 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymeric integration of structure-switching aptamers on transistors for histamine sensing†
Bajramshahe
Shkodra
 ac,
Mattia
Petrelli
a,
Kyung-Ae
Yang
b,
Anna
Tagliaferri
a,
Paolo
Lugli
a,
Luisa
Petti
a and
Nako
Nakatsuka
*c
ac,
Mattia
Petrelli
a,
Kyung-Ae
Yang
b,
Anna
Tagliaferri
a,
Paolo
Lugli
a,
Luisa
Petti
a and
Nako
Nakatsuka
*c
aSensing Technologies Laboratory (STL), Faculty of Engineering, Free University of Bozen-Bolzano, 39100 Bozen, Italy
bCenter for Innovative Diagnostic and Therapeutic Approaches, Department of Medicine, Columbia University, New York 10032, USA
cLaboratory of Biosensors and Bioelectronics, Institute for Biomedical Engineering, ETH Zürich, CH-8092, Switzerland. E-mail: nakatsuka@biomed.ee.ethz.ch
First published on 26th July 2023
Abstract
Aptamers that undergo large conformational rearrangements at the surface of electrolyte-gated field-effect transistor (EG-FETs)-based biosensors can overcome the Debye length limitation in physiological high ionic strength environments. For the sensitive detection of small molecules, carbon nanotubes (CNTs) that approach the dimensions of analytes of interest are promising channel materials for EG-FETs. However, functionalization of CNTs with bioreceptors using frequently reported surface modification strategies (e.g., π–π stacking), requires highly pristine CNTs deposited through methods that are incompatible with low-cost fabrication methods and flexible substrates. In this work, we explore alternative non-covalent surface chemistry to functionalize CNTs with aptamers. We harnessed the adhesive properties of poly-D-lysine (PDL), to coat the surface of CNTs and then grafted histamine-specific DNA aptamers electrostatically in close proximity to the CNT semiconducting channel. The layer-by-layer assembly was monitored by complementary techniques such as X-ray photoelectron spectroscopy, optical waveguide lightmode spectroscopy, and fluorescence microscopy. Surface characterization confirmed histamine aptamer integration into PDL-coated CNTs and revealed ∼5-fold higher aptamer surface coverage when using CNT networks with high surface areas. Specific aptamers assembled on EG-CNTFETs enabled histamine detection in undiluted high ionic strength solutions in the concentration range of 10 nM to 100 μM. Sequence specificity was demonstrated via parallel measurements with control EG-CNTFETs functionalized with scrambled DNA. Histamine aptamer-modified EG-CNTFETs showed high selectivity vs. histidine, the closest structural analog and precursor to histamine. Taken together, these results implied that target-specific aptamer conformational changes on CNTs facilitate signal transduction, which was corroborated by circular dichroism spectroscopy. Our work suggests that layer-by-layer polymer chemistry enables integration of structure-switching aptamers into flexible EG-CNTFETs for small-molecule biosensing.
1 Introduction
Electrolyte-gated field-effect transistors (EG-FETs) are becoming the transducer of choice for many biosensors due to their superior electronic properties and intrinsic signal amplification, which allows the sensitive detection of small molecules.1–4 The formation of an electric double layer at the electrolyte/semiconductor interface of EG-FETs leads to a large gate capacitance, which enables device functionality below ±1 V, a necessary condition when using water-based electrolytes; electrochemical decomposition occurs at 1.23 V.5 Among the channel materials for EG-FETs, carbon nanotubes (CNTs) have demonstrated high potential for sensitive small-molecule biosensing, due to nanometric dimensions comparable to analytes of interest. With diameters that vary from 0.4 nm to 100 nm and a length up to tens of micrometers, CNTs have high aspect-ratios and subsequently high surface areas available for functionalization with bioreceptors.6,7 Furthermore, the solution processability of CNTs enables their integration into diverse substrates: from rigid substrates to flexible foils.6,8 To this point, CNT-based EG-FETs (i.e., EG-CNTFETs) have shown promising results in the field of small-molecule biosensing, demonstrating sensitivities down to pM concentrations in clinically relevant environments.3,9–11To enable sensitive detection of small-molecule targets using EG-CNTFETs, the choice of the biorecognition element is extremely critical: target binding must induce signal transduction within or near the Debye screening length (λD), which is <1 nm for biological environments.1 Structure-switching aptamers have been previously shown to fulfil this requirement.1,4,12–14 Aptamers are artificial single-stranded oligonucleotides designed to recognize targets of interest with high affinity and selectivity through combinatorial in vitro selection.15–18 For certain sequences designed to undergo conformational changes upon target recognition, the negatively charged phosphate backbone moves within or near the λD. This aptamer rearrangement subsequently modulates the semiconductor surface charge density, which is transduced as target-specific signals even in physiologically relevant milieus.
In addition to carefully selecting the bioreceptors that facilitate signal transduction in high ionic milieus, the choice and validation of the optimal functionalization strategy is also a key aspect to take into account. The surface modification is particularly challenging on EG-CNTFETs due to the highly hydrophobic nature of CNTs. Traditional CNT functionalization consists of either covalent or noncovalent surface modification routes.6 Covalent functionalization couples biorecognition elements directly to the oxidized surface of CNTs.19 While this functionalization strategy provides a durable bond, the procedure of generating functional groups on the CNT surfaces (e.g., the use of strong oxidizing agents) can generate defects in the nanotube structures. Therefore, noncovalent functionalization is favorable, as the carbon atoms in the CNTs keep their sp2 hybridization and maintain their optimal electrical properties.8,20 Noncovalent modifications involve physical adsorption of biorecognition elements onto the CNT surface through π–π, hydrophobic, or electrostatic interactions.21,22
The traditional noncovalent functionalization of CNTs is through the usage of 1-pyrenebutyric acid N-hydroxysuccinimide ester (PBASE) as a linker molecule. Here, the benzene rings of PBASE attach to the CNTs due to π–π stacking interactions, while the ester remains free to react with the primary amines of biorecognition elements.6,8 While effective, this functionalization strategy can only be employed on the surface of highly purified CNTs, often directly grown onto substrates using chemical vapor deposition (CVD).23–26 Using CVD, semiconducting CNTs with the necessary high purity level can be integrated into devices. However, this technique suffers from several limitations such as expensive infrastructure compared to other deposition techniques.27 Moreover, the high temperature requirements (from 550 to 1000 °C) for CVD synthesis, hinder the possibility to deposit the CNTs on polymer-based substrates.6 Specific applications including wearable electronics or food safety, require soft and flexible substrates to interface with non-rigid surfaces such as skin and packaging, respectively. An alternative cost-effective and scalable technique compatible with coating polymeric surfaces, involves spray deposition of solution-processed CNTs.28
However, spray deposition results in CNT coatings that lack the necessary purity level for noncovalent surface modification using π–π stacking. To overcome this issue, we investigated a different route of surface modification, harnessing electrostatic and hydrophobic interactions to assemble polymer coatings on CNTs.29 The CNTs were coated with poly-D-lysine (PDL) films that are positively charged to attract the assembly of negatively charged DNA aptamers.30 An advantage of this chemistry, is that polymer-coated EG-CNTFETs have reported improved stability and sensor performance as the CNTs are no longer in direct contact with the electrolyte and are less influenced by solution ions.31,32
Often in CNT-based biosensing reports, surface functionalization is only validated via the electrical characterization (i.e., current vs. voltage characteristics or transfer curves).8,19,23 While the shift in the transistor transfer curves may indicate effective functionalization, observed changes may also be a result of sensor drift over time or altered environmental conditions (e.g., solution evaporation leading to changes in ionic content) as the sensing area is highly sensitive to any changes in charge distribution.33 To this point, the sequential surface assembly of biorecognition elements should be characterized using complementary methods to ensure robust functionalization.
Herein, we characterized the layer-by-layer surface modification of spray-deposited CNTs with PDL and subsequent integration of histamine-specific aptamers (Fig. 1). Importantly, this histamine aptamer was recently isolated through a selection method that produces sequences that undergo substantial structural rearrangements upon target recognition.34 Thus, histamine was used as a model small-molecule target to interrogate whether electrostatically adsorbed aptamers undergoing conformational changes may enable signal transduction at CNT surfaces in high ionic strength environments. In addition, histamine is an important biogenic amine to detect in biomedical diagnostics and food quality assurance sectors.35,36 Taking advantage of the spray deposition method, we assembled the polymer-aptamer film on flexible polyimide substrates, which may be integrated into future wearables or food packaging. The surface assembly was monitored and validated using X-ray photoelectron spectroscopy (XPS), optical waveguide lightmode spectroscopy (OWLS), and fluorescence microscopy.
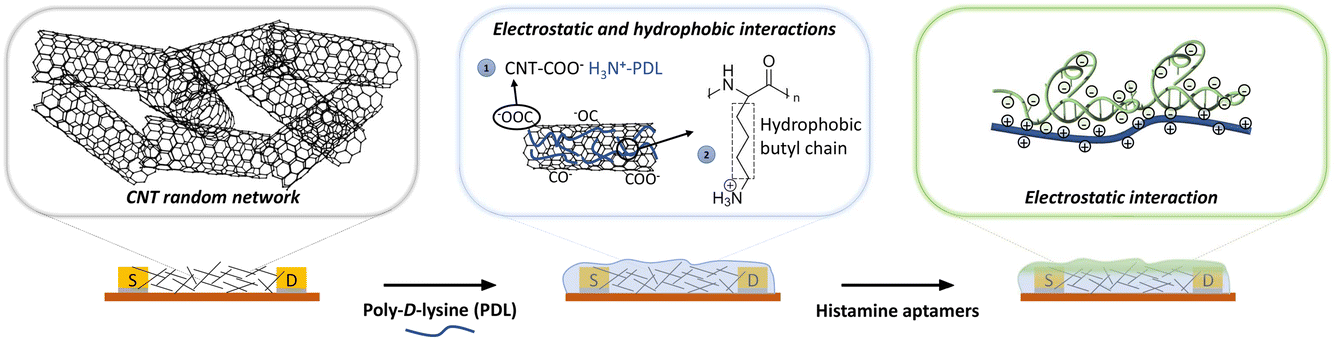 | ||
| Fig. 1 Schematic representation of sequential functionalization on the surface of electrolyte-gated carbon-nanotube (CNT) field-effect transistors. First, the network of CNTs is coated with poly-D-lysine (PDL). The adhesion of PDL on the CNT surface arises from (1) the electrostatic interaction between the negatively charged carbonyl/carboxyl groups on CNTs and protonated amine groups on PDL, and (2) the hydrophobic interactions between the non-polar hydrophobic butyl chain of the PDL and the CNT surface.29 Then, histamine aptamers are assembled on the PDL-coated nanotubes via electrostatic interactions between the highly negatively charged phosphate backbone of the aptamers and the protonated amine groups on the PDL. | ||
This functionalization strategy was then implemented in EG-CNTFET devices. The histamine aptamer-PDL-modified EG-CNTFETs enabled detection of histamine in the dynamic range of 10 nM to 100 μM. Sensors modified with control DNA sequences with the same chemical signature as the histamine aptamer (number and type of bases conserved but the order scrambled) reacted significantly less to histamine, indicating a target-specific response upon aptamer integration. Further, the sensors demonstrated selectivity vs. the histamine precursor, histidine with negligible responses. Circular dichroism spectroscopy confirmed the origin of this selectivity: the histamine aptamer undergoes structure switching only in the presence of histamine, which drives the target-specific sensor responses. Our results suggest that the conformational changes of the histamine aptamer upon target recognition occur within the λD limit at the surface of the EG-CNTFETs, enabling histamine detection in undiluted high ionic solutions.
2 Materials and methods
2.1 Materials
All chemicals were purchased from Sigma-Aldrich, unless otherwise stated. All solutions were prepared using deionized water with a resistivity of 18.2 MΩ cm produced by a Milli-Q system (Millipore, SAS, France). 1× phosphate-buffered saline (1× PBS) with a concentration of 137 mM NaCl, 10 mM phosphate, 2.7 mM KCl and pH 7.4, was used for all experiments, unless otherwise stated. The histamine aptamer (5′-ACG TGC GCC AGA GCG TAT ACC GTT GGA TGA TGG CCC GCG T-3′) with molecular weight 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 532.9 g mol−1 and a melting point of 84.2 °C, and scrambled sequence: (5′-CAA GTT CCT GGA GGG TCG ACG CAC GCG TAA CCG TGT GGC T-3′) with molecular weight 12532.9 g mol−1 and melting point 84.2 °C were purchased from Microsynth AG (Balgach, Switzerland). All DNA solutions (100 μM) were purified via high performance liquid chromatography (HPLC), aliquoted, and stored at −20 °C until use.
532.9 g mol−1 and a melting point of 84.2 °C, and scrambled sequence: (5′-CAA GTT CCT GGA GGG TCG ACG CAC GCG TAA CCG TGT GGC T-3′) with molecular weight 12532.9 g mol−1 and melting point 84.2 °C were purchased from Microsynth AG (Balgach, Switzerland). All DNA solutions (100 μM) were purified via high performance liquid chromatography (HPLC), aliquoted, and stored at −20 °C until use.
2.2 Fabrication of EG-CNTFETs
The device fabrication was performed as described in a previous publication.37 The EG-FET layout consists of gold interdigitated electrodes (IDEs) for the source and drain electrodes (channel length = 50 μm, channel width = 57 mm) and a planar gold gate electrode. A single step of standard negative photolithography was used to pattern the source, drain, and gate electrodes on the same layer, followed by the deposition of 10 nm of titanium (adhesion layer) and 50 nm of gold using electron beam, (SmartBeam EB-4, BeamTec Gmbh, Germany) thermal evaporation (MBRAUN ProVap 5G, equipped with INFICON SQM-160 rate-thickness monitor), and subsequent lift-off.The CNTs were prepared for the spray-deposition process in the form of a water-based solution, in which 0.05% wt CNTs (95% semiconducting, median length 1 μm, average diameter 0.78 nm, Merck KGaA, Darmstadt, Germany) were dispersed using 0.5% wt sodium carboxymethyl cellulose (CMC) as the surfactant, following the preparation protocol defined in our previous work through extensive optimization.37,38 The spray deposition was conducted by an automated system equipped with an industrial air atomizing spray valve (Nordson EFD, USA). The nozzle to substrate distance was fixed at 5 cm, the atomizing pressure was kept at 0.5 bars. The speed of the moving arm was set at 150 mm s−1, and the substrates were heated at 70 °C.37,38
After spraying, to remove the CMC surfactant and to enhance the conductivity of the nanotubes, the devices were immersed in 2.90 M nitric acid (HNO3) for 1 h, at room temperature,39 followed by a water bath for 30 min, and a final drying step on a hot plate at 100 °C for 30 min. The quality of the spray-deposited films was monitored by optical microscopy (Axio Imager, Carl Zeiss Microscopy GmbH, Germany) and atomic force microscopy (Nanosurf CoreAFM, Switzerland).
2.3 Aptamer surface functionalization
The EG-CNTFETs were first functionalized by drop-casting 40 μL PDL (100 μg mL−1 prepared in 1× PBS) onto the CNT channel and incubating for 1 h at room temperature. The excess PDL was removed by rinsing the devices twice with 1× PBS. Histamine aptamers were diluted to 5 μM in 1× PBS and denatured at 95 °C for 5 min and then renatured by cooling to room temperature.40 Then, 20 μL of the histamine aptamers were drop-casted onto the functionalized EG-CNTFETs with PDL and incubated for 1 h at room temperature. After this incubation step, the remaining aptamer excess was removed by rinsing the devices twice with 1× PBS prior to their experimental use.2.4 X-ray photoelectron spectroscopy (XPS)
The XPS spectra were acquired with a Quantera PHI spectrometer, equipped with an Al Kα monochromatic source (1486.6 eV), a hemispherical capacitor electron-energy analyzer, and a 32–channel plate detector. All the spectra were acquired in the fixed-analyzer-transmission mode, using a nominal X-ray beam spot size of either 100 or 200 μm. A low-voltage argon ion gun/electron neutralizer was used during the analysis for charge compensation. The atomic fractions of the detectable elements in the volume probed by the method were estimated using the formula: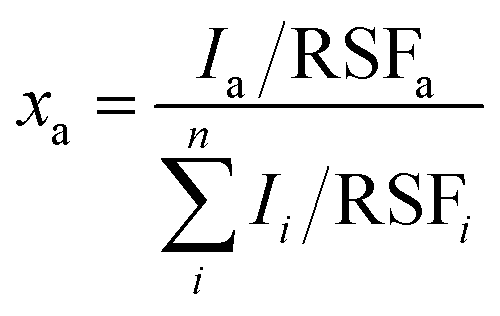 | (1) |
2.5 Optical waveguide lightmode spectroscopy (OWLS)
This method is based on the grating-assisted in-coupling of a He–Ne laser into a planar waveguide and allows direct online monitoring of the optical “dry” mass of macromolecular adsorption. Measurements were performed using a BIOS-I instrument (ASI AG, Zürich, Switzerland) using a flow-through cell (8 × 2 × 1 mm3, PEEK) equipped with a Kalrez O-ring, and planar optical waveguides (8 × 12 mm2) with titanium dioxide coating. Prior to any measurements, waveguides were thoroughly rinsed with isopropyl alcohol followed by deionized water and finally dried using compressed nitrogen. Waveguides were first exposed to 1× PBS buffer solution until a stable baseline was obtained. Then, 100 μg mL−1 PDL solution (prepared in 1× PBS) was injected into the flow cell. To remove the excess PDL, multiple rinsing steps were performed with 1× PBS. After reaching a stable baseline, 5 μM of histamine aptamer solution (prepared in 1× PBS) was injected. Excess aptamers were removed by rinsing with 1× PBS. The adsorbed mass density data were calculated according to de Feijter’s equation: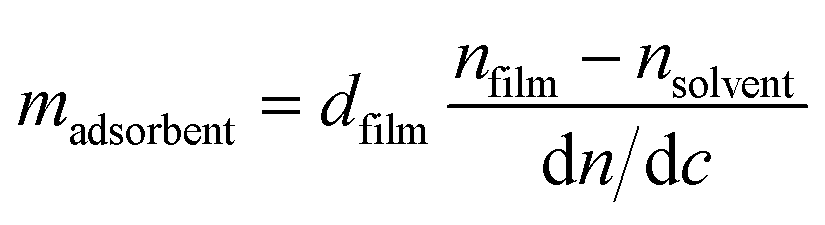 | (2) |
2.6 Fluorescence microscopy
Aptamer functionalization was visualized using a confocal laser scanning microscope (Olympus FluoView 3000). The samples were incubated with SYBR gold dye (ThermoFisher Scientific, MA, U.S.A.) diluted in 1× TE (composed of 10 mM Tris–HCl containing 1 mM EDTA-Na2) buffer for 30 min. Any remaining dye solution was thoroughly rinsed twice with deionized water. Finally, samples were covered with deionized water and fluorescence intensities were measured using an excitation wavelength at 488 nm.2.7 Circular dichroism (CD) spectroscopy
For each CD measurement, 10 μM histamine aptamer or scrambled DNA solutions were prepared in 1× PBS (300 μL total volume). The DNA solutions were heat-treated for 5 min at 95 °C and cooled slowly to room temperature prior to incubation with targets. To interrogate interactions with different molecules, equimolar amounts of the target were incubated with the aptamer prior to measurements. The CD spectroscopic analyses were carried out with a Chirascan Plus (Applied Biophysics Ltd, Leatherhead, UK) at room temperature within 30 min, following target incubation. Quartz cuvettes with a path length of 1.0 mm (Hellma 110-QS) were used. Two scans were acquired per sample with 1 nm resolution, 2.0 nm bandwidth, and a 40 nm min−1 scan rate. Scans were calculated as averages of two instrumental scans and of two replicates per condition.2.8 Electrical characterization
The EG-CNTFET devices were characterized using a probe station connected to a Keysight B1500A semiconductor device parameter analyzer. The devices were placed on the vacuum chuck of the probe station and were taped down to ensure a fixed position. Resistance, transfer, and output characteristics were recorded. To ensure full coverage of the three metallic electrodes (i.e., source, drain, and gate) by 1× PBS, a customized polyethylene chamber was used. The resistance of the devices was measured by sweeping the drain-source voltage, VDS from −0.5 V to 0.5 V and measuring the drain-source current, IDS. The transfer characteristics were recorded sweeping the gate-source voltage, VGS from 0.2 V to −0.8 V, while maintaining the VDS constant at −0.1 V, as previously employed.31,42 The output characteristics were recorded by varying the VDS from 0 V to −0.6 V for different values of VGS (from 0.2 V to −0.8 V, with −0.2 V steps).6 For the real-time measurements, the IDS was continuously recorded while VDS and VGS were held constant at −0.1 V and −0.8 V, respectively.Prior to functionalization, electrical characterization of EG-CNTFETs was performed to minimize device-to-device variability. Only devices having source-drain resistance <10 kΩ were used in this work (Fig. S2a†).37 Transfer characteristics in 1× PBS showed typical p-type behavior, with a ION/IOFF ratio of 630 ± 245 A/A in line with surfactant-based semiconducting CNT devices10,32,42 and a threshold voltage of −0.46 ± 0.05 V, for N = 5 measurements (Fig. S2b†). The output characteristics showed the formation of stable ohmic contacts between the gold IDEs and the CNTs (Fig. S2c†).3 The selected EG-CNTFETs were then functionalized and tested for histamine detection. Prior to histamine sensing measurements, transfer characteristics were taken after each step of functionalization of EG-CNTFETs (Fig. S3†).
When drop-casting the PDL, we ensured full encapsulation of the CNT channel as well as the interdigitated source and drain electrodes (Fig. 1). The EG-CNTFET response was tested both in terms of transfer characteristics (i.e., sweeping the VGS from 0.2 V to −0.8 V) and as a continuous measurement (i.e., constant VGS at −0.8 V). Transfer characteristics (Fig. S4†) or continuous measurements (Fig. S5†) in 1× PBS were recorded for 70 min prior to exposure to the target analyte histamine, to ensure device stability. Further, to ensure solution injection does not affect the device response, 1× PBS was always injected three times prior to histamine injection. The devices required ∼30 min to produce a stable signal prior to exposure to histamine.
2.9 Statistics
Statistics were computed using the program GraphPad Prism (GraphPad Software Inc., San Diego, U.S.A.). All data are reported as mean ± standard error, of the means with P < 0.05 considered statistically significant. Data was analyzed via one-way analysis of variance with post hoc Tukey’s multiple comparisons or unpaired Student’s t-tests.3 Results and discussion
For non-covalent functionalization of CNT-based biosensors, pyrene chemistry using PBASE as a linker molecule is the most widely reported strategy (Fig. S6a†).6 Thus, we originally attempted this functionalization method on the CNTs. The in situ assembly of PBASE was monitored using OWLS (Fig. S6b†) and the surface chemical composition was analyzed via XPS upon PBASE incubation and subsequent aptamer assembly (Fig. S6c and d†). In both techniques, negligible biomolecular assembly on the CNTs was observed. The pyrene strategy was previously employed only on highly pristine CNTs directly grown onto the substrate via CVD.23–26 Herein, we used solution-processed CNTs in CMC surfactant that were spray-deposited on the sensing area, as an alternative low-cost deposition technique compatible with flexible substrates. The inability to assemble PBASE molecules via π–π stacking on the solution-processed CNTs is likely due to impurities on the surface of surfactant-coated CNTs. To this end, we employed an alternative non-covalent electrostatic functionalization method to modify the spray-deposited CNT network with histamine aptamers.The layer-by-layer assembly of the PDL and aptamers on the CNTs was first confirmed by monitoring various elemental peaks in XPS. Fig. 2a shows wide XPS spectra for the three sets of samples: spray-deposited CNTs (blue), PDL-coated CNTs (red), and aptamer-functionalized PDL-CNTs (green). We monitored the carbon C 1s peak at 285.0 eV, the nitrogen N 1s peak at 399.5 eV (Fig. 2b), and the phosphorus P 2p peak at 132.2 eV (Fig. 2c) to track the PDL and aptamer assembly on the CNTs. The energy scales of all the spectra were referred to the adventitious carbon (285.0 eV).
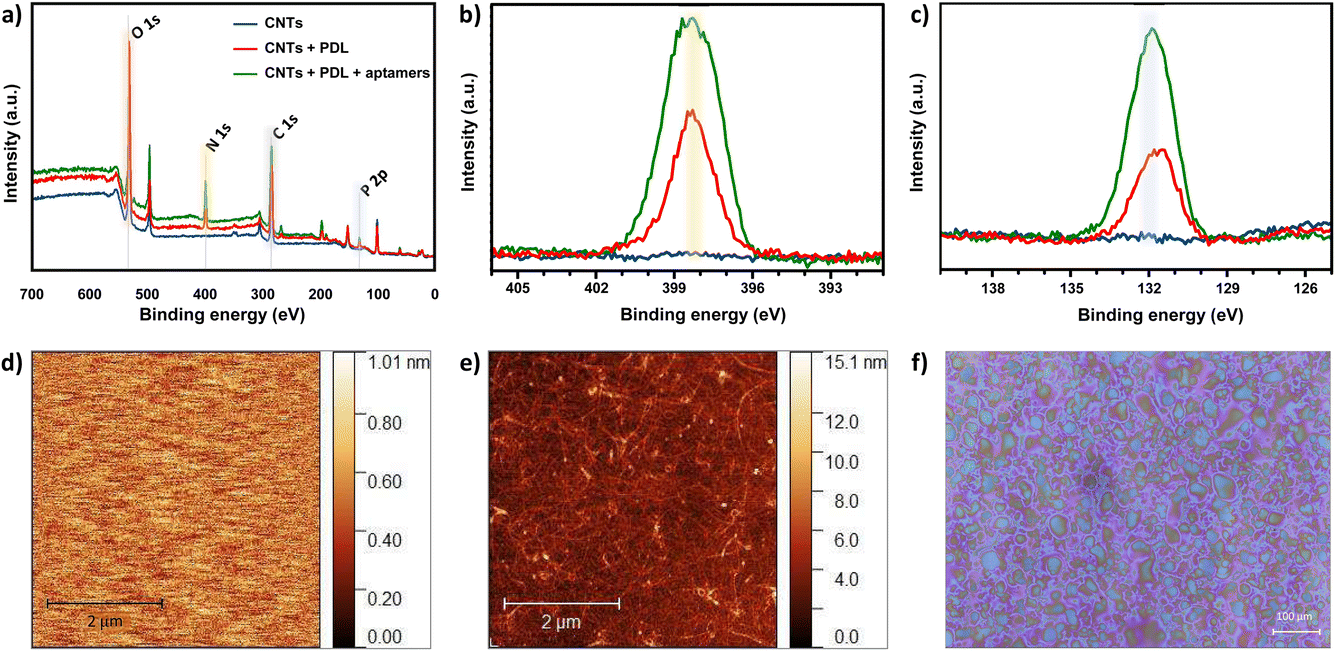 | ||
| Fig. 2 (a) Wide X-ray spectroscopy (XPS) spectra for bare carbon nanotubes (CNTs) (blue), poly-D-lysine (PDL)-coated CNTs (red), and aptamer-modified CNTs (green). Zoom-in of the (b) N 1s peak intensities (yellow highlight) and (c) P 2p peak intensities (blue highlight) that demonstrate the effective layer-by-layer assembly of PDL and aptamers on the CNTs. (d) Representative atomic force microscopy (AFM) image of the bare waveguide and (e) waveguide coated with spray-deposited CNTs. Increased roughness and height indicate coating of substrates with CNTs. (f) Optical microscopy image of the spray-deposited CNTs-carboxymethyl cellulose surfactant ink on the waveguide. The deposited droplets are close to one another and homogeneously cover the waveguide surface indicative of a high quality surface coating. | ||
In all three samples, we observed multiple components in the C 1s peaks. Different contributions to the C 1s peak are assigned as follows: 283.25–283.61 eV to C–C bonding, 283.91–284.84 eV to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and 285.8–287.6 eV to C–O/CO bonding, which appear in all three samples. The new peaks at ∼286 eV assigned to C–N bonding, appear after the addition of each polymer layer due to the C–N bonds present in both PDL and DNA (Fig. S7a–c†).25,43–47
C, and 285.8–287.6 eV to C–O/CO bonding, which appear in all three samples. The new peaks at ∼286 eV assigned to C–N bonding, appear after the addition of each polymer layer due to the C–N bonds present in both PDL and DNA (Fig. S7a–c†).25,43–47
As seen in Fig. 2b, the N 1s peak is absent for bare CNTs as expected. An increase in the N 1s peak is observed for PDL-coated CNTs due to the presence of amine groups. A further increase in the peak is observed upon modifying the surface with aptamers due to the combination of amine groups from the PDL layer and the nitrogenous bases in the aptamer sequence. Similarly, an increase for the P 2p peak intensity is observed when comparing the bare vs. coated CNTs (Fig. 2c). The increase in intensity of the P 2p peak for the PDL-coated CNTs is likely due to the residues of PBS in which the PDL was prepared and dried. The further increase in the P 2p intensity upon addition of the aptamer is due to the DNA phosphate backbone. The increase in the N 1s and P 2P peak heights upon aptamer incubation, confirm the surface functionalization of CNTs with histamine aptamers.
To corroborate the XPS results and to quantify the assembled mass for each polymer layer in situ, OWLS measurements were conducted. For OWLS, glass waveguides are used as substrates to in-couple the laser that enables monitoring of the assembled mass on the surface. Thus, we first ensured reproducible coating of waveguides with CNTs using atomic force microscopy (AFM). The surface of bare (Fig. 2d) vs. CNT-coated (Fig. 2e) waveguides were visualized. The roughness of the bare waveguide extracted from the AFM measurement was Rq = 0.29 ± 0.11 nm (N = 5 measurements). After coating with semiconducting CNTs, the roughness increased to Rq = 1.28 ± 0.14 nm (N = 5 measurements), indicating a higher surface area. Furthermore, to inspect the coating quality, the spray-deposited CNT coating was visualized by optical microscopy.37 The deposited droplets are close to one another and relatively smooth, indicative of a high quality surface coating (Fig. 2f).48
Both bare and CNT-coated waveguides were tested in the OWLS system (Fig. 3a) to investigate the surface area enhancement (and subsequent increase in binding sites for PDL and aptamers) due to the CNT network. Upon achieving a stable baseline in 1× PBS, 100 μg mL−1 PDL solution was injected. A rapid increase in the mass density was observed, to ∼360 ng cm−2 for the CNT-coated waveguide. The schematic of the sequential surface modification on the waveguide and the corresponding binding curve are presented in Fig. 3b. The increase in the mass density for the CNT-coated waveguide is about 5-fold higher compared to the bare waveguide (∼75 ng cm−2, Fig. S8†). This increase in mass density is comparable to the 4-fold increase in surface roughness for the CNT network vs. bare waveguide observed by AFM. Upon ensuring stable PDL layer formation and removing excess PDL by rigorous PBS rinsing, 5 μM histamine aptamers were injected. Aptamer exposure led to an increase in the mass density of ∼300 ng cm−2 for the CNT-PDL-coated waveguide. Alternatively, on the bare waveguide coated with PDL, only ∼70 ng cm−2 of aptamers were assembled, which corresponds to the amount of PDL that was assembled on the bare substrates (Fig. S8†). Multiple PBS rinsing cycles showed negligible changes in mass density, demonstrating the robust integration of aptamers into the PDL layer.
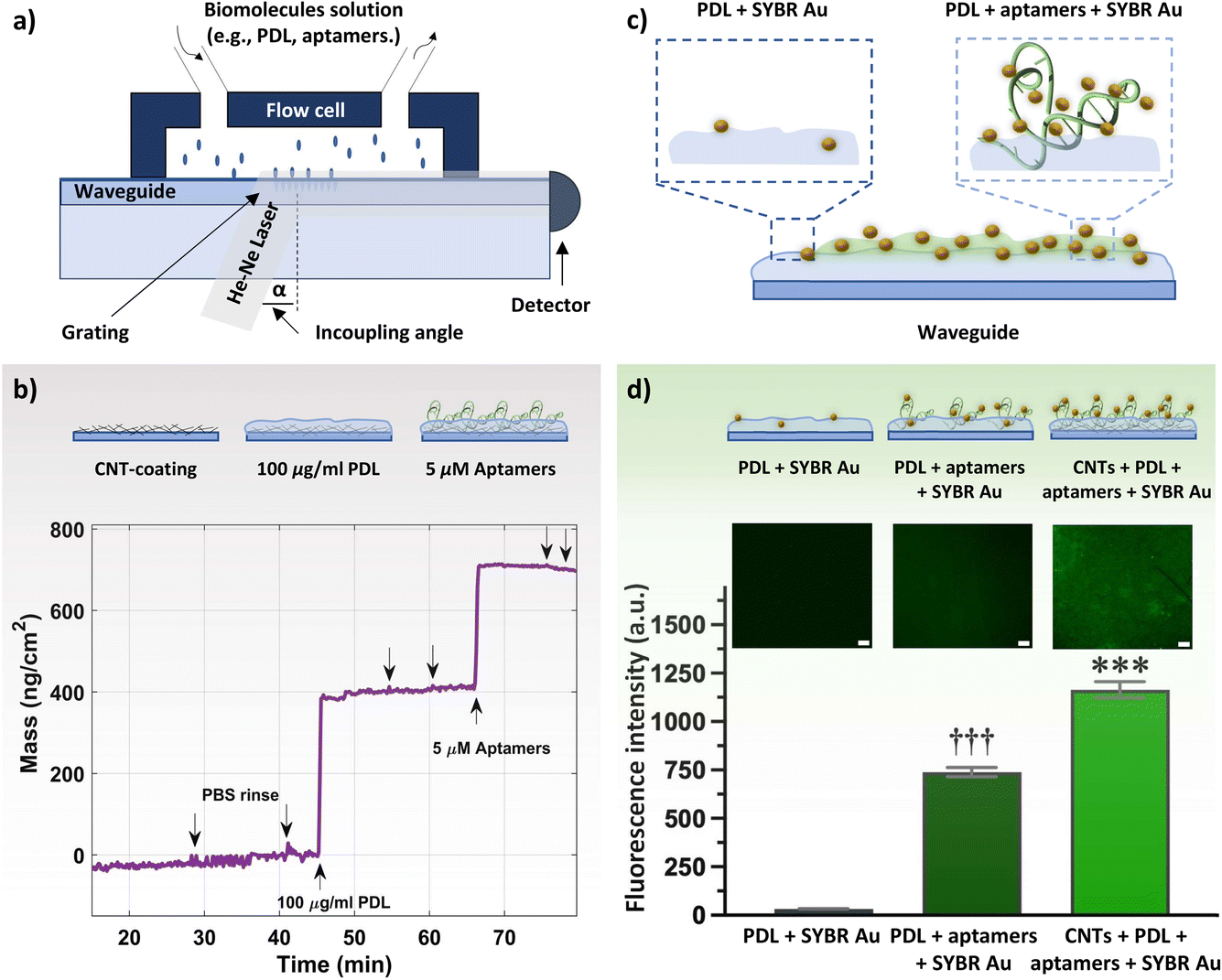 | ||
| Fig. 3 (a) Schematic drawing of the optical waveguide lightmode spectroscopy (OWLS) system, with inserted waveguide. Solutions of biomolecules such as poly-D-lysine (PDL) or aptamers are injected through the inlet into a flow cell to immerse the waveguide. (b) Schematic of the step-by-step modification of waveguides inserted in the OWLS system. The binding curve shows changes in the mass density during the adsorption process of PDL and aptamers on a carbon nanotube (CNT)-coated waveguide. Arrows indicate the time of solution injection. Downward arrows indicate phosphate buffered saline (PBS) injection either to confirm sensor stability or to ensure robust layer formation. (c) Schematic drawing of the of SYBR gold (Au) interaction at the surface of waveguides with histamine aptamers immobilized on a PDL waveguide vs. bare PDL waveguide. (d) Representative fluorescence images obtained by confocal laser scanning microscopy with a scale bar of 100 μm. Relative fluorescence intensities demonstrate maximal SYBR Au fluorescence in the presence of aptamer-PDL-coated CNT networks. Error bars show standard errors of the mean for N = 3 measurements. Group means were significantly different [F(2,12) = 408.6; P < 0.0001; ***P < 0.001 vs. PDL + SYBR Au or PDL + aptamers + SYBR Au; †††P < 0.001 vs. PDL + SYBR Au or CNTs + PDL + aptamers + SYBR Au]. | ||
Fluorescence measurements were conducted to visualize the presence of DNA aptamers on the waveguide surface. To stain the aptamers, SYBR Au, a fluorescent dye for the detection of nucleic acids, was used. This dye exhibits >1000-fold fluorescence enhancement upon binding to single-stranded DNA (Fig. 3c).49 Three samples were incubated with the SYBR Au dye: PDL-coated waveguides (control sample without aptamers), PDL-aptamer-coated waveguides, and CNT-PDL-aptamer-modified waveguides. Schematics of the SYBR Au interactions with the various samples and relative fluorescence intensities are shown in Fig. 3d. The control sample with no aptamer incubation showed negligible fluorescence, indicating minimal interaction of SYBR Au with PDL. The aptamers adsorbed to the PDL-coated bare substrate showed around 740-fold fluorescence enhancement, which was increased to ∼1200-fold fluorescence enhancement for substrates with CNT networks as the initial layer. The fluorescence enhancement of the CNT-PDL-aptamer-coated waveguide is ∼40% higher than only PDL-coated chips, which supports the increase in surface area upon deposition of CNTs observed in the OWLS. Photobleaching that induces permanent fluorescence loss of dyes was performed in a localized area to ensure that the observed fluorescence is occurring due to the presence of aptamers and not due to sample autofluorescence (Fig. S9a and b†).
Taken together, surface characterization via XPS, OWLS, and fluorescence microscopy confirmed histamine aptamer integration into CNT-PDL-coated surfaces. Further, we validated the potential to assemble ∼5-fold higher densities of aptamers by increasing the available surface area using CNT networks. This functionalization strategy was then used to integrate histamine aptamers (Fig. 4a) into EG-CNTFET devices for biosensing.
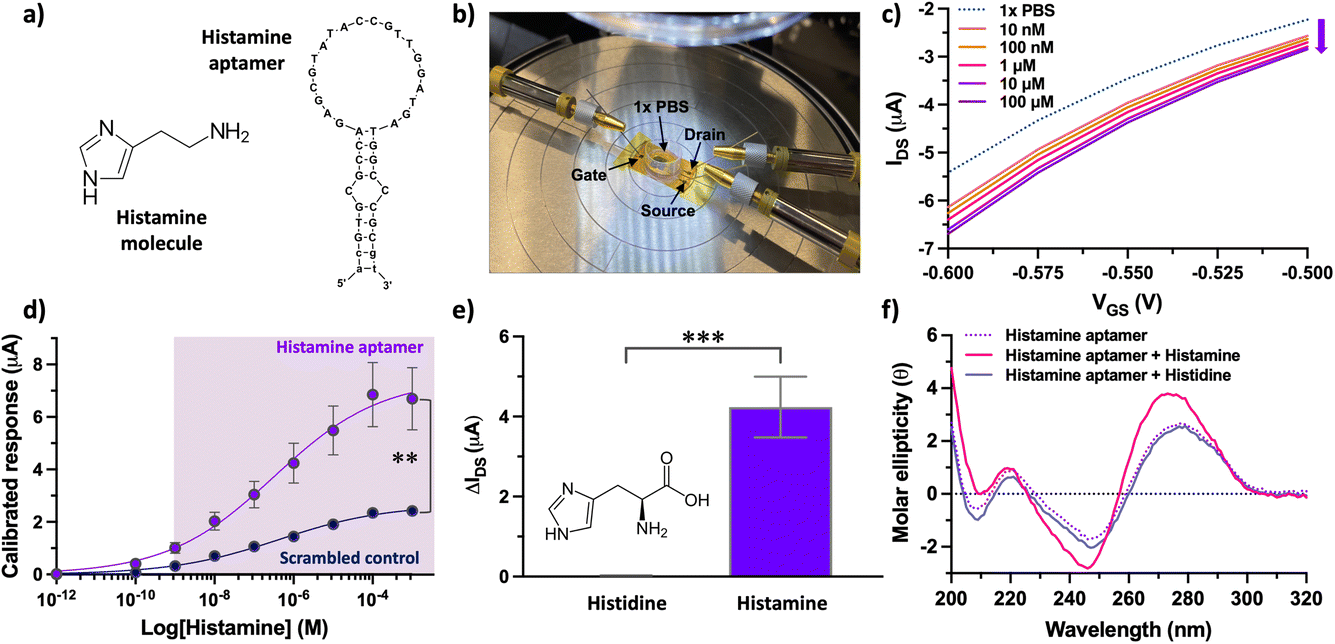 | ||
| Fig. 4 (a) Histamine-specific DNA aptamer (right) that recognizes the histamine molecule (left). (b) A photograph of the aptamer electrolyte-gated carbon nanotube field-effect transistor (EG-CNTFET) biosensor operated with a liquid planar gate. (c) Exposure of histamine aptamer–EG-CNTFETs to histamine in undiluted 1× PBS led to concentration-dependent decreases in the drain-source current (IDS). (d) Calibrated responses of the aptamer–EG-CNTFET biosensors upon exposure to histamine in the concentration range 100 pM to 1 mM. Scrambled control sequences showed statistically lower responses (P < 0.01) upon exposure to histamine at concentrations higher than 10 nM. Error bars represent the standard error of the mean with N = 5 EG-CNTFETs per concentration for the histamine aptamer and N = 3 for the scrambled control. Error bars are too small to be seen for the scrambled control. (e) Histamine aptamer-modified EG-CNTFETs responded minimally to histidine (bar too small to be seen) vs. histamine (N = 5 independent chips per molecule). A two-tailed unpaired t-test indicated group means were significantly different [t(8) = 5.556; P < 0.0001]. (f) Circular dichroism spectroscopy of the histamine aptamer (dotted line) upon target capture showed spectral shifts (solid pink line) while exposure to histidine did not result in spectral changes (solid lavender blue line). | ||
For CNT-based EG-FETs, the stability of the device over time still remains a challenge.50 Therefore, device stability must first be examined to have reliable biosensing data. Joshi et al. have improved EG-CNTFET performance by encapsulating the CNT channel with a polymeric membrane.32 Similarly, Molazemhosseini et al. tackled the stability of EG-CNTFET devices using polymer-wrapped CNTs for the channel material, showing devices with a highly stable response after an hour of stabilization.31 Thus, we hypothesized that insulation of the CNTs by the PDL coating would increase the stability of the devices; the PDL-coated CNT channel is not in direct contact with the electrolyte. On the one hand, covering the CNT channel with a polymeric material leads to a reduction of the conduction properties of the EG-CNTFET devices. On the other hand, the encapsulation provided by the thin PDL layer significantly reduces the modulation of the CNT conduction caused by the ionic species in the electrolyte, leading to overall improved stability of the devices.32 Our objective was to probe this trade-off of channel conductivity vs. stability and to interrogate whether target-specific aptamer structure switching can be transduced at channel surfaces.
Upon reaching a stable baseline, the devices (Fig. 4b) were tested for histamine detection, using a characterization protocol we reported for EG-CNTFET-based sensors in previous work.51,52 Exposure of the aptamer-modified EG-CNTFET sensors to histamine in undiluted 1× PBS, led to concentration-dependent changes in the IDS (Fig. 4c). The changes in the IDS suggested that the structural rearrangements of the aptamers upon histamine recognition were occurring in close proximity to the CNT channel.1,8 The negatively charged backbone of the histamine aptamers may move closer to the sensor surface within or near the λD, increasing the negative charge density in the p-type CNT channel. This rearrangement of charge density induces an increase in the concentration of the charge carriers (holes), and hence an increase (in absolute value) in the drain current of the p-type transistor. This direction of change indicated that we are not merely detecting the charge of the small-molecule analyte, as histamine is positively charged at physiological pH and would lead to charge carrier depletion in the p-type CNT channel.
Nevertheless, to ensure that changes in the IDS arose from the structure-switching aptamers, a control experiment was conducted in parallel. A scrambled sequence was designed, where the same number and type of nucleotides as the specific aptamer were conserved, but the order changed to alter molecular recognition to histamine (Table S1†). While the histamine aptamer-modified CNTFETs detected histamine at concentrations between 10 nM–100 μM, the scrambled control CNTFETs showed a statistically smaller response (P < 0.01) within this sensing regime (Fig. 4d). The non-negligible response of the control sensor that increases with higher histamine amounts is likely due to nonspecific electrostatic interaction of the positively charged histamine molecules with the negatively charged DNA. These results indicate the importance of conducting differential measurements to quantify the extent of specific binding using this surface chemistry.33
Upon confirming sequence specificity of the aptamer-modified CNTFETs, the selectivity vs. histidine, the closest structural analog and precursor to histamine, was tested. Negligible responses to histidine (1 μM) was observed at the surface of histamine-specific sensors (Fig. 4e). To demonstrate that this high selectivity is a result of the target-specific structure switching behavior of the histamine aptamers, we conducted circular dichroism (CD) spectroscopy (Fig. 4f). The intensities and positions of the CD spectral bands indicate the extent of base stacking and dipole coupling in the DNA sequence.53 Thus, this solution-based method enables monitoring of secondary structural rearrangements of aptamers upon target recognition.13 A blue shift was observed upon histamine incubation, which was not observed for histidine. Further, negligible change was observed upon histamine incubation with the control scrambled sequence (Fig. S10†). Thus, the structure-switching behavior of the histamine aptamer is target specific and likely the driving mechanism that enables small-molecule detection at the surface of EG-CNTFETs despite the screening effects of high ionic strength environments.
4 Conclusions
In this work, we harnessed the adhesive properties of PDL, which can coat the surface of CNTs, to graft DNA aptamers in close proximity to the CNT semiconducting channel. The surface chemistry was monitored and corroborated by several analytical techniques. The OWLS and fluorescence measurements not only confirmed the surface chemistry but also showed an enhancement of the surface area due to the deposition of the CNT networks, which resulted in the potential to assemble ∼5-fold higher aptamer densities. The aptamer-modified EG-CNTFETs enabled histamine detection in undiluted 1× PBS in the concentration range of 10 nM to 100 μM. Upon histamine recognition, the specific aptamers underwent substantial conformational rearrangements of negatively charged phosphate backbones (confirmed by CD spectroscopy), which enabled signal transduction in λD-limited, high salt conditions.While this work serves as a mechanistic demonstration of how structure-switching aptamers can transduce target-specific signals when integrated on CNT surfaces through polymer chemistries, there are several remaining challenges. A critical issue using the CNT solution preparation method reported herein, is the resulting mixture of semiconducting and metallic CNTs, which leads to high device-to-device variability. A potential solution to overcome this problem is the use of conjugated polymers for CNT wrapping.54,55 Conjugated polymers offer several benefits such as selective separation of semiconducting vs. metallic CNTs, sorting of nanotubes with specific chiralities and diameters, and even the separation of CNT enantiomers. The use of purified CNTs may improve the sensitivity of the aptamer-integrated EG-CNTFETs in the future.56
A further challenge we anticipate is the use of this surface functionalization strategy in complex biological environments, where charged polymer surfaces are prone to nonspecific adhesion of interferents. This issue may be tackled via polymer chemistries that hinder nonspecific interactions. For example, poly-D-lysine-poly(ethylene glycol) (PDL-PEG) films carry outer PEG layers that repel nonspecific proteins through steric repulsion.30,57 The advantage of this chemistry, is the semi-permeable PDL-PEG coating that permits the passage of DNA near the semiconducting CNT surface to transduce structure-switching dynamics as target binding signals while avoiding biofouling through aversion of nonspecific adsorption. Regardless, we emphasize the importance of conducting differential measurements with control sensors (i.e., with scrambled DNA) when moving to complex environments to ensure quantification of specific interactions.
Our focus herein, was to demonstrate the potential of using polymer surface chemistry for coating spray-deposited CNTs on flexible substrates with aptamers. In addition, we harnessed the structure-switching dynamics of aptamers integrated into the polymer coating to transduce target-specific signals in physiological ionic strength conditions. Novel routes of aptamer integration via tunable polymer chemistries combined with an improved understanding of structure-switching aptamer dynamics at transistor surfaces, will open new doors for flexible EG-CNTFET biosensors.
Author contributions
Shkodra, Bajramshahe: conceptualization, data curation, formal analysis, investigation, software, validation, visualization, writing – original draft. Petrelli, Mattia: data curation, formal analysis, investigation, methodology, software, validation, writing – review & editing. Yang, Kyung-Ae: methodology, investigation, writing – review & editing. Tagliaferri, Anna: investigation, writing – review & editing. Lugli, Paolo: funding acquisition, writing – review & editing. Petti, Luisa: project administration, supervision, writing – review & editing. Nakatsuka, Nako conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, validation, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swiss Federal Laboratories for Materials Science and Technology (Empa) and the Surface Science and Coating Technologies Team for access to the XPS facility and the Laboratory of Organic Chemistry and Chemical Biology (Wennemers Lab) at ETH Zurich for use of the CD spectrometer. The authors also thank members of the Laboratory of Biosensors and Bioelectronics at ETH Zurich – Prof. Janos Vörös – for the helpful scientific discussions and Dr Sophie Girardin and Jens Duru for technical assistance with the fluorescence microscopy measurements. The authors also thank members of the Sensing Technologies Lab, at unibz – Dr Enrico Avancini and Andrea La Monica – for technical assistance. The authors acknowledge ETH Zurich for funding.Notes and references
- N. Nakatsuka, K.-A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim and Y. Yang, et al. , Science, 2018, 362, 319–324 CrossRef CAS PubMed.
- D. Kim, B. Jin, S.-A. Kim, W. Choi, S. Shin, J. Park, W.-B. Shim, K. Kim and J.-S. Lee, Biosensors, 2022, 12, 24 CrossRef CAS PubMed.
- H. Chen, M. Xiao, J. He, Y. Zhang, Y. Liang, H. Liu and Z. Zhang, ACS Sens., 2022, 7(7), 2075–2083 CrossRef CAS PubMed.
- J. Wang, D. Chen, W. Huang, N. Yang, Q. Yuan and Y. Yang, Exploration, 2023, 3, 20210027 CrossRef PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- B. Shkodra, M. Petrelli, M. A. Costa Angeli, D. Garoli, N. Nakatsuka, P. Lugli and L. Petti, Appl. Phys. Rev., 2021, 8, 041325 CAS.
- K. Maehashi, K. Matsumoto, Y. Takamura and E. Tamiya, Electroanalysis, 2009, 21, 1285–1290 CrossRef CAS.
- H. Y. Zheng, O. A. Alsager, B. Zhu, J. Travas-Sejdic, J. M. Hodgkiss and N. O. Plank, Nanoscale, 2016, 8, 13659–13668 RSC.
- G. Elli, S. Hamed, M. Petrelli, P. Ibba, M. Ciocca, P. Lugli and L. Petti, Sensors, 2022, 22, 4178 CrossRef CAS PubMed.
- F. Khosravi, S. M. Loeian and B. Panchapakesan, Biosensors, 2017, 7, 17 CrossRef PubMed.
- W. Shao, G. V. Shurin, X. He, Z. Zeng, M. R. Shurin and A. Star, ACS Appl. Mater. Interfaces, 2022, 14, 1684–1691 CrossRef CAS.
- K. M. Cheung, K.-A. Yang, N. Nakatsuka, C. Zhao, M. Ye, M. E. Jung, H. Yang, P. S. Weiss, M. N. Stojanovic and A. M. Andrews, ACS Sens., 2019, 4, 3308–3317 CrossRef CAS.
- N. Nakatsuka, J. M. Abendroth, K.-A. Yang and A. M. Andrews, ACS Appl. Mater. Interfaces, 2021, 13, 9425–9435 CrossRef CAS.
- C. Zhao, K. M. Cheung, I.-W. Huang, H. Yang, N. Nakatsuka, W. Liu, Y. Cao, T. Man, P. S. Weiss and H. G. Monbouquette, et al. , Sci. Adv., 2021, 7, eabj7422 CrossRef CAS PubMed.
- A. Ruscito and M. C. DeRosa, Front. Chem., 2016, 4, 14 Search PubMed.
- K.-A. Yang, H. Chun, Y. Zhang, S. Pecic, N. Nakatsuka, A. M. Andrews, T. S. Worgall and M. N. Stojanovic, ACS Chem. Biol., 2017, 12, 3103–3112 CrossRef CAS PubMed.
- M. McKeague and M. C. DeRosa, J. Nucleic Acids, 2012, 2012, 748913 Search PubMed.
- A. E. Rangel, A. A. Hariri, M. Eisenstein and H. T. Soh, Adv. Mater., 2020, 32, 2003704 CrossRef CAS PubMed.
- A. Dudina, U. Frey and A. Hierlemann, Sensors, 2019, 19, 3080 CrossRef CAS PubMed.
- S.-J. Wu, N. Schuergers, K.-H. Lin, A. J. Gillen, C. Corminboeuf and A. A. Boghossian, ACS Appl. Mater. Interfaces, 2018, 10, 37386–37395 CrossRef CAS PubMed.
- P. Liu, Y. Jiao, X. Chai, Y. Ma, S. Liu, X. Fang, F. Fan, L. Xue, J. Han and Q. Liu, J. Lumin., 2022, 250, 119084 CrossRef CAS.
- M. Ovais, M. You, J. Ahmad, R. Djellabi, A. Ali, M. H. Akhtar, M. Abbas and C. Chen, TrAC, Trends Anal. Chem., 2022, 153, 116659 CrossRef CAS PubMed.
- N. T. Tung, P. T. Tue, T. Thi Ngoc Lien, Y. Ohno, K. Maehashi, K. Matsumoto, K. Nishigaki, M. Biyani and Y. Takamura, Sci. Rep., 2017, 7, 1–9 CrossRef CAS PubMed.
- R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef CAS PubMed.
- H.-F. Li, C. Wang, L.-F. Liu, N. Xie, M. Pan, P. Wu, X.-D. Wang, Z. Zeng, S. Deng and G.-P. Dai, J. Nanopart. Res., 2020, 22, 1–10 CrossRef.
- A. Palaniappan, W. Goh, D. Fam, G. Rajaseger, C. Chan, B. Hanson, S. Moochhala, S. Mhaisalkar and B. Liedberg, Biosens. Bioelectron., 2013, 43, 143–147 CrossRef CAS PubMed.
- Y. B. Pottathara, Y. Grohens, V. Kokol, N. Kalarikkal and S. Thomas, in Nanomater. Synth., Elsevier, 2019, pp. 1–25 Search PubMed.
- F. C. Loghin, A. Falco, F. Moreno-Cruz, P. Lugli, D. P. Morales, J. F. Salmerón and A. Rivadeneyra, Mater. Lett., 2021, 297, 129939 CrossRef CAS.
- J. Wang, Y. Zhao, F.-X. Ma, K. Wang, F.-B. Wang and X.-H. Xia, J. Mater. Chem. B, 2013, 1, 1406–1413 RSC.
- R. Schlapak, D. Armitage, N. Saucedo-Zeni, W. Chrzanowski, M. Hohage, D. Caruana and S. Howorka, Soft Matter, 2009, 5, 613–621 RSC.
- A. Molazemhosseini, F. A. Viola, F. J. Berger, N. F. Zorn, J. Zaumseil and M. Caironi, ACS Appl. Electron. Mater., 2021, 3, 3106–3113 CrossRef CAS PubMed.
- S. Joshi, V. D. Bhatt, E. Jaworska, M. Becherer, K. Maksymiuk, A. Michalska and P. Lugli, Phys. Status Solidi A, 2018, 215, 1700993 CrossRef.
- A. Frutiger, A. Tanno, S. Hwu, R. F. Tiefenauer, J. Voros and N. Nakatsuka, Chem. Rev., 2021, 121, 8095–8160 CrossRef CAS PubMed.
- K. Yang, N. M. Mitchell, S. Banerjee, Z. Cheng, S. Taylor, A. M. Kostic, I. Wong, S. Sajjath, Y. Zhang and J. Stevens, et al. , Science, 2023, 380, 942–948 CrossRef CAS.
- L. S. J. Ho, R. Fogel and J. L. Limson, Talanta, 2020, 208, 120474 CrossRef.
- B. Shkodra, B. Demelash Abera, G. Cantarella, A. Douaki, E. Avancini, L. Petti and P. Lugli, Biosensors, 2020, 10, 35 CrossRef CAS PubMed.
- B. Shkodra, M. Petrelli, M. A. Costa Angeli, A. K. M. S. Inam, P. Lugli and L. Petti, IEEE Sens. J., 2023, 23(20), 23958–23965 CAS.
- B. Shkodra, M. Petrelli, M. A. Costa Angeli, A. K. M. S. Inam, E. Avancini, N. Münzenrieder, P. Lugli and L. Petti, 2021 IEEE International Conference on Flexible and Printable Sensors and Systems, FLEPS, 2021, pp. 1–4 Search PubMed.
- L. Zhang, M. Rao, J. Kochupurackal, N. Mathews, Y. M. Lam and S. G. Mhaisalkar, MRS Online Proc. Libr., 2012, 1436, 57–62 Search PubMed.
- N. Nakatsuka, A. Faillétaz, D. Eggemann, C. Forró, J. Vörös and D. Momotenko, Anal. Chem., 2021, 93, 4033–4041 CrossRef CAS PubMed.
- S. Lee and J. Vörös, Langmuir, 2005, 21, 11957–11962 CrossRef CAS PubMed.
- S. Joshi, V. D. Bhatt, A. Märtl, M. Becherer and P. Lugli, Biosensors, 2018, 8, 9 CrossRef PubMed.
- X. Deng, L. Lü, H. Li and F. Luo, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS PubMed.
- S. Luo, X. Xu, G. Zhou, C. Liu, Y. Tang and Y. Liu, J. Hazard. Mater., 2014, 274, 145–155 CrossRef CAS PubMed.
- C. S. Kam, T. L. Leung, F. Liu, A. B. Djurišić, M. H. Xie, W.-K. Chan, Y. Zhou and K. Shih, RSC Adv., 2018, 8, 18355–18362 RSC.
- M. Zhao, Y. Cao, X. Liu, J. Deng, D. Li and H. Gu, Nanoscale Res. Lett., 2014, 9, 1–9 CrossRef PubMed.
- X. Zhou, Q. Qu, L. Wang, L. Li, S. Li and K. Xia, J. Nanopart. Res., 2020, 22, 1–13 CrossRef.
- A. Falco, L. Cinà, G. Scarpa, P. Lugli and A. Abdellah, ACS Appl. Mater. Interfaces, 2014, 6, 10593–10601 CrossRef CAS PubMed.
- P. J. Kolbeck, W. Vanderlinden, T. Nicolaus, C. Gebhardt, T. Cordes and J. Lipfert, bioRxiv, 2020, preprint, DOI:10.1101/2020.05.23.112631.
- F. Scuratti, G. E. Bonacchini, C. Bossio, J. M. Salazar-Rios, W. Talsma, M. A. Loi, M. R. Antognazza and M. Caironi, ACS Appl. Mater. Interfaces, 2019, 11, 37966–37972 CrossRef CAS PubMed.
- M. Petrelli, B. Shkodra, M. A. Costa Angeli, A. Scarton, S. Pogliaghi, R. Biasi, P. Lugli and L. Petti, 2022 IEEE Sensors, 2022, pp. 1–4 Search PubMed.
- M. Petrelli, B. Shkodra, A. Falco, M. A. Costa Angeli, S. Vasquez, A. Scarton, S. Pogliaghi, R. Biasi, P. Lugli and L. Petti, Org. Electron., 2023, 122, 106889 CrossRef CAS.
- S. Masiero, R. Trotta, S. Pieraccini, S. De Tito, R. Perone, A. Randazzo and G. P. Spada, Org. Biomol. Chem., 2010, 8, 2683–2692 RSC.
- S. K. Samanta, M. Fritsch, U. Scherf, W. Gomulya, S. Z. Bisri and M. A. Loi, Acc. Chem. Res., 2014, 47, 2446–2456 CrossRef CAS PubMed.
- D. Heimfarth, M. Balcı Leinen, P. Klein, S. Allard, U. Scherf and J. Zaumseil, ACS Appl. Mater. Interfaces, 2022, 14, 8209–8217 CrossRef CAS PubMed.
- S. Z. Bisri, J. Gao, V. Derenskyi, W. Gomulya, I. Iezhokin, P. Gordiichuk, A. Herrmann and M. A. Loi, Adv. Mater., 2012, 24, 6147–6152 CrossRef CAS PubMed.
- B. Akpinar, P. J. Haynes, N. A. Bell, K. Brunner, A. L. Pyne and B. W. Hoogenboom, Nanoscale, 2019, 11, 20072–20080 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fd00123g |
| This journal is © The Royal Society of Chemistry 2024 |