
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering disease analyte response in peptide self-assembly
Sihan
Yu
and
Matthew J.
Webber
 *
*
Department of Chemical & Biomolecular Engineering, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: mwebber@nd.edu
First published on 9th October 2024
Abstract
A need to enhance the precision and specificity of therapeutic nanocarriers inspires the development of advanced nanomaterials capable of sensing and responding to disease-related cues. Self-assembled peptides offer a promising nanocarrier platform with versatile use to create precisely defined nanoscale materials. Disease-relevant cues can range from large biomolecules, such as enzymes, to ubiquitous small molecules with varying concentrations in healthy versus diseased states. Notably, pH changes (i.e., H+ concentration), redox species (e.g., H2O2), and glucose levels are significant spatial and/or temporal indicators of therapeutic need. Self-assembled peptides respond to these cues by altering their solubility, modulating electrostatic interactions, or facilitating chemical transformations through dynamic or labile bonds. This review explores the design and construction of therapeutic nanocarriers using self-assembled peptides, focusing on how peptide sequence engineering along with the inclusion of non-peptidic components can link the assembly state of these nanocarriers to the presence of disease-relevant small molecules.
1. Introduction
The field of nanomedicine emerged from a need to address a number of practical challenges and limitations in traditional pharmaceutical practice.1 Active pharmaceutical agents can be limited in their therapeutic impact by solubility constraints, challenges in physical or chemical stability, suboptimal pharmacokinetic profiles, and dose-limiting toxicity. Carrier materials prepared at the nanoscale present opportunities to preferentially encapsulate these agents as a payload, thereby enhancing solubility, preserving physicochemical stability, reducing systemic exposure, and altering both the circulation half-life and mode of clearance.2 Moreover, the biodistribution of nanoscale drug carriers can be altered by both passive and/or physiological mechanisms as well as active targeting using recognition from antibodies or related biomolecules.3,4 As such, nanomedicine offers a means to increase the therapeutic index, the ratio of the lethal dose (LD50) to the effective dose (ED50), by biasing drug availability and function to sites of need and reducing off-site activity. The preponderance of work in the field of nanomedicine has focused on the development of new cancer therapeutics.5,6 Indeed, the first FDA-approved engineered nanomedicine, Doxil®, was a PEGylated liposomal carrier of the anticancer agent doxorubicin.7 However, the clinical successes of nanomedicine are still somewhat limited and the promise of this field has yet to be fully actualized.8 Beyond opportunities in cancer, a growing body of literature points to promising applications for nanomedicine in treating conditions of increasing prevalence such as diabetes or cardiovascular disease,9,10 while nanotechnologies are a central component of expanding research efforts in immunoengineering.11,12 Recently, a key motivator for continued work in nanomedicine is found in the exceptional efficacy of vaccines based on liposomal nanoparticles and engineered recombinant protein constructs, both of which were instrumental in the global response to the COVID-19 pandemic.13,14In efforts to improve the therapeutic efficacy of nanomedicine, one active area of exploration seeks engineered nanomaterials capable of stimuli-directed therapeutic deployment.15 As with the general approach of biologically targeted nanomedicine, stimuli-responsive materials design offers another tool to improve site-specific action and increase the therapeutic index of active drugs. A variety of disease-relevant indicators may be used as triggers in the design of responsive nanoscale drug delivery systems, including pH, enzymes, glucose, and redox agents.16–19 A general objective of this approach is to use analytes as spatial and/or temporal signals of disease state in order to regulate the availability of a therapeutic agent. Moreover, stimuli-responsive technologies can be integrated as a component of both passively and actively targeted nanomedicine for enhanced functionality or more rapid payload release upon reaching the desired tissue site.
Among various nanomedicine platforms, peptide self-assembly is one versatile approach to design materials at the nanoscale for therapeutic applications.20–23 These assemblies can be engineered into various forms, with differences in the mode of assembly dictating shape and interfacial curvature of the resulting assembly, including spherical nanoparticles and filamentous nanostructures. Peptide-based assemblies have certain inherent benefits in their use as carriers for nanomedicine, including the chemical diversity, biocompatibility, and biodegradability of amino acid building blocks.24,25 Facile synthetic integration of peptide-based affinity motifs enables routes to target these materials to desired sites.26 Moreover, the modularity of molecular-scale design in peptide self-assembly offers opportunities to integrate therapeutic agents directly as components of self-assembling monomers.27,28 The well-defined nanostructures that result from these assemblies can be coupled with specialized motifs that endow a capacity for stimuli-responsive function or afford targeting for more precise therapeutic delivery.26 Integration of stimuli-responsive functionality into the design of self-assembling peptide materials is of particular interest, as the dynamic and non-covalent nature of these assemblies can enable changes in relevant environmental conditions that quickly alter assembly state and drive a more rapid response.29–33
The enclosed review highlights specific uses of peptide self-assembly in conjunction with disease analyte-responsive design for applications in therapeutic delivery (Fig. 1). A number of different tissue-specific or disease-relevant analytes have been explored as spatiotemporal triggers to ensure a therapy is made available at both the place and time of need. In considering only cancer as an example, distinctive conditions within the tumor microenvironment can include an acidic pH, elevated temperature, heightened oxidative potential, and overexpressed proteins and enzymes.34 Other diseases present their own specific biomarkers or analytes of interest that can be used as stimuli to regulate peptide self-assembly. As several reviews have covered the topic of peptide self-assemblies that are responsive to proteins and enzymes,35,36 the present review specifically highlights strategies to engineer peptide self-assemblies that can respond to small molecule (ca., <300 g mol−1) analytes. There are unique challenges in engineering nanomaterials to respond to the presence of often ubiquitous small molecule analytes. For instance, pH-responsive materials (i.e., H+) are engineered to sense and respond to spatially distinct locations within cells, organ systems, or throughout the body.37 Meanwhile, many redox agents (e.g., H2O2), are a hallmark of both healthy and diseased states, acting as signaling molecules in processes for both normal tissue regeneration and underlying inflammatory conditions. Glucose is a ubiquitous analyte present in fluctuating concentrations in both healthy as well as diseased (e.g., diabetic) states; the concentration of glucose in diabetes dictates temporally sensitive therapeutic need.38 As such, engineering peptide assemblies to afford spatiotemporal precision in responding to these small molecule analytes, often including the ability to distinguish concentration of ubiquitous molecules, points to an exciting direction by which to improve the therapeutic efficacy of peptide-based nanomedicine.39 Strategies to engineer peptide self-assembly for response to small molecule analytes with disease relevance will be covered in greater detail in this review.
 | ||
| Fig. 1 Overview of strategies to engineer peptide self-assembly with response to disease-relevant small molecule analytes such as pH (top left), redox species (top right) or glucose (bottom). | ||
2. pH-Responsive peptide self-assembly
While the pH of most physiological regions is near to neutral, certain locations within the body have acidic pH levels, offering a useful stimulus to localize or activate a therapeutic nanocarrier.37 Different regions of the gastrointestinal system have varying degrees of acidic pH, with levels as low as pH 1 in the stomach.40 The tumor microenvironment is characteristically more acidic than normal tissues, a result of the rapid proliferation of cancer cells that elevates glycolysis and lactic acid production.41 Accordingly, unique and regionally specific pH profiles can be used as a trigger to regulate pH-responsive peptide self-assembly for targeted drug delivery.42 This general strategy involves tuning self-assembly properties through the selection of pH-responsive peptide sequences, with optional inclusion of functional groups or labile bonds to enhance pH sensitivity.To design pH-responsive peptide sequences, amino acids with ionizable R-groups are most commonly employed. Of the canonical amino acids, basic amino acids of histidine (H), lysine (K), and arginine (R) can carry a positive charge at a pH level below the pKa of their respective side-chains, while acidic amino acids of glutamic acid (E) and aspartic acid (D) carry a negative charge at a pH level above the pKa of their side-chain carboxylates (Fig. 2A).42 Depending on molecular design, either N-terminal amines or C-terminal carboxylates may also be present and these can display pH-dependent charge according to the respective pKa of these sites. When exposed to an acidic environment, peptide sequences bearing these residues can undergo protonation or deprotonation, leading to alterations in the structure of the assembly. Often, this structural transformation involves modulating electrostatic interactions to promote/disrupt molecular cohesion in β-sheet or coiled-coil motifs.43–48 When integrated in the context of nanomedicine, modulation of these forces can be used as a directive cue to trigger release of a therapeutic or initiate a structural transformation to promote cytotoxicity.49,50 Peptide self-assembly also navigates the phase space between molecular solubility and precipitation; alterations to the charge state of the molecules arising from changes to pH has direct implications on this feature as well. Given that peptide assemblies are densely packed, with each monomer often featuring hydrophobic domains and multivalent charges, the pKa of amino acids can be significantly affected by their proximity to hydrophobic regions, like-charged residues, and hydrogen bonding.51 These effects are well known to cause substantial pKa shifts in proteins, sometimes by several pH units,52 and have been similarly observed for charge-bearing residues in synthetic peptide assemblies.53–56 However, the magnitude of these shifts is highly dependent on molecular design and, as a result, can be difficult to generalize or predict a priori.
 | ||
| Fig. 2 (A) Structures of common basic and acidic amino acids, with their R-group side chain pKa values highlighted and drawn in their charged configuration. The pKa for N- and C-terminal groups are also shown for reference. (B) Schematic illustration of a pH-responsive micelle prepared from an amphiphilic peptide that assembles and disassembles to release anti-tumor drugs within cancer cells. Figure adapted from ref. 57 with permission from Elsevier © 2014. (C) Peptide designed to form pH-sensitive nanoparticles in complexation with nucleic acid payloads. The protonation equilibria and tautomeric forms of the imidazole side chain of histidine offer a pH triggered decomposition of the nanoparticles. Figure adapted from ref. 58 with permission from Royal Society of Chemistry © 2023. (D) Peptide sequence of prodrug FA-EEYSV-NH2 and its properties pH-responsive assembled nanostructure. Figure adapted form ref. 59 with permission from American Chemical Society © 2021. (E) Scheme depicting the pH-responsive disassembly of complexed supramolecular amphiphiles, with TEM images showing the nanostructure at pH 7.4 and 6.2. Figure adapted from ref. 60 with permission from John Wiley and Sons © 2013. | ||
Basic amino acids, and specifically K and R, are frequently incorporated within pH-responsive materials to afford dissociation of peptide assemblies and drug release upon exposure to acidic pH. For example, an amphiphilic peptide bearing a hydrophilic segment consisting of a tumor-targeting sequence and KK as pH-sensor was fused to a hydrophobic segment of six valine (V) residues to enable formation of spherical micelle assemblies to encapsulate drugs (Fig. 2B).57 Upon exposure to a pH 5.0 environment, the micelles disassembled due to increased electrostatic repulsion among protonated lysine residues, leading to accelerated release of an encapsulated chemotherapeutic payload. Though the theoretical pKa for the lysine sidechain would not predict a substantial change in ionization when reduced from a pH value of 7 to 5, it can be assumed that the effective pKa is shifted down as a result of molecular aggregation and self-assembly. A similar effect was observed for an oligopeptide fusing lysine with a leucine (L) segment, L6K4, that was used to encapsulate doxorubicin, with disassembly in the acidic endosomal environment promoting drug release.61 One report demonstrated pH-dependent self-assembly from a telechelic block co-polymer consisting of two poly-L-lysine segments flanking a polypropylene oxide segment.62 Under acidic conditions where the lysine segments were primarily charged, the peptide adopted a random coil and micelle assemblies formed. However, under basic conditions the peptide adopted an α-helix, with the resulting assemblies transitioning to vesicles or disk-like micelles.
With a pKa closer to 6, histidine (H) has a special feature in that it can undergo a charge transition from uncharged at pH 7.4 to charged at even mildly acidic pH. Accordingly, histidine has been used in the design of self-assembled carriers that dissociate to release a drug upon the increased electronic repulsion from charged histidine groups. A recent example developed a highly responsive peptide-based nanoparticle carrier for the controlled release of nucleic acid drugs in the tumor microenvironment, utilizing the pKa of histidine as the trigger (Fig. 2C).58 The peptide included a positively charge block of three K residues on the N-terminal end to enable loading of negatively charged nucleic acid drugs, a pH-sensitive block of three H residues to trigger disassociation of the nanoparticle in an acidic microenvironment, a block of 8 hydrophobic L residues to promote self-assembly in water, and then hydrophilic amino acids and a targeting peptide sequence. Mixing of the peptide with nucleic acid drugs led to nanoparticle formation through a combination of electrostatic complexation and hydrophobic association, but on exposure to an acidic tumor microenvironment the nanoparticles disassembled due to the increased charge repulsion from the histidine residue. Once the nanocarrier was internalized into the acidic endosomal environment of the cancer cell where the pH is around 5.0, additional charging of the histidine segment enabled the nanoparticles to escape the endosome for release of nucleic acid drugs. Another report demonstrated a PEGylated peptide amphiphile with an H6 block flanked by aliphatic and PEG domains; depending on the order of these three domains, either spherical or cylindrical micelles were formed at pH 7.5.63 The cylindrical micelles, in particular, could encapsulate a large amount of a chemotherapeutic for pH-responsive release upon nanostructure disassembly. When explored in a tumor model, the cylindrical assemblies also demonstrated increased tumor accumulation. The pH-responsive charging of a histidine segment has also been used to reversibly conceal and reveal a charged cell-penetrating peptide sequence from micelles prepared from the self-assembly of peptide–polymer conjugates.64 Peptides designed as pH-responsive tissue scaffolds were also reported by inclusion of an H3 segment in the outer portion of a branched amphiphilic peptide.65
The carboxylate groups of E and D instead transition to an uncharged state as pH is decreased, offering a different cue to direct the assembly state of self-assembling peptides. In one example, a peptide prodrug was prepared comprising three segments: a pH-sensitive EE dipeptide linker, a tumor-targeting folic acid (FA) moiety, and an anticancer peptide sequence known as tyroservatide (Fig. 2D).59 In this case, the EE segment not only enhanced water solubility, but also acted as a pH trigger. Under neutral conditions, the EE segment was primarily charged, leading to self-assembly into nanoparticles. However, upon a reduction in pH from 7 to 5, the nanoparticles transitioned into nanofibers due to protonation of the EE side-chain carboxylates. Another report encapsulated drug into the core of a peptide amphiphile bearing a segment containing ten consecutive E residues.66 This peptide formed micelles under neutral conditions but became insoluble in the acidic environment of the endosome to release an encapsulated chemotherapeutic within the cell.
Including both acidic and basic amino acids enables the interplay of charged groups under varying pH conditions, tuning electrostatic interactions to dictate assembly state. In many reports, peptides with a mix of both charged groups could form stable assemblies, or even hydrogels, under roughly neutralized charge conditions but disassembled to release a payload when a drop in pH altered the electrostatics of the assembly.67–71 One naturally derived amyloid peptide was modified on its N- and C-termini with K and D, respectively, and demonstrated the ability to form pH-dependent β-sheets and nanostructures.72 When coupled with an oil–water microfluidic droplet generator, this peptide was able to form microcapsules that could be used for the encapsulation and pH-triggered burst release of encapsulated therapeutic payloads. Zwitterions are net-neutral motifs carrying both positive and negative charges; though no native amino acids have zwitterionic side-chains, a pH-responsive polypeptide assembly was also demonstrated from synthetic side-chains bearing zwitterionic groups.73
Electrostatic interactions between oppositely charged groups can also be used for non-covalent fusion of assembly motifs, leading to pH-responsive building blocks for material self-assembly. In one example, a hydrophobic poly-L-leucine terminated with a C-terminal glutamic acid (i.e., two –COOH groups) was mixed with a poly-L-lysine dendrimer (Fig. 2E).60 Electrostatic interactions at neutral pH resulted in formation of a non-covalent amphiphile between the two compounds that could assemble into a nanoparticle; under acidic conditions the electrostatic interactions between these two groups were disrupted, leading to release of the encapsulated payload. A non-covalent amphiphile approach has also been shown for a peptide–PEG conjugate bearing a poly-L-homoarginine segment in complex with carboxylate-containing polyaromatic hydrophobic group, wherein the complex was disrupted under acidic pH conditions to drive disassembly.74 Poly-L-glutamic acid has also been conjugated to PEG, with a self-assembling amphiphile arising upon electrostatic complex formation between the negatively charged glutamic acid block and positively charged doxorubicin.75 Upon exposure to the acidic environment of the endosome, these electrostatic interactions were disrupted, causing disassembly and intracellular drug release.
Charge balance can also be tuned in peptide self-assembly through concealing charged amino acids and revealing these under a pH stimulus. In one example, a pH-responsive hydrogel was formed in situ from a peptide bearing both K and E residues and fused to a drug, methotrexate; the amines of each K residue were modified with an acid-responsive 2,3-dimethylmaleic anhydride (DA) moiety.76 Under weakly acidic conditions, the amide bond between DA and the lysine side chain was hydrolyzed to expose the charged amine groups. This peptide could then transform from a clear solution at pH 7.4 to a hydrogel at pH 6.5 due to balance in charge between the newly exposed K residues and the E residues on the peptide. Another report demonstrated the use of anhydride compounds to mask lysine residues with labile anionic groups, resulting in a highly negative pro-gelator peptide.77 However, upon exposure to an acidic stimuli, the lysine residues were unmasked resulting in assembly of the net-neutral oligopeptide gelator.
Bonds that are pH-sensitive can also be used to link a self-assembling peptide to a therapeutic, for triggered disassembly and/or release under application of a pH stimulus (Fig. 3A). For example, a recent report conjugated a self-assembling peptide to an immune adjuvant, mannan, using a pH-sensitive imine linkage to afford controlled release upon bond rupture (Fig. 3B).78 Other reports have shown self-assembling peptide–drug conjugates through the use of pH-sensitive labile bonds like hydrazones or esters linking the drug to the peptide.79–84 In many of these cases, the drug serves a dual purpose as both the therapeutic compound being delivered as well as an active driver of self-assembly, given the drug moieties are typically hydrophobic and able to participate in ordered (e.g., π–π) interactions. Linkers such as hydrazones, imines, and acetals exhibit increased lability at lower pH, with faster hydrolysis rates under acidic conditions, while ester linkages can undergo hydrolysis through either acid- or base-catalyzed mechanisms and are also susceptible to degradation by native esterases.85 The pH conditions that facilitate hydrolysis, as well as the hydrolysis rate, can vary depending on the type of linker, its hydration level within the assembled structure, and other steric or electronic factors entailed in the design of the peptide–drug conjugate. This general approach to prepare nanoscale assemblies through peptide–drug conjugation has been termed “one component nanomedicine” and offers an opportunity to take advantage of prodrug chemistry and facilitate quantitative and reproducible drug loading in self-assembled peptide nanostructures.27
 | ||
| Fig. 3 (A) Scheme of peptide-drug conjugates linked by pH-sensitive bonds, enabling triggered disassembly or release in response to a pH stimulus. Some exemplary pH-labile bonds are shown. (B) Illustration depicting O-Man (blue) and R-Man (red) encapsulated within a lysine-bearing hydrogel. The aldehyde groups on O-Man form imine bonds with the amine groups on the peptide fibers, while the hydroxyl groups on R-Man are unable to form these same bonds. Figure adapted from ref. 78 with permission from American Chemical Society © 2023. | ||
The use of pH-responsive peptide assemblies for targeted drug delivery presents a promising strategy for localizing therapeutic action in specific environments, such as the tumor microenvironment or regions of the gastrointestinal tract. By designing peptides with ionizable amino acids, these assemblies can be triggered to self-assemble or disassemble in response to pH changes, facilitating drug release, inducing structural changes crucial for therapeutic function, or enabling pH-sensitive hydrolysis of covalent bonds. However, challenges remain in predicting pKa shifts within complex, densely packed assemblies, as molecular design heavily influences ionization behavior. Additionally, fine-tuning the stability of peptide assemblies across relevant physiological pH gradients remains difficult. Future opportunities lie in improving predictive models for pH responsiveness and expanding peptide designs to more precisely control assembly, disassembly, and drug release in specific physiological conditions.
3. Redox-responsive peptide self-assembly
The presence of redox species offers another cue to regulate peptide self-assembly for uses in spatially controlled nanomedicine. Triggers of interest include multiple different reactive oxygen species (ROS) as well as native reducing agents like glutathione (GSH). Certain physiological locations, and even subcellular compartments like endosomes,86 have elevated levels of these analytes that contribute antioxidant function and also underpin aspects of the innate immune system. In terms of disease relevance, the aberrant metabolism of cancer cells is correlated with an increased production of hydrogen peroxide (H2O2) and superoxide radicals,87,88 offering useful triggers in the design of new cancer therapeutics.89 Regions of inflammation, injury, or tissue damage are likewise characterized by elevated production of ROS.90 Redox analytes therefore offer a useful cue to control peptide self-assembly for spatially controlled assembly or disassembly in order to localize a therapeutic payload.91Whereas pH can reversibly modulate peptide self-assembly through tuning the extent of electrostatic repulsion or molecular cohesion in the assembly, responsiveness to redox species more often involves rupture of a covalent bond. Many approaches in this regard have explored use of disulfide bonds that can be cleaved in reducing environments, including when exposed to environments with high GSH.92,93 Early work in the field of peptide self-assembly explored use of cysteine residues to promote intermolecular disulfide formation to enhance fiber rigidity.94 Conversely, an oligopeptide monomer cyclized by disulfide bonds between terminal cysteine groups was shown to form an extended β-sheet hydrogelator upon exposure to reductive environments (Fig. 4A).95 In another work, disulfide formation between cysteine residues was shown to stabilize a folded β-hairpin structure to drive fibrillar assembly and hydrogelation,96 while disulfide formation also increased the stiffness of hydrogels prepared from cysteine-modified multidomain peptides compared to their reduced form.97 Light-activated formation of disulfide bonds between hydrophobically modified oligopeptides under conditions of redox cycling was also shown to yield a variety of different self-assembled states.98
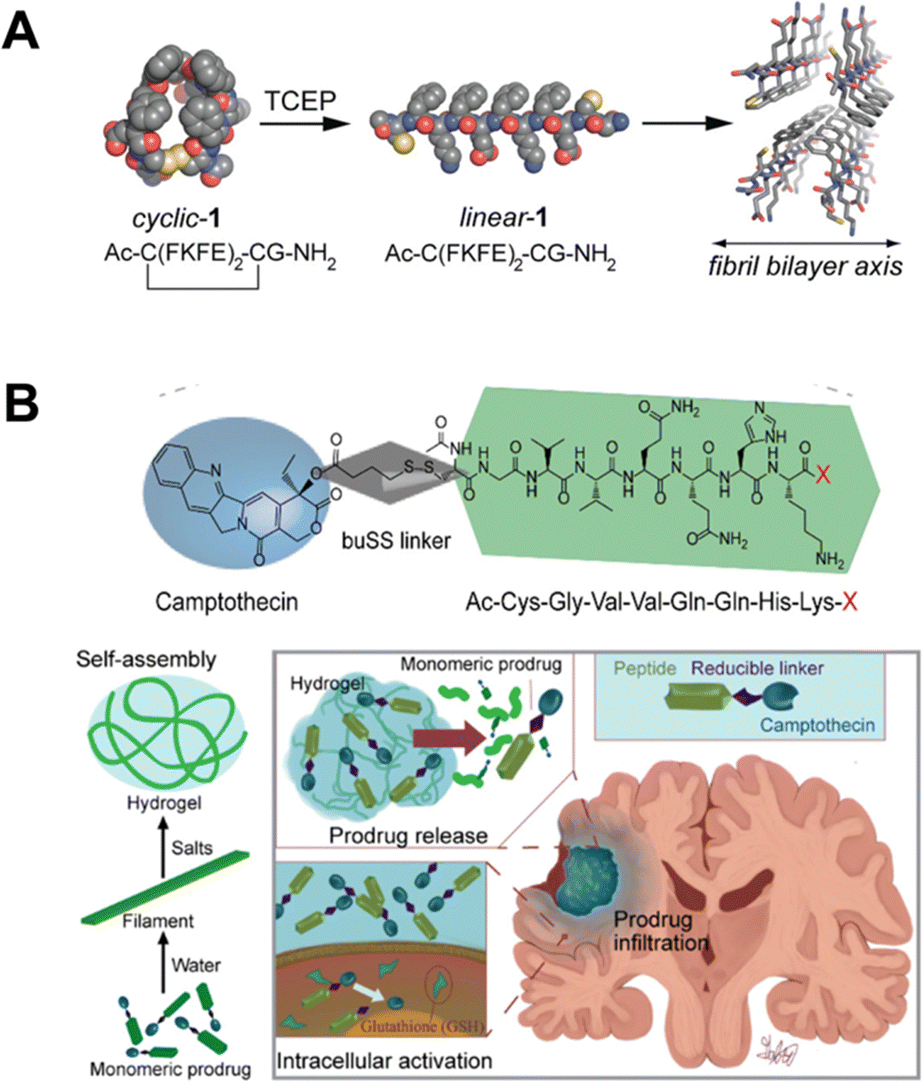 | ||
| Fig. 4 (A) A reductive trigger to induce a conformational switch from cyclic to linear and self-assembling peptides. Figure adapted from ref. 95 with permission from American Chemical Society © 2010. (B) Chemical structure of a designed camptothecin (CPT)-based self-assembling prodrug (top) along with its mechanism of action for controlled drug release in response to intracellular trigger following injection into the site of a glioblastoma. Figure adapted from ref. 99 with permission from Elsevier © 2020. | ||
Self-assembling peptide–drug conjugates have also been prepared by fusing certain anticancer drugs, such as camptothecin (CPT), to a β-sheet-forming peptide sequence using a disulfide linker.100,101 CPT is a very hydrophobic drug capable of chiral packing, offering a driving force for self-assembly in water.102 In one example, the peptides assembled into diverse morphologies, including nanofilaments and nanotubes, according to the number of CPT units (1, 2, or 4) attached to the peptide.100 The formation of these nanostructures concealed the CPT and shielded the disulfide linker from rapid degradation. However, at high GSH concentrations the linker was ruptured to release free CPT for chemotherapeutic function.100,103 A related approach also attached a single CPT to a different β-sheet-forming peptide sequence to self-assemble into filamentous nanofibers that formed hydrogels (Fig. 4B).99 Injection of these hydrogels into the site of glioblastoma resection resulted in a significant enhancement in post-surgical survival. Combination of hydrogel-forming CPT-modified peptides with checkpoint inhibitors for immune therapy also demonstrated anti-tumor function with improved survival by combining pharmaceutical and immune therapies.104 In a related approach, the delivery of a potent STING agonist from CPT-linked peptide gelators also demonstrated promise for intratumoral delivery of both agents to better treat cancer and afford immune memory upon rechallenging with tumors.105 Though most work in this area has used CPT, the general approach is modular for the integration of other disulfide-linked drugs such as paclitaxel.106
The formation and rupture of redox-active bonds, like disulfides, can also enable reversible assembly and disassembly of peptide building blocks. In one example, an arginine-rich oligopeptide building block was demonstrated to form a phase-separated coacervate upon disulfide formation between terminal cysteine residues when exposed to the oxidizing conditions of H2O2.107 Formation of this disulfide bond increased the effective molecular weight of the arginine-rich peptide, allowing it to form a complex coacervate when mixed with a multivalent anion. However, when exposed to reducing agents like GSH, the disulfide linkage between the oligopeptides was ruptured leading to dissolution of the coacervate phase; the active agent, tissue plasminogen activator, was released as the coacervate dissolved.
The thioether residue of methionine offers another native redox-responsive moiety that can be included in peptide self-assembly.108 Under oxidizing conditions, methionine (Met) can be sequentially converted into hydrophilic methionine sulfoxide (MetO) and sulfone (MetO2). The increase in hydrophilicity of a methionine block under oxidizing conditions was used to prepare polypeptide vesicles,109 taking advantage of the transition from α-helix to random coil in the poly-methionine block upon oxidation to the sulfoxide.110 Toward functional use of this approach, a methionine-modified oligopeptide was shown to self-assemble into nanoribbons; when these ribbons were co-assembled with a monomer bearing a photosensitizer generating oxidative conditions under light irradiation, the nanoribbons transformed into nanoparticles driven by methionine oxidation (Fig. 5A).111 The oxidized nanoparticle form showed enhanced tumor penetration and improved antitumor therapeutic efficacy. Methionine can also be used alongside pH-responsive motifs, such as peptides bearing carboxylate moieties, to enable multi-stimuli-responsive functionality, as demonstrated in the reversible supramolecular polymerization of a peptide-modified discotic amphiphile.112
 | ||
| Fig. 5 (A) Chemical structure of a methionine-containing hexapeptide, as well as its oxidized form (top). The redox state of the peptide controls its assembly into nanoribbons or nanoparticles. The hexapeptide can be co-assembled with derivatives bearing agents for photodynamic or chemotherapeutic treatment. The formed structures undergo a morphological shift driven by in situ ROS generation, promoting tumor penetration and enhancing the combined efficacy of photodynamic and chemotherapeutic treatment. Figure adapted from ref. 111 with permission from Elsevier © 2021. (B) Schematic representation showing the self-assembly of boronate-containing peptides to form a nanofiber network, with H2O2-triggered gel degradation. Figure adapted from ref. 113 with permission from Springer Nature © 2014. | ||
In an analogous mechanism to methionine oxidation, non-native amino acids or functional groups bearing selenoethers can be inserted into self-assembling peptides to take advantage of the increased hydrophilicity upon oxidative conversion to selenoxide motifs.114–117 Peptide assemblies have also been designed for orthogonal self-sorting and self-assembly by combining a peptide that forms nanostructures under oxidizing conditions and disulfide formation with another peptide that forms nanostructures under reducing conditions when a selenoxide group is converted to its selenoether.118 Depending on the nature of the nanostructure formed, different cellular organelles could be targeted to promote cell death. In another example of switchable self-assembly via redox inputs, an oligopeptide containing selenomethionine was demonstrated to form nanoparticles when this side-chain was oxidized into its selenoxide form by H2O2, but formed nanoribbons upon GSH reduction.119 The more cationic nanoribbons preferentially targeted the negatively charged mitochondrial membrane, where the higher levels of H2O2 drive nanoribbon disassembly, thus offering an organelle-specific targeting approach.
Phenylboronic acids (PBA) and phenylboronic esters are sensitive to oxidation by H2O2.120 One commonly explored strategy in the context of H2O2-responsive nanostructures has used self-immolative PBA motifs that undergo bond rearrangement and release of an intermediate linker species upon reaction with H2O2.121 Modification with a self-immolative PBA group offered a redox-controlled method to achieve self-assembly of a diphenylalanine peptide that resulted upon H2O2 exposure and PBA removal (Fig. 5B).113,122 This chemistry was also attached to an oligopeptide and, upon H2O2 exposure, the self-immolation of the boronate yielded an intermediate that could further rearrange through an O,N-acyl shift to form a self-assembling peptide.123 This reaction cascade was shown to occur inside of a living cell, leading to nanofiber formation that promoted apoptosis. This same approach to engineering a reaction cascade involving PBA self-immolation and a subsequent O,N-acyl shift was also used to facilitate intracellular self-assembly of a metallopeptide conjugate.124
Redox-responsive peptides provide an exciting approach to regulate peptide self-assembly by utilizing environmental redox cues, such as reactive oxygen species and reducing agents like glutathione. Through the incorporation of redox-sensitive functionalities, such as disulfide bonds or methionine residues, peptides can undergo reversible assembly and disassembly in response to oxidative or reductive environments. This has enabled the design of self-assembling structures that can dynamically respond to the redox state of their surroundings. However, a significant challenge lies in predicting the stability and responsiveness of these assemblies, as redox-triggered bond cleavage and structural transformations depend heavily on the molecular context and redox conditions. Additionally, ensuring precise control over the self-assembly process across different biological environments remains difficult. Future opportunities include refining the design of redox-sensitive motifs to achieve more predictable and tunable self-assembly, as well as integrating multi-stimuli responsive systems to further enhance the functionality and adaptability of peptide-based assemblies.
4. Glucose-responsive peptide self-assembly
Blood glucose dysregulation and chronic hyperglycemia is a characteristic feature of diabetes, making glucose an important disease-relevant small molecule analyte to use in the design of responsive therapeutics.125 The vision of this approach is to treat diabetes by mimicking the glucose-sensing capabilities of a healthy endocrine system, which regulates blood sugar through insulin and glucagon signaling. Glucose-responsive materials would therefore autonomously sense real-time blood glucose levels and release the appropriate hormone to restore blood glucose control.126 Enzymatic actuation from glucose oxidase (GOx), an enzyme that converts glucose into useful secondary stimuli of pH (via gluconic acid) and H2O2, offers one commonly used glucose-sensing approach in materials design.38,127 Accordingly, approaches to endow glucose response in peptide self-assembly using GOx have similar design rationale to systems designed to respond to acidic pH or the presence of H2O2, as discussed in the previous two sections. Meanwhile, other approaches to design glucose-responsive materials have integrated glucose-binding PBA motifs; in addition to being redox-responsive, PBAs are able to bind reversibly to cis-1,2 diol species (like glucose) at pH levels at or above the pKa of the boronate, forming a tetrahedral boronate ester bearing a negative charge.125 While this charge stabilization and concomitant electrostatic modulation are the primary means of glucose response, PBA-based glucose binders are simultaneously responsive to oxidation by H2O2, as mentioned in the preceding section.The combination of GOx with charge-bearing amino acid residues can be used to induce glucose-responsive sol–gel transitions in self-assembling peptides, according to the pH-dependent charge state of the specific amino acids used.128,129 Oligopeptides designed to self-assemble through β-sheet formation, and which contain basic side-chains like lysine, arginine, and ornithine, have been explored alongside GOx encapsulation for glucose-responsive insulin release.130,131 Under physiological conditions in the absence of glucose, no pH stimulus is generated by GOx and the materials formed stable hydrogels. However, as glucose is introduced into the system, its conversion into gluconic acid by GOx results in a reduction to pH leading to increased electrostatic repulsion and hydrogel disassembly. This mechanism worked for two distinct cationic β-sheet hydrogelator motifs, leading to glucose-responsive release of encapsulated insulin in both cases.130,131 Glucose sensing by GOx has also been combined with self-immolation of a phenylboronic ester motif to enable glucose-responsive oligo-phenylalanine peptide self-assembly actuated by GOx, facilitating a gel-to-sol transition in the presence of glucose as the H2O2 byproduct of glucose conversion by GOx drives immolation of the boronate.113
The inclusion of PBA motifs on peptide-based materials more often leverages the stabilized negative charge arising from PBA–glucose binding to facilitate an electrostatic transition dictating assembly or aggregation state of the material. PBA motifs bind to a variety of cis-1,2 and cis-1,3 diols, making their glucose binding non-specific; additionally, their typical glucose-binding affinities, on the order of 10 M−1, can limit glucose recognition under normal physiological concentrations of approximately 4–10 mM;132,133 in spite of some successful demonstrations of function, there remain opportunities to improve this sensing mechanism. In designing self-assembling materials, one commonly employed route has modified polypeptides, such as polylysine, with PBA motifs to enable electrostatic modulation of the material upon glucose binding. The preparation of electrostatic complex assemblies between PBA-modified cationic polymers, like polylysine, and negatively charged insulin has thus been used as an approach to prepare glucose-responsive materials for insulin delivery.134–137 These materials can also be used to facilitate self-assembly into particulates that are then further fabricated into microneedles for insulin delivery, with glucose-responsive solubilization of the materials dictated by electrostatic modulation (Fig. 6A).138 The resulting microneedles prepared from these self-assembled particles demonstrated blood glucose correction in a diabetic mouse model. PBA motifs have also been integrated as terminal charge-bearing groups on peptide amphiphile gelators, enabling dual-responsive materials capable of pH- or glucose-dependent release.139
 | ||
| Fig. 6 (A) Formation of self-assembled glucose-responsive particulates for insulin loading. Glucose binding to pendant PBA groups leads to particulate disassembly and insulin release. Figure adapted from ref. 138 with permission from Royal Society of Chemistry © 2021. (B) Schematic of peptide self-assembly/disassembly regulated by actuation from GOx, converting glucose levels into a pH stimulus. In the presence of glucose, materials maintained a self-assembled state to encapsulate a glucagon payload, but in the absence of glucose the materials disassociated to release the drug. Figure adapted from ref. 140 with permission from American Chemical Society © 2021. | ||
Different self-assembling peptides have also been designed to respond to the absence of glucose, targeting intervention in a low blood glucose (hypoglycemia) emergency through the release of a glucagon hormone to correct blood glucose. The first example of this approach combined GOx within a peptide amphiphile of the sequence C10-V2A2E2; reduced microenvironmental pH arising from the conversion of glucose to gluconic acid led to stabilized hydrogels in the presence of glucose (Fig. 6B).140 This approach resulted in the release of encapsulated glucagon that was inversely related to the amount of glucose in the environment, and showed protection against the worst effects of hypoglycemia in a mouse model of insulin overdose. Subsequent works demonstrated similar performance using a multi-domain peptide terminated with a PBA group that formed an uncommon nanocoil morphology.141 In the presence of glucose, these nanocoils were extended to form an entangled hydrogel network capable of encapsulating glucagon, with the gels dissolving in the absence of glucose to release the glucagon payload. This platform also demonstrated glucose correction in a mouse model of hypoglycemia from insulin overdose. Another approach demonstrated formation of a liquid–liquid phase-separated droplet upon mixing of a PBA-terminated cationic peptide amphiphile and the negatively charged glucagon under normal glucose conditions.142 However, under low glucose conditions this droplet phase dissolved to release the glucagon payload, again correcting blood glucose in a mouse model.
Peptide self-assembly triggered by the presence of glucose has also been used to develop new glucose sensors. In one example, oligopeptides terminated with PBA groups and modified with fluorescent dyes were demonstrated to self-assemble into nanoparticles upon exposure to glucose; enhanced and concentration-dependent fluorescence of the pendant dyes afforded a means to quantify glucose in physiological conditions.143 Photoluminescent glucose sensing has also been achieved using peptide-based gelators to encapsulate GOx and quantum dots.144
Glucose-responsive peptide self-assembly offers an innovative strategy for developing materials that can respond to changes in glucose levels, with the potential to improve diabetes management. By integrating glucose-sensing elements into peptide designs, self-assembly can be triggered by glucose-induced changes in pH, oxidation state, or glucose binding. For example, GOx-mediated conversion of glucose to gluconic acid lowers pH, leading to hydrogel disassembly and insulin release, while PBA motifs enable glucose binding to modulate electrostatic interactions that control peptide assembly. However, challenges remain, such as the toxicity of GOx and its H2O2 byproduct, as well as the non-specific binding of PBA to other diols and its limited affinity for glucose binding under physiological conditions. Additionally, fine-tuning the balance between stability and responsiveness of these assemblies across varying glucose concentrations is difficult. Future opportunities lie in optimizing glucose recognition to increase specificity and developing more modular and dual-responsive peptide systems for enhanced therapeutic control and glucose sensing.
5. Conclusions
Through a combination of molecular design and amino acid selection, materials prepared from self-assembled peptides can be designed to exhibit stimuli-responsive function directed by disease-relevant analytes. This general design paradigm points to an approach to integrate peptide-based drug carriers with disease sensing, linking therapeutic deployment to spatially relevant cues. Most commonly, the charge and/or hydrophilicity of self-assembling peptide monomers is altered by the presence of these analytes, while in other cases the presence of selectively labile bonds can mediate a chemical transformation driving changes to the propensity for self-assembly. The tailorability of this general approach, beginning from sequence-defined and discrete molecular-scale building blocks, offers versatile functionality. Key properties of peptides, including biocompatibility derived from their natural origin and inherent degradability, further support use of these materials as versatile therapeutic nanocarriers. In the context of the general goals of nanomedicine, this approach therefore presents a useful toolbox to prepare functional carrier materials. Moving forward, the field of peptide self-assembly faces remaining challenges alongside unexplored opportunities. Relative to polymeric or lipid-based nanoparticles, the body of research in the use of self-assembled peptides as functional drug carriers remains in its infancy. As this field continues to progress, care must be taken to further understand and characterize the biocompatibility of these materials, including an appreciation for possible immunogenicity that has been demonstrated in tangential applications using this same materials design paradigm to prepare materials for immune stimulation in the context of vaccination.145,146 Stability and predictable performance in vivo are other key properties that must be more fully characterized. In addition, the complexities of the assembly landscape, and inherent dependence of environmental parameters, must be more clearly understood. Addressing these various challenges through continued research and exploration will be key to unlocking the full therapeutic potential of analyte-responsive self-assembled peptide materials as a component of the growing nanomedicine arsenal.Compared to traditional nanomedicine approaches, peptide self-assembly offers unique advantages in terms of tunability, biocompatibility, and responsiveness to analytes. Unlike polymeric nanoparticles, peptide-based materials can be designed with precise molecular sequences of discrete building blocks, enabling highly specific assembly pathways and fine-tuned interactions with disease-relevant cues, such as pH, redox species, and glucose levels. This sequence-defined nature allows for greater customization of functional properties, such as selective degradability and targeted release, often with enhanced biocompatibility due to their biological origins. However, peptide assemblies face challenges in achieving the same level of stability and robustness in vivo as traditional nanocarriers. While peptide-based systems are highly sensitive to environmental changes, this also makes them more prone to premature disassembly or inconsistent performance under normal physiological fluctuations. Additionally, the field of peptide self-assembly is still relatively young, and further research is required to fully understand their assembly mechanisms, immunogenicity, and long-term behavior in therapeutic applications compared to more established nanomedicine platforms.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MJW gratefully acknowledges funding support from the Helmsley Charitable Trust (2019PG-T1D016 and 2102-04994), American Diabetes Association Pathway Accelerator Award (1-19-ACE-31), Breakthrough T1D (5-CDA-2020-947-A-N), and a National Science Foundation CAREER award (BMAT, 1944875).References
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- Y. Shi, R. van der Meel, X. Chen and T. Lammers, Theranostics, 2020, 10, 7921–7924 CrossRef PubMed.
- F. X. Gu, R. Karnik, A. Z. Wang, F. Alexis, E. Levy-Nissenbaum, S. Hong, R. S. Langer and O. C. Farokhzad, Nano Today, 2007, 2, 14–21 CrossRef.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958–962 CrossRef CAS PubMed.
- Y. (chezy) Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef PubMed.
- W. C. W. Chan, Acc. Chem. Res., 2017, 50, 627–632 CrossRef CAS PubMed.
- R. M. DiSanto, V. Subramanian and Z. Gu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 548–564 CAS.
- B. Hu, K. O. Boakye-Yiadom, W. Yu, Z.-W. Yuan, W. Ho, X. Xu and X.-Q. Zhang, Adv. Healthcare Mater., 2020, 9, e2000336 CrossRef PubMed.
- M. S. Goldberg, Nat. Rev. Cancer, 2019, 19, 587–602 CrossRef CAS PubMed.
- A. Cifuentes-Rius, A. Desai, D. Yuen, A. P. R. Johnston and N. H. Voelcker, Nat. Nanotechnol., 2021, 16, 37–46 CrossRef CAS PubMed.
- Y. H. Chung, V. Beiss, S. N. Fiering and N. F. Steinmetz, ACS Nano, 2020, 14, 12522–12537 CrossRef CAS PubMed.
- S. Friedrichs and D. M. Bowman, Nat. Nanotechnol., 2021, 16, 362–364 CrossRef CAS PubMed.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2017, 2 Search PubMed.
- M. J. Webber, Bioeng. Transl. Med., 2016, 1, 252–266 CrossRef CAS PubMed.
- M. Caldorera-Moore and N. A. Peppas, Adv. Drug Delivery Rev., 2009, 61, 1391–1401 CrossRef CAS PubMed.
- Y. Lu, W. Sun and Z. Gu, J. Controlled Release, 2014, 194, 1–19 CrossRef CAS PubMed.
- A. S. Braegelman and M. J. Webber, Theranostics, 2019, 9, 3017–3040 CrossRef CAS PubMed.
- H. Wang, M. Monroe, F. Leslie, C. Flexner and H. Cui, Trends Pharmacol. Sci., 2022, 43, 510–521 CrossRef CAS PubMed.
- M. J. Sis and M. J. Webber, Trends Pharmacol. Sci., 2019, 40, 747–762 CrossRef CAS PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- V. B. Kumar, B. Ozguney, A. Vlachou, Y. Chen, E. Gazit and P. Tamamis, J. Phys. Chem. B, 2023, 127, 1857–1871 CrossRef CAS PubMed.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- T. Li, X.-M. Lu, M.-R. Zhang, K. Hu and Z. Li, Bioact. Mater., 2022, 11, 268–282 CAS.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- H. Su, J. M. Koo and H. Cui, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Chem. Soc. Rev., 2017, 46, 6638–6663 RSC.
- Y. Zhou, Q. Li, Y. Wu, X. Li, Y. Zhou, Z. Wang, H. Liang, F. Ding, S. Hong, N. F. Steinmetz and H. Cai, ACS Nano, 2023, 17, 8004–8025 CrossRef CAS PubMed.
- M. Delfi, R. Sartorius, M. Ashrafizadeh, E. Sharifi, Y. Zhang, P. De Berardinis, A. Zarrabi, R. S. Varma, F. R. Tay, B. R. Smith and P. Makvandi, Nano Today, 2021, 38, 101119 CrossRef CAS PubMed.
- Y. Wang, X. Zhang, K. Wan, N. Zhou, G. Wei and Z. Su, J. Nanobiotechnol., 2021, 19, 253 CrossRef CAS PubMed.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- M. Abbas, Q. Zou, S. Li and X. Yan, Adv. Mater., 2017, 29, 1605021 CrossRef PubMed.
- N. M. Anderson and M. C. Simon, Curr. Biol., 2020, 30, R921–R925 CrossRef CAS PubMed.
- Z. Liu, J. Guo, Y. Qiao and B. Xu, Acc. Chem. Res., 2023, 56, 3076–3088 CrossRef CAS PubMed.
- B. J. Kim and B. Xu, Bioconjugate Chem., 2020, 31, 492–500 CrossRef CAS PubMed.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS PubMed.
- M. A. VandenBerg and M. J. Webber, Adv. Healthcare Mater., 2019, 8, e1801466 CrossRef PubMed.
- Y. Song, Z. Zhang, Y. Cao and Z. Yu, ChemBioChem, 2023, 24, e202200497 CrossRef CAS PubMed.
- D. F. Evans, G. Pye, R. Bramley, A. G. Clark, T. J. Dyson and J. D. Hardcastle, Gut, 1988, 29, 1035–1041 CrossRef CAS PubMed.
- E. Boedtkjer and S. F. Pedersen, Annu. Rev. Physiol., 2020, 82, 103–126 CrossRef CAS PubMed.
- Z. Li, Y. Zhu and J. B. Matson, ACS Appl. Bio Mater., 2022, 5, 4635–4651 CrossRef CAS PubMed.
- Y. Zimenkov, S. N. Dublin, R. Ni, R. S. Tu, V. Breedveld, R. P. Apkarian and V. P. Conticello, J. Am. Chem. Soc., 2006, 128, 6770–6771 CrossRef CAS PubMed.
- A. Aggeli, M. Bell, L. M. Carrick, C. W. G. Fishwick, R. Harding, P. J. Mawer, S. E. Radford, A. E. Strong and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619–9628 CrossRef CAS PubMed.
- A. Ghosh, M. Haverick, K. Stump, X. Yang, M. F. Tweedle and J. E. Goldberger, J. Am. Chem. Soc., 2012, 134, 3647–3650 CrossRef CAS PubMed.
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- B. Apostolovic and H.-A. Klok, Biomacromolecules, 2008, 9, 3173–3180 CrossRef CAS PubMed.
- W. Zhang, L. Yu, T. Ji and C. Wang, Front. Chem., 2020, 8, 549 CrossRef CAS PubMed.
- S. Yamamoto, K. Nishimura, K. Morita, S. Kanemitsu, Y. Nishida, T. Morimoto, T. Aoi, A. Tamura and T. Maruyama, Biomacromolecules, 2021, 22, 2524–2531 CrossRef CAS PubMed.
- S. Mafe, Chem. Phys., 2004, 296, 29–35 CrossRef CAS.
- D. W. Urry, S. Q. Peng, T. M. Parker, D. C. Gowda and R. D. Harris, Angew. Chem., Int. Ed. Engl., 1993, 32, 1440–1442 CrossRef.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS PubMed.
- R. Wakabayashi, A. Higuchi, H. Obayashi, M. Goto and N. Kamiya, Int. J. Mol. Sci., 2021, 22, 3459 CrossRef CAS PubMed.
- S. Zarzhitsky, H. Edri, Z. Azoulay, I. Cohen, Y. Ventura, A. Gitelman and H. Rapaport, Biopolymers, 2013, 100, 760–772 CrossRef CAS PubMed.
- J. Liang, W.-L. Wu, X.-D. Xu, R.-X. Zhuo and X.-Z. Zhang, Colloids Surf., B, 2014, 114, 398–403 CrossRef CAS PubMed.
- Z. Wang, X. Zhang, M. Han, X. Jiao, J. Zhou, X. Wang, R. Su, Y. Wang and W. Qi, J. Mater. Chem. B, 2023, 11, 8974–8984 RSC.
- D. Wang, Z. Fan, X. Zhang, H. Li, Y. Sun, M. Cao, G. Wei and J. Wang, Langmuir, 2021, 37, 339–347 CrossRef CAS PubMed.
- X. Xu, Y. Li, H. Li, R. Liu, M. Sheng, B. He and Z. Gu, Small, 2014, 10, 1133–1140 CrossRef CAS PubMed.
- Z. Gong, X. Liu, J. Wu, X. Li, Z. Tang, Y. Deng, X. Sun, K. Chen, Z. Gao and J. Bai, Nanotechnology, 2020, 31, 165601 CrossRef CAS PubMed.
- J. G. Ray, S. S. Naik, E. A. Hoff, A. J. Johnson, J. T. Ly, C. P. Easterling, D. L. Patton and D. A. Savin, Macromol. Rapid Commun., 2012, 33, 819–826 CrossRef CAS PubMed.
- T. J. Moyer, J. A. Finbloom, F. Chen, D. J. Toft, V. L. Cryns and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14746–14752 CrossRef CAS PubMed.
- B.-X. Zhao, Y. Zhao, Y. Huang, L.-M. Luo, P. Song, X. Wang, S. Chen, K.-F. Yu, X. Zhang and Q. Zhang, Biomaterials, 2012, 33, 2508–2520 CrossRef CAS PubMed.
- B. F. Lin, K. A. Megley, N. Viswanathan, D. V. Krogstad, L. B. Drews, M. J. Kade, Y. Qian and M. V. Tirrell, J. Mater. Chem., 2012, 22, 19447–19454 RSC.
- C. Chang, P. Liang, L. Chen, J. Liu, S. Chen, G. Zheng and C. Quan, J. Biomater. Sci., Polym. Ed., 2017, 28, 1338–1350 CrossRef CAS PubMed.
- P. Moitra, K. Kumar, P. Kondaiah and S. Bhattacharya, Angew. Chem., Int. Ed., 2014, 53, 1113–1117 CrossRef CAS PubMed.
- F. Raza, Y. Zhu, L. Chen, X. You, J. Zhang, A. Khan, M. W. Khan, M. Hasnat, H. Zafar, J. Wu and L. Ge, Biomater. Sci., 2019, 7, 2023–2036 RSC.
- J. Li, Z. Wang, H. Han, Z. Xu, S. Li, Y. Zhu, Y. Chen, L. Ge and Y. Zhang, Chin. Chem. Lett., 2022, 33, 1936–1940 CrossRef CAS.
- Y. Zhao, H. Yokoi, M. Tanaka, T. Kinoshita and T. Tan, Biomacromolecules, 2008, 9, 1511–1518 CrossRef CAS PubMed.
- M. Altman, P. Lee, A. Rich and S. Zhang, Protein Sci., 2000, 9, 1095–1105 CrossRef CAS PubMed.
- U. Shimanovich, A. Levin, D. Eliaz, T. Michaels, Z. Toprakcioglu, B. Frohm, E. De Genst, S. Linse, K. S. Åkerfeldt and T. P. J. Knowles, Small, 2021, 17, e2007188 CrossRef PubMed.
- N. Liu, J. Han, X. Zhang, Y. Yang, Y. Liu, Y. Wang and G. Wu, Colloids Surf., B, 2016, 145, 401–409 CrossRef CAS PubMed.
- K. Praveen, S. Das, V. Dhaware, B. Pandey, B. Mondal and S. S. Gupta, ACS Appl. Bio Mater., 2019, 2, 4162–4172 CrossRef CAS PubMed.
- M. Li, W. Song, Z. Tang, S. Lv, L. Lin, H. Sun, Q. Li, Y. Yang, H. Hong and X. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 1781–1792 CrossRef CAS PubMed.
- J. Zhang, W. Lin, L. Yang, A. Zhang, Y. Zhang, J. Liu and J. Liu, Biomater. Sci., 2022, 10, 854–862 RSC.
- A. S. Carlini, W. Choi, N. C. McCallum and N. C. Gianneschi, Adv. Funct. Mater., 2021, 31, 2007733 CrossRef CAS PubMed.
- B. H. Pogostin, G. Saenz, C. C. Cole, E. M. Euliano, J. D. Hartgerink and K. J. McHugh, Bioconjugate Chem., 2023, 34, 193–203 CrossRef CAS PubMed.
- M. J. Webber, J. B. Matson, V. K. Tamboli and S. I. Stupp, Biomaterials, 2012, 33, 6823–6832 CrossRef CAS PubMed.
- M. J. Sis, D. Liu, I. Allen and M. J. Webber, Biomacromolecules, 2024, 25, 4482–4491 CrossRef CAS PubMed.
- M. J. Sis, Z. Ye, K. La Costa and M. J. Webber, ACS Nano, 2022, 16, 9546–9558 CrossRef CAS PubMed.
- J. B. Matson, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2012, 8, 3586–3595 RSC.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 47, 7962–7964 RSC.
- Y. Gao, Y. Kuang, Z.-F. Guo, Z. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS PubMed.
- F. Seidi, R. Jenjob and D. Crespy, Chem. Rev., 2018, 118, 3965–4036 CrossRef CAS PubMed.
- N. Kaludercic, S. Deshwal and F. Di Lisa, Front. Physiol., 2014, 5, 285 Search PubMed.
- S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1–3 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794–798 CAS.
- G. T. Wondrak, Antioxid. Redox Signaling, 2009, 11, 3013–3069 CrossRef CAS PubMed.
- S. J. Forrester, D. S. Kikuchi, M. S. Hernandes, Q. Xu and K. K. Griendling, Circ. Res., 2018, 122, 877–902 CrossRef CAS PubMed.
- B. Hu, Z. Lian, Z. Zhou, L. Shi and Z. Yu, ACS Appl. Bio Mater., 2020, 3, 5529–5551 CrossRef CAS PubMed.
- Z. Jiang and S. Thayumanavan, Isr. J. Chem., 2020, 60, 132–139 CrossRef CAS PubMed.
- D. Yang, W. Chen and J. Hu, J. Phys. Chem. B, 2014, 118, 12311–12317 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2010, 132, 9526–9527 CrossRef CAS PubMed.
- H. Dong, M. Wang, S. Fan, C. Wu, C. Zhang, X. Wu, B. Xue, Y. Cao, J. Deng, D. Yuan and J. Shi, Angew. Chem., Int. Ed., 2022, 61, e202212829 CrossRef CAS PubMed.
- L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS PubMed.
- P. Roth, R. Meyer, I. Harley, K. Landfester, I. Lieberwirth, M. Wagner, D. Y. W. Ng and T. Weil, Nat. Synth., 2023, 2, 980–988 CrossRef.
- P. Schiapparelli, P. Zhang, M. Lara-Velazquez, H. Guerrero-Cazares, R. Lin, H. Su, R. W. Chakroun, M. Tusa, A. Quiñones-Hinojosa and H. Cui, J. Controlled Release, 2020, 319, 311–321 CrossRef CAS PubMed.
- A. G. Cheetham, P. Zhang, Y.-A. Lin, L. L. Lock and H. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS PubMed.
- A. G. Cheetham, Y.-C. Ou, P. Zhang and H. Cui, Chem. Commun., 2014, 50, 6039–6042 RSC.
- H. Wang, H. Su, T. Xu and H. Cui, Angew. Chem., Int. Ed., 2023, 62, e202306652 CrossRef CAS PubMed.
- H. Su, P. Zhang, A. G. Cheetham, J. M. Koo, R. Lin, A. Masood, P. Schiapparelli, A. Quiñones-Hinojosa and H. Cui, Theranostics, 2016, 6, 1065–1074 CrossRef CAS PubMed.
- F. Wang, H. Su, D. Xu, M. K. Monroe, C. F. Anderson, W. Zhang, R. Oh, Z. Wang, X. Sun, H. Wang, F. Wan and H. Cui, Biomaterials, 2021, 279, 121182 CrossRef CAS PubMed.
- F. Wang, H. Su, D. Xu, W. Dai, W. Zhang, Z. Wang, C. F. Anderson, M. Zheng, R. Oh, F. Wan and H. Cui, Nat. Biomed. Eng., 2020, 4, 1090–1101 CrossRef CAS PubMed.
- F. Wang, Q. Huang, H. Su, M. Sun, Z. Wang, Z. Chen, M. Zheng, R. W. Chakroun, M. K. Monroe, D. Chen, Z. Wang, N. Gorelick, R. Serra, H. Wang, Y. Guan, J. S. Suk, B. Tyler, H. Brem, J. Hanes and H. Cui, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2204621120 CrossRef CAS PubMed.
- J. Wang, M. Abbas, Y. Huang, J. Wang and Y. Li, Commun. Chem., 2023, 6, 1–8 CrossRef PubMed.
- H. L. Schenck, G. P. Dado and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 12487–12494 CrossRef CAS.
- A. R. Rodriguez, J. R. Kramer and T. J. Deming, Biomacromolecules, 2013, 14, 3610–3614 CrossRef CAS PubMed.
- S.-I. Aiba, N. Minoura and Y. Fujiwara, Makromol. Chem., 1982, 183, 1333–1342 CrossRef CAS.
- N. Song, Z. Zhou, Y. Song, M. Li, X. Yu, B. Hu and Z. Yu, Nano Today, 2021, 38, 101198 CrossRef CAS.
- D. Spitzer, L. L. Rodrigues, D. Straßburger, M. Mezger and P. Besenius, Angew. Chem., Int. Ed., 2017, 56, 15461–15465 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed.
- X. Miao, W. Cao, W. Zheng, J. Wang, X. Zhang, J. Gao, C. Yang, D. Kong, H. Xu, L. Wang and Z. Yang, Angew. Chem., Int. Ed., 2013, 52, 7781–7785 CrossRef CAS PubMed.
- Z. Huang, Q. Luo, S. Guan, J. Gao, Y. Wang, B. Zhang, L. Wang, J. Xu, Z. Dong and J. Liu, Soft Matter, 2014, 10, 9695–9701 RSC.
- G. Wu, C. Ge, X. Liu, S. Wang, L. Wang, L. Yin and H. Lu, Chem. Commun., 2019, 55, 7860–7863 RSC.
- C. Ge, J. Zhu, G. Wu, H. Ye, H. Lu and L. Yin, Biomacromolecules, 2022, 23, 2647–2654 CrossRef CAS PubMed.
- X. Liu, M. Li, J. Liu, Y. Song, B. Hu, C. Wu, A.-A. Liu, H. Zhou, J. Long, L. Shi and Z. Yu, J. Am. Chem. Soc., 2022, 144, 9312–9323 CrossRef CAS PubMed.
- H. Wang, Y. Song, W. Wang, N. Chen, B. Hu, X. Liu, Z. Zhang and Z. Yu, J. Am. Chem. Soc., 2024, 146, 330–341 CrossRef CAS PubMed.
- H. G. Kuivila and A. G. Armour, J. Am. Chem. Soc., 1957, 79, 5659–5662 CrossRef CAS.
- J. L. M. Jourden, K. B. Daniel and S. M. Cohen, Chem. Commun., 2011, 47, 7968–7970 RSC.
- M. Ikeda, T. Tanida, T. Yoshii and I. Hamachi, Adv. Mater., 2011, 23, 2819–2822 CrossRef CAS PubMed.
- M. Pieszka, S. Han, C. Volkmann, R. Graf, I. Lieberwirth, K. Landfester, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2020, 142, 15780–15789 CrossRef CAS PubMed.
- Z. Zhou, K. Maxeiner, P. Moscariello, S. Xiang, Y. Wu, Y. Ren, C. J. Whitfield, L. Xu, A. Kaltbeitzel, S. Han, D. Mücke, H. Qi, M. Wagner, U. Kaiser, K. Landfester, I. Lieberwirth, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2022, 144, 12219–12228 CrossRef CAS PubMed.
- Y. Xiang, B. Su, D. Liu and M. J. Webber, Adv. Ther., 2024, 7, 2300127 CrossRef CAS.
- M. J. Webber, Diabetes, 2024, 73, 1032–1038 CrossRef PubMed.
- A. R. Mohanty, A. Ravikumar and N. A. Peppas, Regener. Biomater., 2022, 9, rbac056 CrossRef CAS PubMed.
- X.-D. Xu, B.-B. Lin, J. Feng, Y. Wang, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Macromol. Rapid Commun., 2012, 33, 426–431 CrossRef CAS PubMed.
- J. Rodon Fores, M. L. Martinez Mendez, X. Mao, D. Wagner, M. Schmutz, M. Rabineau, P. Lavalle, P. Schaaf, F. Boulmedais and L. Jierry, Angew. Chem., Int. Ed., 2017, 56, 15984–15988 CrossRef CAS PubMed.
- M. Fu, C. Zhang, Y. Dai, X. Li, M. Pan, W. Huang, H. Qian and L. Ge, Biomater. Sci., 2018, 6, 1480–1491 RSC.
- X. Li, M. Fu, J. Wu, C. Zhang, X. Deng, A. Dhinakar, W. Huang, H. Qian and L. Ge, Acta Biomater., 2017, 51, 294–303 CrossRef CAS PubMed.
- Y. Xiang, S. Xian, R. C. Ollier, S. Yu, B. Su, I. Pramudya and M. J. Webber, J. Controlled Release, 2022, 348, 601–611 CrossRef CAS PubMed.
- W. L. A. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 CrossRef CAS PubMed.
- J. Zhang, X. Wei, W. Liu, Y. Wang, A. R. Kahkoska, X. Zhou, H. Zheng, W. Zhang, T. Sheng, Y. Zhang, Y. Liu, K. Ji, Y. Xu, P. Zhang, J. Xu, J. B. Buse, J. Wang and Z. Gu, Nat. Biomed. Eng., 2023 DOI:10.1038/s41551-023-01138-7.
- S. Xian, Y. Xiang, D. Liu, B. Fan, K. Mitrová, R. C. Ollier, B. Su, M. A. Alloosh, J. Jiráček, M. Sturek, M. Alloosh and M. J. Webber, Adv. Mater., 2024, 36, e2308965 CrossRef PubMed.
- Y. Wang, P. Chen, W. Liu, X. Wei, J. Zhang, X. Wei, Y. Liu, L. Rao, S. Zhang, J. Yu, X. Ye, J. Wang and Z. Gu, Nano Today, 2023, 51, 101937 CrossRef CAS.
- J. Wang, Z. Wang, G. Chen, Y. Wang, T. Ci, H. Li, X. Liu, D. Zhou, A. R. Kahkoska, Z. Zhou, H. Meng, J. B. Buse and Z. Gu, ACS Nano, 2021, 15, 4294–4304 CrossRef CAS PubMed.
- D. Shen, H. Yu, L. Wang, X. Chen, J. Feng, Q. Zhang, W. Xiong, J. Pan, Y. Han and X. Liu, J. Mater. Chem. B, 2021, 9, 6017–6028 RSC.
- N. Tao, G. Li, M. Liu, W. Gao and H. Wu, Tetrahedron, 2017, 73, 3173–3180 CrossRef CAS.
- S. Yu, S. Xian, Z. Ye, I. Pramudya and M. J. Webber, J. Am. Chem. Soc., 2021, 143, 12578–12589 CrossRef CAS PubMed.
- S. Yu, Z. Ye, R. Roy, R. R. Sonani, I. Pramudya, S. Xian, Y. Xiang, G. Liu, B. Flores, E. Nativ-Roth, R. Bitton, E. H. Egelman and M. J. Webber, Adv. Mater., 2024, 36, e2311498 CrossRef PubMed.
- S. Yu, W. Chen, G. Liu, B. Flores, E. L. DeWolf, B. Fan, Y. Xiang and M. J. Webber, J. Am. Chem. Soc., 2024, 146, 7498–7505 CrossRef CAS PubMed.
- P. K. Mehta, L. N. Neupane and K.-H. Lee, Sens. Actuators, B, 2023, 375, 132913 CrossRef CAS.
- J. H. Kim, S. Y. Lim, D. H. Nam, J. Ryu, S. H. Ku and C. B. Park, Biosens. Bioelectron., 2011, 26, 1860–1865 CrossRef CAS PubMed.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed.
- J. S. Rudra, T. Sun, K. C. Bird, M. D. Daniels, J. Z. Gasiorowski, A. S. Chong and J. H. Collier, ACS Nano, 2012, 6, 1557–1564 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |