes competes with the desorption of hydrogen. Evidently, the desorption of hydrogen is a key step in the storage cycle and, therefore, the desorption of Pd–H complexes may inhibit the beneficial effect of Pd doping.
We have proposed,16 as a way to overcome this difficulty, to attach the Pd atoms and clusters to defects of the graphene layer, for instance to graphene vacancies. Pd atoms and clusters attach strongly to the vacancies, where they get firmly anchored to the graphene layer. Preliminary results on the adsorption of molecular hydrogen on a single Pd atom saturating a graphene vacancy16 or a vacant site in the wall of a carbon nanotube,17 CNT, were quite promising. In this paper we have investigated the effect that anchoring small Pd clusters on graphene vacancies has on the adsorption/desorption of molecular hydrogen. This effect cannot be inferred from previous studies on pristine graphene. We have performed simulations, using the Density Functional Formalism, investigating the different adsorption channels, molecular and dissociative, of hydrogen on Pd clusters anchored on graphene vacancies, and the competition between those channels as a function of the number of adsorbed molecules. We have also studied the desorption step and the competition between desorption of hydrogen and desorption of Pd–H complexes. In Section 2 we present the key features of the Density Functional Formalism used in our computer simulations. Section 3 presents the results and we finish with some Conclusions in Section 4.
2 Theoretical model
We investigated the adsorption of hydrogen on a Pd6 cluster anchored on a graphene vacancy using the Density Functional Formalism (DFT). The graphene layer is considered here as a model of the graphitic walls of nanoporous carbon materials, and the vacancies simulate defects of the walls. As we have shown in previous works,15,18 palladium exhibits a strong tendency to aggregate and form three dimensional clusters on the surface of graphene. The clusters grow preferentially in the neighborhood of vacancies of the graphene layer, where they attach strongly. The DFT calculations were performed with the DACAPO code.19 The code implements the supercell methodology. The graphene layer is represented by a supercell containing 5 × 5 hexagonal unit cells, each of which contains two C atoms (see Fig. 1). Thus the supercell size in the X direction is 12.33 Å. In the Z direction the supercell is taken to be large enough (14 Å) to avoid interactions between the images of the graphene layer in different supercells. The interactions of the valence electrons with the nuclear cores are described through Vanderbilt ultrasoft pseudopotentials.20 A basis set of plane waves is used to expand the wave functions and the electronic density, with cutoff values of 350 eV and 1000 eV, respectively, for good convergence. We considered four k points in the first Brillouin zone, following the Monkhorst–Pack scheme.21 Since the supercells used in the calculations are quite large, this selection is sufficient to guarantee convergence in the cohesive energies better than 10 meV. The generalized gradient approximation of Perdew and Wang (GGA-PW91)22 for the exchange–correlation functional is employed. An extensive search on the possible adsorption sites of the hydrogen molecules and of the hydrogen atoms of the dissociated molecule on the Pd6 cluster has been performed. The search included the adsorption of hydrogen molecules and of hydrogen atoms on the vertices, edges and faces of the Pd6 cluster. Then, all the structures, the graphene layer with the vacant site, the Pd6 on the graphene monovacancy and the molecular and dissociated hydrogen adsorbed on the supported palladium clusters, were fully optimized until the forces acting on all the atoms were smaller than 0.05 eV Å−1.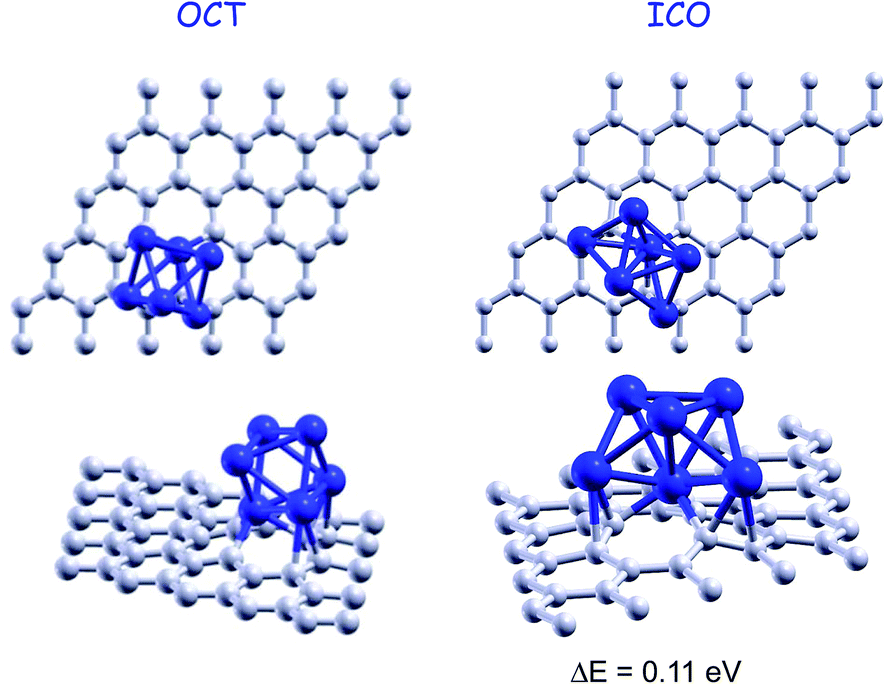 | ||
| Fig. 1 Top and side views of the octahedral structure, OCT, and the icosahedral-type structure, ICO, of Pd6 adsorbed on a graphene vacancy. The OCT structure binds to the graphene vacancy with an energy of 5.62 eV. The adsorbed ICO structure is 0.11 eV higher in energy than the OCT structure. | ||
3 Results
We investigated the mechanisms of adsorption of hydrogen on Pd6 clusters bound to a graphene vacancy. Pd6 clusters are taken as representatives for clusters in the small size range. The lowest energy octahedral, OCT, structure of free Pd6 clusters experiences only a minor distortion upon deposition on a graphene layer with a vacancy, although it adsorbs with a substantial energy of 5.62 eV (see Fig. 1). Pd6 rests supported on one of its triangular faces. One of the Pd atoms of this face is sitting above the center of the vacancy, saturating the dangling bonds of the three C atoms around the vacant site. The other two Pd atoms are above two C–C bonds around the vacancy. The graphene layer distorts about the vacancy and the adsorbed Pd cluster. The C atoms in direct contact with the Pd-saturated vacancy get out of plane (in the direction of the Pd cluster) about 0.5–0.7 Å and the next shell of C atoms (counted from the vacancy) about 0.3–0.4 Å. The magnetic moment μ = 2μB of the free Pd6 cluster is quenched down to zero upon adsorption on the vacancy. This is in contrast with Pd6 deposited on pristine graphene, which retains the magnetic moment of the free cluster. Molecular hydrogen may adsorb on the Pd6 clusters anchored on graphene vacancies following two adsorption modes: (i) molecular adsorption, in which the molecule is slightly activated but the hydrogen atoms remain bound to each other and (ii) dissociative adsorption, in which the molecule is broken and each hydrogen atom chemisorbs independently to the Pd cluster. These two adsorption modes were also found for hydrogen adsorption on palladium clusters supported on pristine graphene. We have studied in detail the competition between the two adsorption channels for palladium clusters anchored to a graphene vacancy as a function of the hydrogen content, that is, as additional hydrogen molecules adsorb into the clusters. The adsorption energies of the successively attached hydrogen molecules to the Pd clusters are defined as| Enthad(H2) = E[(n − 1)H2 + Pd6 on Gvac] + E(H2) − E[nH2 + Pd6 on Gvac], | (1) |
3.1 Molecular adsorption of hydrogen
The preferred site for the molecular adsorption of H2 is on top of one of the Pd atoms which is not in direct contact with the graphene surface. The same behaviour was found for hydrogen adsorption on Pd6 deposited on pristine graphene. The adsorption energy is 0.74 eV, somewhat higher than the adsorption energy, 0.56 eV, on Pd6 supported on pristine graphene. The hydrogen molecule is slightly activated and the H–H distance becomes 0.86 Å. A slightly smaller activation, with H–H distances of 0.79–0.80 Å, is found for hydrogen molecules adsorbed on single Pd atoms saturating graphene or CNT vacancies.16,17 The H–H distance of the free H2 molecule is 0.75 Å. The H–H bond is weakened but not broken. One hydrogen molecule can adsorb on top of any of the Pd atoms of the cluster, except on top the Pd atom which is saturating the vacancy. The adsorption energies are quite similar, about 0.7 eV, for the three Pd atoms which are not in direct contact with the graphene layer, and are substantially reduced to 0.3–0.4 eV for the two Pd atoms directly in contact with the graphene surface.Fig. 2 shows the structures of the successive adsorption of molecular hydrogen on the OCT structure of Pd6 anchored on a graphene vacancy. With the first molecule adsorbed, a second hydrogen molecule adsorbs preferentially on top of a different Pd atom, with a practically identical adsorption energy, 0.73 eV. Adsorption of a second molecule on top of the same Pd atom as the first one leads to a smaller adsorption energy of 0.44 eV. A somewhat lower adsorption energy, 0.53 eV, than the first two molecules is found for adsorption of the third hydrogen molecule on top of the remaining Pd atom which is not in direct contact with the graphene surface. On the other hand, adsorption on one of the occupied vertices leads to a smaller adsorption energy, 0.40 eV. The fourth and fifth hydrogen molecules can still be adsorbed on the two Pd atoms which are not occupied by previous hydrogen molecules, the two Pd atoms supported on the graphene layer, with adsorption energies of 0.47 and 0.34 eV, respectively. However the fourth molecule has a slightly higher adsorption energy, 0.50 eV, for adsorption on one of the vertices which is not in direct contact with the graphene surface, although it is already occupied by another H2 molecule. Then the fifth molecule adsorbs on one of the Pd atoms in contact with the graphene surface with an energy of 0.36 eV, and a sixth molecule may adsorb on the remaining Pd atom in contact with graphene with an energy of 0.34 eV. All these results are summarized in Table 1. The distance between the hydrogen molecules and the corresponding Pd atoms where they are attached is quite constant, about 1.7–1.8 Å, for the first 6 hydrogen molecules adsorbing onto the Pd6 cluster supported on the graphene vacancy. This distance is about 10% smaller than the distance of 2.0 Å between the hydrogen molecule and a single Pd atom saturating a graphene16 or a CNT17 vacancy. This indicates a stronger interaction of the hydrogen molecules with the Pd clusters than with the Pd atoms saturating carbon vacancies.
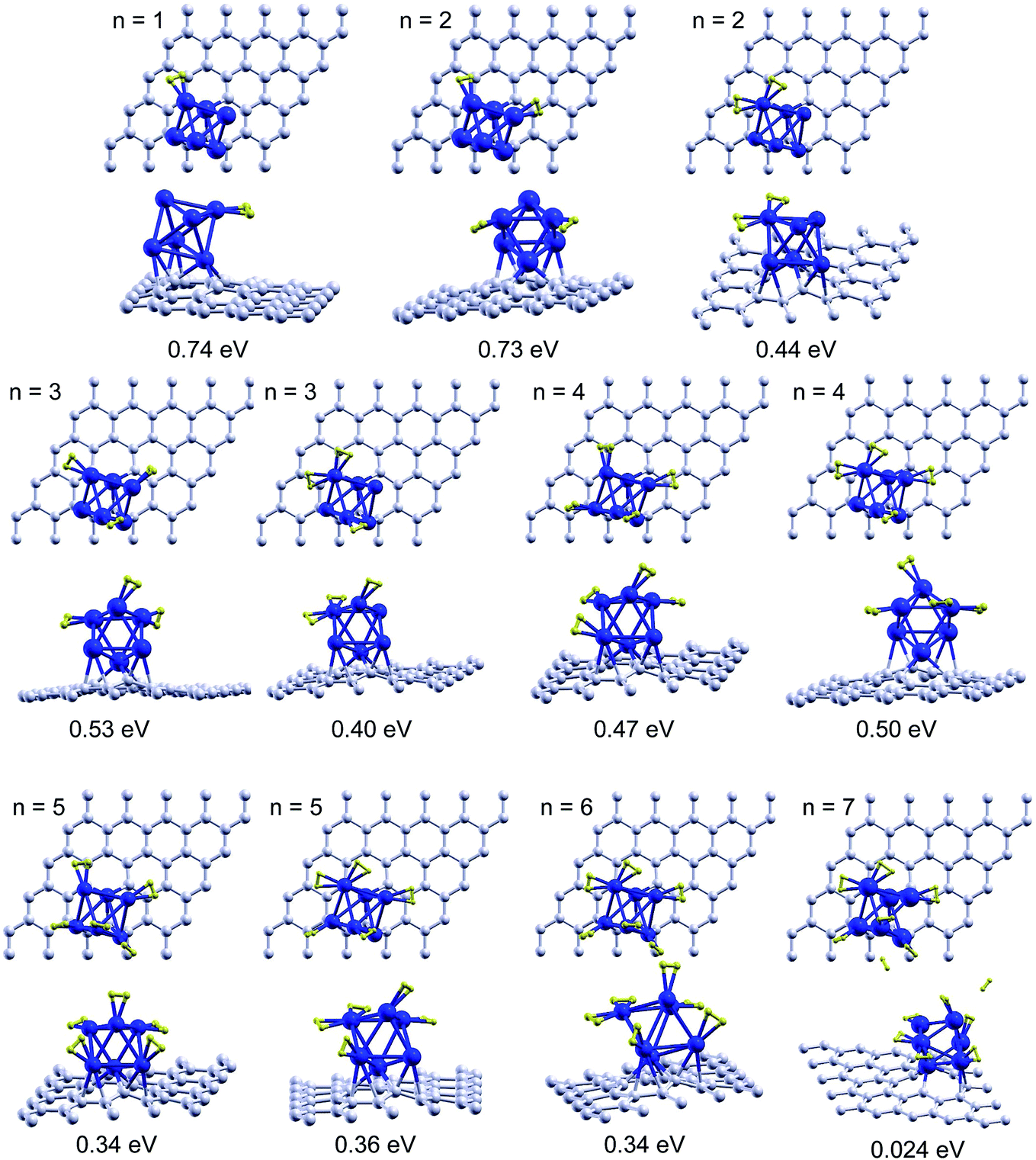 | ||
| Fig. 2 Top and side views of successive molecular adsorption of hydrogen on the OCT structure of Pd6 anchored on a graphene vacancy. The reported energies are the adsorption energies of the last hydrogen molecule calculated using eqn (1). | ||
| n | Different vertices | Two in same vertex |
|---|---|---|
| Enthad | Enthad | |
| 1 | 0.74* | |
| 2 | 0.73* | 0.44 |
| 3 | 0.53* | 0.40 |
| 4 | 0.47 | 0.50* |
| 5 | 0.34 | 0.36* |
| 6 | 0.34* |
Direct adsorption of six hydrogen molecules seems to be the saturation limit on Pd6 anchored on a vacancy. Because of the steric effects produced by the presence of the supporting graphene layer and by the previously bound hydrogen molecules, the seventh molecule cannot attach directly to the Pd cluster. This molecule will begin forming a second hydrogen shell around the Pd cluster, at a substantially longer distance, 3.2 Å, than the hydrogen molecules of the first layer, and with quite a small adsorption energy of the order of a few tens of meV. This adsorption strength is similar to that found for the second hydrogen layer covering pure carbon nanotubes.23 We are not investigating this second layer because molecules having such small adsorption energies are not relevant for hydrogen storage at normal temperatures.
3.2 Dissociative chemisorption of hydrogen
The dissociative chemisorption channel is preferred over the molecular adsorption of hydrogen. The hydrogen molecule dissociates and the two hydrogen atoms adsorb on a face and an edge of the octahedron, respectively. The chemisorption energy of 1.03 eV is substantially larger than the molecular adsorption energy. The adsorption of the hydrogen atoms on two faces of the octahedron is slightly less stable, by only 0.05 eV (see Fig. 3). Those structures, however, do not correspond to the lowest energy configuration. The dissociative chemisorption of hydrogen induces a structural transition in the palladium cluster from the octahedral structure to the structure of an incomplete pentagonal bipyramid in which one of the atoms of the pentagonal base is missing. One of the apex atoms of the bipyramid saturates the vacant site of graphene and the two base Pd atoms next to the empty base position attach to two C–C bonds, respectively, around the vacancy. We will refer to this Pd6 structure as the ICO structure, because it can be viewed as part of an icosahedron (see Fig. 1). We find a distortion of the graphene layer around the ICO structure of Pd6 similar to that about the OCT structure. The ICO structure of the free Pd6 cluster has a cohesive energy 0.29 eV lower (less stable) than the octahedral structure. Upon adsorption on a graphene vacancy the ICO structure gains some stability but it is still 0.11 eV less stable than the supported octahedron. It is the dissociative adsorption of hydrogen that prompts the structural change. The two hydrogen atoms attach to the two non-adjacent triangular faces at the side of the bipyramid whose apex atom is not the one saturating the vacancy (upper side of the bipyramid, for further reference). The hydrogen chemisorption energy, 1.25 eV per molecule, is 0.22 eV higher than the adsorption energy of the dissociated molecule on the octahedral structure. The chemisorbed structures are shown in Fig. 3. The dissociative chemisorption of hydrogen is also the preferred adsorption channel on Pd6 supported on pristine graphene, with an energy of 1.30 eV. However, in this case the chemisorption of hydrogen does not induce a structural change in the Pd cluster.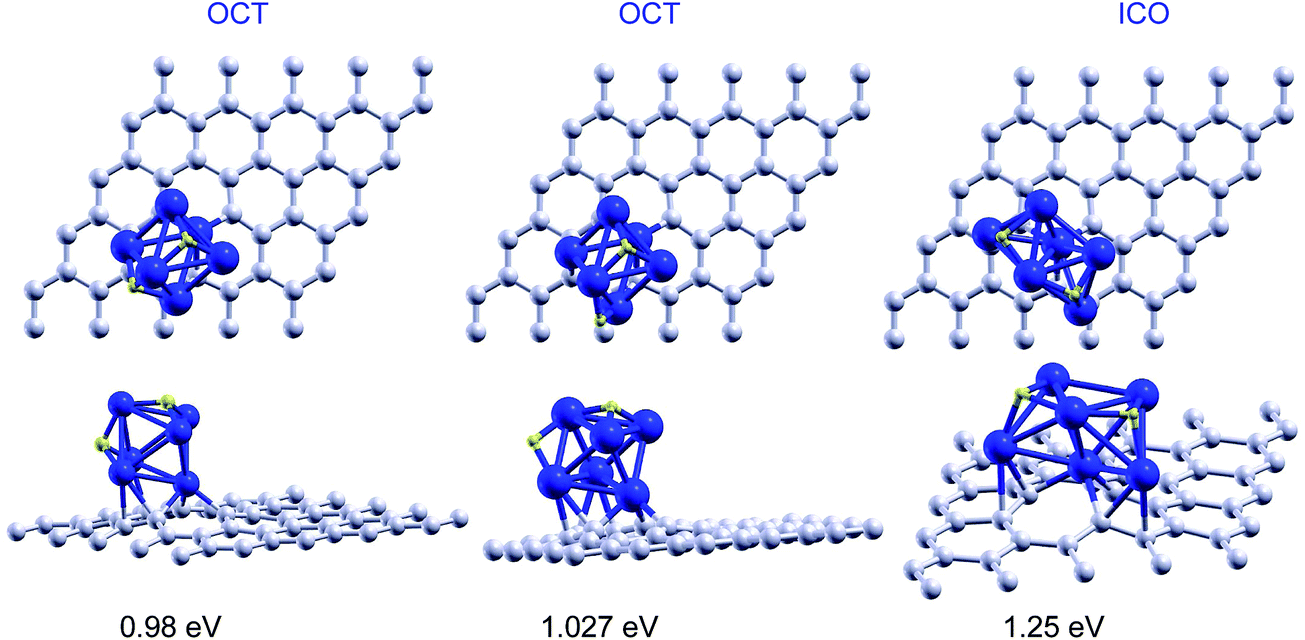 | ||
| Fig. 3 Top and side views of the dissociative chemisorption of one hydrogen molecule on the OCT and ICO structures of Pd6 anchored on a graphene vacancy. The reported energies are the adsorption energies calculated using eqn (1) for n = 1. | ||
3.3 Competition between dissociative chemisorption and molecular adsorption of hydrogen
We investigated the adsorption of several hydrogen molecules, and the competition between dissociative chemisorption and molecular adsorption. We started with the lowest energy ICO structure of Pd6 supported on a graphene vacancy with one dissociatively chemisorbed hydrogen molecule, and investigated the adsorption of additional molecules. The structures are shown in Fig. 4. A second hydrogen molecule adsorbs preferentially on the upper side of the bipyramid following the dissociative chemisorption channel, with an adsorption energy of 0.90 eV. This energy is reduced slightly with respect to the chemisorption of the first molecule. One of the hydrogen atoms becomes attached to the remaining face of the upper side of the bipyramid, and the second atom to one Pd–Pd bond of the bipyramid base, and one of the previously attached H atoms moves a little from a face position to above a Pd–Pd bond. This molecule may also attach as a nondissociated molecule on top of one of the Pd atoms of the base not in direct contact with the graphene layer, with almost the same energy of 0.87 eV. For adsorption of a third molecule, the two adsorption channels are almost degenerate in energy. The adsorption energies of a third molecule on Pd6 with two dissociated hydrogen molecules are 0.61 eV (molecular adsorption) and 0.58 eV (chemisorption). The dissociative channel is only 30 meV less stable than the molecular channel, which is within the limits of accuracy of our calculations. The dissociative chemisorption of the third molecule changes the adsorption site of one H atom, pushing it out of face. Then, finally, one H atom is attached to a face and the remaining H atoms attach to different Pd–Pd bonds. A fourth molecule cannot dissociate on Pd6 with three dissociated hydrogen molecules. The adsorption energy is negative, −0.27 eV, which means that this is not a bound state with respect to a desorbed hydrogen molecule. Instead, the fourth molecule attaches as a molecule to one of the Pd atoms of the cluster with an adsorption energy of 0.51 eV. Almost degenerate in energy, although slightly more stable, 0.52 eV, is the configuration with two dissociated molecules and two non-dissociated adsorbed molecules. The successive molecular adsorption of the fifth and sixth hydrogen molecules on the Pd6 with three dissociated and one non-dissociated adsorbed hydrogen molecules takes place with adsorption energies of 0.52 eV and 0.42 eV, respectively. The adsorption energies drop dramatically down to 0.16 eV, 0.10 eV, and 0.09 eV, respectively, for the successive adsorption of three more hydrogen molecules. Adsorption energies below 0.2 eV are of little interest for the storage of hydrogen at room temperature. In summary, Pd6 is able to chemisorb dissociatively up to three hydrogen molecules, and to adsorb, with reasonably high energies, three non-dissociated additional molecules. These results are summarized in the columns labelled as ICO of Table 2. The transition from stable dissociative chemisorption to molecular adsorption takes place when all the favorable sites of the upper side of the bipyramid (faces and Pd–Pd bonds not in direct contact with the graphene surface and not involving the Pd atom saturating the vacancy) have been filled. Note that due to steric effects it is not possible to fill out all the favorable faces and edges. For instance, the adsorption of H atoms on two edges of a triangle prevents the adsorption of a H atom on the face. | ||
| Fig. 4 Top and side views of successive adsorption of hydrogen on the ICO structure of anchored Pd6 with one dissociatively chemisorbed hydrogen molecule. Both the molecular and the dissociative channels are shown from n = 2 to n = 4, and only the molecular channel is shown for n = 5–9. The adsorption energies of the last hydrogen molecule are calculated using eqn (1). | ||
| n | ICO dissociative channel Enthad | ICO molecular channel Enthad | OCT dissociative channel Enthad | OCT molecular channel Enthad |
|---|---|---|---|---|
| n dissociated | 1 molecular | n dissociated | 1 molecular | |
| 1 | 1.25* | 1.03* | 0.74 | |
| 2 | 0.90* | 0.87 | 0.81* | 0.78 |
| 3 | 0.58 | 0.61* | 0.65* | 0.62 |
| 4 | −0.27 | 0.51 | 0.03 | 0.56* |
| (n − 2) molecular | ||||
| 5 | 0.52* | |||
| (n − 3) molecular | (n − 3) molecular | |||
| 6 | 0.52* | 0.48* | ||
| 7 | 0.42* | 0.50* | ||
| 8 | 0.16* | |||
| 9 | 0.10* | |||
| 10 | 0.09* | |||
For completeness, we also studied the competition between dissociative and molecular adsorption on the octahedral structure, OCT, of the anchored Pd6 cluster with one dissociatively chemisorbed hydrogen molecule. The dissociative channel is preferred for the adsorption of the second hydrogen molecule, with an adsorption energy of 0.81 eV, whereas the molecular adsorption energy is 0.78 eV. The third hydrogen molecule adsorbs on Pd6 with similar energies of 0.65 eV and 0.62 eV for the dissociative and the molecular channels, respectively. In contrast to the ICO case, the fourth molecule can dissociate on the octahedral Pd6 cluster with three dissociated hydrogen molecules, but the adsorption energy is very small, 0.03 eV, with respect to the desorbed molecule. However the fourth molecule can attach as a non-dissociated molecule with an energy of 0.56 eV. The fifth and sixth hydrogen molecules can also attach as non-dissociated molecules with adsorption energies of 0.48 eV and 0.50 eV, respectively. A seventh molecule cannot attach directly to the Pd cluster and begins to form a second layer at a larger distance and with a much lower adsorption energy. Those structures are shown in Fig. 5 and the results are summarized in the columns labelled as OCT of Table 2.
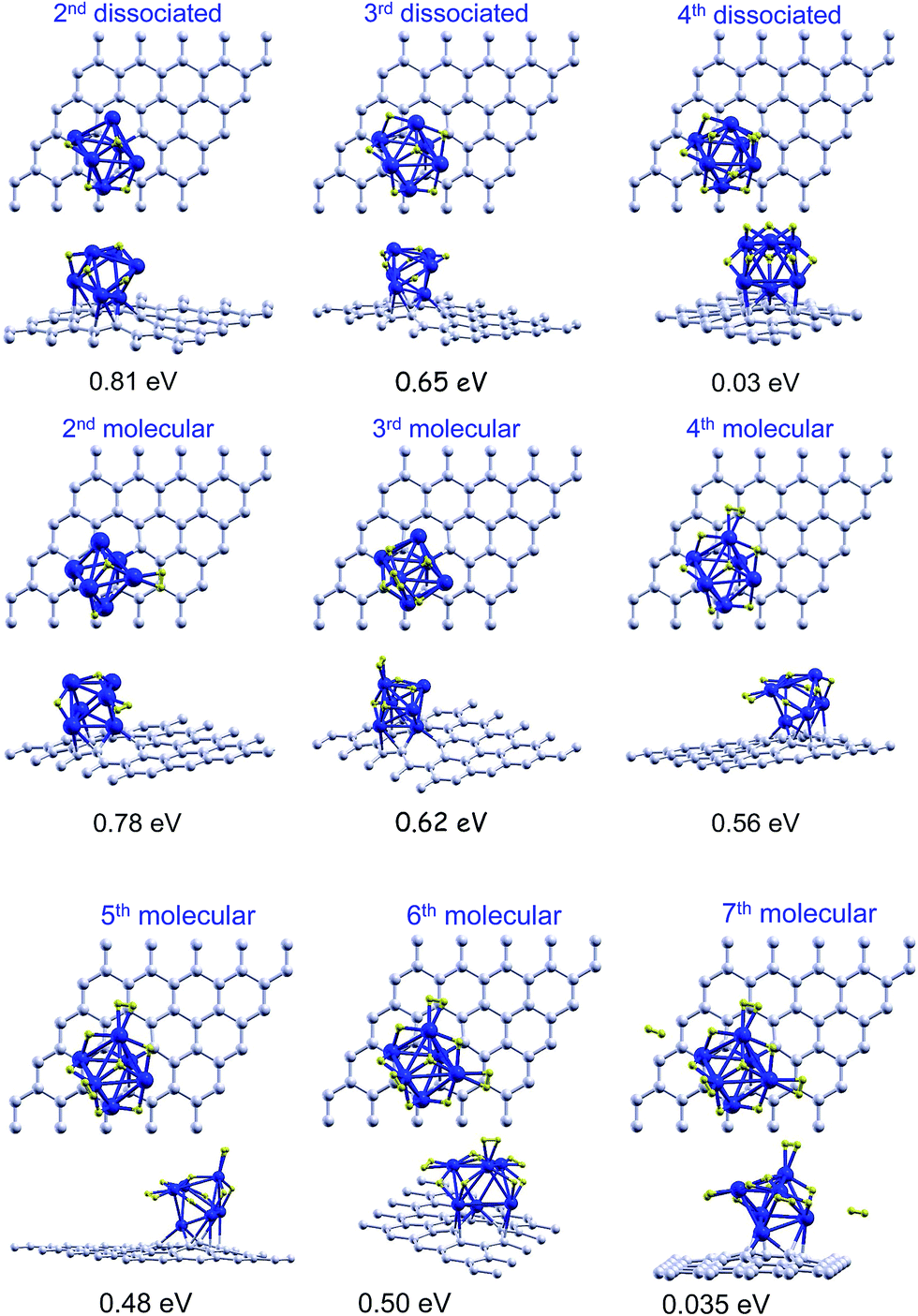 | ||
| Fig. 5 Top and side views of successive adsorption of hydrogen on the OCT structure of anchored Pd6 with one dissociatively chemisorbed hydrogen molecule. Both the molecular and the dissociative channels are shown from n = 2 to n = 4, and only the molecular channel is shown for n = 5–7. The adsorption energies of the last hydrogen molecule are calculated using eqn (1). | ||
From our results we conclude that the successive adsorption of hydrogen on Pd clusters anchored on graphene vacancies does not depend much on the structure of the Pd clusters, since a similar behaviour has been found for Pd6 in the ICO and the OCT structures. Once the Pd cluster is saturated with dissociated hydrogen molecules, it is still able to adsorb more hydrogen following the molecular channel. Therefore, the Pd clusters anchored on graphene vacancies retain their capacity to adsorb hydrogen. Clearly, the number of adsorbed hydrogen molecules in the supported clusters is smaller than that for free gas phase clusters24 due to the steric effects of the graphene layer. However, gas phase clusters are not appropriate models for hydrogen storage or other material applications. Pd doping contributes to an enhancement of the amount of hydrogen stored in a Pd-doped porous carbon material.
Further, the so-called spillover mechanism25 has been proposed as responsible for the enhancement. According to this mechanism, molecules adsorbed on a first surface are transported to a second surface that does not adsorb the molecules under the same conditions. Our results on the adsorption and saturation with hydrogen of supported Pd clusters correspond to the first step of the spillover mechanism. The transfer mechanism of hydrogen attached to Pd towards the graphene layer, where the adsorption energies of hydrogen are less than 100 meV, remains to be elucidated. It is also interesting to notice that in order to use the stored hydrogen as a fuel, molecular hydrogen has to be catalytically dissociated in the anode of the hydrogen fuel cell. Palladium and other transition metals are usually employed as catalysts in this dissociation step. Our results on the dissociation of H2 on supported palladium clusters provide a first hint of the role played by Pd clusters in the anode of hydrogen fuel cells.
3.4 Bonding between hydrogen and the Pd cluster
To investigate the nature of the bonding between hydrogen and the Pd cluster, both in the molecular and the dissociative channels, we calculated the electronic density difference between the system with adsorbed hydrogen and the two separated subsystems, formed by the Pd cluster anchored to the graphene vacancy on one hand and the free hydrogen molecule or free hydrogen atoms, in the proper positions, on the other. Fig. 6 shows the electronic density difference for the molecular adsorption of one, two and three hydrogen molecules on the OCT Pd6. There is an increase (yellow surface in the figure) of electronic density in the region between the hydrogen molecule and the Pd atom to which the molecule is attached, which indicates a covalent-type of bonding between the hydrogen molecule and the Pd atom. Moreover, some polarization of the electronic density is also apparent. It is interesting to notice that the same type of bonding is observed for an increasing number of hydrogen molecules attached to different Pd atoms and also in the case of two hydrogen molecules attached to the same Pd atom. This indicates that the hydrogen molecules bind independently to the Pd cluster. The top panels of Fig. 7 show the electronic density difference for the dissociative chemisorption of hydrogen on the ICO and the OCT structures of Pd6. Here, each hydrogen atom is embedded in a region of positive electronic density difference, that is, each hydrogen builds up an excess of electronic density around it. This type of bonding is typical of metal hydrides and could be interpreted as an ionic type of bonding. A similar behaviour is found in the case of two dissociatively chemisorbed hydrogen molecules on ICO Pd6 (see lower left panel of Fig. 7). Both bonding types can be observed simultaneously in the case of two adsorbed hydrogen molecules, one dissociatively chemisorbed and the second adsorbed as a non-dissociated molecule (see lower right panel of Fig. 7). This indicates that the two adsorption channels take place independently of each other, and do not involve the complete Pd cluster but only locally involve the nearby Pd atoms . It is also notable that the graphene layer does not participate actively in the adsorption of hydrogen on the anchored Pd clusters.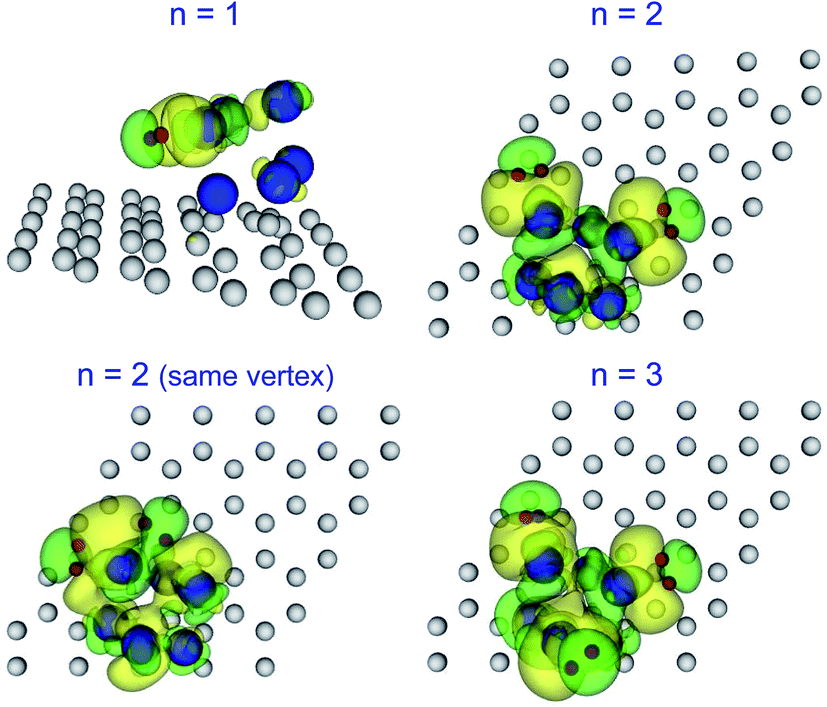 | ||
| Fig. 6 Electronic density difference between the system with adsorbed hydrogen and the separated subsystems, formed by the Pd cluster anchored to the graphene vacancy on one hand and the free hydrogen molecule (in the proper position) on the other, for the molecular adsorption of n = 1–3 hydrogen molecules on the OCT structure of Pd6. The yellow isosurfaces correspond to positive values of the electronic density difference and the green isosurfaces to negative values. Red, blue and grey balls represent H, Pd and C atoms, respectively. | ||
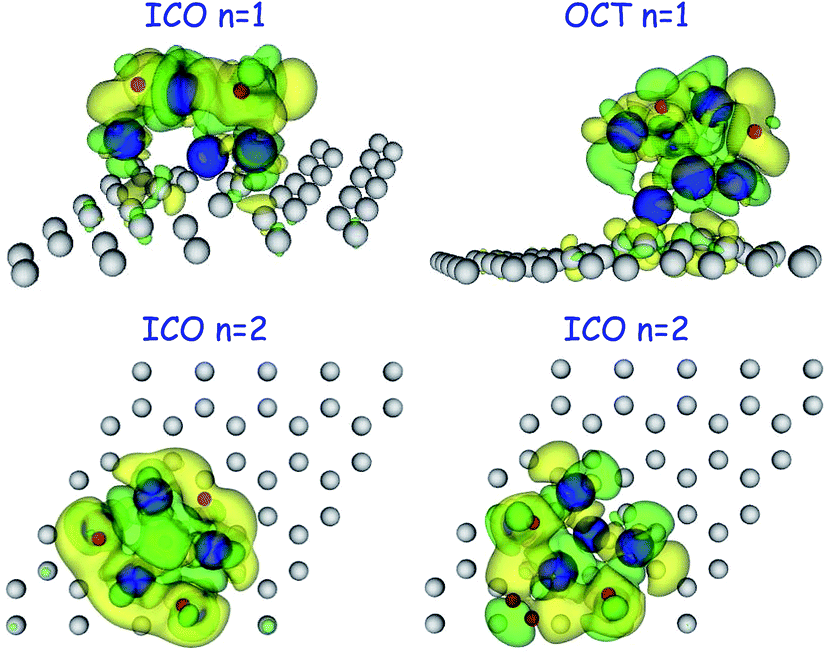 | ||
| Fig. 7 Electronic density difference between the system with adsorbed hydrogen and the separated subsystems formed by the Pd cluster anchored to the graphene vacancy on one hand and the free hydrogen atoms or the free molecule (in the proper position) on the other. Top panels are the dissociative chemisorption of one hydrogen molecule on the ICO (left) and the OCT (right) structure of Pd6. The lower panels are the adsorption of two hydrogen molecules: on the left, the two molecules are chemisorbed; on the right, one molecule is chemisorbed and the other adsorbed as a non-dissociated molecule. Yellow isosurfaces correspond to positive values of the electronic density difference and green isosurfaces to negative values. Red, blue and grey balls represent H, Pd and C atoms, respectively. | ||
Since the adsorption and dissociation of hydrogen is mainly due to local interactions with the palladium clusters, we expect similar adsorption and dissociation mechanisms on Pd clusters anchored on graphene vacancies and on Pd clusters deposited on pristine graphene. The hydrogen dissociation barriers in the latter are about 0.3 eV (ref. 15) and, therefore, similar values are expected for the dissociation of hydrogen on Pd clusters anchored on graphene vacancies.
3.5 Desorption of hydrogen
Let us now focus on the desorption step of the hydrogen storage cycle. The desorption energy of hydrogen is equal to the adsorption energy. This energy was compared with the energy required to desorb the Pd6–H2 complexes for both the molecular adsorption and the dissociative chemisorption channels. The desorption energy of the Pd6–H2 complex from the graphene layer with a vacancy in the case of molecular adsorption on the OCT structure of Pd6 is 5.66 eV. Similar energies, 5.62 and 5.89 eV, are found for desorption of the Pd6–H2 complex when hydrogen is dissociatively chemisorbed on the OCT and ICO structures of Pd6, respectively. All those energies are much higher than the desorption energies of the H2 molecule: 0.74 eV in the case of molecular adsorption on OCT Pd6, and 1.03 and 1.25 eV for the cases of dissociative chemisorption on the OCT and ICO structures of Pd6, respectively. Therefore in the desorption step of the storage cycle, only hydrogen will be released because the energies required to release the Pd–H complexes are too high. This is in contrast with the desorption behaviour found for Pd clusters deposited on pristine graphene. There, the desorption energies of the Pd–H complexes are similar to the desorption energies of hydrogen molecules (roughly between 0.5 and 1 eV) and, therefore, desorption of the complexes competes with the desorption of hydrogen, thus spoiling the beneficial effect of the Pd dopant on the storage capacity of the nanoporous carbon materials. Here, the Pd clusters are firmly anchored to a graphene vacancy (5.62 eV), which prevents the desorption of the Pd–H complexes. As a conclusion we find that anchoring Pd6 to a graphene vacancy retains the capabilities of the cluster to adsorb hydrogen but completely inhibits the possibility of desorption of the Pd–H complexes. A similar behaviour was found for Pd atoms anchored on graphene vacancies. This solves the problem of desorption of Pd–H complexes that was present in the case of Pd atoms and clusters supported on pristine graphene.4 Conclusions
Hydrogen adsorption on nanoporous carbon materials is one of the most promising technologies for hydrogen storage. In this paper we have investigated the adsorption, dissociation and desorption of hydrogen from Pd6 clusters anchored on graphene vacancies, on the basis of the Density Functional Formalism. A graphene layer with a vacancy was used as a model of the defective pore walls of nanoporous carbons and the Pd6 cluster was considered as representative of small-size Pd clusters. The effect of vacancies on hydrogen adsorption cannot be assessed from studies of Pd clusters supported on pristine graphene. Pd6 anchored on a vacancy may attach directly up to six hydrogen molecules with adsorption energies in the range of 0.3–0.7 eV. Six adsorbed molecules saturate the cluster and, due to steric effects, additional hydrogen molecules cannot attach directly to the Pd cluster and will begin to form a second hydrogen shell at a larger distance from the cluster and very weakly bound to it. The dissociative chemisorption of hydrogen is preferred over the molecular adsorption for adsorption from one to three hydrogen molecules. The dissociative adsorption energies are in the range of 0.6–1.2 eV. The six atoms of three hydrogen molecules saturate all the favorable sites of the Pd cluster for chemisorption: the faces and Pd–Pd bonds not in direct contact with the graphene layer and not involving the Pd atom which saturates the graphene vacancy. However, not all those faces and edges can be simultaneously filled due to steric effects. Three additional hydrogen molecules can be adsorbed as non-dissociated molecules onto the Pd cluster with three chemisorbed molecules, with adsorption energies of 0.5 eV. The desorption of hydrogen takes place with desorption energies in the range of 0.3–1.2 eV. Furthermore, since the Pd cluster is firmly anchored to the graphene vacancy, desorption of the Pd–H complexes requires much higher energies, by a factor of 5, than the desorption of hydrogen. Then, the desorption step of the storage cycle involves only desorption of hydrogen. Therefore, anchoring the Pd clusters to vacancies is very promising because (i) the anchored Pd clusters retain their capacity for adsorbing hydrogen, and (ii) it completely prevents the desorption of Pd–H complexes, which was a problem in the case of Pd clusters supported on pristine graphene. Our results on the dissociative adsorption channel are also of interest for the catalytic dissociation process in the anode of the hydrogen fuel cells.Acknowledgements
This work was supported by MICINN of Spain (Grant MAT2011-22781) and Junta de Castilla y León (Grant VA050U14).References
- DOE, Multi-Year Research, Development and Demonstration Plan: Planned Program Activities for 2005-2015. Technical Plan–Storage. Updated April 2009, http://www1.eere.energy.gov/hydrogenandfuelcells/mypp/pdfs/storage.pdf, 2009.
- P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 CrossRef CAS.
- Y. Gogotsi, R. K. Dash, G. Yushin, T. Yildirim, G. Laudisio and J. E. Fischer, J. Am. Chem. Soc., 2005, 127, 16006–16007 CrossRef CAS PubMed.
- A. Linares-Solano, M. Jordá-Beneyto, D. L.-C. M. Kunowsky, F. Suárez-García and D. Cazorla-Amorós, in Carbon Materials: Theory and Practice, ed. P. G. A. P. Terzyk and P. Kowalczyk, Research Signpost, Kerala, India, 2008, pp. 245–281 Search PubMed.
- I. Cabria, M. J. López and J. A. Alonso, Carbon, 2007, 45, 2649–2658 CrossRef CAS PubMed.
- I. Cabria, M. J. López and J. A. Alonso, Int. J. Hydrogen Energy, 2011, 36, 10748–10759 CrossRef CAS PubMed.
- J. S. Arellano, L. M. Molina, A. Rubio and J. A. Alonso, J. Chem. Phys., 2000, 112, 8114–8119 CrossRef CAS PubMed.
- J. A. Alonso, J. S. Arellano, L. M. Molina, A. Rubio and M. J. López, IEEE Trans. Nanotechnol., 2004, 3, 304–310 CrossRef.
- B. K. Pradhan, G. U. Sumanasekera, K. W. Adu, H. E. Romero, K. A. Williams and P. C. Eklund, Phys. B, 2002, 323, 115–121 CrossRef CAS.
- G. E. Froudakis, J. Phys.: Condens. Matter, 2002, 14, R453 CrossRef CAS.
- C. I. Contescu, K. van Benthem, S. Li, C. S. Bonifacio, S. J. Pennycook, P. Jena and N. C. Gallego, Carbon, 2011, 49, 4050–4058 CrossRef CAS PubMed.
- M. J. López, I. Cabria and J. A. Alonso, J. Chem. Phys., 2011, 135, 104706 CrossRef PubMed.
- I. López-Corral, E. Germán, M. A. Volpe, G. P. Brizuela and A. Juan, Int. J. Hydrogen Energy, 2010, 35, 2377–2384 CrossRef PubMed.
- I. López-Corral, E. Germán, A. Juan, M. A. Volpe and G. P. Brizuela, J. Phys. Chem. C, 2011, 115, 4315–4323 Search PubMed.
- I. Cabria, M. J. López, S. Fraile and J. A. Alonso, J. Phys. Chem. C, 2012, 116, 21179–21189 CAS.
- M. J. López, I. Cabria and J. A. Alonso, J. Phys. Chem. C, 2014, 118, 5081–5090 Search PubMed.
- I. López-Corral, J. D. Celis, A. Juan and B. Irigoyen, Int. J. Hydrogen Energy, 2012, 37, 10156–10164 CrossRef PubMed.
- I. Cabria, M. J. López and J. A. Alonso, Phys. Rev. B, 2010, 81, 035403 CrossRef.
- Dacapo, see http://wiki.fysik.dtu.dk/dacapo for a description of the total energy code, based on the density functional theory, 2009.
- D. Vanderbilt, Phys. Rev. B, 1990, 41, R7892 CrossRef.
- H. Monkhorst and J. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- J. P. Perdew and Y. Wang, Phys. Rev. B, 1992, 45, 13244 CrossRef.
- I. Cabria, M. J. López and J. A. Alonso, Comput. Mater. Sci., 2006, 35, 238–242 CrossRef CAS PubMed.
- C. Zhou, S. Yao, J. Wu, R. C. Forrey, L. Chen, A. Tachibana and H. Cheng, Phys. Chem. Chem. Phys., 2008, 10, 5445–5451 RSC.
- W. Conner and J. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |