Adventures in corrole features by electrospray ionization mass spectrometry studies
Bernardo A. Iglesias
*,
Joana F. B. Barata
,
Catarina I. V. Ramos
,
M. Graça Santana-Marques
,
M. Graça P. M. S. Neves
and
José A. S. Cavaleiro
*
Department of Chemistry and QOPNA, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: bernardopgq@gmail.com; jbarata@ua.pt; c.ramos@ua.pt; santana.marques@ua.pt; gneves@ua.pt; jcavaleiro@ua.pt; Tel: +351 234370717
First published on 18th February 2014
Abstract
In this short review the importance of electrospray mass spectrometry in corrole chemistry is highlighted. ESI-MS and ESI-MS/MS are depicted as important tools for the identification and characterization of novel corrole derivatives. Metallocorrole chemistry and differentiation of corrole isomers via ESI-MS/MS and/or TWIM-MS were chosen as two focal points.
 Bernardo A. Iglesias | Bernardo A. Iglesias completed his BSc at the University of Santa Maria, Brazil in 2008 and his PhD at the University of São Paulo, Brazil in the Institute of Chemistry (2012) under the supervision of Prof. Koiti Araki. He joined José Cavaleiro's lab as a post-doctoral fellow to research corrole chemistry applications. He currently is a researcher at the University of Aveiro, Portugal in Organic Chemistry. |
 Joana F. B. Barata | Joana F. B. Barata received her BSc degree (2001) from University of Aveiro, Portugal. She obtained her MSc degree (2004) in Chemistry – Natural Products and Food Chemistry and her PhD degree (2009) in Chemistry at the University of Aveiro, both under the supervision of Prof. José Cavaleiro and Prof. Maria Graça Neves. Since 2010 she has been working with Prof. Tito Trindade, Prof. J. Cavaleiro and Prof. M. G. Neves as a post-doctoral research fellow. She has been awarded with the “Dow Portugal Prize” (2001) and PYC prize – Portuguese young chemists award (2010). Her research interests are the chemistry of corrole macrocycles and its application as nanomaterials. |
 Catarina I. V. Ramos | Catarina I. V. Ramos received her MSc in Biomolecular Advanced Methods and PhD in Chemistry, from the University of Aveiro, Portugal. Currently, she is a researcher in the Mass Spectrometry Centre of the University of Aveiro. Her current research interests concern the application of mass spectrometry to the study of non-covalent interactions of nucleic acids with neutral and multicharged porphyrinoid macrocycles. |
 M. Graça Santana-Marques | M. Graça Santana Marques is a retired professor of the University of Aveiro. Her research interests include the use of mass spectrometry in organic and inorganic chemistry, namely in the study of non-covalent interactions of coordination compounds and porphyrinoid macrocycles with oligonucleotides. |
 M. Graça P. M. S. Neves | Maria da Graça P. M. S. Neves is Associated Professor with Habilitation at the Department of Chemistry, University of Aveiro (UA). She obtained her Habilitation (Agregação) and PhD at UA, her Masters degree at UMIST, Manchester, UK and her degree (Licenciatura) in Chemistry at University of Lourenço Marques, Mozambique. Her research interests are centered on the synthesis functionalization and potential applications of tetrapyrrolic macrocycles like porphyrins, corroles and phthalocyanines. |
 José A. S. Cavaleiro | José. S. Cavaleiro got his BSc studies at Coimbra University, Portugal and his PhD degree at the Robert Robinson Laboratories, Liverpool University, UK. His academic career started at Coimbra University and later continued at Lourenço Marques (Mozambique) and Aveiro (Portugal) Universities. Since 1986 he has been Professor of Chemistry at the Aveiro University. José A. S. Cavaleiro is the recipient of several prizes (e.g., Parke-Davis prize, Liverpool University, 1973; Ferreira da Silva prize, Portuguese Chemical Society, 2004; Spanish-Portuguese prize, Royal Spanish Chemical Society, 2010). His research interests are centered on the synthesis, reactivity and potential applications of porphyrin derivatives, with 410 scientific publications. |
Introduction
The pioneering studies developed initially by Fisher1 and later by Woodward2 and other groups on porphyrins and by Johnson and Kay3,4 on corroles are responsible for the exciting chemistry associated with these two types of macrocycles.Today, the importance of porphyrin derivatives is not only limited to their central role in vital functions, like respiration and photosynthesis, but also to their applications in several fields such as medicine,5 catalysis6 and new electronic materials.7
Although corrole chemistry is still far from being as well explored as porphyrin chemistry, corroles are now considered an independent group of macrocycles within the larger family of tetrapyrrolic compounds. This is mainly due to the advances made on the synthetic methodologies of these macrocycles,8–13 in particular to the huge effort devoted to their functionalization,14 to obtain high value corroles with potential applications in several fields.
In fact, the interest in these compounds has increased significantly over the past decade, and currently, the popularity of corroles is, in some cases, approaching that of porphyrins.15–26
Corroles have properties that are different from those of their porphyrin counterparts. These properties are due to their corrin-like structural carbon skeleton, which is the cause of their lower symmetry,27 increased ring tension, due to the direct β–β pyrrole connection, tautomeric isomerism of the core28 and coordination with trivalent metal cations.29 In terms of electronic structure, corroles are aromatic macrocycles with an 18π-electron conjugated system similar to that of porphyrins,30 which is able to coordinate effectively metal ions in high oxidation states. Thus the smaller trianionic corrolato ligand has a greater ability to stabilize metal centres in higher oxidation states and open-shell electron configurations than the larger dianionic porphyrinato ligand.31
The establishment of efficient synthetic routes for tetrapyrrolic macrocycles has been an important objective over the years. Since the first synthesis reported by Rothemund32 in 1935 concerning the non-natural meso-substituted β-free porphyrin 1 (Fig. 1), a large number of synthetic methodologies have been reported. This progress in porphyrin chemistry had no parallel situation in the case of corroles until recently. The real boost in the chemistry of these compounds started in 1999 with the development, by Gross9 and Paolesse,10 of simple and efficient synthetic methodologies leading to meso-substituted corroles 2 (Fig. 1). Based on these synthetic approaches, new progress was made by other research groups. In particular, Gryko's group was able to refine the experimental conditions giving access to meso-substituted A3- and trans-A2B-corroles.11–13 Other synthetic methodologies have been reported introducing different approaches such as microwave irradiation,33 heterogeneous acid catalysts34 and ionic liquids as solvent.35
 | ||
| Fig. 1 General structures of meso-tetra(aryl)porphyrins 1 and meso-tri(aryl)corroles 2. | ||
The development of synthetic pathways leading to meso-tri(aryl)corroles has been accompanied by a considerable effort in the establishment of strategies for their functionalization and several conventional procedures are now well established. These involve, for example, halogenation,36–42 sulfonation/chlorosulfonation,43–46 nitration/amination,46–53 nucleophilic aromatic substitution,17,53,54 cycloaddition55–60 and metal catalysed reactions.61–72 Concerning porphyrins it can be said, with some certainty, that virtually any kind of substituted porphyrin can now be synthesized, just by a “one-pot” reaction, or by multiple synthetic steps depending on the substituent.
Several techniques contributed to the structural characterization of these macrocycles, namely NMR, XRD, absorption and emission spectroscopy, cyclic voltammetry and mass spectrometry. In particular, mass spectrometry has played an important role as a versatile tool in the characterization of such compounds.
Tetrapyrrolic macrocycles, in particular the porphyrins, have been studied by mass spectrometry using different ionization techniques. Initially, electron ionization (EI) was used, but unwanted gas-phase decomposition73 led to the use of alternative ionization techniques. Nevertheless, an important diagnostic cleavage of the porphyrin macrocycle was reported under electron impact.74 Chemical ionization (CI) was also used, and for some porphyrins and metalloporphyrins, formation of the corresponding porphyrinogens was observed.75,76 This feature led to the development of a method for pyrrole sequencing by CI.77 Field desorption (FD) was used to characterize porphyrins from mixtures of natural sources as early as 1975;78 a similar procedure was applied for the characterization of metallophthalocyanins.79 FD and other ionization techniques were also used to analyse the oxidation products of corroles.80 Desorption ionization techniques using a liquid matrix, such as fast atom bombardment (FAB) and liquid secondary ion mass spectrometry (LSIMS), were also used, for instance to characterize metallated phthalocyanines,81 and porphyrins,82,83 and also free base chlorins and bacterichlorins.84 More recently, matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI) have been increasingly used in the characterization of tetrapyrrolic macrocycles. MALDI has been applied to study porphyrins, azaporphyrins, phthalocyanines, multiporphyrin arrays,85,86 products from the photolysis of chlorins and bacteriochlorins,87,88 and corrole derivatives.89 ESI has been used in the case of free base and metallated, neutral and cationic porphyrins90–99 and also in the study of the interactions of cationic porphyrins with nucleotides.100–102
Besides ESI, other less common atmospheric pressure ionization techniques, such as atmospheric pressure photoionization (APPI) and atmospheric pressure chemical ionization (APCI), have been used, namely for metallochlorins,88 metalloporphyrins103 and corrole derivatives.80,104
Notwithstanding its advantages and in contrast to the extensive use of MS techniques in porphyrin chemistry, the role of MS in corrole characterization is still very limited. Most of the reported papers on this topic describe the use of ESI-MS in the characterization of corroles, especially via accurate mass measurements,58,89,105–115 although a few studies of their gas-phase chemistry, using this technique, have been published.54,80,104,116–121 These included the structural differentiation of corrole isomers by techniques such as TWIM-MS (travelling wave ion mobility mass spectrometry)118–120 and the investigation of their gas-phase coordination chemistry.117 MALDI has been less used, as for instance in the radical induced fragmentation of amino acid ester corroles,89 along with ESI, and in the corrole titrations with fluoride and cyanide anions.25,26
In this review the use of ESI-MS in the analysis of corrole derivatives will be highlighted, mainly their gas-phase coordination chemistry and the differentiation of isomers via ESI-MSn (n > 2) and ESI-TWIM-MS. Relevant related studies with porphyrins will be also reported on a comparison basis.
Mass spectrometry techniques used to study tetrapyrrolic macrocycles
The first studies on tetrapyrrolic macrocycles by mass spectrometry used ionization techniques such as electron ionization (EI) and chemical ionization (CI). Although both techniques are still used, they can only be applied to the analysis of volatile, thermally stable compounds, because the first step of these ionization processes is the vaporization of the analyte achieved by heating. In the case of EI, once in the gas-phase, the neutral analyte, M, interacts with a homogeneous beam of electrons (typically at 70 electron volts energy), causing its ionization, usually as radical cations M+˙, and fragmentation. The latter usually follows predictable pathways giving rise to fragment ions, which convey structural information about the analyte. In the case of CI, a reagent gas (for instance, ammonia or methane) is partially ionized by an electron beam and then reacts with the unionized reagent gas, usually, to form protonated reagent gas molecules. The ionization of the vaporized analyte is due to proton transfer from the reagent, with formation of the protonated analyte molecules, [M + H]+, and is therefore a much lower energy process than EI, so that fragmentation is greatly reduced. As both EI and CI require volatilization of the analyte, these techniques can only be used for compounds with molecular masses up to, and around, 500 Da.The analysis of polar and non-volatile analytes was initially achieved by FD. In this technique, a high-potential electric field is applied to an emitter (generally a filament), to which the analyte is directly applied as a thin film or as small crystals, leading to the formation of ionized molecules of the analyte. The process produces radical cations M+˙ and less often, protonated molecules [M + H]+, both with low internal energies, thus the mass spectra produced by FD show few fragment ions or no fragment ions at all. FD has largely been supplanted by newer ionization techniques, namely, FAB, LSIMS, ESI, MALDI and others.
In the case of FAB and LSIMS, the analyte is mixed with a non-volatile matrix and then is bombarded, under vacuum, in the case of FAB with a high energy beam of atoms (Ar, Xe) and in the case of LSIMS by accelerated primary ions (Ar+, Xe+, Cs+). Common matrices include glycerol, thioglycerol and 3-nitrobenzyl alcohol. With the introduction of FAB and LSIMS less volatile and more labile tetrapyrrolic compounds could be analysed, which include compounds with molecular masses up to 6000 Da. Depending on the matrix and on the analyte either M+˙ or [M + H]+ ions, or both, can be formed by FAB and LSIMS.
MALDI is a two-step ionization process, in which a laser beam is used to desorb the analyte, co-crystallized with a matrix. The matrix absorbs radiation at the laser wavelength, but not the analyte. The first step is the ablation of the upper layer of the matrix, producing a hot plume containing several species, such as neutral and ionized matrix molecules, including protonated species, matrix clusters and others. In the second step, the analyte molecules are ionized, usually by protonation, forming the [M + H]+ ions. The increase in molecular mass range when MALDI is used is significant when compared with FAB and LSIMS: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 300000 Da, depending on the analyzer used. In all the above mentioned methods, the ionization of the analyte usually gives rise to mono-charged ionic species.
000 to 300000 Da, depending on the analyzer used. In all the above mentioned methods, the ionization of the analyte usually gives rise to mono-charged ionic species.
In the case of ESI, a solution of the analyte is introduced through a capillary, at atmospheric pressure, into a small-diameter tip held at potential of approximately 3000 V, and is dispersed into a fine aerosol of charged droplets. These droplets pass down potential and pressure gradients towards the analyzer. The evaporation of the solvent from the charged droplets is usually assisted by a nebulizer gas. The size of charged droplets decreases until they become unstable, and form more stable droplets by coulombic fission. This process of desolvation and fission is repeated until desolvated ions are produced. The typical solvents used in electrospray ionization are prepared by mixing water with volatile organic compounds (methanol or acetonitrile). ESI can produce ions from macromolecules with low internal energy overcoming their propensity to fragment when ionized. ESI is advantageous over other ionization processes (for instance MALDI) since it may produce multiple charged ions, usually [M + nH]n+ effectively extending the mass range of the analyzer up to the MDa range. The development of ESI also allowed the effective coupling of mass spectrometry with flow separation techniques such as HPLC (high pressure liquid chromatography).
Following the implementation of electrospray as an ionization technique, other atmospheric pressure ionization methods have been developed, such as APCI and APPI.
APCI is an ionization method analogous to chemical ionization (CI), although it occurs at atmospheric pressure. Similarly to CI, it is not suitable for the analysis of thermally labile compounds. It has a strong resemblance to electrospray ionization (ESI) and as such is also commonly used in conjunction with HPLC. Unlike ESI, for which the ionization is brought about through the potential difference between a spray capillary (needle) and sampling cone, along with rapid desolvation, in APCI the analyte solution is introduced into a pneumatic nebulizer and desolvated in a heated quartz tube, before being submitted to electrical discharge (corona discharge), creating ions. The corona discharge replaces the electron filament in CI, producing primary ions by electron ionization. The primary ions collide with the vaporized solvent molecules to form secondary reactant gas ions. The latter undergo repeated collisions with the analyte molecules, leading to the formation of thermalysed analyte ions, with high ionization efficiency. Very few fragment ions are formed and the mass spectra show predominantly the protonated analyte molecules, [M + H]+, although other adduct ions can be also observed.
Another atmospheric pressure technique is APPI. Similarly to ESI, the liquid solution is vaporized with the help of a nebulizing gas, such as nitrogen, but then it enters in an ionization chamber at atmospheric pressure. There, the mixture of solvent and sample molecules is exposed to photons of ultraviolet light, usually from a krypton lamp (10 eV). This specific energy is enough to ionize the analyte molecules, but not high enough to ionize air components and other surrounding molecules. The photons also excite the solvent molecules and, as the process occurs at atmospheric pressure, these solvent molecules collide with molecules of the analyte. A small fraction of these collisions result in the ionization of the analyte and both the radical cations, M+˙ and the protonated analyte molecules [M + H]+ can be formed.
To increase the ionization yield of the analyte molecules, dopant-assisted APPI (APPI-d) can be also used. The dopant molecules (usually toluene) are in a large excess relative to the analyte molecules and are directly ionized by the UV photons. The ionized dopant molecules collide with the neutral analyte molecules giving rise to the radical cations M+˙ and the protonated analyte molecules [M + H]+.
Mass spectrometry analysis of corroles by ESI-MS
The first report of ESI-MS/MS applied to corroles was the study of 5,10,15-tris(pentafluorophenyl)corrole (TFPC) 3, Fig. 2, present as an impurity of 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin (TFPP).115 | ||
| Fig. 2 Representative structure of 5,10,15-tris(pentafluorophenyl)corrole (TFPC) 3 and its respective metal complexes 3A, 3B and 3C. | ||
The product ion spectra of the protonated [M + H]+ (positive ion mode) and deprotonated [M − H]− (negative ion mode) of TFPC showed consecutive losses of hydrogen fluoride molecules with formation of direct o-phenyl-to-β-linkages, generating fused five-membered fragment ions, similar to those already observed for other porphyrins and porphyrinoids, namely TFPP, its neutral and cationic pyrrolidine-fused chlorin and isobacteriochlorin derivatives84 and meso-tetra(heptafluoropropyl)porphyrin (TFHP).99
Compared to some of the above mentioned compounds, TFPC and TFPP possess simpler structures that enable HF molecule elimination to occur as the only significant loss. The mechanism of this elimination was proposed to be that of a nucleophilic aromatic substitution, common for the pentafluorophenyl group. Computational studies of the relative energies of the ions formed by HF molecule elimination indicated the occurrence of cooperative interactions between o-phenyl-to-β-linkages, which occurred in a regioselective unidirectional mode. A good correlation could also be established between the calculated relative energies for each species resulting from HF molecule losses and the relative abundances of its product ions obtained in the negative ion mode. Thus, the number of HF molecule eliminations observable in the negative ion mode spectra was recognized as a reliable tool for the identification of the number of pentafluorophenyl groups attached to a macrocycle.
Although the HF molecule eliminations were observed both in the positive and negative ion modes, the authors found that the fragmentation patterns of deprotonated TFPC and TFPP were richer and better defined when using ESI in the negative ion mode.115
In the specific case of TFPC 3, three consecutive HF molecule eliminations, one from each pentafluorophenyl substituent, lead to species 4A shown in Fig. 3. Similarly, for TFPP, four consecutive HF eliminations led to species 4B presented in the same figure. The authors proposed that similar HF eliminations with formation of o-phenyl-to-β-linkages were expected to occur for meso-pentafluorophenyl substituted porphyrinic and porphyrinoid compounds. They also suggested that ESI-MS/MS could act as a strategic pointer for the bulk synthesis of novel porphyrinoid structures.115
 | ||
| Fig. 3 Structures of fused fragment ions for TFPC (4A) and TFPP (4B).115 | ||
ESI is an ionization technique for which solution-phase information is retained into the gas-phase, to the point that very weak non-covalent interactions can be observed. It is thus interesting to compare the reported gas-phase behavior of the [M + H]+ ions of TFPC and TFPP and of their [M − H]− counterparts, with their solution acid–base properties. Like porphyrins, corrole inner-core nitrogen atoms can be both protonated and deprotonated, but neutral corroles have an unusually high N–H acidity compared with neutral porphyrins.122 Moreover, solution formation of mono-protonated porphyrins [M + H]+ is not a dominant process, as the doubly-protonated species are formed very rapidly.
The reported advantages of the use of ESI in the negative ion mode115 may be explained in the case of TFPC by its easy deprotonation. On the other hand, when the positive ion mode is used, protonated TFPP is expected to be less stable than protonated TFPC. Therefore, TFPC ions are expected to be more stable than their analogous TFPP species both in the positive and negative ion modes.
The observed similarity of the gas-phase behavior of protonated TFPC and TFPP in the positive ion mode and of their deprotonated counterparts in the negative ion mode is apparently at odds with their solution properties. A probable explanation is that although solution stability can be directly related to the gas-phase abundance of the [M + H]+ and [M − H]− ions of TFPC and TFPP, their structural similarity may be the cause of their analogous behavior after mass selection and collisional activation.
Danikiewicz, Gryko and collaborators80 used electrospray and three other ionization techniques, field desorption (FD), atmospheric pressure ionization (APPI) and atmospheric pressure chemical ionization (APCI), to monitor the chemical processes occurring in solutions containing the meso-substituted free base corroles 5–10 with electron withdrawing, electron donating and sterically hindered groups (Fig. 4).
 | ||
| Fig. 4 Structures of different corroles studied by Danikiewicz, Gryko and co-workers.80 | ||
The authors were especially interested in assessing the purity of the compounds but they also wished to determine the structure of their light- and oxygen-induced oxidation products. The stability of the corroles in a range of solvents (methanol, acetonitrile, ethyl acetate, dichloromethane and hexane) was tested by acquiring their mass spectra immediately after dilution, and 30 minutes and 1 hour later. The solutions were kept under ambient light in contact with air. Contrary to what was expected, it was found that the stability of corroles 5–10 in solvents that contained the highest amount of dissolved oxygen was rather high, whereas for acetonitrile solutions, the ions corresponding to oxidation products were formed with the highest relative abundances. Using the acetonitrile solutions, corrole 10 was found to be the most stable due to the electron withdrawing effect of the pentafluorophenyl groups in the meso positions (Fig. 4). Corroles 5 and 8 bearing the sterically hindered substituents bis-mesityl and tris-trimethoxyphenyl, respectively, were found to lead to the most abundant oxidation products.
The oxidized species of corrole 5,15-bis(mesityl)-10-(4-cyanophenyl)corrole 5 were isolated and characterized by NMR, IR, and identified by their product ion spectra and accurate mass measurements. Based on these data, the authors suggested the following structures for the oxidation products: the isocorrole 11A and the two biliverdin-like isomers 11B (major) and 11C (Fig. 5).
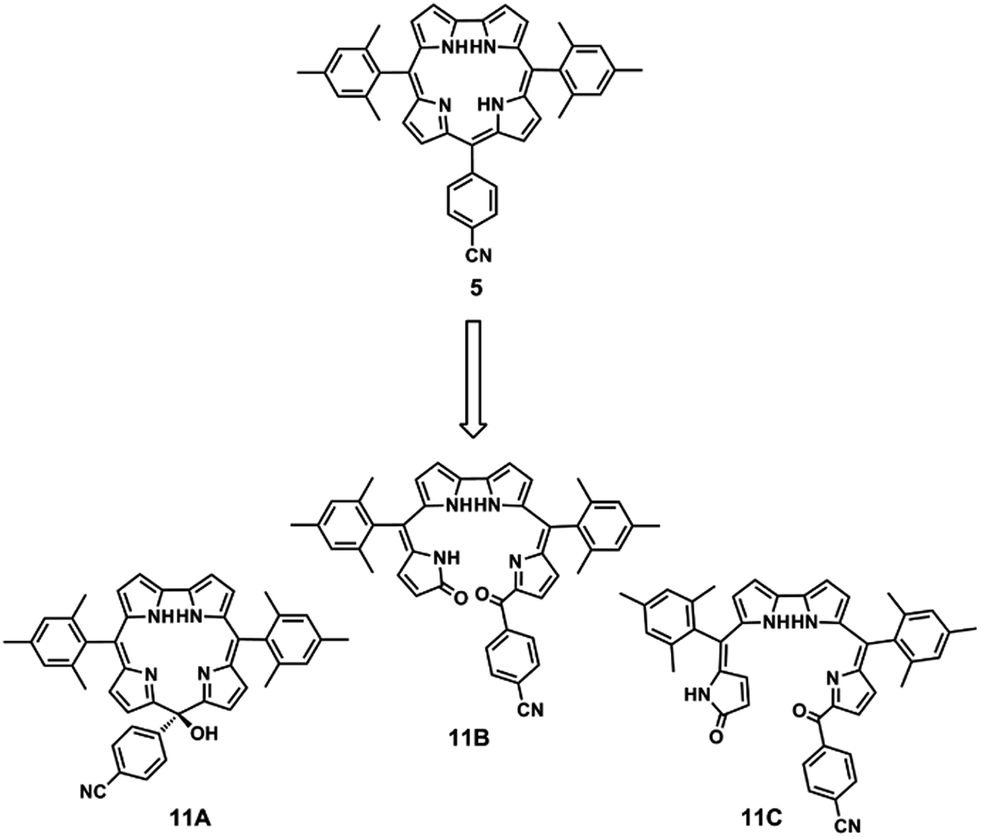 | ||
| Fig. 5 Structures of oxidized species identified by Danikiewicz, Gryko and co-workers.80 | ||
Field desorption (FD) was found to be the most suitable ionization method to evaluate the purity degree of corroles, whereas oxidation was always induced by electrospray ionization even when the corrole samples were pure.
Danikiewicz, Gryko, Lewtak and Swider104 selected porphyrins 12–17, corroles 5, 8, 18–22 and the cyanocobalamins 23 and 24 (see structures in Fig. 4 and 6) to evaluate the ionization efficiency of three atmospheric pressure ionization techniques, ESI, APCI and APPI using toluene as a dopant (APPI-d) in order to acquire useful information for the design of future MS or liquid chromatography-MS experiments. The relative sensitivity of the above mentioned ionization techniques was assessed both in the negative and positive ion modes by evaluating the relation between the instrumental response (represented by peak area) and the analyte concentration.
 | ||
| Fig. 6 Structures of different tetrapyrrolic macrocycles studied by Danikiewicz, Gryko and co-workers.104 | ||
In the positive ion mode, the highest sensitivity was achieved for the free-base porphyrins (12 and 16) and corroles (5, 8, 18 and 19) when using ESI, as well as for the cyanocobalamin 23 and its derivative 24. For the same free-base compounds APCI and APPI-d gave similar results although a slightly better sensitivity was achieved for the free-base porphyrins when using APPI-d. A different situation was observed for cyanocobalamin 23 and its derivative 24, where higher sensitivity was observed for APCI than for APPI-d. For metalloporphyrins 13–15 and 17, APCI and APPI-d provided better sensitivity than ESI. For metallocorroles 20–22 no common pattern concerning the sensitivity of the API methods was found.104
When compared with the positive ion mode, a higher sensitivity was observed for all corroles, in the negative ion mode, for all the ionization techniques. In the case of the free base corroles, 18 and 19, this feature is explained by their higher acidity122 when compared, for instance, with the free base porphyrins 12 and 16. The higher ability of metallocorroles to form anions, which has led to their trial as anion sensors,25,123 may explain the higher sensitivity observed.
In the negative mode ESI proved to be much more sensitive than APCI and APPI-d techniques. For the metalloporphyrins, with the exception of zinc complexes 13 and 17, APPI-d was the most sensitive ionization method, in the negative ion mode, although for all the studied porphyrins, sensitivity in this mode was lower than in the positive ion mode.
For both ion modes, ESI was shown to be the best ionization technique for cyanocobalamins, free-base corroles and porphyrins, whereas for metallocorroles and metalloporphyrins, APPI-d proved to be the best one.
When compared with porphyrins, corroles have a higher tendency to generate anions by electron capture in mass spectrometry sources. On the other hand, electron capture is a practical technique for the generation of peptide radicals. The special redox properties of corroles, namely their high tendency to generate anions by electron capture in electrospray sources, were used by Denekamp and Rabkin89 for the gas-phase generation of anion radicals in amino acid esters corroles 25–36 (Fig. 7), in order to induce the initiation of homolytic cleavages at the α-carbon of the amino acid ester chain.
 | ||
| Fig. 7 Structures of substituted Cu(III)-corrolates with amino acid esters studied by Denekamp and co-workers.89 | ||
In a first approach, complexes of 5,10,15-tris(phenyl)corrole (TPC) with Fe, Mn, Co, Ni, Cr, Pd, Ag and Cu were mass analysed by MALDI and ESI, and cation and anion radical formation was observed for all of them, with both techniques.89 A charge derivatization reagent must be stable upon collisional activation, thus MS/MS experiments were conducted for all of the above mentioned metallocorroles. The most stable ions were, as expected, the negatively charged copper and nickel complexes. Due to synthetic limitations, only the copper complex was used. Moreover the latter is a d8 complex that upon electron capture becomes an open shell d9 species required to initiate homolytic processes. Derivatization was applied on a variety of amino acid esters, affording complexes that were easily ionized into stable radical anions. The derivatized amino acids were collisionally induced to dissociate, forming a series of fragments, some of which indicate cleavage at the α-carbon with the involvement of the alkyl side chain as desired.
The various anion radicals can be formed from two main groups of compounds: the first group, which includes the esters of alanine, histidine, leucine, isoleucine, phenylalanine, methionine, tyrosine and valine, can lose a radical moiety, giving rise to a closed-shell product ion; the second group, which includes the esters of tryptophan, serine, threonine and glutamic acid can eliminate a closed-shell neutral species, giving radical product ions. The exception was lysine, which did not afford informative side chain fragmentations.
Almost all the product ion spectra of the M−˙ ions of the derivatized amino acids showed fragments corresponding to the cleavage at the α-carbon either with or without the loss of the alkoxy ester moiety. The cleavage of N–Cα bonds occurred after carbonyl hydrogen abstraction and the active species for radical induced fragmentation were proposed to be located at the carbonyl that leaves off the R–Cα alkyl chain or at the corrole backbone.
Gas-phase coordination chemistry of metalloporphyrinoids
The formation of metalloporphyrinoids in the source was studied by FAB mass spectrometry for meso-tetra(aryl)porphyrins 37–40 and for the meso-tetra(phenyl)chlorin 41 (Fig. 8).83,84 In this study the authors used divalent salts of Mg, Fe, Co, Ni, Cu, Zn, Cd and Pb and a trivalent salt of Fe(III). The formation of metalloporphyrin and metallochlorin ions was observed for all the compounds tested, except for the Mg(II), Fe(II), Fe(III) and Ni(II) salts. The analysis of the relative abundances of the PH3+ and MetP+˙ ions formed from mixtures of the free base, PH2, and of the corresponding metallated compounds, MetP, indicated that metalloporphyrin formation under FAB was predominantly a solution process, as the PH3+ ions were formed with higher relative abundances than the corresponding MetP+˙ ions, indicating that desorption of the former was easier.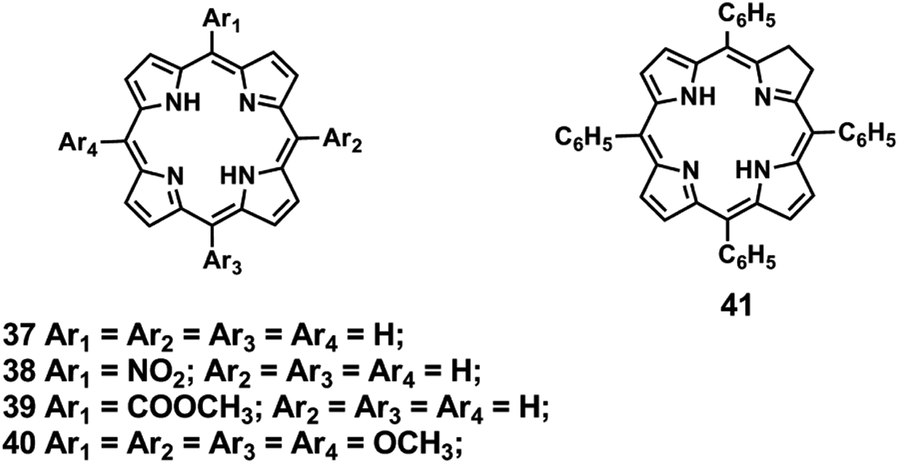 | ||
| Fig. 8 Structures of meso-tetra(aryl)porphyrins 37–40 and of meso-tetra(phenyl)chlorin 41.83,84 | ||
UV-visible absorption spectroscopy was also used to study their formation in solution and similar results to those obtained by FAB were attained, showing that the interactions with the matrix did not play a predominant role in metalloporphyrin formation under FAB. Differences in the metal counter ion were also found to influence the overall metalloporphyrin formation. A comparison with solution chemistry behaviour through the use of static solution coordination parameters did not lead to a complete explanation of the observed results, showing that kinetic parameters had to be considered.
In the case of metallocorroles, their formation by electrospraying solutions of TFPC 3 (Fig. 2) and divalent and trivalent metal cations in their acetate salt form and also by desorption of these solutions from liquid matrices was investigated.117 The isotopic patterns of the metallocorrole ions formed by these processes were compared to those of the corresponding metallocorrole ions formed from previously synthesized metallocorroles. The authors selected divalent metal acetates such as Mn(II), Co(II), Cu(II), Zn(II) and also the trivalent Mn(III) acetate, in order to investigate the influence of the oxidation state of the metal ion on metallocorrole formation in the source, and, concurrently, if oxidation of the divalent ions could occur during the ionization process.
This allowed not only a comparison of solution and gas-phase coordination chemistry, but also the acquisition of information on the ionization processes either in the case of liquid secondary ion mass spectrometry (LSIMS) or electrospray mass spectrometry (ESI-MS).
When using LSIMS the molecular ion peaks corresponding to the metallocorrole ions MC+˙, of the tested metal salts were obtained. From the analysis of the experimental and calculated isotopic patterns it was established that M(III)C+˙ were formed for all salts except when using Zn(II) acetate. The isotopic patterns, both experimental and calculated, for the synthesized metallocorrole 3A, 3B and 3C ions (Fig. 2) were also obtained and were identical to those of the corresponding ions formed in the source.
When using ESI spectra in the positive ion mode the formation of the M(III)C+˙ ions, when Mn(II), Co(II) and Cu(II) acetates were used, was predominant. However, in the presence of Zn(II) acetate, the ion corresponding to M(II)C+ was formed with low relative abundance, whereas the M(II)CH+˙ ion was much more abundant. When using Mn(III) acetate the formation of metallocorrole ions was not detected. The acquired data pointed to oxidation of the metal centre in the source during the formation of metallocorrole positive ions in the case of divalent metal cations.
When the studies were performed by ESI in the negative ion mode, the M(II)C− ions were observed for TFPC 3 with Mn(II), Co(II), Cu(II) and Zn(II) acetate. In the ESI spectra of the metallocorroles 3A, 3B and 3C synthesized by classical procedures, ions with the same m/z values as the ions formed in the source were observed (Fig. 2).
Based on the obtained data a mechanism for the formation of the metallocorrole ions M(III)C+˙ either by LSIMS or electrospray in the positive ion mode was proposed. It involved a stepwise proton-to-metal substitution (eqn (a)–(d)), assisted by the acetate ions, followed by oxidation (eqn (e)–(g)). In the case of ESI in the negative ion mode only the M(II)C− ions were detected, as further oxidation was prevented by the negative needle potential.117

For the metallocorroles 3A, 3B and 3C synthesized under classical conditions, the formation of the M(III)C+˙ and M(III)CH+ ions, in the positive ion mode, must occur by electron removal and protonation of the neutral corroles, respectively. In the negative ion mode, electron attachment with formation of the M(III)C−˙ radical anions was observed.
The complex ionization mechanism proposed can be further justified if the electrolytic nature of electrospray processes, put forward two decades ago124–126 and generally accepted, despite some controversy,127 is taken into account. A study on the electrochemistry of meso-substituted free-base corroles, including TFPC,128 has shown that the prevailing mechanisms for the oxidation and reduction of free base H3C corroles are the formation of species such as H2C−, [H2C˙]2−, H2C˙, H2C+, [H2C]3− [H3C˙]+, [H3C˙]−, H4C+, H4C˙ and [H4C˙]2+, depending on the solvents used, portraying a multistep process involving protonation, deprotonation and one electron transfer.
The product ion spectra of the metallocorrole ions formed in the source and from previously synthesized metallocorroles were identical for each metal centre. The main fragmentation pathway for the metallocorrole ions was via elimination of one to three HF molecules.
Corrole isomer differentiation by ESI-MS
Isomer differentiation using ESI-MS/MS can be accomplished if the product ions of the isomers, positional, diastereoisomers and others, have different m/z values or if they have the same m/z values, but different relative abundances.93,121 In general, isomer differentiation by ESI-MS/MS is not straightforward and it is often based on the formation of adducts, complexes or derivatized species, either in the source129,130 or before the introduction into the mass spectrometer and in many instances, multiple stage mass analysis MSn (n > 2) is required.131–133Two β-(aminomethyl)gallium(III) TFPC isomeric derivatives 42A and 42B (Fig. 9) were successfully differentiated, using ESI in the positive and in the negative ion modes.116
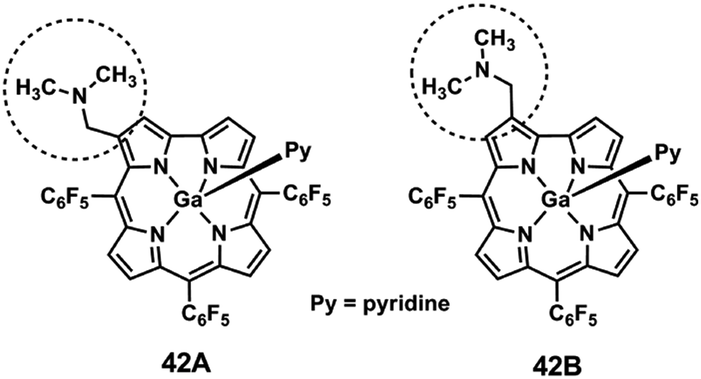 | ||
| Fig. 9 Structures of studied β-(aminomethyl)gallium(III) derivatives.116 | ||
This was accomplished in the positive ion mode through two types of diagnostic ions: the first formed by loss of a neutral dimethylamine molecule [(M-py) − NH(CH3)2]+, corresponding to the base peak in the mass spectra, and the second, the [(M-py) − NH(CH3)2 + H2O]+ ion resulting from an unusual addition of one water molecule to the former. Accurate mass measurements, multistage mass spectrometry ESI-MSn (n > 2) and energy-resolved MS, were also used to investigate the formation and structure of the above mentioned diagnostic ions. In the negative ion mode, isomer discrimination was achieved through the fragmentation of the methoxide adduct ions [(M-py) + CH3O]−.
In the positive ion mode, the product ion spectra of the fragment ions formed by dimethylamine molecule elimination, [(M-py) + H − NH(CH3)2]+, showed losses of HF molecules for the two isomers, but the relative abundances of the fragment ions formed by the loss of one HF molecule, [(M-py) + H − NH(CH3)2 − HF]+, are quite different, indicating the formation of two isomeric ions with different stabilities.
Two different structures for the ions formed by HF loss were proposed: a six-membered ring 44 (Scheme 1, top) and a five-membered ring 46 (Scheme 1, bottom) similar to previously described structures.84,99,115
 | ||
| Scheme 1 | ||
The observed difference in stabilities was found to be consistent with data obtained from semi-empirical calculations. The calculated formation enthalpy values for the ions [(M-py) + H]+ and [(M-py) + H − NH(CH3)2]+ were similar for both isomers, but the calculated formation enthalpies for the [(M-py) + H − NH(CH3)2 − HF]+ ions, shown in Scheme 1, were different: for the [(M-py) + H − NH(CH3)2 − HF]+˙ structure, shown at the top, the calculated formation enthalpy is −230.6 kcal mol−1 for 44, whereas for the isomeric structure below, 46, the formation enthalpy is −197.5 kcal mol−1. Isomer differentiation could also be achieved through the MS3 spectra corresponding to the sequence [(M-py) + H]+ → [M-py + H − NH(CH3)2]+ → product ions.
In the negative ion mode, differentiation can be achieved through the product ion spectra of the [(M-py) + CH3O]− ions, (represented as M′−) formed from methanol used as the eluent. The differences between the two spectra are self-evident: the base peak corresponds to the [M′ − (CH3)2N − HF]−˙ ions for one of the isomers and to the [M′ − (CH3)2N]−˙ ions for the other (Fig. 10).
 | ||
| Fig. 10 Product ion spectra of the ions [(M-py) + CH3O]− (A – isomer 42A and B – isomer 42B). * – precursor ion; × − [M′ − (CH3)2N]−˙; • – [M′ − (CH3)2N˙ − HF]−˙; ○ – [M′ − (CH3)2N˙ − CH3O˙]−; ◆ – [M′ − (CH3)2N˙ − 2HF]−˙; ■ – [M′ − (CH3)2N˙ − HF − CH3O˙]−; ✶ − [M′ − (CH3)2N˙ − 2HF − CH3O˙]− and ✦ – [M′ − (CH3)2N˙ − 3HF − CH3O˙]−. (Reproduced/adapted from ref. 116). | ||
Besides the HF losses, the main fragment ions are formed by losses of dimethylamino radicals (CH3)2N˙, by themselves, or as joint losses with HF molecules and methoxide radicals, CH3O˙.
The formation of the [M′ − (CH3)2N − HF]−˙ ions can be explained by a mechanism involving the formation of a six-membered fused ring for isomer A (corrole 42A), similar to the one proposed for their positive ion counterpart. As this process is not available in isomer B (corrole 42B) loss of a (CH3)2N˙ radical is the preferred process. ESI-MS/MS, in combination with NMR analysis, was also used to differentiate the two free-base galactopyranose-tris(pentafluorophenyl)corrole positional isomers 47A and 47B (Fig. 11).54
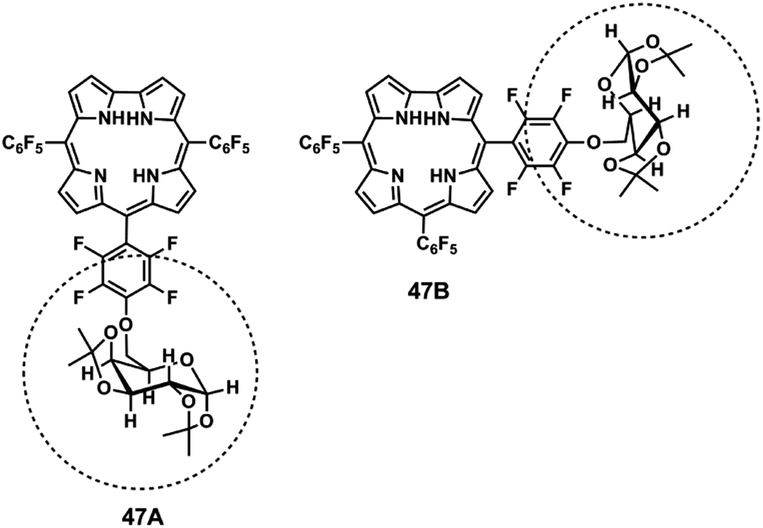 | ||
| Fig. 11 Structure of galactopyranose-tris(pentafluorophenyl)corrole positional isomers.54 | ||
The product ion spectra of the [M + H]+ ions of the isomeric pair 47A, 47B, formed by electrospray ionization, showed fragment ions with the same m/z values. However the two most abundant product ions, [M + H − (Gal-H)]+ and [M + H − (Gal-H) − HF]+, were formed with different relative abundances for each isomer. The first ion was formed by cleavage of the C–O bond and migration of one hydrogen to the oxygen of the ether bond by a mechanism already described for glyco-tetrakis(pentafluorophenyl)porphyrin derivatives.134,135 The second ion resulted from a joint loss of the former fragment and a HF molecule. Loss of HF occurs with formation of direct o-phenyl-to-β-linkages as discussed above.
Recently, the techniques of ion mobility spectrometry (IMS) and ion mobility spectrometry coupled to mass spectrometry (IM-MS) are being used with success in isomer differentiation.118–120 Ion mobility mass spectrometry, which is becoming increasingly popular, provides high sensitivity, specificity, and analysis times on millisecond time range. Besides the usual mass and charge parameters, the separation in IM-MS is also based on analyte shape (measured by its collision cross section), which affects its interactions with gases throughout a drift tube.136–140 More recently the technique of TWIM-MS was developed. Unlike conventionally IMS, in which an electric field is applied continuously, TWIM uses continuous transient voltage pulses.141
Eberlin and collaborators119 used this technique in the positive ion mode to study mixtures containing the isomeric meta/para and adj/opp ruthenated meso-(pyridyl)porphyrins 48–59 (Fig. 12). The more compact meta positional isomers were found to display shorter drift times than the para isomers, and the separation was substantially increased for the multiply charged species. The isomers displayed very close drift times both in CO2 and N2 and TWIM-MS was unable to differentiate these porphyrin isomers.
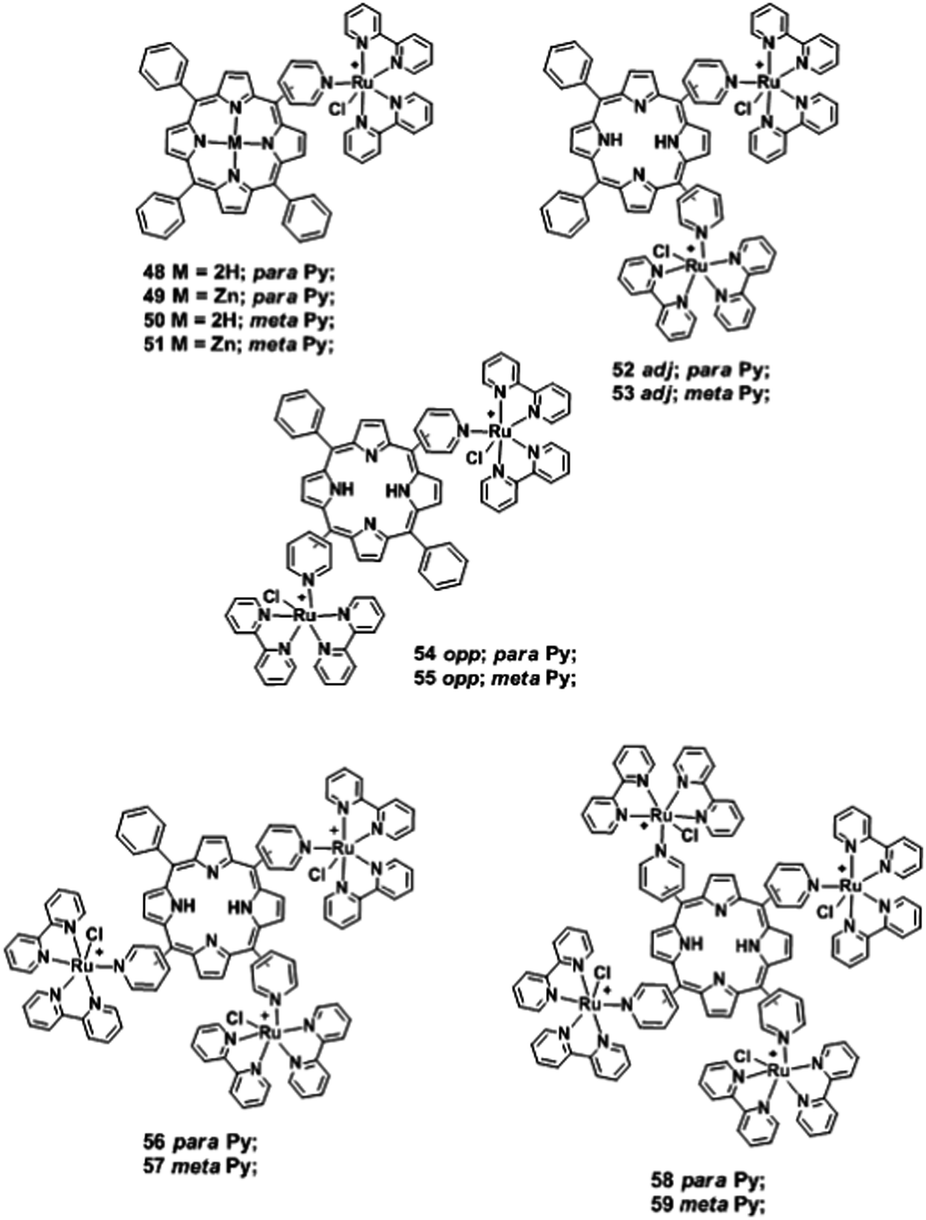 | ||
| Fig. 12 Structures of ruthenium(II)-porphyrinato complexes 48–59.119 | ||
Eberlin and co-workers120 also used the ESI coupled to TWIM analysis to investigate the protonation and deprotonation sites of meso-tetra(pyridyl) and meso-tetrakis(carboxyphenyl)porphyrins 60–62 (Fig. 13), both in the positive and negative ion modes. In the positive ion mode, for the meso-tetra(pyridyl)porphyrins 60 and 61, two different protonation sites were identified: the first, on one of the inner nitrogens and the second, on one of the N-pyridyl substituents. A similar behaviour was observed in the negative ion mode for meso-tetrakis(carboxyphenyl)porphyrin 62, where two “de-protomers” were identified, the first resulting from deprotonation of an inner nitrogen and the second from a carboxyl substituent.
 | ||
| Fig. 13 Structures of meso-tetra(pyridyl)porphyrins 60, 61 and meso-tetrakis(carboxyphenyl)porphyrin 62.120 | ||
Corrole 3 and four of its isomers 63–66 (Fig. 14) with subtle structural changes were also studied by Eberlin and collaborators118 using travelling wave ion mobility mass spectrometry (TWIM-MS) and collision induced dissociation (CID) of electrosprayed ions.
 | ||
| Fig. 14 Structures of corrole 3 isomers studied by Eberlin and co-workers.118 | ||
In this work, the relative gas-phase mobilities of the five protonated isomers were evaluated by TWIM-MS using two drift gases, N2 and CO2. When N2 was used, corroin 65 was the isomer with the higher mobility followed by norrole 66, N(2)-confused corrole 64 and N(4)-confused corrole 63, with similar drift times and finally by corrole 3 (Fig. 15). When CO2, with higher polarizability and mass, was used as drift gas, corroin 65 presented a much higher mobility and the mobility of corrole 3 was higher than that of N(4)-confused corrole 63 (Fig. 15).
 | ||
| Fig. 15 TWIM drift time graphical plots obtained for the protonated isomers analysed: N(4)-confused corrole 63, N(2)-confused corrole 64, corroin 65, norrole 66 and corrole 3, respectively. (Reproduced/adapted from ref. 118). | ||
As the charge is the same for all the protonated isomers and the calculation of the collision cross sections using density functional theory calculation (DFT) led to a tight range of values, ion-induced dipole interactions were responsible for the different mobilities observed, especially when CO2 was used as drift gas. DFT calculations showed that corroin 65 possesses the most compact tridimensional structure, as expected from its shorter drift times (Fig. 15).
Sequential losses of HF molecules were observed in the product ion spectra of the protonated molecules of these isomers, as observed before by Furuta and collaborators.113 N(4)-confused corrole 63 showed a unique and diagnostic extensive loss of a NH3 molecule, which allowed its rapid differentiation from the other isomers.
Conclusions
The importance of electrospray ionization mass spectrometry in the analysis of corrole derivatives is well illustrated by the range of examples discussed in this review. Fragmentation and degradation mechanisms, identification of new compounds and macrocycle structure characterization were some of the areas where ESI-MS played an important role. The increase in the number of publications in this field is also a pointer of the importance of this technique. On the other hand, the still limited number of publications on specific aspects of corrole gas-phase chemistry shows that novel applications of this technique can still be developed.It is predictable that ESI, especially when used with TWIM analyzers, will be the technique of choice for the analysis, not only of corrole isomers, but of corrole-based complex structures, such as corrole–porphyrin dimers, corrole adducts with biomolecules and other supramolecular structures, either covalently linked or resulting from solution self-assembly.
Previous studies of the adducts of G-quadruplex DNA and cationic corrole isomers by circular dichroism (CD) and temperature melting assays showed that these compounds are G-quadruplex stabilizers and thus potential candidates for new anticancer drugs. It is probable that ESI-TWIM-MS will be used in the future for the design and development of corrole based anticancer drugs, including the implementation of screening techniques.
Besides ESI, other recently developed ionization techniques, such as easy ambient sonic spray ionization (EASI-MS), which can be used directly from layer chromatography (TLC) plates, are extremely promising for the identification of products from corrole syntheses.
Acknowledgements
We would like to thank Fundação para a Ciência e a Tecnologia (FCT, Portugal), the European Union, QREN, FEDER, COMPETE, for funding the Organic Chemistry Research Unit (QOPNA) (project PEst-C/QUI/UI0062/2013; FCOMP-01-0124-FEDER-037296). J. F. B. Barata and C. I. V. Ramos also thank Fundação para a Ciência e Tecnologia (FCT) for the grants SFRH/BPD/63237/2009 and SFRH/BPD/85902/2012, respectively. B. A. Iglesias also thanks Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq – Brazil) for the grant 200802/2012-7.Notes and references
- H. Fisher and K. Zeile, Liebigs Ann. Chem., 1929, 468, 98–116 CrossRef PubMed.
- R. B. Woodward, W. A. Ayer, J. M. Beaton, F. Bickelhaupt, R. Bonnett, P. Buchschacher, G. L. Closs, H. Dutler, J. Hannah, F. P. Hauck, S. Itô, A. Langemann, E. Le Goff, W. Leimgruber, W. Lwowski, J. Sauer, Z. Valenta and H. Voltz, J. Am. Chem. Soc., 1960, 82, 3800–3802 CrossRef CAS.
- A. W. Johnson and R. Price, J. Chem. Soc., 1960, 1649–1653 RSC.
- A. W. Johnson and I. T. Kay, J. Chem. Soc., 1965, 1620–1629 RSC.
- R. K. Pandey and G. Zheng, Porphyrins as Photosensitizers in Photodynamic Therapy, in The Porphyrin Handbook, Academic Press, San Diego, 2000, vol. 6, pp. 157–225 Search PubMed.
- T. Aida and S. Inoue, Metalloporphyrins as Catalysts for Precision Macromolecular Synthesis, in The Porphyrin Handbook, Academic Press, San Diego, 2000, vol. 6, pp. 133–155 Search PubMed.
- J.-H. Chou, M. E. Kosal, H. S. Nalwa, N. A. Rakow and K. S. Suslick, Applications of Porphyrins and Metalloporphyrins to Materials Chemistry, in The Porphyrin Handbook, Academic Press, San Diego, 2000, vol. 6, pp. 43–131 Search PubMed.
- (a) S. Nardis, D. Monti and R. Paolesse, Mini-Rev. Org. Chem., 2005, 2, 355–374 CrossRef CAS; (b) R. Paolesse, Synlett, 2008, 2215–2230 CrossRef CAS PubMed.
- Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427–1429 CrossRef CAS.
- R. Paolesse, L. Jaquinod, D. J. Nurco, S. Mini, F. Sagone, T. Boschi and K. M. Smith, Chem. Commun., 1999, 1307–1308 RSC.
- D. T. Gryko and B. Koszarna, Org. Biomol. Chem., 2003, 1, 350–357 CAS.
- B. Koszarna and D. T. Gryko, J. Org. Chem., 2006, 71, 3707–3717 CrossRef CAS PubMed.
- D. T. Gryko, Eur. J. Org. Chem., 2002, 1735–1743 CrossRef CAS.
- J. F. B. Barata, C. I. M. Santos, M. G. P. M. S. Neves, M. A. F. Faustino and J. A. S. Cavaleiro, in Synthesis and Modification of Porphyrinoids, Topics in Heterocyclic Chemistry, Springer-Verlag, Berlin, 2014, vol. 33, pp. 79–141 Search PubMed.
- I. Aviv-Harel and Z. Gross, Chem.–Eur. J., 2009, 15, 8382–8394 CrossRef CAS PubMed.
- L. Flamigni and D. T. Gryko, Chem. Soc. Rev., 2009, 38, 1635–1646 RSC.
- I. Aviv and Z. Gross, Chem. Commun., 2007, 1987–1999 RSC.
- K. Kurzatkowska, E. Dolusic, W. Dehaen, K. Sieroń-Stołtny, A. Sieroń and H. Radecka, Anal. Chem., 2009, 81, 7397–7405 CrossRef CAS PubMed.
- J.-M. Barbe, G. Canard, S. Brandès and R. Guilard, Angew. Chem., Int. Ed., 2005, 44, 3103–3106 CrossRef CAS PubMed.
- M. Tasior, D. T. Gryko, J. Shen, K. M. Kadish, T. Becherer, H. Langhals, B. Ventura and L. Flamigni, J. Phys. Chem. C, 2008, 112, 19699–19709 CAS.
- Z. Gross and H. B. Gray, Adv. Synth. Catal., 2004, 346, 165–170 CrossRef CAS PubMed.
- Z. Okun, L. Kupershmidt, T. Amit, S. Mandel, O. Bar-Am, M. B. H. Youdim and Z. Gross, ACS Chem. Biol., 2009, 4, 910–914 CrossRef CAS PubMed.
- J. F. B. Barata, A. L. Daniel-da-Silva, M. G. P. M. S. Neves, J. A. S. Cavaleiro and T. Trindade, RSC Adv., 2013, 3, 274–280 RSC.
- C. I. M. Santos, E. Oliveira, J. C. Menezes, J. F. B. Barata, M. A. F. Faustino, V. Ferreira, J. A. S. Cavaleiro, M. G. P. M. S. Neves and C. Lodeiro, Tetrahedron, 2013 DOI:10.1016/j.tet.2013.07.022 , in press.
- C. I. M. Santos, E. Oliveira, J. F. B. Barata, M. A. F. Faustino, J. A. S. Cavaleiro, M. G. P. M. S. Neves and C. Lodeiro, J. Mater. Chem., 2012, 22, 13811–13819 RSC.
- C. I. M. Santos, E. Oliveira, J. Fernandez-Lodeiro, J. F. B. Barata, S. Santos, M. A. F. Faustino, J. A. S. Cavaleiro, M. G. P. M. S. Neves and C. Lodeiro, Inorg. Chem., 2013, 52, 8564–8572 CrossRef CAS PubMed.
- (a) N. S. Hush, J. M. Dyke, M. L. Williams and I. S. Woolsey, J. Chem. Soc., Dalton Trans., 1974, 395–399 RSC; (b) A. Ghosh and K. Jynge, Chem.–Eur. J., 1997, 3, 823–833 CrossRef CAS PubMed.
- Y. B. Ivanova, V. A. Savva, N. Z. Mamardashvili, A. S. Starukhin, T. H. Ngo, W. Dehaen, W. Maes and M. M. Kruk, J. Phys. Chem. A, 2012, 116, 10683–10694 CrossRef CAS PubMed.
- J. L. Sessler and S. J. Weghorn, Expanded, Contracted & Isomeric Porphyrins, in Tetrahedron Organic Chemistry Series, Pergamon, New York, 1997, vol. 18, pp. 11–120 Search PubMed.
- H. Palmer, in Molecular electronic structures of transition metal complexes I, Structure and Bonding Series, Springer, Berlin, 2012, vol. 142, pp. 49–90 Search PubMed.
- J. A. S. Cavaleiro, A. C. Tomé and M. G. P. M. S. Neves, in Handbook of Porphyrin Science, World Scientific, Singapore, 2010, vol. 2, pp. 193–294 Search PubMed.
- P. Rothemund, J. Am. Chem. Soc., 1935, 57, 2010–2011 CrossRef CAS.
- J. P. Collman and R. A. Decréau, Tetrahedron Lett., 2003, 44, 1207–1210 CrossRef CAS.
- P. Kumari and S. M. S. Chauhan, J. Heterocycl. Chem., 2008, 45, 779–783 CrossRef CAS PubMed.
- H. Y. Zhan, H. Y. Liu, H. J. Chen and H. F. Jiang, Tetrahedron Lett., 2009, 50, 2196–2199 CrossRef CAS PubMed.
- A. Mahammed, M. Botoshansky and Z. Gross, Dalton Trans., 2012, 41, 10938–10940 RSC.
- J. Vestfrid, M. Botoshansky, J. H. Palmer, A. C. Durrell, H. B. Gray and Z. Gross, J. Am. Chem. Soc., 2011, 133, 12899–12901 CrossRef CAS PubMed.
- R.-B. Du, C. Liu, D.-M. Shen and Q.-Y. Chen, Synlett, 2009, 2701–2705 CAS.
- S. Nardis, F. Mandoj, R. Paolesse, F. R. Fronczek, K. M. Smith, L. Prodi, M. Montalti and G. Battistini, Eur. J. Inorg. Chem., 2007, 2345–2352 CrossRef CAS PubMed.
- J. H. Palmer, M. W. Day, A. D. Wilson, L. M. Henling, Z. Gross and H. B. Gray, J. Am. Chem. Soc., 2008, 130, 7786–7787 CrossRef CAS PubMed.
- G. Golubkov, J. Bendix, H. B. Gray, A. Mahammed, I. Goldberg, A. J. DiBilio and Z. Gross, Angew. Chem., Int. Ed., 2001, 40, 2132–2134 CrossRef CAS.
- S. Nardis, G. Pomarico, F. Mandoj, F. R. Fronczek, K. M. Smith and R. Paolesse, J. Porphyrins Phthalocyanines, 2010, 14, 752–757 CrossRef CAS PubMed.
- A. Mahammed, I. Goldberg and Z. Gross, Org. Lett., 2001, 3, 3443–3446 CrossRef CAS PubMed.
- A. Mahammed and Z. Gross, J. Porphyrins Phthalocyanines, 2010, 14, 911–923 CrossRef CAS.
- Z. Gross and A. Mahammed, J. Porphyrins Phthalocyanines, 2002, 6, 553–555 CrossRef CAS.
- I. Saltsman, A. Mahammed, I. Goldberg, E. Tkachenko, M. Botoshansky and Z. Gross, J. Am. Chem. Soc., 2002, 124, 7411–7420 CrossRef CAS PubMed.
- M. Stefanelli, M. Mastroianni, S. Nardis, S. Licoccia, F. R. Fronczek, K. M. Smith, W. Zhu, Z. Ou, K. M. Kadish and R. Paolesse, Inorg. Chem., 2007, 46, 10791–10799 CrossRef CAS PubMed.
- S. Nardis, M. Stefanelli, P. Mohite, G. Pomarico, L. Tortora, M. Manowong, P. Chen, K. M. Kadish, F. R. Fronczek, G. T. McCandless, K. M. Smith and R. Paolesse, Inorg. Chem., 2012, 51, 3910–3920 CrossRef CAS PubMed.
- M. Mastroianni, W. Zhu, M. Stefanelli, S. Nardis, F. R. Fronczek, K. M. Smith, Z. Ou, K. M. Kadish and R. Paolesse, Inorg. Chem., 2008, 47, 11680–11687 CrossRef CAS PubMed.
- M. Stefanelli, F. Mandoj, M. Mastroianni, S. Nardis, P. Mohite, F. R. Fronczek, K. M. Smith, K. M. Kadish, X. Xiao, Z. Ou, P. Chen and R. Paolesse, Inorg. Chem., 2011, 50, 8281–8292 CrossRef CAS PubMed.
- J.-M. Barbe, G. Canard, S. Brandès and R. Guilard, Eur. J. Org. Chem., 2005, 4601–4611 CrossRef CAS PubMed.
- J. P. Collman and R. A. Decréau, Org. Lett., 2005, 7, 975–978 CrossRef CAS PubMed.
- T. Hori and A. Osuka, Eur. J. Org. Chem., 2010, 2379–2386 CrossRef CAS PubMed.
- T. A. F. Cardote, J. F. B. Barata, M. A. F. Faustino, A. Preuß, M. G. P. M. S. Neves, J. A. S. Cavaleiro, C. I. V. Ramos, M. G. O. Santana-Marques and B. Röder, Tetrahedron Lett., 2012, 53, 6388–6392 CrossRef CAS PubMed.
- J. F. B. Barata, A. M. G. Silva, M. A. F. Faustino, M. G. P. M. S. Neves, A. C. Tomé, A. M. S. Silva and J. A. S. Cavaleiro, Synlett, 2004, 1291–1293 CAS.
- L. S. H. P. Vale, J. F. B. Barata, C. I. M. Santos, M. G. P. M. S. Neves, M. A. F. Faustino, A. C. Tomé, A. M. S. Silva, F. A. A. Paz and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2009, 13, 358–368 CrossRef CAS.
- C. I. M. Santos, E. Oliveira, J. F. B. Barata, M. A. F. Faustino, J. A. S. Cavaleiro, M. G. P. M. S. Neves and C. Lodeiro, Inorg. Chim. Acta, 2013 DOI:10.1016/j.ica.2013.09.049 , in press.
- L. S. H. P. Vale, J. F. B. Barata, M. G. P. M. S. Neves, M. A. F. Faustino, A. C. Tomé, A. M. S. Silva, F. A. A. Paz and J. A. S. Cavaleiro, Tetrahedron Lett., 2007, 48, 8904–8908 CrossRef CAS PubMed.
- F. D'Souza, R. Chitta, K. Ohkubo, M. Tasior, N. K. Subbaiyan, M. E. Zandler, M. K. Rogacki, D. T. Gryko and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 14263–14272 CrossRef CAS PubMed.
- K. Lewandowska, B. Barszcz, J. Wolak, A. Graja, M. Grzybowski and D. T. Gryko, Dyes Pigm., 2013, 96, 249–255 CrossRef CAS PubMed.
- T. H. Ngo, F. Puntoriero, F. Nastasi, K. Robeyns, L. Van Meervelt, S. Campagna, W. Dehaen and W. Maes, Chem.–Eur. J., 2010, 16, 5691–5705 CrossRef CAS PubMed.
- W. Maes, T. H. Ngo, J. Vanderhaeghen and W. Dehaen, Org. Lett., 2007, 9, 3165–3168 CrossRef CAS PubMed.
- A. Scrivanti, V. Beghetto, U. Matteoli, S. Antonaroli, A. Marini, F. Mandoj, R. Paolesse and B. Crociani, Tetrahedron Lett., 2004, 45, 5861–5864 CrossRef CAS PubMed.
- S. Berg, K. E. Thomas, C. M. Beavers and A. Ghosh, Inorg. Chem., 2012, 51, 9911–9916 CrossRef CAS PubMed.
- M. König, L. M. Reith, U. Monkowius, G. Knör, K. Bretterbauer and W. Schöefberger, Tetrahedron, 2011, 67, 4243–4252 CrossRef PubMed.
- T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073–14075 CrossRef CAS PubMed.
- L. M. Reith, M. Koenig, C. Schwarzinger and W. Schöefberger, Eur. J. Inorg. Chem., 2012, 4342–4349 CrossRef CAS PubMed.
- L. M. Reith, M. Stiftinger, U. Monkowius, G. Knör and W. Schöefberger, Inorg. Chem., 2011, 50, 6788–6797 CrossRef CAS PubMed.
- M. Bröring, M. Funk and C. Milsmann, J. Porphyrins Phthalocyanines, 2009, 13, 107–113 CrossRef.
- I. Nigel-Etinger, A. Mahammed and Z. Gross, Catal. Sci. Technol., 2011, 1, 578–581 CAS.
- M. Tasior, D. T. Gryko, J. Shen, K. M. Kadish, T. Becherer, H. Langhals, B. Ventura and L. Flamigni, J. Phys. Chem. C, 2008, 112, 19699–19709 CAS.
- H.-Y. Zhan, H.-Y. Liu, J. Lu, A.-Z. Wang, L.-L. You, H. Wang, L.-N. Ji and H.-F. Jiang, J. Porphyrins Phthalocyanines, 2010, 14, 150–157 CrossRef CAS.
- A. G. Smith and P. B. Farmer, Biomed. Mass Spectrom., 1982, 9, 111–114 CrossRef CAS PubMed.
- J. D. Laycock, J. A. Ferguson, R. A. Yost, J. M. E. Quirke, A. Rohrer, R. Ocampo and H. Callot, J. Mass Spectrom., 1997, 32, 978–983 CrossRef CAS.
- P. Sundararaman, E. J. Gallegos, E. W. Baker, J. R. B. Slayback and M. R. Johnston, Anal. Chem., 1984, 56, 2552–2556 CrossRef CAS.
- G. J. Shaw, G. Eglinton and J. M. E. Quirke, Anal. Chem., 1981, 53, 2014–2020 CrossRef CAS.
- G. J. Van Berkel, G. L. Glish, S. A. McLuckey and A. Albert, Anal. Chem., 1990, 62, 786–793 CrossRef CAS.
- N. Evans, D. E. Games, A. H. Jackson and S. A. Matlin, J. Chromatogr. A, 1975, 115, 325–333 CrossRef CAS.
- V. Sugumaran, V. Kagdiyal, R. Kumar, R. Sarin, A. K. Gupta, A. S. Sarpal and B. Basu, Pet. Sci. Technol., 2011, 30, 278–289 CrossRef CAS.
- P. Swider, A. Nowak-Krol, R. Voloshchuk, J. P. Lewtak, D. T. Gryko and W. Danikiewicz, J. Mass Spectrom., 2010, 452, 1443–1451 CrossRef PubMed.
- R. B. Freas and J. E. Campana, Inorg. Chem., 1984, 23, 4654–4658 CrossRef CAS.
- J. B. Carlson and P. Vouros, J. Mass Spectrom., 1996, 31, 1403–1408 CrossRef.
- M. R. M. Domingues, M. G. O. Santana-Marques and A. J. F. Correia, Int. J. Mass Spectrom. Ion Processes, 1997, 165–166, 551–559 CrossRef.
- R. A. Izquierdo, C. M. Barros, M. G. O. Santana-Marques, A. J. F. Correia, A. M. G. Silva, A. C. Tomé, A. M. S. Silva, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Rapid Commun. Mass Spectrom., 2004, 18, 2601–2611 CrossRef CAS PubMed.
- M. Schafer and H. Budzikiewicz, J. Mass Spectrom., 2001, 36, 1062–1068 CrossRef CAS PubMed.
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, J. Porphyrins Phthalocyanines, 1999, 3, 283–291 CrossRef CAS.
- A. Kasselouri, O. Bourdon, D. Demore, J. C. Blais, P. Prognon, G. Bourg-Heckly and J. Blais, Photochem. Photobiol., 1999, 70, 275–279 CrossRef CAS.
- H.-P. Lassalle, N. Lourette, B. Maunit, J.-F. Muller, F. Guillemin and L. Bezdetnaya-Bolotine, J. Mass Spectrom., 2005, 40, 1149–1156 CrossRef CAS PubMed.
- C. Denekamp and E. Rabkin, J. Am. Soc. Mass Spectrom., 2007, 18, 791–801 CrossRef CAS PubMed.
- M. R. M. Domingues, P. Domingues, M. G. P. M. S. Neves, A. C. Tomé and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2009, 13, 524–527 CrossRef CAS.
- C. I. V. Ramos, M. G. O. Santana-Marques, A. J. F. Correia, V. V. Serra, J. P. C. Tomé, A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Am. Soc. Mass Spectrom., 2007, 18, 762–768 CrossRef CAS PubMed.
- C. I. V. Ramos, M. G. O. Santana-Marques, A. J. F. Correia, J. P. C. Tomé, C. M. Alonso, A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mass Spectrom., 2008, 43, 806–813 CrossRef CAS PubMed.
- R. A. Izquierdo, C. M. Barros, M. G. O. Santana-Marques, A. J. F. Correia, E. M. P. Silva, F. Giuntini, M. A. F. Faustino, J. P. C. Tomé, A. C. Tomé, A. M. S. Silva, M. G. P. M. S. Neves, J. A. S. Cavaleiro, A. F. Peixoto, M. M. Pereira and A. A. C. C. Pais, J. Am. Soc. Mass Spectrom., 2007, 18, 218–225 CrossRef CAS PubMed.
- G. J. Van Berkel, S. A. McLuckey and G. L. Glish, Anal. Chem., 1992, 3, 235–242 CAS.
- V. E. Vandell and P. A. Limbach, J. Mass Spectrom., 1998, 33, 212–220 CrossRef CAS.
- I. Batinic-Haberle, R. D. Stevens and I. Fridovich, J. Porphyrins Phthalocyanines, 2000, 4, 217–227 CrossRef CAS.
- J. Witowska-Jarosz, L. Gorski, E. Malinowska and M. Jarosz, J. Mass Spectrom., 2002, 37, 1236–1241 CrossRef CAS PubMed.
- D. M. Tomazela, F. C. Gozzo, I. Mayer, F. M. Engelmann, K. Araki, H. E. Toma and M. N. Eberlin, J. Mass Spectrom., 2004, 39, 1161–1167 CrossRef CAS PubMed.
- K. S. F. Lau, M. Sadilek, G. E. Khalil, M. Gouterman and C. Bruckner, J. Am. Soc. Mass Spectrom., 2005, 16, 1915–1920 CrossRef CAS PubMed.
- E. M. P. Silva, C. I. V. Ramos, P. M. R. Pereira, F. Giuntini, M. A. F. Faustino, J. P. C. Tomé, A. C. Tomé, A. M. S. Silva, M. G. O. Santana-Marques, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2012, 16, 101–113 CrossRef CAS.
- C. I. V. Ramos, C. M. Barros, A. M. Fernandes, M. G. O. Santana-Marques, A. J. F. Correia, J. P. C. Tomé, M. C. T. Carrilho, M. A. F. Faustino, A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mass Spectrom., 2005, 40, 1439–1447 CrossRef CAS PubMed.
- C. I. V. Ramos, J. P. C. Tomé and M. G. O. Santana-Marques, J. Mass Spectrom., 2012, 47, 173–179 CrossRef CAS PubMed.
- A. Rosell-Melé and J. R. Maxwell, Rapid Commun. Mass Spectrom., 1996, 10, 209–213 CrossRef.
- P. Swider, J. P. Lewtak, D. T. Gryko and W. Danikiewicz, J. Mass Spectrom., 2013, 48, 1116–1124 CrossRef CAS PubMed.
- D. T. Gryko, Chem. Commun., 2000, 2243–2244 RSC.
- A. E. Meier-Callahan, H. B. Gray and Z. Gross, Inorg. Chem., 2000, 39, 3605–3607 CrossRef CAS.
- T. Ding, E. A. Aleman, D. A. Modarelli and C. J. Ziegler, J. Phys. Chem. A, 2005, 109, 7411–7417 CrossRef CAS PubMed.
- J. P. Collman, M. Kaplun and R. A. Decréau, Dalton Trans., 2006, 554–559 RSC.
- T. H. Ngo, W. V. Rossom, W. Dehaen and W. Maes, Org. Biomol. Chem., 2009, 7, 439–443 CAS.
- M. Bröring, C. Milsmann, S. Ruck and S. Kohler, J. Organomet. Chem., 2009, 694, 1011–1015 CrossRef PubMed.
- W. Schöfberger, F. Lengwin, L. M. Reith, M. List and G. Knör, Inorg. Chem. Commun., 2010, 13, 1187–1190 CrossRef PubMed.
- S. H. Kim, H. Park, M. S. Seo, M. Kubo, T. Ogura, J. Klajn, D. T. Gryko, J. S. Valentine and W. Nam, J. Am. Chem. Soc., 2010, 132, 14030–14032 CrossRef CAS PubMed.
- M. Toganoh, Y. Kawabe and H. Furuta, J. Org. Chem., 2011, 76, 7618–7622 CrossRef CAS PubMed.
- O. G. Tsay, B.-K. Kim, T. L. Lu, J. Kwak and D. G. Churchill, Inorg. Chem., 2013, 52, 1991–1999 CrossRef CAS PubMed.
- K. S. F. Lau, M. Sadilek, M. Gouterman, G. E. Khalil and C. Bruckner, J. Am. Soc. Mass Spectrom., 2006, 17, 1306–1314 CrossRef CAS PubMed.
- C. I. V. Ramos, M. G. O. Santana-Marques, A. J. F. Correia, J. F. B. Barata, A. C. Tomé, M. G. P. M. S. Neves, J. A. S. Cavaleiro, P. E. Abreu, M. M. Pereira and A. A. C. C. Pais, J. Mass Spectrom., 2012, 47, 516–522 CrossRef CAS PubMed.
- J. B. F. Barata, C. M. Barros, M. G. O. Santana-Marques, M. G. P. M. S. Neves, M. A. F. Faustino, A. C. Tomé, A. J. F. Correia and J. A. S. Cavaleiro, J. Mass Spectrom., 2007, 42, 225–232 CrossRef CAS PubMed.
- M. Fasciotti, A. F. Gomes, F. C. Gozzo, B. A. Iglesias, G. F. Sá, R. J. Daroda, M. Toganoh, H. Furuta, K. Araki and M. N. Eberlin, Org. Biomol. Chem., 2012, 10, 8396–8402 CAS.
- P. M. Lalli, B. A. Iglesias, D. K. Deda, H. E. Toma, G. F. Sá, R. J. Daroda, K. Araki and M. N. Eberlin, Rapid Commun. Mass Spectrom., 2012, 26, 263–268 CrossRef CAS PubMed.
- P. M. Lalli, B. A. Iglesias, H. E. Toma, G. F. Sá, R. J. Daroda, J. C. S. Filho, J. E. Szulejko, K. Araki and M. N. Eberlin, J. Mass Spectrom., 2012, 47, 712–719 CrossRef CAS PubMed.
- C. I. V. Ramos, M. G. O. Santana-Marques, R. F. Enes, A. C. Tomé, J. A. S. Cavaleiro and M. Nogueras, J. Mass Spectrom., 2009, 44, 911–919 CrossRef CAS PubMed.
- A. Mahammed, J. J. Weaver, H. B. Gray, M. Abdelas and Z. Gross, Tetrahedron Lett., 2003, 44, 2077–2079 CrossRef CAS.
- L. Lvova, C. Di Natale, A. D'Amico and R. Paolesse, J. Porphyrins Phthalocyanines, 2009, 13, 1168–1178 CrossRef CAS.
- A. T. Blades, M. G. Ikonomou and P. Kebarle, Anal. Chem., 1991, 63, 2109–2114 CrossRef CAS.
- G. J. Van Berkel and F. Zhou, Anal. Chem., 1995, 67, 2916–2923 CrossRef CAS.
- G. J. Van Berkel, in The Electrolytic Nature of Electrospray, Electrospray Ionization Mass Spectrometry: Fundamentals, Instrumentation, and Applications, ed. R. B. Cole, John Wiley & Sons, New York, 1997, pp. 65–105 Search PubMed.
- J. F. De la Mora, G. J. Van Berkel, C. G. Enke, R. B. Cole, M. Martinez-Sanchez and J. B. Fenn, J. Mass Spectrom., 2000, 35, 939–952 CrossRef CAS.
- J. Shen, J. Shao, Z. Ou, W. E. B. Koszarna, D. T. Gryko and K. M. Kadish, Inorg. Chem., 2006, 45, 2251–2265 CrossRef CAS PubMed.
- H. Tsunematsu, H. Ikeda, H. Hanazono, M. Inagaki, R. Isobe, R. Higuchi, Y. Goto and M. Yamamoto, J. Mass Spectrom., 2003, 38, 188–195 CrossRef CAS PubMed.
- K. P. Madhusudanam, J. Mass Spectrom., 2006, 41, 1096–1104 CrossRef PubMed.
- H. Perreault and C. E. Costello, J. Mass Spectrom., 1999, 34, 184–197 CrossRef CAS.
- S. P. Gaucher and J. A. Leary, Anal. Chem., 1998, 70, 3009–3014 CrossRef CAS.
- X. Zhu and T. Sato, Rapid Commun. Mass Spectrom., 2007, 21, 191–198 CrossRef CAS PubMed.
- L. M. O. Lourenço, J. P. C. Tomé, M. R. M. Domingues, P. Domingues, P. J. Costa, V. Félix, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Rapid Commun. Mass Spectrom., 2009, 23, 3478–3483 CrossRef PubMed.
- M. R. M. Domingues, P. Domingues, A. Reis, A. J. F. Correira, J. P. C. Tomé, A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mass Spectrom., 2004, 39, 158–167 CrossRef CAS PubMed.
- U. Schwarz, M. Vonderach, M. Kappes, R. Kelting, K. Brendle and P. Weis, Int. J. Mass Spectrom., 2013, 339–340, 24–33 CrossRef CAS PubMed.
- A. B. Kanu, P. Dwivedi, M. Tam, L. Matz and H. H. Hill Jr, J. Mass Spectrom., 2008, 43, 1–22 CrossRef CAS PubMed.
- C. M. Benton, C. K. Lim, C. Moniz and D. J. L. Jones, Rapid Commun. Mass Spectrom., 2012, 26, 480–486 CrossRef CAS PubMed.
- L. Ahonen, M. Fasciotti, G. B. Gennäs, T. Kotiaho, R. J. Daroda, M. N. Eberlin and R. Kostiainen, J. Chromatogr. A, 2013, 1310, 133–137 CrossRef CAS PubMed.
- G. R. Hilton and J. L. P. Benesch, J. R. Soc. Interface, 2012, 9, 801–816 CrossRef CAS PubMed.
- M. Fasciotti, P. M. Lalli, G. Heerdt, R. A. Steffen, Y. E. Corilo, G. F. Sá, R. J. Daroda, F. A. M. Reis, N. H. Morgon, R. C. L. Pereira, M. N. Eberlin and C. F. Klitzke, Int. J. Ion Mobility Spectrom., 2013, 16, 117–132 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |