
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeville rebooted – practical N2O5 synthesis†
Lee E.
Edwards
 a,
Benson M.
Kariuki
a,
Matthew
Didsbury
b,
Christopher D.
Jones
c and
Thomas
Wirth
*a
a,
Benson M.
Kariuki
a,
Matthew
Didsbury
b,
Christopher D.
Jones
c and
Thomas
Wirth
*a
aCardiff University, School of Chemistry, Cardiff, CF10 3AT, UK. E-mail: wirth@cf.ac.uk
bBAE Systems Land UK, Glascoed, Usk, Monmouthshire NP15 1XL, UK
cFalcon Project Ltd, The Magazine Area, Westcott Venture Park, Aylesbury, Bucks HP18 0XB, UK
First published on 9th May 2024
Abstract
Dinitrogen pentoxide (N2O5), the anhydride of nitric acid, was synthesised by Henri Étienne Sainte-Claire Deville in Paris in 1849 using silver nitrate and chlorine gas. Herein, we revisit, optimise, and modify Deville's method using photocatalysis to enable a safe, clean, practical, and reproducible alternative for N2O5 synthesis in quantitative yields. Moreover, it is predicted that the modifications can accommodate an industrial scale-up, but the silver chloride generated must be recycled.
Nitro compounds are used extensively in the pharmaceutical, agricultural and energetic materials industries.1 Dinitrogen pentoxide is of great interest to the energetic materials community as it has its own specific qualities for the nitration of organic compounds.2–5 It is a much milder nitrating agent and has proven to be far more selective than mixed or concentrated nitric acid.2 N2O5 has the potential of reducing considerable amounts of waste and side products traditionally associated with energetic materials manufacture while improving safety at the same time. However, N2O5 is not commercially available due to its thermal instability. N2O5 readily sublimes with a vapour pressure of 1 atm at 33 °C and has a half-life of 10 days at 0 °C and 10 hours at 20 °C as it decomposes through releasing NO2 and oxygen: N2O5 → 2 NO2 + O2.6–8 Therefore, it is necessary to synthesise N2O5 on demand. In this context we revisited the work of Deville, the original discoverer of N2O5.9,10 It is relevant to the current work to appreciate Deville's original methodology to understand why the method has been left to history. We report on our modifications and enhancements and provide images of N2O5 crystals in Fig. 1 and 2 synthesised by our improved method. The improved method is reproducible and used routinely and safely within our laboratories.
 | ||
| Fig. 1 Single N2O5 crystal cluster from the current work. This cluster was 1.2 cm across at its longest point. | ||
 | ||
| Fig. 2 A collection of N2O5 crystal clusters. Note that the clusters are usually up to 1–1.5 cm across. | ||
In the 1840s Deville was working on metal chloride analysis and characterisation. During this time, he discovered dinitrogen pentoxide and reported its synthesis in 1849.9 Deville used a U-shaped tube filled with silver nitrate as the reaction vessel. Dry chlorine gas was passed through the silver nitrate at 95 °C and the reaction to N2O5 and solid silver chloride occurred. Some N2O5 decomposition was evident as NO2 and its liquid dimer N2O4 was condensing immediately after the exit from the silver nitrate at the cooling section in the second U-shaped tube which was at a temperature of around 0 °C (Fig. 3).
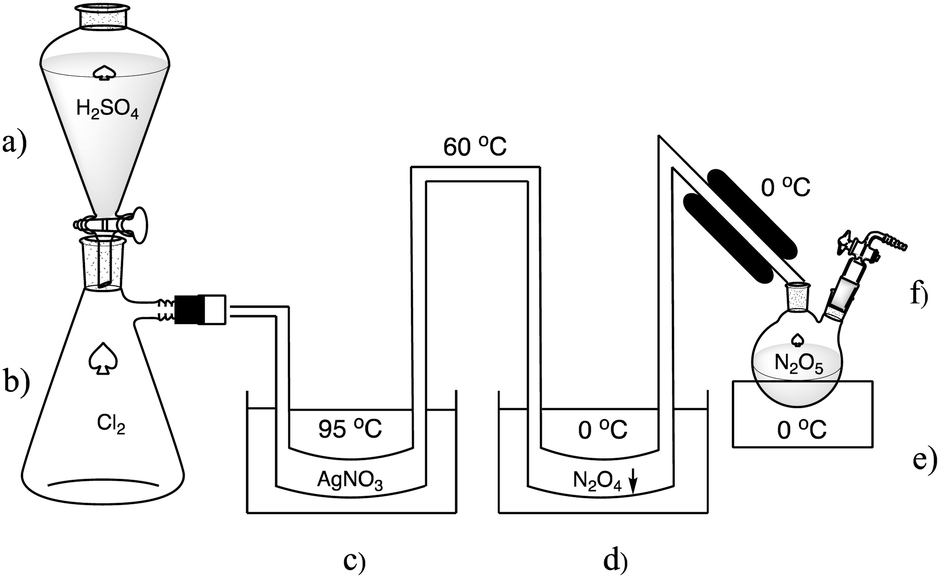 | ||
| Fig. 3 Deville's experimental apparatus for the synthesis of N2O5: (a) H2SO4 for Cl2 desiccant and displacement. (b) Chlorine gas. (c) Water bath at 95 °C to heat AgNO3 at the bottom of the U-shaped tube. (d) Cold trap at 0 °C for NO2 condensation. (e) Cold trap at 0 °C for crystallising N2O5. (f) Vent for O2 and unreacted Cl2. A more detailed diagram based on Deville's description is shown in the ESI† (Fig. S1). | ||
Deville stated that the silver nitrate was primed by drying at 180 °C while being purged with carbon dioxide. The system was attached to a glass bottle containing the chlorine gas. The bottle had a system to drop feed concentrated sulfuric acid that both dried and displaced the chlorine through a valve. The chlorine had to pass through calcium hypochlorite for further drying and chlorine enrichment and finally through pumice which acted as a frit. The chlorine was then introduced by the displacement with sulfuric acid at a rate of 2–3 mL min−1. The chlorine gas passed through the silver nitrate, and some reacted to form N2O5 and AgCl. The N2O5 and unreacted chlorine was then passed through the cooled tubing and the N2O5 finally condensed into crystal clusters of approximately 1 cm size which were subsequently collected in glass vials sealed with heat in a CO2 atmosphere for storage. It was noted that thermal decomposition often resulted in gas pressure destruction of theses vials. However, Deville’ system of N2O5 synthesis was able run continuously for days at a time with just the sulfuric acid, water bath and fuel for the heater having to be maintained. It is our understanding that Deville’ method is the only one to date describing crystals of translucent N2O5 similar to the size and clarity shown in Fig. 1 and 2. All other methods of N2O5 synthesis since 1849 use cold traps that rapidly precipitate the N2O5 as much finer crystals or synthesised in solvents such as HNO3 or trifluoroacetic anhydride. While Deville's method was successful in generating N2O5, there was also the issue of chlorine, and NOx being constantly vented, and the conversion of Cl2 and AgNO3 to AgCl and N2O5 was not efficient.
After Deville’ discovery, the most widely used method of synthesising N2O5 is the dehydration of nitric acid with phosphorous pentoxide (P4O10) (Fig. 4, eq. 1).11 This method is further illustrated in the ESI† (Fig. S2).7 HNO3 is added dropwise to P2O5 that has a supply of ozonised oxygen to oxidise any evolved NO2/N2O4 to N2O5. The N2O5 sublimes and is condensed collected as a fine white solid in a cold trap at −78 °C.7 Yields for this method are typically around 50%. Another method that is challenging to reproduce is the dehydration of nitric acid with trifluoroacetic anhydride.12 The most popular method of N2O5 synthesis is the direct ozonolysis of N2O4 (Fig. 4, eq. 2). Again, the N2O5 is condensed after the reaction and collected as a fine white solid in a cold trap at −78 °C. Here the yield is reported to be quantitative as indicated by the red stream of NO2/N2O4 becoming immediately colourless when mixing with ozone.13,14 However, this method is completely reliant upon a supply of NO2/N2O4, and this can pose significant challenges. However, ozonolysis of nitric oxide has also been reported with a yield of greater than 99%.15 Ozone has been used frequently in N2O5 synthesis, and this is likely due to the ease at which ozone can oxidise NO2/N2O4 to N2O5. Other examples include bubbling ozone through picryl pyridinium nitrate or catalytic amounts of picric acid in fuming nitric acid solutions at temperatures up to 65 °C. The N2O5 sublimes and is subsequently collected in a cold trap at −78 °C with yields reported of 87% and 50%, respectively.16 N2O5 has also been synthesised from FNO2 and lithium nitrate in quantitative yield, albeit only on mmol scale and temperatures of −190 °C (Fig. 4, eq. 3).17 Dinitrogen pentoxide is electrochemically synthesised on a large scale through the oxidation of N2O4 to N2O5 in nitric acid which was first reported in 1983. Work in this area is significant, with several patents and publications, including electrolysis of N2O4 in a dual cell setup with a membrane (Fig. 4, eq. 4). The investigators quote yields of >30% N2O5 by mass in nitric acid.18,19 The mixture of N2O5 in nitric acid provides a separate and unique option for nitration reactions.20 But also this electrochemical approach requires N2O4 with the above highlighted challenges.
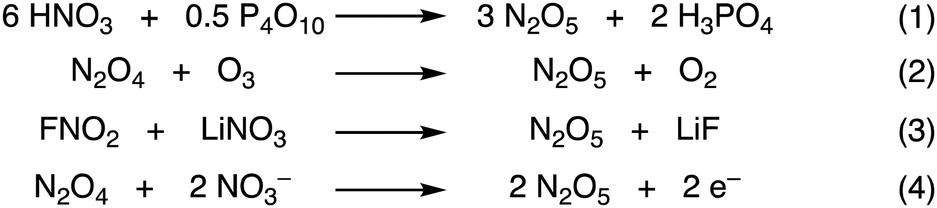 | ||
| Fig. 4 Other methods for the synthesis of N2O5. | ||
Herein we describe our investigations to simplify the Deville method, by removing the NO2/N2O4 contamination issue and by operating at lower temperatures through the utilisation of additional photocatalysis.
Assessing the practicalities Deville’ method, it became clear that there was a requirement to create a closed system with gas/vacuum access through a valve. This simplified the operation while ensuring the protection of N2O5. The AgNO3 had to be heated in this system, while the N2O5 reaction product had to be condensed at a temperature that would not condense NO2(g) to N2O4(l). Furthermore, the cooling should be well above the temperature to freeze N2O4(s) (–11.1 °C). Finally, the temperature of the condenser had to be above 0 °C to prevent condensation if exposed to atmosphere. These requirements were all met with a Chemglass Sublimator (full details of this device are in the ESI†). This apparatus is built of 2 halves. The upper half contains the cold finger with the glass pipework to enable a flow of coolant from a chiller, and a sealed valve to enable evacuation of the sublimator to a full vacuum, then the subsequent addition of the chlorine for the reaction. The lower half of the sublimator contains AgNO3 as a fine powder, and a small magnetic stirrer. The two halves have a Viton seal between them, and the assembly is clamped together and evacuated and sealed. The sublimator is then positioned on a hotplate/stirrer and cooled to 4 °C. Dry chlorine (0.8 bar) is introduced via a septum on the sublimator and the opened valve. This is the point where our modifications to Deville’ method diverge, and they will be described separately. Firstly, the AgNO3 was heated in a sand bath to 60 °C. After 72 hours there was no reaction. The temperatures were subsequently increased until a successful reaction at 95 °C. This was the temperature that Deville reported for heating the AgNO3 before immediately cooling the outward flow of gas to 60 °C in an attempt to control the decomposition of N2O5 which was noted as being considerable. After cooling the AgNO3 to room temperature the formation of crystalline N2O5 was observed. It should be noted that the reaction takes 72 hours at 95 °C to convert all AgNO3 to AgCl. The N2O5 was characterised by single crystal XRD which was identical to the structure described in the literature.9,21 The full conversion of AgNO3 to complete and single phase AgCl was confirmed by powder XRD. Both results are graphically illustrated in Fig. S11, S12 and S13 (ESI†), respectively, with the full details in the ESI.† Further confirmation of full conversion was provided by solubility examination. AgNO3 is highly soluble in water whereas AgCl is practically insoluble. The full mass of AgCl was always recovered from water by filtration and drying. The yield was >95% after titration of the recovered material in dichloromethane as described in the literature.2 Furthermore, confirmation of the N2O5 product was provided by a repeat of the work of Maksimowski et al. with the selective formation of mono- and dinitrotoluene with 1, 2 or 3 equivalents of N2O5 from toluene.2
These results demonstrated that this safe, and much simplified modification to Deville's method could be used to enable dinitrogen pentoxide chemistry to those who previously found access a roadblock due to the previously acknowledged requirement for either NO2/N2O4 or an ozone generator, or both. However, at this point it was acknowledged that 72 hours and thermal decomposition was an issue (Fig. 5).
 | ||
| Fig. 5 The apparatus post reaction and with the hotplate temperature at 95 °C. This temperature is too high for N2O5 to condense on the cold finger. The condensation occurs after the hotplate is switched off. Note the deep brown colour from the N2O5 thermal decomposition to NO2 and O2. Deville experienced this decomposition and had to accommodate it with additional cooling steps and traps. | ||
Experience during these experiments highlighted the photocatalytic properties of AgNO3. The decomposition of silver nitrate when exposed to light to silver, NO2 and oxygen is well known and documented and has been used to create silver nanoparticles,22 and its main use for many years was in photography. However, the undesirable product of this decomposition was NO2 so this, at first, seemed detrimental. The literature photochemistry of dinitrogen pentoxide is sparse and is centred entirely on the cycle of N2O5 in the upper atmosphere.23,24 The thesis of P. Steele Connell did shed some light on the possibility of photocatalysis using Deville's method.24 Steele Connell stated that N2O5 absorbs in the UV range of 210–385 nm, and that in the presence of nothing but N2O5, light will reversibly dissociate N2O5 to NO2 and NO3 with the equilibrium much faster on the recombination. But, if NO is introduced, it will react with the NO3 to irreversibly generate 2 NO2 which will deplete the N2O5 recombination partner. For the current work, with the tightly controlled environment of the Chemglass sublimator with just silver nitrate and chlorine present, there would be no chance of side reactions. This led to a further successful modification of Deville's method.
The experiment was performed with the following modifications (Fig. 6). After the addition of the chlorine to the sublimator, a UVITEC Cambridge UV Lamp was placed in front of the apparatus irradiating the lower part of the sublimator containing the AgNO3. The wavelength of 365 nm was selected as the literature23 reports the generation of N2O3 from N2O5 at shorter wavelengths, and Steele Connell stated that the upper absorption limit is 385 nm. The apparatus was then placed in a light proof box, with the AgNO3 being heated at 65 °C. After 18 hours, the cold finger had deposits of clusters on N2O5 formed mainly around the bottom of the cold finger and on the opposite side to the UV light, so the decomposition hypothesis proved to be correct. Furthermore, there was no visible NO2 decomposition product present. Subsequent analysis was carried out as previously described and demonstrated full conversion of AgNO3 to AgCl and N2O5 in >95% yield. Running the reaction for longer led to N2O5 decomposition. An additional experiment at 50 °C with radiation at 365 nm converted ∼50% AgNO3 to AgCl, but there was a significant amount of N2O5 decomposition in the form of NO2. The method at 65 °C with UV irradiation at 365 nm has now been used routinely numerous times with reproducible results. As most of the chlorine is consumed during the reaction, the only other side products are O2 and AgCl. The latter can be recycled by reduction to silver with H2O2 and NaOH and then nitrating with nitric acid.25 The only waste being sodium chloride which seems to be an acceptable waste product considering other waste such as H2SO4/oleum and fuming HNO3 generated during traditional nitration reactions.
 | ||
| Fig. 6 The apparatus with the UV modification to the Deville method. Note that the colour has no NO2 character and that the N2O5 condensers immediately after it has been produced. The UV lamp (left side) has been partially blocked to only irradiate the lower part of the sublimator with the silver nitrate. | ||
In conclusion, we have revisited and rebooted Deville's distinguished method by providing modifications to simplify, enhance the safety, performance, repeatability of N2O5 synthesis in yields >95%. The optimum conditions for a full conversion, quantitative yield and the minimum of decomposition were heating the AgNO3 to 65 °C, with the cold finger at 4 °C and the AgNO3 irradiated with UV light at 365 nm with a reaction time of 18 hours. The full details of the experimental and apparatus are in the ESI,† including details on the process of N2O5 recovery. We foresee no barriers to scaling up this method to kg scale with an appropriately sized apparatus.
We acknowledge funding from Llywodraeth Cymru (Smart Expertise grant 82502) and Cardiff University. L. E. E. and T. W. thanks BAES (Glascoed) and Falcon Project Ltd (Aylesbury) for funding. We also thank Cardiff School of Chemistry Workshops and Facilities (Steve Morris, Mark Reece, Luigi De Rose, Richard Webb, Robert Jenkins) for their support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Noriega, J. Cardoso-Ortiz, A. López-Luna, M. D. R. Cuevas-Flores and J. A. Flores De La Torre, Pharmaceuticals, 2022, 15, 717 CrossRef CAS PubMed.
- P. Maksimowski, A. Nastała and W. Tomaszewski, Cent. Eur. J. Energ. Mater., 2022, 19, 424–437 CrossRef CAS PubMed.
- M. B. Talawar, R. Sivabalan, B. G. Polke, U. R. Nair, G. M. Gore and S. N. Asthana, J. Hazard. Mater., 2005, 124, 153–164 CrossRef CAS PubMed.
- A. K. Kharchenko, R. V. Fauziev, M. N. Zharkov, I. V. Kuchurov and S. G. Zlotin, RSC Adv., 2021, 11, 25841–25847 RSC.
- D. Fischer, T. M. Klapötke and J. Stierstorfer, Chem. Commun., 2016, 52, 916–918 RSC.
- F. Daniels and A. C. Bright, J. Am. Chem. Soc., 1920, 42, 1131–1141 CrossRef CAS.
- C. C. Addison and N. Logan, Developments in Inorganic Nitrogen Chemistry, Ed. C. B. Colburn, Elsevier, Amsterdam, 1973, vol. 2, pp. 27–69 Search PubMed.
- F. Daniels and E. G. Johnson, J. Am. Chem. Soc., 1921, 43, 53–71 CrossRef CAS.
- M. H. Deville, C. R. Acad. Bulg. Sci. Paris, 1849, 28, 257 Search PubMed.
- J. Gay, Henri Sainte-Claire Deville: sa vie et ses travaux, Gautheir-Villars et Fils, Imprimieurs Libraries, Paris, 1889 Search PubMed.
- N. S. Gruenhut, M. Goldfrank, M. L. Cushing and G. V. Gaesar, in Inorganic syntheses, ed. L. F. Audrieth, McGraw-Hill, New York, 1950, vol. III, pp. 78–81 Search PubMed.
- J. H. Robson, J. Am. Chem. Soc., 1955, 77, 107–108 CrossRef CAS.
- P. A. Guye, US Pat., 1348873, 1920 Search PubMed.
- J. C. Harris and H. B. Trebellas, Inorg. Synth., 1967, 9, 83–88 CrossRef.
- N. I. Kobozev, M. Temkin and S. J. Freiberg, Gen. Chem., 1933, 3, 534 CAS.
- K. Okon, Biul. Wojsk. Akad. Tech., 1958, 7, 3 CAS.
- W. W. Wilson and K. O. Christie, Inorg. Chem., 1987, 26, 1631–1633 CrossRef CAS.
- J. E. Harrar and R. K. Pearson, J. Electrochem. Soc., 1983, 130, 108 CrossRef CAS.
- Q. Wang, M. Su, X. Zhang, L. Wang, J. Wang and Z. Mi, Electrochim. Acta, 2007, 52, 3667–3672 CrossRef CAS.
- H. Feuer and A. T. Nielsen, Nitro Compounds, Purdue University Press, West Lafayette, Indiana, 1990, pp. 268–365 Search PubMed.
- E. Grison, K. Eriks and J. L. de Vries, Acta Cryst., 1950, 3, 290–294 CrossRef CAS.
- F. Naaz, U. Farooq, M. A. M. Khan and T. Ahmad, ACS Omega, 2020, 5, 26063–26076 CrossRef CAS PubMed.
- T. G. Koch and J. R. Sodeau, J. Chem. Soc., Faraday Trans., 1996, 92, 2347–2351 RSC.
- P. Steele Connell, The photochemistry of dinitrogen pentoxide, University of California, Lawrence Berkeley Laboratory, 1979 Search PubMed.
- P. C. Hsu, Z. Chiba, B. J. Schumacher, L. C. Murguia and M. G. Adamson, Recovery of silver from waste silver chloride for the MEO system, US Dept. of Energy, Technical report, 1996 DOI:10.2172/250506.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of N2O5; original Deville method; optimised methods with pictures; methods of N2O5 recovery. See DOI: https://doi.org/10.1039/d4cc01403k |
| This journal is © The Royal Society of Chemistry 2024 |