DOI:
10.1039/D5SC02449H
(Edge Article)
Chem. Sci., 2025,
16, 12510-12518
Carbene-stabilized 6,12-diboraanthanthrenes: unveiling the multistage redox properties of polycyclic aromatic hydrocarbons featuring electron-rich boron centers†
Received
1st April 2025
, Accepted 5th June 2025
First published on 5th June 2025
Abstract
Electronic manipulation of boron centers in polycyclic aromatic hydrocarbon (PAHs) frameworks often leads to unique redox and photophysical properties. Herein, we report the first isolation and redox investigation of carbene-stabilized 6,12-diboraanthanthrenes 3/4 with electron-rich boron centers. Combining experimental and theoretical studies confirms that 3/4 exhibits a closed-shell singlet ground state and strong global aromaticity. Oxidation of 3/4 with TEMPO led to the oxidized product 5. Furthermore, the reversible one or two-electron oxidation process for 3/4 has been confirmed by cyclic voltammetry. Sequential oxidation of 3/4 with AgSbF6 results in the isolable radical monocations 6/7, and dications 8/9. The radical character of 6/7 is confirmed by multi-line EPR spectra, with the hyperfine coupling splitting mainly attributed to the two boron nuclei. As rare diboron-doped dicationic PAHs, 8/9 display bright yellowish fluorescence. Reduction of 3/4 with 2 equivalents of KC8 leads to the formation of the dianionic species 10/11, where the carbene ligands are initially reduced and subsequently reconstructed, confirming the electron-rich nature of boron centers.
Introduction
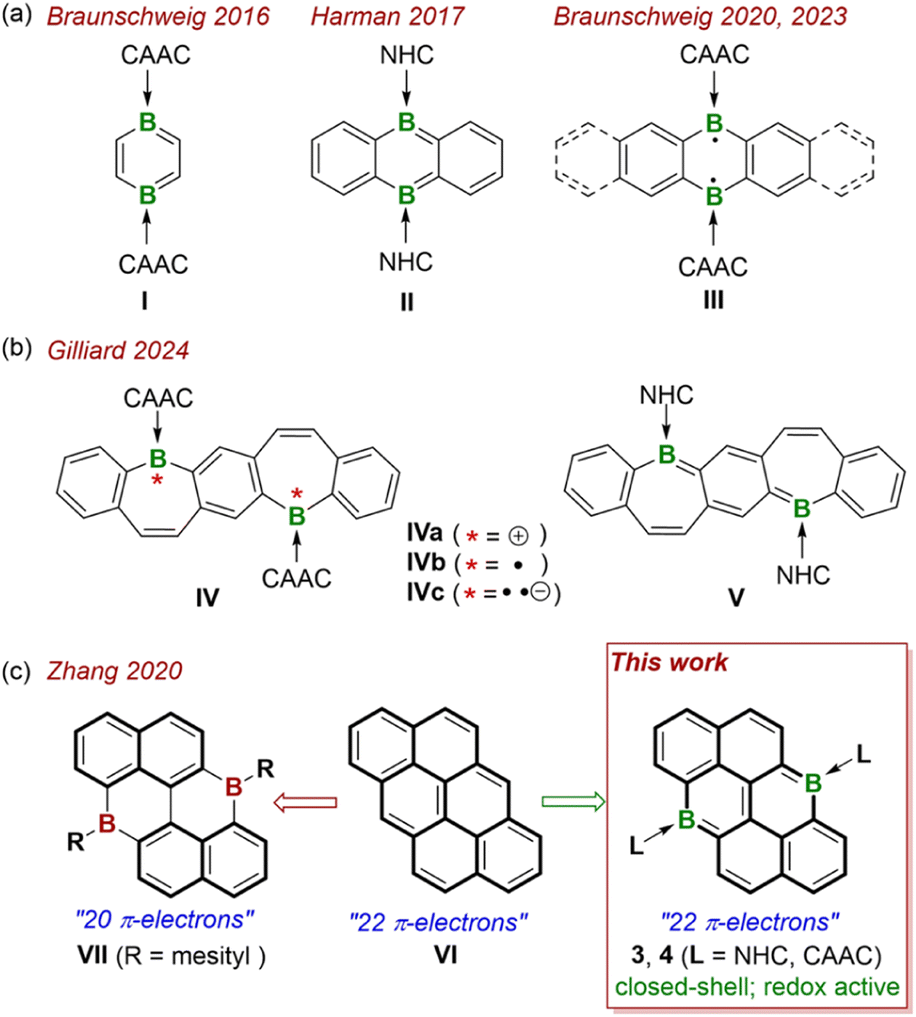
The design of molecules with multiple redox properties plays a crucial role in advancing various fields, such as energy storage,1 electrochromic devices,2 catalysis,3 and information storage.4 Among them, boron-doped polycyclic aromatic hydrocarbons (PAHs), combined with the unique electronic properties of PAHs with the functionality imparted by boron doping, have been considered as one promising class of material.5 Typically, the most well-studied redox properties of such systems are primarily modulated by utilizing the empty pz orbitals of boron centers as π-electron acceptors, which likely makes the system more prone to reduction and resistant to oxidation.6 Meanwhile, the integration of a Lewis acidic boron unit (R–B, R = anionic substituent) into PAHs leads to an obvious reduction in the number of π-electrons compared to their all carbon-based counterparts and hampers the fine regulation of their electronic properties. As a result, obtaining boron-doped PAHs that maintain an isoelectronic structure with their all-carbon analogues is highly desirable, as it not only expands the synthetic scope of boron-doped PAHs with fantastic electronic structures but also provides valuable insights into their redox–active properties and potential applications of these materials. To address this situation, the incorporation of boron units (L → B, L = neutral ligand), which are considered isosteric and isoelectronic to carbon units (R–C), has emerged as an effective approach for constructing bora-cyclic compounds and developing new types of boron-doped PAHs.7 With the distinct σ-donating and π-accepting abilities, diamino-N-heterocyclic carbenes (NHCs) and cyclic(alkyl)(amino) carbenes (CAACs) have proven to be excellent ligands for the stabilization and electronic manipulation of such boron-doped organic systems.8 The Braunschweig group conducted systematic studies on the CAAC-stabilized 1,4-diborabenzene I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 and a laterally extended linear diboraacenes III,10 with the two boron atoms positioned at the para position of the six-membered ring (Fig. 1a). These studies revealed that the elongation of the acenes, the positioning of the boron centers, and the orientation of the CAAC ligands collectively influence their electronic properties, resulting in either closed-shell or open-shell biradical characters. Meanwhile, the Harman group reported the isolation of NHC-stabilized 9,10-diboraanthracene II,11 shows a completely closed-shell electronic structure. Notably, the Gilliard group has made significant contributions to this field, and comprehensively investigated the redox properties of the boron-doped PAHs.7,12 Specifically, by incorporating two boron units (L → B) into a single PAH skeleton, they reported NHC/CAAC-stabilized bis(9-boraphenanthrene)13 and fused bis-borepin IV and V,14 disclosing that their biradical character could be finely regulated by tuning the steric and electronic features of carbene ligands (Fig. 1b). Remarkably, CAAC-stabilized bis-borepin with multiple redox-states, including the dication IVa, neutral biradical IVb, and dianion IVc have been isolated, revealing the advantage of this approach in the stabilization of multiple charged redox species of the diboron doped PAHs. Despite these studies greatly advanced this research field,7 the research area is still in its infancy and the diversity of the PAHs skeletons, especially with larger π-electronic systems, is quite limited. Therefore, more specific species are required to gain a deeper understanding of the relationship between electronic structures, redox properties, and the positioning/types of boron units (L → B) within PAHs, which is essential for the development of new materials with tailored properties for various applications.
9 and a laterally extended linear diboraacenes III,10 with the two boron atoms positioned at the para position of the six-membered ring (Fig. 1a). These studies revealed that the elongation of the acenes, the positioning of the boron centers, and the orientation of the CAAC ligands collectively influence their electronic properties, resulting in either closed-shell or open-shell biradical characters. Meanwhile, the Harman group reported the isolation of NHC-stabilized 9,10-diboraanthracene II,11 shows a completely closed-shell electronic structure. Notably, the Gilliard group has made significant contributions to this field, and comprehensively investigated the redox properties of the boron-doped PAHs.7,12 Specifically, by incorporating two boron units (L → B) into a single PAH skeleton, they reported NHC/CAAC-stabilized bis(9-boraphenanthrene)13 and fused bis-borepin IV and V,14 disclosing that their biradical character could be finely regulated by tuning the steric and electronic features of carbene ligands (Fig. 1b). Remarkably, CAAC-stabilized bis-borepin with multiple redox-states, including the dication IVa, neutral biradical IVb, and dianion IVc have been isolated, revealing the advantage of this approach in the stabilization of multiple charged redox species of the diboron doped PAHs. Despite these studies greatly advanced this research field,7 the research area is still in its infancy and the diversity of the PAHs skeletons, especially with larger π-electronic systems, is quite limited. Therefore, more specific species are required to gain a deeper understanding of the relationship between electronic structures, redox properties, and the positioning/types of boron units (L → B) within PAHs, which is essential for the development of new materials with tailored properties for various applications.
 |
| | Fig. 1 (a) Neutral 1,4-diborabenzene I and its lateral extended acenes II and III. (b) Bis-borepin IV with various redox states and dibora-quinone V. (c) All-carbon anthanthrene VI, boron-doped anthanthrene VII and 6,12-diboraanthanthrenes 3 and 4 (this work). | |
Anthanthrene VI, a well-known prototypical nonlinear PAHs, has demonstrated great potential for use as a semiconductor or luminescent material (Fig. 1c).15 By incorporating main group elements such as N,16 chalcogen elements (O, S, Se)17 into the anthanthrene structure, a series of heteroatom-doped anthanthrenes have been successfully synthesized, significantly expanding the scope of anthanthrene-based PAHs. Significantly, the electron-deficient boron atom has also been successfully introduced into the anthanthrene framework, leading to the formation of VII with excellent electroluminescent properties (Fig. 1c).18 However, compared to the all-carbon anthanthrene VI, VII has two fewer π-electrons in its central skeleton, complicating a direct comparison of the boron effect in terms of bonding, electronic structure, and physical/chemical properties. To address this challenge, neutral 6,12-diboraanthanthrenes 3 and 4 are proposed as isoelectronic and isostructural boron analogues of VI. Meanwhile, 3 and 4 can also be considered as the vertical extended 9,10-diboraanthracenes II and III (Fig. 1c). Herein, we report the experimental and theoretical studies of 3 and 4, revealing that these compounds exhibit a closed-shell ground state and considerable aromaticity over the central framework. The subsequent oxidation/reduction reactions were investigated comprehensively to disclose their unique multistage redox behaviors. The successful isolation and detailed characterization of the corresponding oxidized products monocationic radicals (5, 6) and dications (7, 8), as well as the reduced species (9, 10) demonstrate the electron-rich character of the boron centers within this unique framework, providing deeper insights into the electronic structure and reactivity of these novel boron-doped PAHs.
Results
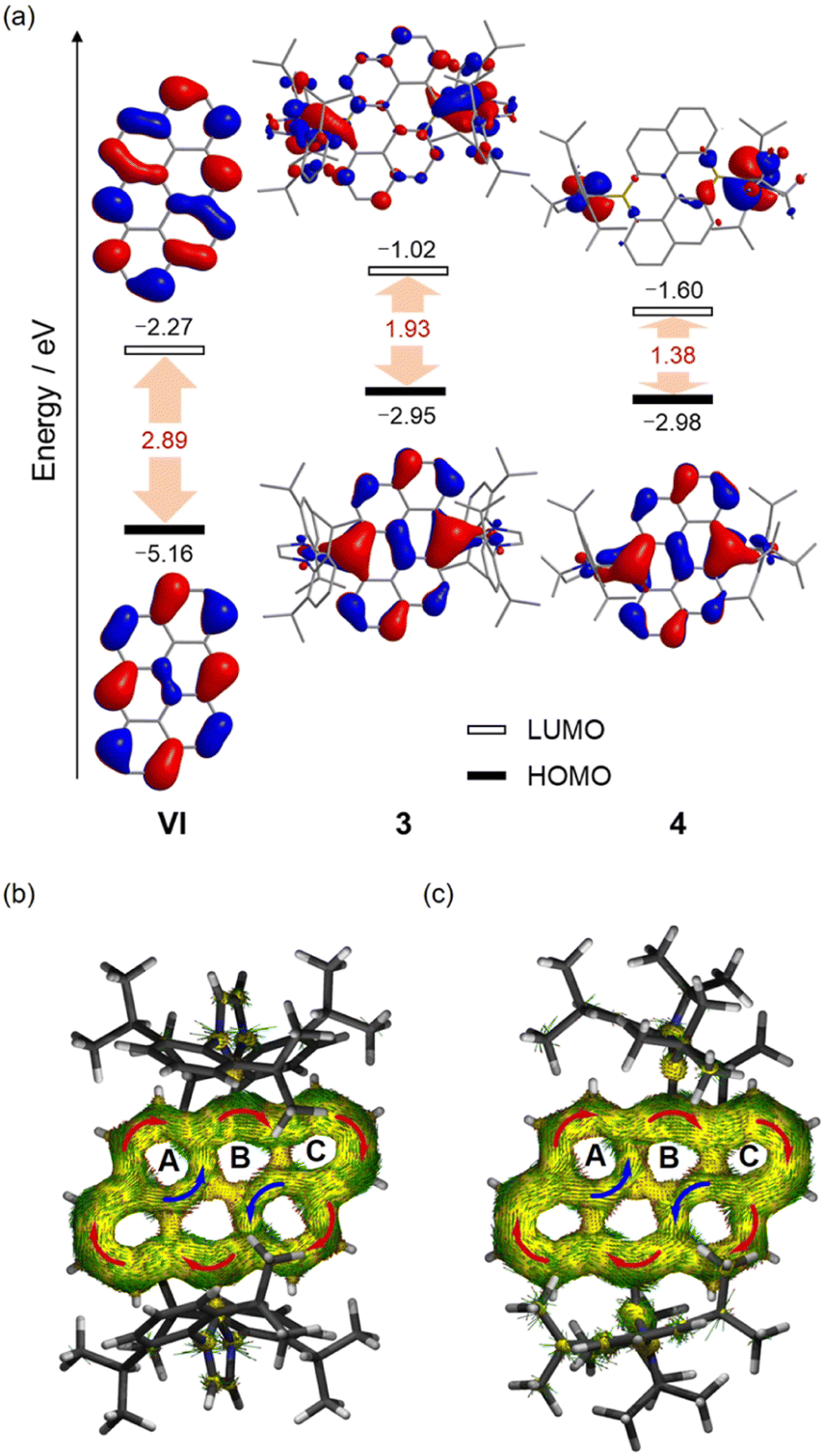
Initially, we attempted to isolate the bromide-substituted boron species 2 directly by following the reported procedure,18a but achieving the desired purity of 2 proved challenging and impeded the subsequent preparation of 3 and 4. As an alternative approach, the air-stable hydroxyl-substituted diboron 1 was first isolated and purified by simple hydrolysis of the crude 2, and treated with 2 equivalents of BBr3 to yield 2 in high purity (Scheme 1). Next, one-pot reactions of 2 with 2 equivalents of carbene ligands (N-heterocyclic carbene (NHC) or cyclic(alkyl)(amino) carbene (CAAC)) and 2 equivalents of potassium graphite (KC8) straightforwardly afforded 3 or 4 in high yields. 3 and 4 are thermally stable at room temperature, both in the solid state and in solution, but rapidly decomposed upon exposure to air or moisture. The solubility of 3 and 4 is quite poor in common organic solvents, such as benzene, toluene, THF, etc. However, after careful selection of the deuterated solvents, satisfactory multiple NMR spectra for 3 were obtained using DMF-d7 as the solvent (Fig. S4–S6†). In contrast, the extremely poor solubility of 4 hindered the collection of satisfactory solution NMR spectra. Thus, its solid-state NMR spectrum was collected instead (Fig. S13–S15†). In the 1H NMR spectrum of 3, a singlet at 8.33 ppm is observed, corresponding to the protons of the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– units of NHCs, indicating its high symmetric geometry in solution. The 11B NMR spectrum displays a broad singlet at 22.88 ppm, comparable to these (20.1–24.4 ppm) of reported II and NHC-stabilized bis(9-boraphenanthrene).11,13 In the solid-state NMR spectra of 4, the 11B NMR resonance appears around 19.54 ppm, close to that of 3 (Fig. S12 and S15†). To further confirm the closed-shell character of 4, its solid-state EPR spectrum was collected, and no noticeable signal was observed (Fig. S44†). Single-crystal X-ray diffraction studies further confirmed the generation of 3 and 4 (Fig. 2). Both molecules bear high symmetry with the central hexacyclic rings displaying a perfect planar geometry. The bond lengths of B1–C11 for 3 and 4 are 1.590(3) Å and 1.610(3) Å, respectively, which are compared with those of the reported species II (1.5922(19) Å),11 NHC-stabilized bis(9-boraphenanthrene) (1.604(3) Å)13 and fused bis-borepin (1.6058(16) Å),14a but longer than those (1.525(3)–1.545(5) Å) of the reported diradical species IVb,14aIII,10b and CAAC-stabilized bis(9-boraphenanthrene),13 confirming the single bond character of B1–C11 bonds. The endocyclic B1–C1 and B1–C8′ bonds for 3 (1.485(4) Å, 1.515(4) Å) and 4 (1.491(4) Å, 1.517(4) Å) are shorter than those of the typical B–C single bonds,19 but comparable with those (1.521(2) Å, 1.5153(19) Å) of II, revealing their partial double bond character. In the UV-vis absorption spectra, the lowest-energy absorption peaks are observed around 656 nm and 880 nm for 3 and 4, respectively, which are significantly red-shift compared with that (432 nm) of all-carbon anthanthrene VI (Fig. 3). The maximum absorption of 3 primarily corresponds to the electronic transition from the HOMO → LUMO (96%), while that of 4 corresponds to the HOMO → LUMO (85%) and the HOMO → LUMO + 2 (5.7%) transitions, as predicted by time-dependent density functional theory (TD-DFT) calculations (Fig. S51, S52, Tables S9 and S10†). Note that the well-separated locations of the HOMOs and LUMOs in 3 and 4 imply that these transitions likely correspond to intramolecular charge transfer (ICT), which is distinct from VI (Fig. 4). The trend in the absorption shift among the VI, 3 and 4 is also consistent with the narrowing HOMO–LUMO gaps due to the doping of the boron centers.10a
CH– units of NHCs, indicating its high symmetric geometry in solution. The 11B NMR spectrum displays a broad singlet at 22.88 ppm, comparable to these (20.1–24.4 ppm) of reported II and NHC-stabilized bis(9-boraphenanthrene).11,13 In the solid-state NMR spectra of 4, the 11B NMR resonance appears around 19.54 ppm, close to that of 3 (Fig. S12 and S15†). To further confirm the closed-shell character of 4, its solid-state EPR spectrum was collected, and no noticeable signal was observed (Fig. S44†). Single-crystal X-ray diffraction studies further confirmed the generation of 3 and 4 (Fig. 2). Both molecules bear high symmetry with the central hexacyclic rings displaying a perfect planar geometry. The bond lengths of B1–C11 for 3 and 4 are 1.590(3) Å and 1.610(3) Å, respectively, which are compared with those of the reported species II (1.5922(19) Å),11 NHC-stabilized bis(9-boraphenanthrene) (1.604(3) Å)13 and fused bis-borepin (1.6058(16) Å),14a but longer than those (1.525(3)–1.545(5) Å) of the reported diradical species IVb,14aIII,10b and CAAC-stabilized bis(9-boraphenanthrene),13 confirming the single bond character of B1–C11 bonds. The endocyclic B1–C1 and B1–C8′ bonds for 3 (1.485(4) Å, 1.515(4) Å) and 4 (1.491(4) Å, 1.517(4) Å) are shorter than those of the typical B–C single bonds,19 but comparable with those (1.521(2) Å, 1.5153(19) Å) of II, revealing their partial double bond character. In the UV-vis absorption spectra, the lowest-energy absorption peaks are observed around 656 nm and 880 nm for 3 and 4, respectively, which are significantly red-shift compared with that (432 nm) of all-carbon anthanthrene VI (Fig. 3). The maximum absorption of 3 primarily corresponds to the electronic transition from the HOMO → LUMO (96%), while that of 4 corresponds to the HOMO → LUMO (85%) and the HOMO → LUMO + 2 (5.7%) transitions, as predicted by time-dependent density functional theory (TD-DFT) calculations (Fig. S51, S52, Tables S9 and S10†). Note that the well-separated locations of the HOMOs and LUMOs in 3 and 4 imply that these transitions likely correspond to intramolecular charge transfer (ICT), which is distinct from VI (Fig. 4). The trend in the absorption shift among the VI, 3 and 4 is also consistent with the narrowing HOMO–LUMO gaps due to the doping of the boron centers.10a
 |
| | Scheme 1 Synthetic routes to 3 and 4 (Photographs of the solutions of 3 and 4 in o-difluorobenzene (o-DFB) under room light). NHC = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene, CAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene. | |
 |
| | Fig. 2 Molecular structures of 3: top view (a) and side view (b), and 4: top view (c) and side view (d). Thermal ellipsoids are set at the 50% probability. Hydrogen atoms and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (°). 3: B1–C11: 1.590(3), B1–C1: 1.485(4), C1–C10: 1.456(3), C10–C10′: 1.431(4), C10′-C9′: 1.443(3), C9′-C8′: 1.444(3), C8′-B1: 1.515(4); C11–B1–C1: 120.6(2), C1–B1–C8′: 119.4(2), C11–B1–C8′: 120.0(8). 4: B1–C11: 1.610(3), B1–C1: 1.491(4), C1–C10: 1.460(3), C10–C10′: 1.435(4), C10′-C9′: 1.442(3), C9′-C8′: 1.440(3), C8′-B1: 1.517(4); C11–B1–C1: 122.8(2), C1–B1–C8′: 117.8(2), C11–B1–C8′: 118.4(2). | |
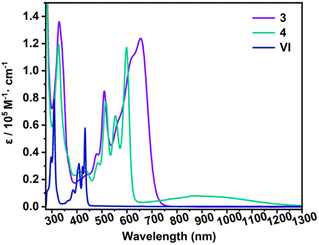 |
| | Fig. 3 UV-vis absorption spectra of 3, 4 and all-carbon anthanthrene (VI) in o-difluorobenzene. | |
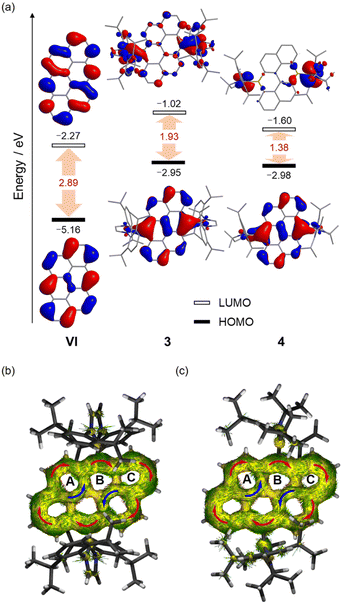 |
| | Fig. 4 Plots of the HOMO and LUMO of 3, 4 and all-carbon anthanthrene VI (hydrogen atoms are omitted for clarity, isovalue = 0.03) (a), and the ACID plots (π-electron only) of 3 (b) and 4 (c) (the external magnetic field is applied orthogonal to the plane, and the red and blue arrows indicate the clockwise (diamagnetic) and counter-clockwise (paramagnetic) current flow, respectively). | |
Given their remarkable electronic structures, we performed detailed theoretical calculations on three electronic states (closed-shell singlet (CS), open-shell singlet (OS), and triplet (T)) at the (U)B3LYP/6-311G(d,p) level. However, attempts to optimize 3 and 4 as open-shell singlet species (OS) resulted in structures that collapsed into closed-shell singlet (3/4-CS) electronic configurations. The calculated singlet–triplet energy gaps ΔECS–T are −17.6 kcal mol−1 for 3 and −4.8 kcal mol−1 or 4, respectively. The optimized structure of 3/4-CS matches well with the X-ray data, while the triplet structures of 3/4-T deviate significantly from the experimental observations (Table S8†). These findings, supported by both experimental and theoretical studies, confirm that 3 and 4 exhibit closed-shell singlet ground states. Moreover, for a deeper understanding of the electronic effect of the substitution of carbon units (R–C) with boron units (L → B), the molecular orbitals (MOs) for 3, 4 and their hydrocarbon anologue VI were calculated for comparison (Fig. 5a). Compared to VI, the locations of the HOMOs of 3 and 4 are very similar, primarily located on the central hexacyclic framework, with minimal contribution from the p orbitals of the Ccarbene centers, but noticeably the energy of HOMOs of 3 and 4 is much higher than that of VI, implying that 3 and 4 are more prone to oxidation reactions. In contrast to that of VI, the LUMOs of 3 and 4 are mostly located on the carbene ligands, with slight extension to the central framework. Collectively, the HOMO–LUMO (H–L) gaps of 3 (1.93 eV) and 4 (1.38 eV) are significantly smaller than that of VI, indicating that incorporating boron atoms into organic molecules is an effective strategy for narrowing their HOMO–LUMO gaps, primarily by raising the energy of the HOMO.10a Meanwhile, the narrower H–L gap of 4 was confirmed in comparison to that of 3, showing similar trends observed in II and III with the substitution of NHCs by CAACs.10a,b,11 Wiberg bond index (WBI) values of the B1–C11 bonds are 0.89 (3) and 0.88 (4), indicating the single bond character for the B–C bonds. In contrast, WBI values larger than 1 for the B1–C1 (1.17 (3), 1.17 (4)) and B1–C8′ (1.05(3), 1.04(4)) bonds support the delocalization of π-electrons over the central framework. To assess their aromaticity, we conducted the nucleus-independent chemical shift (NICS)20 and anisotropy of the induced current density (ACID)21 calculations. All the six-membered rings in the central framework exhibit moderate to strong aromaticity as indicated by the negative NICS(1)zz values [3: −9.3 ppm (A), −25.21 ppm (B), −32.55 ppm (C); 4: −7.13 ppm (A), −24.21 ppm (B), −31.23 ppm (C)]. The ACID plots show diatropic ring currents around the periphery of the central frameworks, suggesting their global aromatic character (Fig. 4b and c). Meanwhile, the broken paratropic ring currents (depicted in blue) of rings A in 3 and 4 imply a diminished aromatic character, consistent with their relatively small NICS(1)zz value for these rings. Additionally, based on the ACID and NICS analysis of anthanthrene VI, a similar aromatic character to 3 and 4 is confirmed (Fig. S50†), demonstrating that the π-electrons formally supplied by the two boron centers are well delocalized over the entire central framework to maintain the considerable aromaticity. Notably, 3 and 4 represent rare examples of neutral diboron-doped PAHs, where the boron centers contribute π-electrons rather than simply providing empty p orbitals, as is typical in conjugated systems.10a,11,14a
 |
| | Fig. 5 (a) Reversible oxidation reactions of 3 and 4. (b) Cyclic voltammogram of 7 and 8 in DCM/0.1 M Bu4NPF6 measured at a glass carbon (GC) working electrode with a scanning rate of 100 mV s−1. | |
In consideration of their unique electronic structures of 3 and 4, particularly the electron–rich properties of the boron centers, we further investigated their redox reactivity. Oxidation of 3 or 4 with two equivalents of TEMPO (2,2,6,6tetramethylpiperidin-1-oxyl) led to the formation of the borinic acid ester 5, which was fully characterized by multiple NMR spectra and X-ray crystallography (Scheme 2). The 1H NMR spectrum confirms the loss of the carbene ligands and the formation of the anti and syn-atropisomers in a static ratio of 83:17 based on the orientation of TMP (2,2,6,6-tetramethylpiperidiyl) substituents.22 The 11B NMR spectrum of 5 displays a broad singlet at 39.21 ppm, indicating its tricoordinate chemical environment. The anti-rotamer of 5 was further characterized by single-crystal X-ray diffraction analysis, where the TMP substituents are oriented in trans to each other and the boron centers adopt a planar trigonal geometry. The B–O bonds (1.3672(19) Å) are shorter than typical single bonds but comparable with those (1.3636(16) Å) of the B–O bond in reported borinic acid ester.22
 |
| | Scheme 2 Oxidation reactions of 3 or 4 with TEMPO, and the molecular structure of anti-5. | |
Moreover, controlled oxidation reactions of 3 and 4 with one or two equivalents of silver hexafluoroantimonate (AgSbF6) afforded the corresponding air- and moisture-sensitive radical monocations 6 and 7 as dark brown solids, or dications 8 and 9 as red solids, in moderate to high yields (Fig. 5a and S46†). Note that these three redox states could be interconverted upon the addition of specific amounts of AgSbF6 or KC8, as confirmed by cyclic voltammetry (CV) measurements (Fig. 5b). Due to the poor solubility of 3 and 4, their dicationic species, 8 and 9 were chosen for electrochemical analysis. The electrochemical behavior of 8 and 9 reveals two reversible reduction waves with half-wave potentials of E1/2 = −1.22 V and −2.01 V for 8, and −1.20 V and −1.98 V for 9, relative to the ferrocene/ferrocenium (Fc/Fc+) redox couple. The two observed redox processes could be assigned to the formation of the corresponding one- and two-electron-reduced species. In their solid-state structures, the trigonal-planar geometries of the boron centers are confirmed, with the sum of the bond angles around each boron atom being close to 360°, indicative of sp2 hybridization (Fig. 6). The radicals 6, 7, and dication 8 feature nearly planar hexacyclic rings, while dication 9 exhibits a noticeable bent geometry with two boron-containing six-membered rings adopting boat conformations. The Ccarbene–B bond distances (1.588(4) Å (6), 1.598(6) Å (7), 1.600(7) Å (8), and 1.590(6) Å (9)) are comparable with those of 3 and 4, suggesting similar bond situations as a boron–carbon single bond. The slight elongation of the endocyclic C–B lengths from 3/4 to 6/7 to 8/9, supported by the decreasing WBI values, is consistent with the gradual reduction in π-electron density, leading to weaker bonding interactions (Fig. S66 and Table S19†).12c Meanwhile, DFT calculations reveal that the frontier molecular orbital for the SOMOs of 6/7, LUMOs of 8/9, and the HOMOs of 3/4 are nearly identical with negligible contributions from the carbene ligands, suggesting that the stepwise oxidations predominantly occur on the central hexacyclic frameworks (Fig. S53, S54, S57 and S58†).
 |
| | Fig. 6 Molecular structures of radical cations 6 (a), 7 (b), and dications 8 (c), 9 (d). Thermal ellipsoids are set at the 50% probability. Hydrogen atoms, counteranions, and solvent molecules are omitted for clarity. | |
The electron paramagnetic resonance (EPR) spectra of 6 and 7, measured in o-difluorobenzene (o-DFB) at room temperature exhibit a multi-line structure centered at g = 2.0035 (6), g = 2.0034 (7) (Fig. 7). These spectra have been successfully simulated, revealing that the hyperfine coupling splitting is mainly attributed to two boron nuclei (6: a(10B) = 3.28 MHz, a(11B) = 6.56 MHz; 7: a(10B) = 3.33 MHz, a(11B) = 6.67 MHz) (Fig. 7), which are comparable with that of radical cation of II,11 confirming the presence of an unpaired electron on the central hexacyclic rings. The calculated singly occupied molecular orbitals (SOMOs) of 6 and 7 are primarily dominated by the π-type orbitals over the central hexacyclic rings (Fig. S53 and S54†). The spin density is predominantly delocalized across the central hexacyclic framework with significant contributions (6: 45.2%, 7: 43.4%) from the two boron atoms, while the spin density on the carbene ligands is neglectable (Fig. 7, Tables S11 and S12†). Note that 6 and 7 represent the rare isolable cyclic diboron-containing radical cations,10b,11,23 The UV-vis absorption spectra show the maximum absorption bands around 880 nm and 870 nm for 6 and 7 (Fig. S45†), respectively. These bands can primarily be assigned to the SOMO → αLUMO (91%) electronic transitions, which correspond to the D0 → D1 excitation, as determined from the TD-DFT calculations (Fig. S55, S56, Table S13 and S14†).
 |
| | Fig. 7 Spin density distribution of 6 (a) and 7 (b) at UB3LYP/6-311G(d, p) level (isovalue = 0.002, H atoms omitted for clarity), and experimental CW X-band (black trace) and simulated (red trace) EPR spectra of 6 (c) and 7 (d) in o-difluorobenzene at room temperature. | |
The dication species 8 and 9 display similar UV-vis and fluorescence spectra, with a slight red-shift observed for 8 compared to 9 (Fig. 8). In o-difluorobenzene solutions, 8 and 9 exhibit lowest-energy absorption peaks at 577 nm (ε = 1.35 × 104 M−1 cm−1) and 561 nm (ε = 0.97 × 104 M−1 cm−1), and emit yellowish fluorescence at 563 nm and 553 nm (8: Φ = 0.25, τ = 19.5 ns; 9: Φ = 0.16, τ = 17.9 ns), respectively, which are red-shifted compared to the fluorescence peak (515 nm) of R–B unit doped diboraanthanthrene.18a TD-DFT calculations reveal that the lowest energy absorption bands for 8 and 9 arise from an allowed vertical S0 → S1 electronic transition, primarily corresponding to the HOMO → LUMO (95%) electronic transition (Tables S15 and S16†). Structural optimizations of the singlet excited state (S1) for 8 and 9 were performed to better understand their fluorescence properties. The locations of the excited-state HOMO (ES-HOMO) and the excited-state LUMO (ES-LUMO) over the central frameworks confirm that the emission behaviours originate from a local excited-state (LE) transition (Fig. S63 and S65†).
 |
| | Fig. 8 Normalized UV-vis absorption (solid lines) and fluorescence spectra (λex = 490 nm, dashed lines) of 8 and 9 in o-difluorobenzene (inset: photographs of cuvettes containing 8 and 9 in o-difluorobenzene under room light (left) and UV light (right). | |
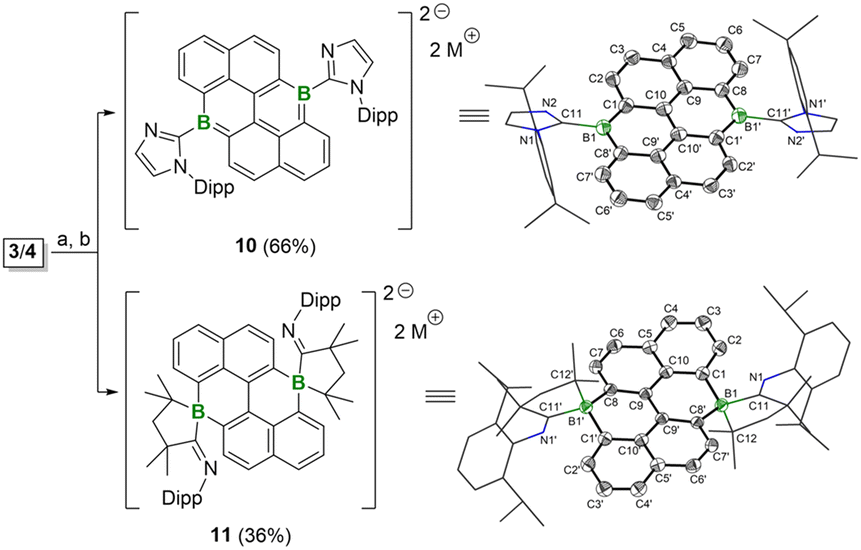
To gain a complete understanding of the redox behaviors of 3 and 4, their reduction reactions were further investigated in detail. A reduction of 3 or 4 with one equivalent of KC8 led to the unidentified mixture. However, when 3 and 4 were subjected to the two equivalents of KC8, after workup, the dianion species 10 and 11 were successfully isolated (Fig. 9). The 11B{1H} NMR spectra show a singlet for 10 and 11 at 28.06 ppm and −8.03 ppm, corresponding to the tri- or tetra-coordinate boron centers, respectively. The solid-state structures of 10 and 11 further confirmed their distinct coordination environments for the boron centers (Fig. 9). For 10, the central hexacyclic framework is planar and the boron atoms are positioned in a trigonal geometry. The bond lengths for B1–C1 and B1–C8′ are 1.489(5) Å and 1.533(5) Å, which are comparable to those of 3, suggesting their partial BC double bond character. Thus, 10 can be described as the diboron-doped dianionic anthanthrene. Similar activation of the NHC ligand has also been observed during the reduction process of NHC-stabilized borafluorene and bis-borepin.14a,24 In contrast, the much longer bond distances of B1–C1 (1.602(2) Å) and B1–C8′ (1.619(2) Å) in 11 are observed, indicating their B–C single bond character. Each boron center in 11 is coordinated by four carbon atoms, forming a spirocyclic structure with five- and six-remembered rings. Based on those results, the reduction of 3 and 4 likely proceeds on the carbene ligands, driven by the highly electron-rich nature of the boron centers and the relatively electron-deficient carbene carbons. Indeed, the calculated molecular orbitals (MOs) of 3 and 4 reveal that the lowest unoccupied molecular orbitals (LUMOs) are predominantly located on the carbene ligands (Fig. 4a). Accordingly, plausible mechanisms for the generation of 10 and 11 are proposed (Scheme S1†). Initially, one-electron reduction occurred on each Ccarbene center and was followed by the formation of the CcarbeneN double bond with the concurrence of the homolytic cleavage of the N–Cdipp single bonds of 3 to form 10, and the homolytic cleavage of N–C(Me)2 single bonds of 4 to result in the generation of CN(–dipp) double bonds and formation of the tetracoordinate boron species 11. Thus, the activation of the carbene ligands during the reduction process further supports the electron-rich nature of the boron center in 3 and 4, which is completely distinct from that of the Lewis acidic boron units doped PAHs.
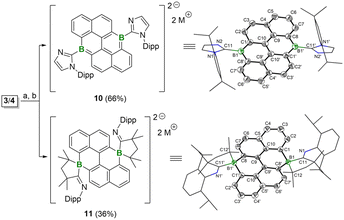 |
| | Fig. 9 Reduction reactions of 3 and 4. Conditions: (a) 2 equiv. KC8, THF, −78 °C to r.t.; (b) 2 equiv. [2.2.2]cryptand; M = [K(cryptand)]. Molecular structures of 10 and 11 (thermal ellipsoids are set at the 50% probability. Hydrogen atoms, countercations, and solvent molecules are omitted for clarity). | |
Conclusions
In this work, the first isolated carbene-stabilized 6,12-diboraanthanthrenes 3 and 4 have been synthesized and fully characterized. The closed-shell singlet states of 3 and 4 were confirmed as indicated by their NMR-active spectra, the planar geometry of the central hexacyclic frameworks, and the significant electron delocalization over the frameworks supported by DFT calculations. Their aromaticity was evaluated by the ACID and NICS calculations, showing an aromaticity comparable to that of all-carbon anthanthrene. Reactions of 3/4 with TEMPO resulted the oxidized product 5. Furthermore, their multistage redox behaviours were investigated in detail. One- or two-electron oxidation reactions of 3 and 4 with AgSbF6 lead to radical mono-cations 6 and 7, and dications 8 and 9. A joint experimental and theoretical study indicates that the unpaired electron in 6 and 7 is delocalized across the central π-systems and largely distributed over the two boron centers. 8 and 9 exhibit strong orange-yellowish fluorescence attributed to π → π* transitions, representing rare examples of dicationic diboron-doped PAHs. Two-electron reduction reactions of 3 and 4 resulted in C–N bond cleavage of the carbene ligands, leading to the dianionic species 10 and 11, these observations during the redox processes reveal the electron-rich nature of the boron centers in 3 and 4. This work clearly demonstrates that substituting carbon with boron, while maintaining the same number of π-electrons, significantly alters the electronic structure of PAHs, offering new insights into electronic manipulation via a boron unit (L → B)-doped strategy, as opposed to the traditional Lewis-acidic boron-doping method. This approach enhances the development of boron-doped PAHs with diverse redox behaviours and fascinating optical and magnetic properties.
Data availability
Data supporting this study have been included in the ESI.† Crystallographic data for 3–11 have been deposited at the Cambridge Crystallographic Data Centre under CCDC nos. CCDC 2413197 (3), 2413201 (4), 2413196 (5), 2413203 (6), 2413198 (7), 2413204 (8), 2413202 (9), 2413200 (10), 2413199 (11) and can be obtained from https://www.ccdc.cam.ac.uk.
Author contributions
B. S. designed the project. Y. W. and T. S. carried out the experiments. B. S., L. Y. and G. Z. did the theoretical calculations. B. S. and Y. W. drafted the paper. All authors discussed the results and contributed to the preparation of the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Natural Science Foundation of China (22471207), the “1000-Plan program” of Shaanxi Province, the Qin Chuang Yuan Program of Shaan Xi Province (QCYRCXM-2022-28), and the startup funds from Xi'an Jiaotong University. We thank Dr Chang Huang, Gang Chang, and Pei Zhou at the Instrument Analysis Center of Xi'an Jiaotong University for their help in NMR and X-ray crystallographic tests. We thank Lingbing Kong (Shandong University) and Yuanting Su (Soochow University) for their valuable suggestions on the manuscript.
Notes and references
-
(a) J. Luo, B. Hu, M. Hu, Y. Zhao and T. L. Liu, Status and prospects of organic redox flow batteries toward sustainable energy storage, ACS Energy Lett., 2019, 4, 2220–2240 CrossRef CAS;
(b) P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Opportunities and challenges for organic electrodes in electrochemical energy storage, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed;
(c) G. Kwon, Y. Ko, Y. Kim, K. Kim and K. Kang, Versatile redox-active organic materials for rechargeable energy storage, Acc. Chem. Res., 2021, 54, 4423–4433 CrossRef CAS PubMed;
(d) X. Fang, Z. Li, Y. Zhao, D. Yue, L. Zhang and X. Wei, Multielectron organic redoxmers for energy-dense redox flow batteries, ACS Mater. Lett., 2022, 4, 277–306 CrossRef CAS;
(e) K. Hatakeyama-Sato and K. Oyaizu, Redox: organic robust radicals and their polymers for energy conversion/storage devices, Chem. Rev., 2023, 123, 11336–11391 CrossRef CAS;
(f) M. Shoaib, P. Vallayil, N. Jaiswal, P. Iyapazham Vaigunda Suba, S. Sankararaman, K. Ramanujam and V. Thangadurai, Advances in redox flow batteries – A comprehensive review on inorganic and organic electrolytes and engineering perspectives, Adv. Energy Mater., 2024, 14, 2400721 CrossRef CAS.
- T. Jarosz, K. Gebka, A. Stolarczyk and W. Domagala, Transparent to black electrochromism—the “holy grail” of organic optoelectronics, Polymers, 2019, 11, 273 CrossRef.
-
(a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed;
(b) N. A. Romero and D. A. Nicewicz, Organic photoredox catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed;
(c) H. W. Moon and J. Cornella, Bismuth redox catalysis: an emerging main-group platform for organic synthesis, ACS Catal., 2022, 12, 1382–1393 Search PubMed;
(d) X. Wu, Y. Chang and S. Lin, Titanium radical redox catalysis: Recent innovations in catalysts, reactions, and modes of activation, Chem, 2022, 8, 1805–1821 Search PubMed.
-
(a) J. S. Lindsey and D. F. Bocian, Molecules for charge-based information storage, Acc. Chem. Res., 2011, 44, 638–650 Search PubMed;
(b) N. R. Erickson, C. D. Holstrom, H. M. Rhoda, G. T. Rohde, Y. V. Zatsikha, P. Galloni and V. N. Nemykin, Tuning electron-transfer properties in 5,10,15,20-tetra(1'-hexanoylferrocenyl)porphyrins as prospective systems for quantum cellular automata and platforms for four-bit information storage, Inorg. Chem., 2017, 56, 4716–4727 CrossRef;
(c) D. T. Payne, W. A. Webre, Y. Matsushita, N. Zhu, Z. Futera, J. Labuta, W. Jevasuwan, N. Fukata, J. S. Fossey, F. D'Souza, K. Ariga, W. Schmitt and J. P. Hill, Multimodal switching of a redox-active macrocycle, Nat. Commun., 2019, 10, 1007 CrossRef PubMed.
-
(a) Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, Doped graphene sheets as anode materials with superhigh rate and large capacity for lithium ion batteries, ACS Nano, 2011, 5, 5463–5471 Search PubMed;
(b) S. Osumi, S. Saito, C. Dou, K. Matsuo, K. Kume, H. Yoshikawa, K. Awaga and S. Yamaguchi, Boron-doped nanographene: Lewis acidity, redox properties, and battery electrode performance, Chem. Sci., 2016, 7, 219–227 RSC.
-
(a) J. F. Araneda, W. E. Piers, M. J. Sgro and M. Parvez, Bronsted acid-catalyzed skeletal rearrangements in polycyclic
conjugated boracycles: a thermal route to a ladder diborole, Chem. Sci., 2014, 5, 3189–3196 RSC;
(b) V. M. Hertz, M. Bolte, H. W. Lerner and M. Wagner, Boron-containing polycyclic aromatic hydrocarbons: facile synthesis of stable, redox-active luminophores, Angew. Chem., Int. Ed., 2015, 54, 8800–8804 Search PubMed;
(c) N. Baser-Kirazli, R. A. Lalancette and F. Jakle, Enhancing the acceptor character of conjugated organoborane macrocycles: a highly electron-deficient hexaboracyclophane, Angew. Chem., Int. Ed., 2020, 59, 8689–8697 Search PubMed;
(d) L. Yuan, J. Guo, Y. Yang, K. Ye, C. Dou and Y. Wang, Polycyclic π-system with bilayer assembly and multi-redox activity, CCS Chem., 2023, 5, 876–884 CrossRef CAS;
(e) J. Guo, K. Zhang, Y. Wang, H. Wei, W. Xiao, K. Yang and Z. Zeng, Fully-fused boron-doped olympicenes: modular synthesis, tunable optoelectronic properties, and one-electron reduction, Chem. Sci., 2023, 14, 4158–4165 Search PubMed;
(f) S. E. Prey, J. Gilmer, S. V. Teichmann, L. Čaić, M. Wenisch, M. Bolte, A. Virovets, H.-W. Lerner, F. Fantuzzi and M. Wagner, Synthesis, bridgehead functionalization, and photoisomerization of 9,10-diboratatriptycene dianions, Chem. Sci., 2023, 14, 5316–5322 RSC;
(g) S. E. Prey, C. Herok, F. Fantuzzi, M. Bolte, H. W. Lerner, B. Engels and M. Wagner, Multifaceted behavior of a doubly reduced arylborane in B–H-bond activation and hydroboration catalysis, Chem. Sci., 2023, 14, 849–860 Search PubMed;
(h) S. E. Prey and M. Wagner, Threat to the throne: can two cooperating boron atoms rival transition metals in chemical bond activation and catalysis?, Adv. Synth. Catal., 2020, 363, 2290–2309 Search PubMed.
- K. K. Hollister, K. E. Wentz and R. J. Gilliard, Redox- and charge-state dependent trends in 5, 6, and 7-membered boron heterocycles: a neutral ligand coordination chemistry approach to boracyclic cations, anions, and radicals, Acc. Chem. Res., 2024, 57, 1510–1522 CrossRef CAS PubMed.
-
(a) M. F. Silva Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, N-Heterocyclic carbene stabilized boryl radicals, Angew. Chem., Int. Ed., 2016, 56, 1135–1140 CrossRef PubMed;
(b) Y. Su and R. Kinjo, Boron-containing radical species, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik, C. Claes, W. C. Ewing, I. Krummenacher, K. Lubitz and C. Schneider, Neutral diboron analogues of archetypal aromatic species by spontaneous cycloaddition, Angew. Chem., Int. Ed., 2016, 55, 11271–11275 CrossRef CAS PubMed.
-
(a) M. Dietz, M. Arrowsmith, K. Drepper, A. Gartner, I. Krummenacher, R. Bertermann, M. Finze and H. Braunschweig, Structure and electronics of a series of CAAC-stabilized diboron-doped acenes from 1,4-diboranaphthalene to 6,13-diborapentacene, J. Am. Chem. Soc., 2023, 145, 15001–15015 Search PubMed;
(b) C. Saalfrank, F. Fantuzzi, T. Kupfer, B. Ritschel, K. Hammond, I. Krummenacher, R. Bertermann, R. Wirthensohn, M. Finze, P. Schmid, V. Engel, B. Engels and H. Braunschweig, cAAC-stabilized 9,10-diboraanthracenes-acenes with open-shell singlet biradical ground states, Angew. Chem., Int. Ed., 2020, 59, 19338–19343 CrossRef CAS;
(c) M. Dietz, M. Arrowsmith, A. Gartner, K. Radacki, R. Bertermann and H. Braunschweig, Harnessing the electronic differences between CAAC-stabilised 1,4-diborabenzene and 9,10-diboraanthracene for synthesis, Chem. Commun., 2021, 57, 13526–13529 RSC.
- J. W. Taylor, A. McSkimming, C. F. Guzman and W. H. Harman, N-Heterocyclic carbene-stabilized boranthrene as a metal-free platform for the activation of small molecules, J. Am. Chem. Soc., 2017, 139, 11032–11035 CrossRef CAS PubMed.
-
(a) K. K. Hollister, W. Yang, R. Mondol, K. E. Wentz, A. Molino, A. Kaur, D. A. Dickie, G. Frenking, S. Pan, D. J. D. Wilson and R. J. Gilliard, Isolation of stable borepin radicals and anions, Angew. Chem., Int. Ed., 2022, 61, e202202516 CrossRef CAS;
(b) W. Yang, K. E. Krantz, L. A. Freeman, D. A. Dickie, A. Molino, G. Frenking, S. Pan, D. J. D. Wilson and R. J. Gilliard, Persistent borafluorene radicals, Angew. Chem., Int. Ed., 2020, 59, 3850–3854 Search PubMed;
(c) C.-L. Deng, K. K. Hollister, A. Molino, B. Y. E. Tra, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard, Unveiling three interconvertible redox states of boraphenalene, J. Am. Chem. Soc., 2024, 146, 6145–6156 Search PubMed;
(d) W. Yang, K. E. Krantz, L. A. Freeman, D. A. Dickie, A. Molino, A. Kaur, D. J. D. Wilson and R. J. Gilliard, Stable borepinium and borafluorenium heterocycles: a reversible thermochromic “switch” based on boron–oxygen interactions, Chem.–Eur. J., 2019, 25, 12512–12516 CrossRef CAS PubMed.
- S. K. Sarkar, K. K. Hollister, A. Molino, A. D. Obi, C.-L. Deng, B. Y. E. Tra, B. M. Stewart, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard, Bis(9-boraphenanthrene) and its stable biradical, J. Am. Chem. Soc., 2023, 145, 21475–21482 Search PubMed.
-
(a) K. K. Hollister, A. Molino, N. Jones, V. V. Le, D. A. Dickie, D. S. Cafiso, D. J. D. Wilson and R. J. Gilliard, Unlocking biradical character in diborepins, J. Am. Chem. Soc., 2024, 146, 6506–6515 CrossRef CAS;
(b) K. K. Hollister, A. Molino, V. V. Le, N. Jones, W. J. Smith, P. Müller, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard, Pentacyclic fused diborepinium ions with carbene- and carbone-mediated deep-blue to red emission, Chem. Sci., 2024, 15, 14358–14370 RSC.
-
(a) L. Zhang, B. Walker, F. Liu, N. S. Colella, S. C. B. Mannsfeld, J. J. Watkins, T.-Q. Nguyen and A. L. Briseno, Triisopropylsilylethynyl-functionalized dibenzo[def,mno]chrysene: a solution-processed small molecule for bulk heterojunction solar cells, J. Mater. Chem., 2012, 22, 4266–4268 Search PubMed;
(b) B. K. Shah, D. C. Neckers, J. Shi, E. W. Forsythe and D. Morton, Photophysical properties of anthanthrene-based tunable blue emitters, J. Phys. Chem. A, 2005, 109, 7677–7681 CrossRef CAS PubMed;
(c) Y. Geng, C. Yi, M. P. Bircher, S. Decurtins, M. Cascella, M. Grätzel and S.-X. Liu, Anthanthrene dye-sensitized solar cells: influence of the number of anchoring groups and substitution motif, RSC Adv., 2015, 5, 98643–98652 RSC;
(d) J.-B. Giguère, N. S. Sariciftci and J.-F. Morin, Polycyclic anthanthrene small molecules: semiconductors for organic field-effect transistors and solar cells applications, J. Mater. Chem. C, 2015, 3, 601–606 Search PubMed;
(e) J.-B. Giguère and J.-F. Morin, Synthesis and optoelectronic properties of 6,12-bis(amino)anthanthrene derivatives, J. Org. Chem., 2013, 78, 12769–12778 CrossRef PubMed.
- A. Rieche, W. Rudolph and R. Seifert, Über dinaphthylendiimin und dehydrodinaphthylendiimin, Ber. Dtsch. Chem. Ges. B, 1940, 73, 343–350 CrossRef.
-
(a) Z. Liu, W. Han, J. Lan, L. Sun, J. Tang, C. Zhang and J. You, Molecular engineering of chalcogen-embedded anthanthrenes viaperi-selective C–H activation: fine-tuning of crystal packing for organic field-effect transistors, Angew. Chem., Int. Ed., 2022, 62, e202211412 CrossRef PubMed;
(b) T. Kamei, M. Uryu and T. Shimada, Cu-catalyzed aerobic oxidative C–H/C–O cyclization of 2,2′-binaphthols: practical synthesis of PXX derivatives, Org. Lett., 2017, 19, 2714–2717 Search PubMed;
(c) D. Stassen, N. Demitri and D. Bonifazi, Extended O-doped polycyclic aromatic hydrocarbons, Angew. Chem., Int. Ed., 2016, 55, 5947–5951 Search PubMed;
(d) N. Kobayashi, M. Sasaki and K. Nomoto, Stable peri-xanthenoxanthene thin-film transistors with efficient carrier injection, Chem. Mater., 2009, 21, 552–556 Search PubMed;
(e) N. Lv, M. Xie, W. Gu, H. Ruan, S. Qiu, C. Zhou and Z. Cui, Synthesis, properties, and structures of functionalized peri-xanthenoxanthene, Org. Lett., 2013, 15, 2382–2385 CrossRef CAS;
(f) O. Matuszewska, T. Battisti, R. R. Ferreira, N. Biot, N. Demitri, C. Mézière, M. Allain, M. Sallé, S. Mañas-Valero, E. Coronado, E. Fresta, R. D. Costa and D. Bonifazi, Tweaking the optoelectronic properties of S-doped polycyclic aromatic hydrocarbons by chemical oxidation, Chem.–Eur. J., 2023, 29, e202203115 Search PubMed.
-
(a) Y. Xia, M. Zhang, S. Ren, J. Song, J. Ye, M. G. Humphrey, C. Zheng, K. Wang and X. Zhang, 6,12-Dihydro-6,12-diboradibenzo[def,mno]chrysene: a doubly boron-doped polycyclic aromatic hydrocarbon for organic light emitting diodes by a one-pot synthesis, Org. Lett., 2020, 22, 7942–7946 Search PubMed;
(b) Y. Xia, J. Zou, T. Fan, T. Li, J. Zhang and Y. Ding, 7-mesityl-7H-dinaphtho[1,8-bc:1′,2′-e]borinine: a boron-doped polycyclic aromatic hydrocarbon for organic light-emitting diodes by Si–B exchange, Tetrahedron Lett., 2022, 108, 154134 CrossRef CAS.
- R. Borthakur and V. Chandrasekhar, Boron-heteroelement (B–E; E = Al, C, Si, Ge, N, P, As, Bi, O, S, Se, Te) multiply bonded compounds: recent advances, Coord. Chem. Rev., 2021, 429, 213647 CrossRef CAS.
-
(a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-independent chemical shifts: a simple and efficient aromaticity probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 Search PubMed;
(b) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Nucleus-independent chemical shifts (NICS) as an aromaticity criterion, Chem. Rev., 2005, 105, 3842–3888 Search PubMed.
- D. Geuenich, K. Hess, F. Kohler and R. Herges, Anisotropy of the induced current density (ACID), a general method to quantify and visualize electronic delocalization, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- M. Dietz, M. Arrowsmith and H. Braunschweig, CAAC-stabilised 9,10-diboraanthracene: an electronically and structurally flexible platform for small-molecule activation and metal complexation, Dalton Trans., 2024, 53, 449–453 Search PubMed.
-
(a) X. Xie, C. J. Adams, M. A. M. Al-Ibadi, J. E. McGrady, N. C. Norman and C. A. Russell, A polycyclic borazine radical cation: [1,2-B2{1,2-(MeN)2C6H4}2]+, Chem. Commun., 2013, 49, 10364–10366 Search PubMed;
(b) Y. Su, Y. Li, R. Ganguly and R. Kinjo, Crystalline boron-linked tetraaminoethylene radical cations, Chem. Sci., 2017, 8, 7419–7423 Search PubMed.
- K. E. Wentz, A. Molino, S. L. Weisflog, A. Kaur, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard, Stabilization of the elusive 9-carbene-9-borafluorene monoanion, Angew. Chem., Int. Ed., 2021, 60, 13065–13072 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Guijiang
Zhou
,
Guijiang
Zhou