DOI:
10.1039/D3PY00605K
(Paper)
Polym. Chem., 2023,
14, 4169-4181
Synthesis of ABx and ABxC poly(ester-ether) polymers: polymer sequences and effects of Bx and BxC units on thermal properties†
Received
28th May 2023
, Accepted 15th August 2023
First published on 15th August 2023
Abstract
Ring-opening copolymerization (ROCOP) of cyclic anhydrides and epoxides has been under intensive investigation as a potential alternative approach for synthesizing tunable polyesters due to a wide range of monomer scopes compared to those of conventional cyclic esters such as lactides or lactones. While most catalytic systems focus on the alternate insertions of cyclic anhydrides (A) and epoxides (B) giving alternating (AB)n polyesters, the synthesis of ABx poly(ester-ether) polymers having controllable Bx ether sequences has recently gained significant interest for potential tunable functions. Herein, an ABx poly(ester-ether) with tunable ether linkages (x = 2.3–5.2) was synthesized conveniently from the ROCOP of cyclic anhydrides (succinic anhydride, phthalic anhydride, 1,8-naphthalic anhydride, terpene-based cyclic anhydride, maleic anhydride and diglycolic anhydride) and epoxides (cyclohexene oxide, cyclopentene oxide and propylene oxide) using commercially available tin(II) 2-ethylhexanoate (SnOct2) as the catalyst. Exceptionally high purity of ABx polymers up to 99% was achieved. Kinetic studies revealed the following rate law: rate = kp[SA][CHO]2[SnOct2]. The versatility of SnOct2 was demonstrated and allowed the ABx polymers to integrate with biodegradable polylactides and polyurethanes in one pot. THF or 1,4-dioxane (C) could be copolymerized as the third component in one pot giving a novel ABxC poly(ester-ether) with the highest selectivity of up to 74% for THF and 11% for 1,4-dioxane. The presence of Bx or BxC units was found to significantly accelerate the degradation rates of the polymers compared to conventional alternating (AB)n polyesters.
Introduction
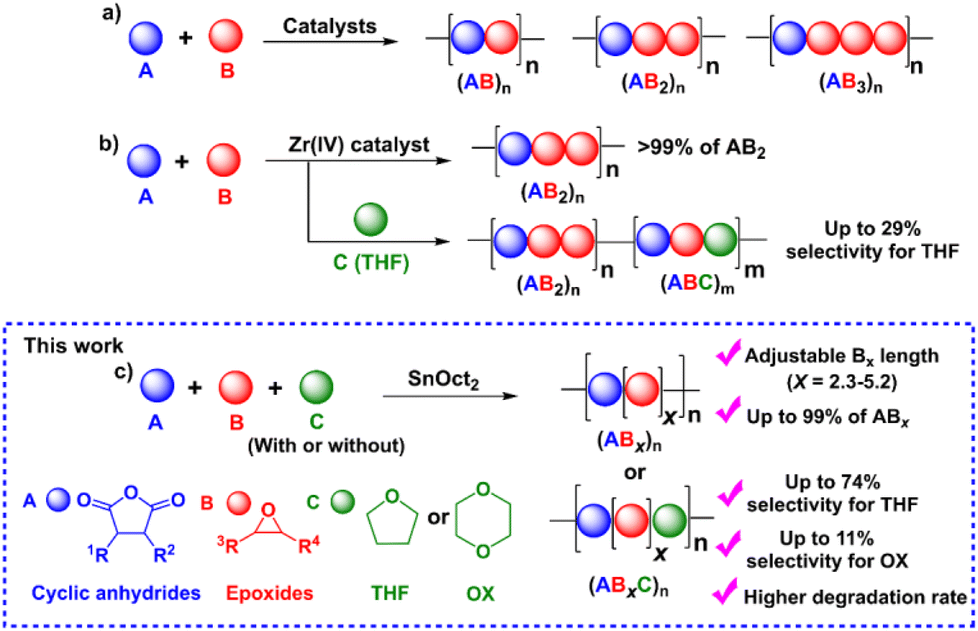
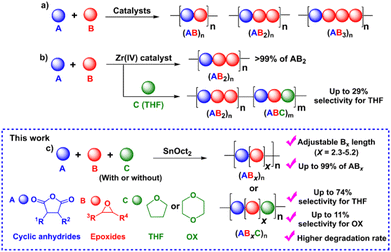
Aliphatic polyesters are among the most extensively studied classes of alternative materials owing to their potential biodegradability, biocompatibility, and low toxicity when used in biomedical and environmental applications.1–4 They are commonly synthesized via the ring-opening polymerization (ROP) of cyclic esters such as lactides and lactones.5,6 However, their physical and mechanical properties are limited due to the narrow range of monomer scopes and difficulty of post-polymerization functionalization from the lack of functional groups on the polymer backbones.5,6 To address these drawbacks, the ring-opening copolymerization (ROCOP) of cyclic anhydrides (A) and epoxides (B) giving (AB)n polyesters via alternate insertions of A and B units (Scheme 1a) has attracted great attention. Due to a wide range of epoxides and cyclic anhydrides, the resulting polymers could lead to many backbone structures, functions, and properties.5–12 The initial development of catalytic systems for ROCOP of cyclic anhydrides and epoxides involved simple metal complexes such as metal alkoxides,13,14 metal halides15 and free amines16 affording polyesters with various degrees of ether linkages. Later development has led to several highly efficient catalysts that provide perfectly alternating polyesters based on ligated metal complexes such as those of Al, Cr, Co, Fe, Zn, and Mg.5,6,17–19 Lewis-base cocatalysts are normally required in many catalytic systems for efficient ROCOP.5,6 The concept of multinuclear metal complexes was also investigated giving excellent cooperative effects of multiple metals in ROCOP.18,20–25 While alternating (AB)n polymers have been extensively explored, we recently reported the synthesis of a novel (ABB)n polymer sequence with up to 70% purity using a Schiff base tin(II) complex as a catalyst.26 The new polymer sequence ABB contains double insertions of epoxides that can improve the thermal properties of the polymers. Other tin(II) catalysts such as Sn(OMe)2,27 amine Schiff-base tin(II),28 and bis (amidinate)tin(II)29 were also reported revealing preferences for multiple insertions of epoxides leading to poly(ester-ether). Trinuclear Co(III) complexes were shown to promote consecutive epoxide insertions resulting in polymers having single-glass transition and tunable properties.30 Recently, a new Zr(IV) complex was shown to copolymerize cyclic anhydride (A), epoxide/oxetane (B), and THF (C) giving a new sequence of >99% ABB or ABB/ABC polymers when C is present (Scheme 1b).31
 |
| | Scheme 1 ROCOP of cyclic anhydrides (a), epoxides (b), and cyclic ethers (c). | |
Although ester-ether linkages are undesirable according to several reports, the existence of ester-ether linkages in polymer chains could potentially lead to new types of polymeric materials with added tunable functions and properties compared to conventional alternating polyesters and also provide additional advantages in several applications.32–34 For example, the hydrolytic degradation of block copolymers of polyethylene glycol (PEG) and polyesters can be accelerated by the hydrophilic part of PEG.35–38 This hydrolytic degradation behavior is very important for drug delivery systems to determine the amount of released products to targets.35,38 Williams and co-workers recently synthesized a tapered and miscible block copolymer of poly(ester-ran-ether)-b-PLA from L-lactide, propylene oxide, and maleic anhydride. The obtained tapered block copolymer was blended with commercial PLA giving materials with improved toughness and elongation at break without affecting other properties.27 The ABB/ABC poly(ester-alt-ether) from phthalic anhydride (A), butylene oxide/oxetane (B), and THF (C) was shown to have a faster hydrolysis rate compared to the conventional (AB)n polyester.31 The copolymer containing both (AB)n and (ABx)m sequences could be used as miscibility-promoting agents in mixed polyether/polyester systems for adjusting the phase-separation behavior as reported by Lu and co-workers.30 Due to these promising properties of poly(ester-ether) polymers, the investigations of polymer structures and the control of (AB)n and (ABx)m contents have gained increasing attention and are extremely significant for determining the properties of these polymers in numerous applications.
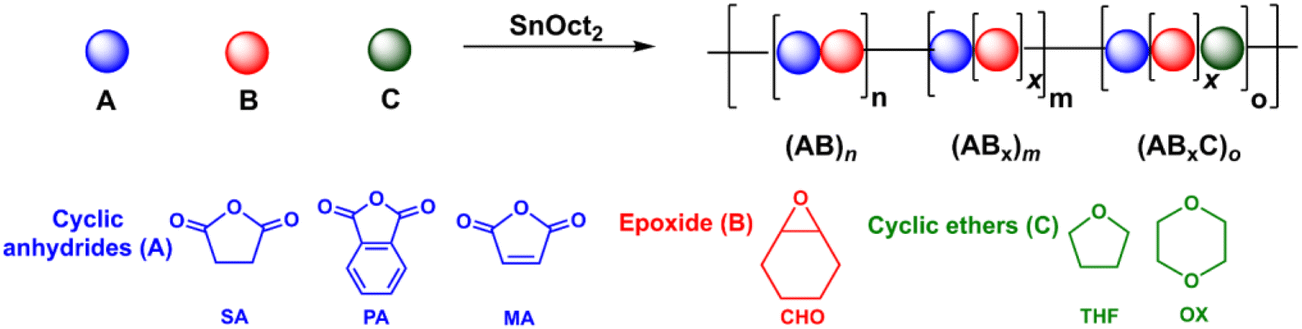
Despite increasing interest in ABx polymers, the discovery of catalytic systems that can produce a high percentage of ABx with controllable ether units is still lacking. Based on the successful findings mentioned above, it is evident that tin(II) complexes are extremely appealing for promoting multiple insertions of epoxides. While ligated tin(II) complexes, or other ligated metal complexes, are attractive, they are generally inconvenient for conventional and industrial applications due to their multi-step synthesis and high moisture-sensitivity. Therefore, we turned our attention to a commercially available simple complex of tin(II) 2-ethylhexanoate (SnOct2) because SnOct2 has rarely been explored for ROCOP39 but it has been widely adopted for ring-opening polymerization (ROP) of lactides, lactones, and cyclic carbonates due to its high performance, low price, easy handling, low toxicity and good stability at high temperature.40,41 In addition, SnOct2 contains carboxylates that could function similar to other metal carboxylates already demonstrated as efficient catalysts for ROCOP.42–45 Herein, SnOct2 was investigated in the ROCOP of cyclic anhydrides (A), epoxides (B), and cyclic ethers (C) with the aim being to control multiple insertions of the B units and to demonstrate the versatility of the catalyst toward other renowned polymeric compounds, such as polylactides and polyurethanes (Scheme 1c).
Experimental
Materials
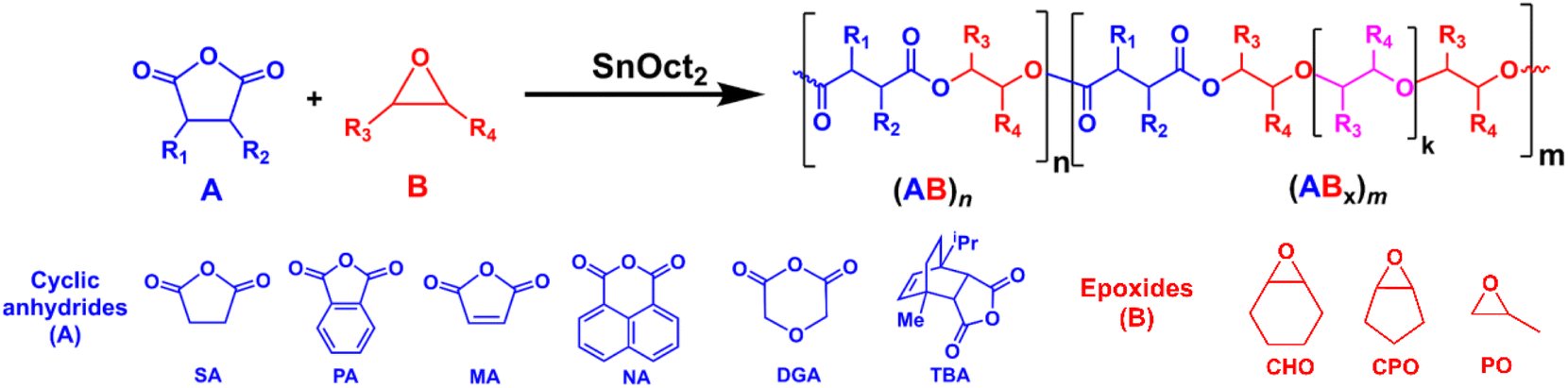
All manipulations were carried out in a glovebox or by using standard Schlenk techniques under a nitrogen atmosphere. Toluene and tetrahydrofuran were dried using a solvent purification system (MB SPS-800, MBRAUN). Cyclohexene oxide (CHO, TCI), cyclopentene oxide (CPO, TCI), propylene oxide (PO, TCI), 1,4-dioxane (OX, Fisher Chemical), and benzyl alcohol (BnOH, AJAX FINECHEM) were dried over calcium hydride, and distilled under vacuum before use. Maleic anhydride (MA, TCI), phthalic anhydride (PA, TCI), diglycolic anhydride (DGA, ACROS) and succinic anhydride (SA, ACROS) were purified by sublimation under vacuum three times before use. 1,8-Naphthalic anhydride (NA, TCI) was purified by recrystallization from hot chloroform, dried under vacuum, and stored in a glovebox. Tin(II) 2-ethylhexanoate (SnOct2, TCI) was dried under vacuum overnight and stored in a glovebox. L-Lactide (L-LA, TCI) was recrystallized from toluene and purified by sublimation under vacuum three times before use. Benzoic acid (BA, MERCK) and 1,4-benzendimethanol (BDM, TCI) were dried under vacuum at 50 °C overnight before use. 4,4′-Diphenylmethane diisocyanate (MDI, TCI) was used as received. Terpene-based cyclic anhydride (TBA) was synthesized according to the literature procedure.11 Other chemicals were obtained from commercial suppliers and used as received.
Measurements
1H, COSY, 13C and DOSY NMR spectra were recorded on a Bruker AVANCE III HD-600 MHz spectrometer and referenced to the protio impurity of commercial chloroform-d (CDCl3, δ 7.26 ppm) as the internal standard. Mass spectrometry was carried out using a Bruker data analysis Esquire-LC mass spectrometer (APCI mode). GPC analyses were carried out on a Malvern Viscotek TDAmax double detector device equipped with a refractive index detector and a viscometer with three 300 mm × 8.0 mm ID columns packed with a porous styrene-divinylbenzene copolymer. GPC columns were eluted using tetrahydrofuran at a flow rate of 1.0 mL min−1 at 35 °C. The universal calibration curve was calibrated with polystyrene standards ranging from 1200 to 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 amu. Molecular weights and dispersities were calculated using the refractometric (RI) method. Differential scanning calorimetry (DSC) measurements were performed on a PerkinElmer DSC-8500 instrument to analyze the glass-transition temperature (Tg) of polymers over a temperature range from 0 °C to 100 °C, −60 °C to 100 °C and 0 °C to 200 °C at a heating rate of 10 °C min−1 and a gas flow rate of 20 mL min−1 under a nitrogen atmosphere. Tg and enthalpy values from the second heating curves were calculated to remove the thermal history of the samples.
000 amu. Molecular weights and dispersities were calculated using the refractometric (RI) method. Differential scanning calorimetry (DSC) measurements were performed on a PerkinElmer DSC-8500 instrument to analyze the glass-transition temperature (Tg) of polymers over a temperature range from 0 °C to 100 °C, −60 °C to 100 °C and 0 °C to 200 °C at a heating rate of 10 °C min−1 and a gas flow rate of 20 mL min−1 under a nitrogen atmosphere. Tg and enthalpy values from the second heating curves were calculated to remove the thermal history of the samples.
General polymerization procedures
Solution polymerization.
The following procedure is for ROCOP of SA and CHO using an SA:CHO:SnOct2 mole ratio of 50:100:1 with an initial CHO concentration of 1.00 M and a temperature of 80 °C. Polymerization using other monomer ratios or temperatures can be carried out similarly. SA (0.100 g, 1.00 mmol), CHO (0.1960 g, 2.00 mmol), SnOct2 (8.0 mg, 20 μmol), and dry toluene (added to a total solution volume of 2.00 mL) were added into a Schlenk flask. The content of the flask was stirred and it was placed in a preheated oil bath at 80 °C. At the desired time, a small amount of crude mixture was withdrawn from the polymerization reaction for conversion analysis by NMR. The polymer residue was dried under vacuum to remove the solvent. Subsequently, the crude mixture was dissolved in a small amount of dichloromethane and precipitated with excess cold methanol. The precipitated polymer was collected and dried under vacuum for characterization by NMR, GPC, mass spectroscopy analysis, and DSC.
Neat polymerization.
SA (0.1651 g, 1.65 mmol), CHO (0.970 g, 9.90 mmol) and SnOct2 (13.4 mg, 33 μmol), were placed in a Schlenk flask with a magnetic stirrer at an SA:CHO:SnOct2 mole ratio of 50:300:1. The reaction mixture was stirred in a preheated oil bath at 80 °C. At the desired time, a small amount of sample was withdrawn from the polymerization reaction for conversion analysis by the NMR technique. The polymer was dissolved in a small amount of dichloromethane and precipitated with excess cold methanol. The precipitated polymer was collected and dried under vacuum for characterization by NMR, GPC, mass spectroscopy analysis, and DSC.
Terpolymerization with THF or 1,4-dioxane.
The one-pot ROCOP of CHO, SA and THF using an SA:CHO:THF:SnOct2 mole ratio of 50:150:150:1 was conducted at 80 °C in a Schlenk flask using SA (0.1650 g, 1.65 mmol), CHO (0.4850 g, 4.95 mmol), THF (0.3569 g, 4.95 mmol) and SnOct2 (13.4 mg, 33 μmol). The reaction was monitored by NMR for conversion analysis. The polymer residue was dried under vacuum to remove the solvent. Subsequently, the crude mixture was dissolved in a small amount of dichloromethane and precipitated with excess cold methanol. The precipitated polymer was collected and dried under vacuum for characterization by NMR, GPC, and DSC. Polymerizations using 1,4-dioxane or other monomer ratios were carried out similarly.
Sequential terpolymerization.
Terpolymerization was conducted using the sequential addition method. Firstly, the copolymerization of MA and CHO using an MA:CHO:SnOct2 mole ratio of 50:300:1 with an initial CHO concentration of 2.00 M at 80 °C was performed. To a Schlenk flask MA (65.4 mg, 0.667 mmol), CHO (0.392 g, 4.00 mmol), SnOct2 (5.4 mg, 13 μmol), and dry toluene (to obtain a total solution volume of 2.00 mL) were added. At the desired time, a small amount of crude mixture was withdrawn from the polymerization reaction for measuring the conversion and molecular weight using the NMR technique and GPC, respectively. After MA was fully consumed (about 1 h), 50 equivalents of L-LA (0.0961 g, 0.667 mmol) were subsequently added to the reaction. The polymerization reached a high monomer conversion of 85% in 4 h. The crude product was dried under vacuum, dissolved in a small amount of dichloromethane, and poured into excess cold methanol. The precipitated polymer was collected and dried under vacuum for characterization by NMR, GPC and DSC.
The synthesis of polyurethane.
The ROCOP of CHO and SA using a CHO:SA:SnOct2:BDM mole ratio of 1200:200:1:20 with an initial CHO concentration of 2.00 M at 80 °C was performed to produce the copolymer diol. SA (0.0667 g, 0.667 mmol), CHO (0.392 g, 4.00 mmol), SnOct2 (1.4 mg, 3.3 μmol), BDM (9.2 mg, 67 μmol), and dry toluene (added to a total solution volume of 2 mL) were added into a Schlenk flask. After SA was completely consumed, MDI (0.0167 g, 66.6 μmol) was added to the reaction mixture and the reaction was left for 24 h. The crude product was dried under vacuum, dissolved in a small amount of dichloromethane, and poured into excess cold methanol. The precipitated polymer was collected and dried under vacuum for characterization by NMR, GPC and IR.
Hydrolysis of copolymers.
Hydrolysis of the copolymers was performed under basic conditions. The polymer (0.0300 g) was dissolved in 2.0 mL of tetrahydrofuran and then 1.0 mL of 7.0 M NaOH aqueous solution was subsequently added. The reaction mixture was heated at 80 °C for 3 days. The aqueous phase was acidified with a 4.0 M HCl solution. The hydrolyzed components were further extracted with dichloromethane (3 × 10 mL). The combined organic phase was dried over sodium sulfate, filtered, and concentrated under vacuum. The composition of the resulting mixtures was analyzed by atmospheric pressure chemical ionization (APCI) mass spectrometry.
Polymer degradation experiments.
All polymer samples were purified by precipitation in excess cold methanol 3 times prior to the degradation study. A polymer sample (70 mg) was added to an aqueous NaOH solution (5.0 M, 7.00 mL). The reaction was sealed, heated at 70 °C and stirred at 700 rpm. The polymer samples for GPC analysis were obtained by cooling the reaction to RT, cutting a small piece from the coagulated polymer (approximately 5 mg), washing it with water, and dissolving it in THF. The degradation was monitored by GPC analysis to obtain the molecular weight Mn. After 4 days, all polymers completely disappeared except for poly(CHO-alt-SA), which was still intact.
Calculations of polymer compositions.
The compositions of AB, ABx, and ABxC sequences in the copolymers were calculated as shown in the ESI† with some modifications from the previous report.31
Results and discussion
Synthesis of the ABx poly(ester-ether)
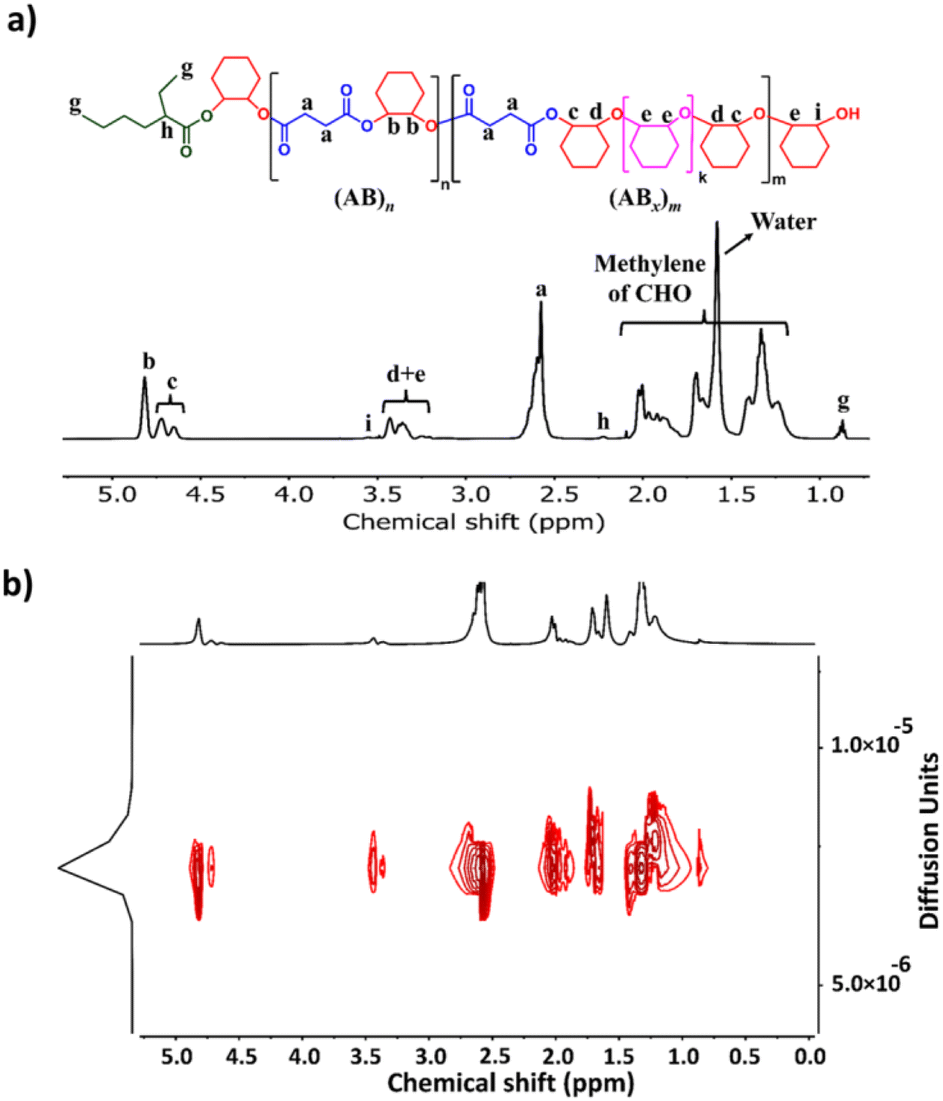
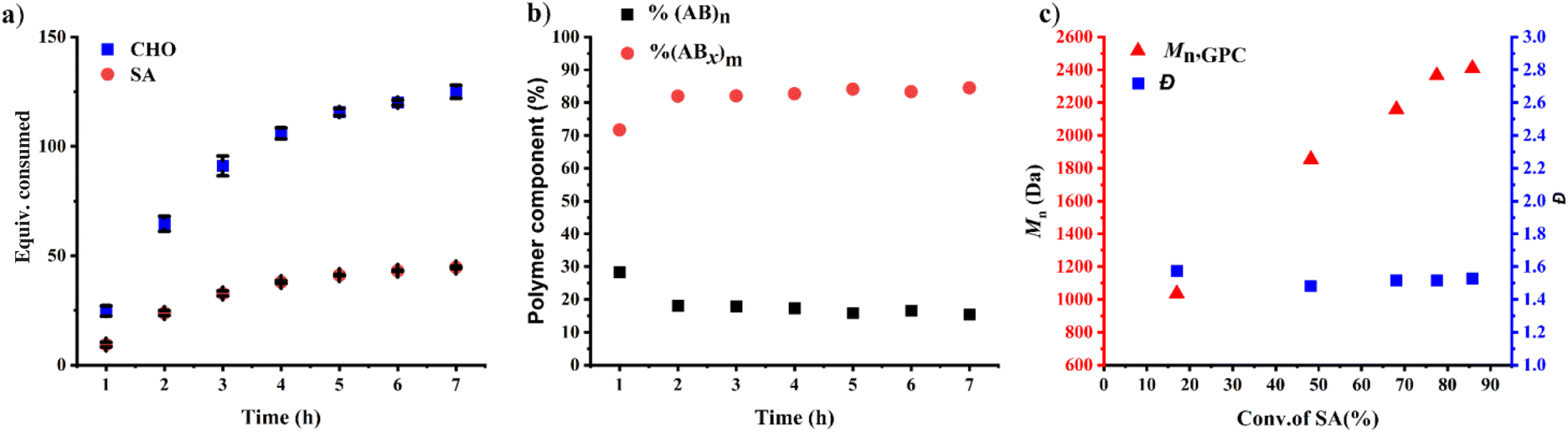
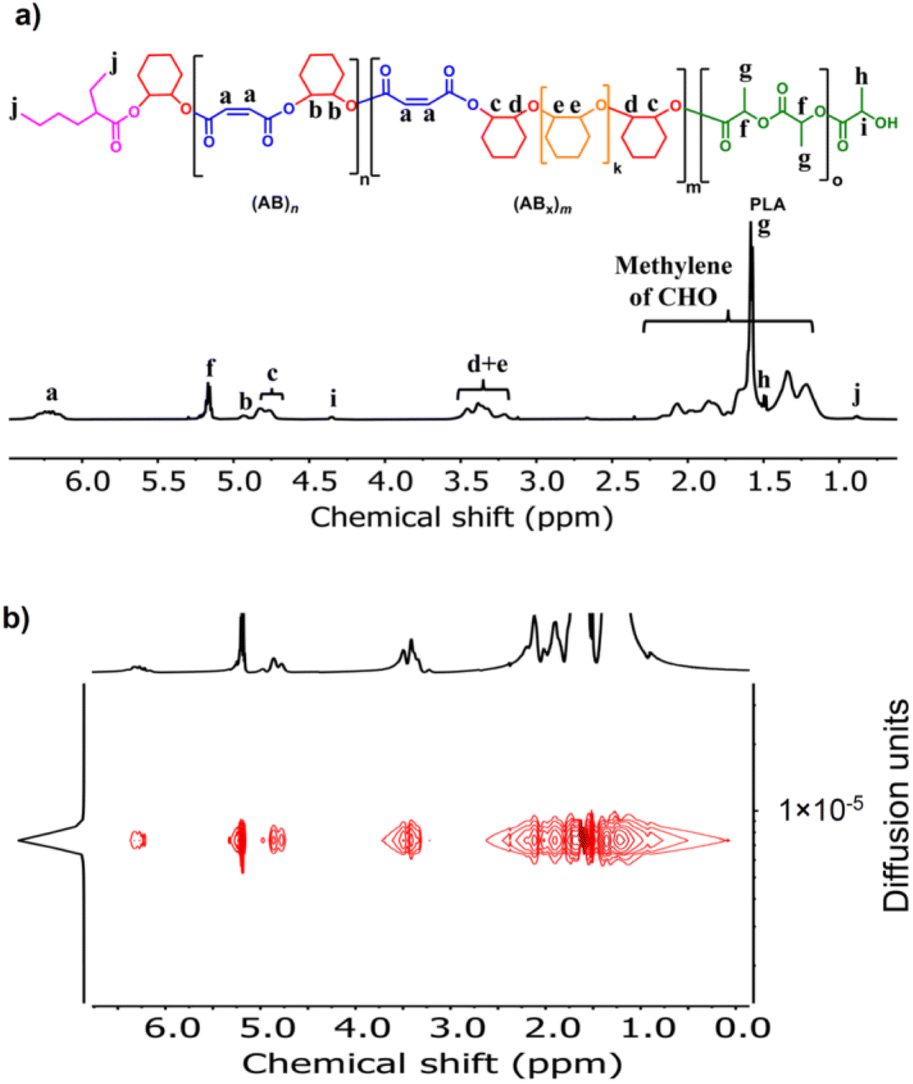
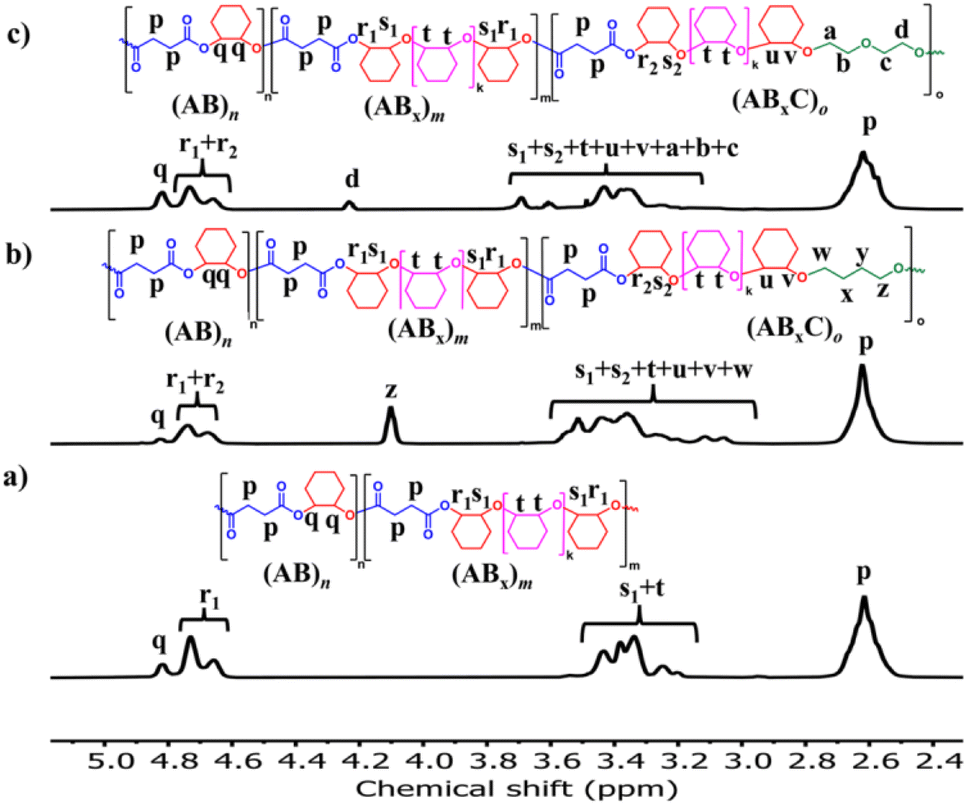
The initial ROCOP of succinic anhydride (SA) and cyclohexene oxide (CHO) was tested using SnOct2 as a catalyst at an [SA]:[CHO]:[SnOct2] ratio of 50:100:1 in toluene at 110 °C (Table 1, entry 1). Although alcohols are commonly added as an initiator for ROP of cyclic esters, we did not add any alcohols here because SnOct2 already contains carboxylates capable of initiating the ROCOP. After 4 h, the NMR analysis of the crude mixture showed conversions of >99% for SA (DP = 50) and 81% for CHO (DP = 81) clearly suggesting multiple insertions of CHO (DPCHO/DPSA > 1). Because CHO is a symmetric molecule, the 1H NMR spectrum of the methine protons in CHO moieties in the copolymer is rather simple and very informative. We reported a thorough analysis of AB and ABx sequences earlier (A = SA and B = CHO) where the methine protons of CHO moieties appeared at 4.82 ppm for alternating (AB)n sequences, at 4.65–4.75 ppm for ABx sequences, and at 3.2–3.5 ppm for Bx sequences.26 The integration of these peaks can be used to directly calculate the amount of each sequence (see the ESI†). From these NMR assignments, the 1H NMR spectrum of the purified copolymer (Fig. 1a) revealed a copolymer (Mn = 4.8 kDa, Đ = 1.46) consisting of 58% of perfectly alternating copolymer (AB)n observed at 4.82 ppm (protons b) and 42% of multiple CHO insertions or (ABx)m observed at 4.65–4.75 ppm (protons c). The average number (xav) of B units in (ABx)m sequences (excluding AB sequences) can be directly calculated from the 1H NMR spectrum from the ratio of all CHO units in the ABx sequence (protons c + d + e) divided by capping CHO units (protons c) and found to be 2.5 (see the ESI† for detailed calculations of polymer compositions and xav). 2-Ethylhexanoate was observed as an end group in the NMR spectrum (Fig. 1a) and as a major series in the MALDI-TOF spectrum (Fig. S5†). The diffusion-ordered spectroscopy (DOSY) NMR spectrum exhibited a single diffusion coefficient, indicating that (AB)n and (ABx)m units were established on the same polymer chains (Fig. 1b). Lowering the polymerization temperature from 110 °C to 80 and 60 °C took a longer time to complete (10 h and 31 h, respectively) but can increase the ABx polymer content from 42 to 56 and 60%, respectively (Table 1, entries 1–3). The xav values also slightly increased from 2.5 to 2.6 and 2.8, respectively. The polymerization temperature of 80 °C was then chosen for further polymerization studies because it provided a good balance of high amounts of the ABx polymer within reasonable polymerization times. It is to be noted that, after SA was completely consumed, the polymerization appeared to stop although some CHO still remained suggesting that long chain (CHO)n polyether could not be produced by the SnOct2 catalyst under these conditions (Fig. S1†). To confirm this result, homopolymerization of CHO by SnOct2 was carried out under the same conditions at 80 °C. Only a trace amount of poly(CHO) was detected after 24 h (Fig. S2†). Therefore, this result indicated that long blocks of CHO were unfavorable in the presence of the tin catalyst and that the presence of SA was mandatory for the copolymerization to continue. This could be a result of the increasing steric hindrance of the longer (CHO)n blocks as demonstrated earlier.26
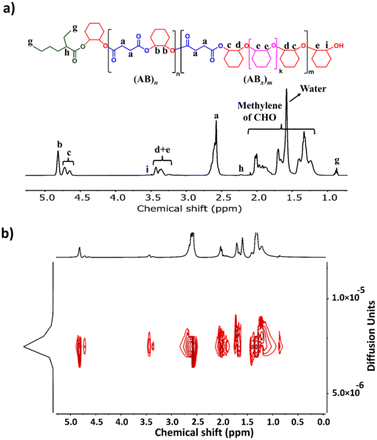 |
| | Fig. 1 (a) 1H NMR and (b) DOSY spectra (600 MHz, CDCl3, 30 °C) of poly(ester-ether) (Table 1, entry 1). | |
Table 1 ROCOP of cyclic anhydrides and epoxides catalyzed by SnOct2
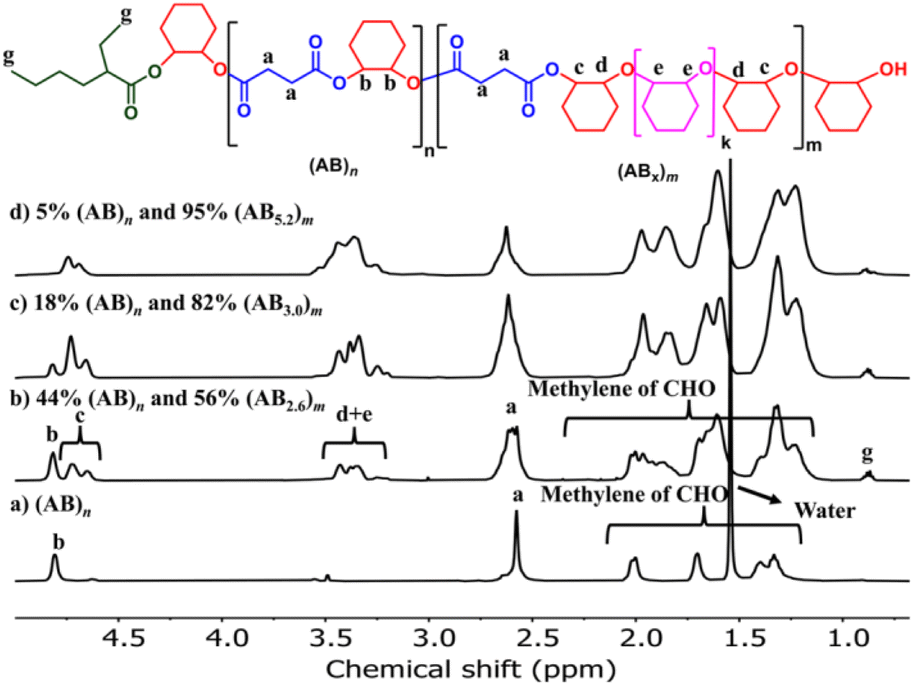
The effects of monomer concentrations and feed ratios were subsequently investigated as shown in Table 1 affording polymers having molecular weights of 2.1–4.3 kDa and moderate dispersity values of around 1.5 (entries 2 and 4–6). As the CHO ratios (B) increased from 100 equiv. to 300 and 600 equiv. (Table 1, entries 2, 4 and 5), the amount of ABx polymer increased from 56 to 67, and 75%, respectively. The xav values also increased from 2.6 to 3.0 and 3.4 indicating longer CHO units in the polymer chains as the amount of CHO increased. The existence of AB and ABx sequences are confirmed by the presence of b protons in the 1H NMR spectrum at 4.82 ppm for AB sequences (see Fig. 2a for comparison), and the presence of c protons at 4.65–4.75 ppm for ABx sequences as shown in Fig. 2b.26 When initial CHO concentrations were doubled from 1.0 M to 2.0 M (entries 4 vs. 6), the amount of ABx polymer significantly increased from 67 to 82% while the xav values remained constant at 3.0 (Fig. 2c). Complete hydrolysis of the obtained copolymer in entry 6 was also carried out under aqueous basic conditions at 80 °C (Fig. S30†). The hydrolyzed compositions were subsequently determined by APCI mass spectrometry (Fig. S31†) revealing a distribution of Bx fragments with x = 2–4 with x = 3 as the majority, in line with the observed xav = 3 from entry 6. A kinetic study run in triplicate under the conditions of entry 6 (Fig. 3a and Table S1†) revealed that the consumption ratios of CHO compared to SA were in a narrow range of 2.64–2.80 since the beginning of the polymerization indicating a greater preference for multiple insertions of CHO from the start. With a consumption ratio of less than 6 (initial [CHO]:[SA] = 6), the remaining [CHO] will be in excess compared to [SA] throughout the polymerization. A plot of polymer composition vs. time (Fig. 3b) revealed that the %AB and %ABx in the copolymer are in a narrow range of around 17% and 83%, respectively. Therefore, the excess CHO does not change polymer compositions during the polymerization. The molecular weight from GPC also increased linearly with increasing monomer consumption with dispersities of around 1.5 (Fig. 3c and Table S2†). The molecular weights were also determined using NMR by end-group integration. The Mn,NMR values were higher than those from GPC. The difference is possibly due to Mn,GPC being obtained in reference to the polystyrene standard. Nevertheless, both molecular weights obtained from NMR and GPC methods progressed linearly along the same increasing trend with increasing SA conversion. The low molecular weight could be from impurities from cyclic anhydrides46,47 and some protic impurities from commercial SnOct2 that can undergo chain transfer causing low Mn.40,41 The amount of ABx polymer can be increased impressively to 95% with only 5% of the alternate AB polymer when the polymerization is carried out in neat CHO (Table 1, entry 7 and Fig. 2d). The xav value increased to 5.2 indicating longer polyether segments in the polymer chains. Therefore, the amount of ABx polymer and the polyether chain length can be controlled by tuning the A:B ratios, monomer concentrations, and polymerization temperature.
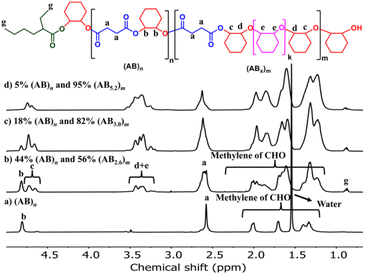 |
| | Fig. 2
1H NMR spectra (600 MHz, CDCl3, 30 °C) of the copolymer from ROCOP of SA and CHO having different AB and ABx components. | |
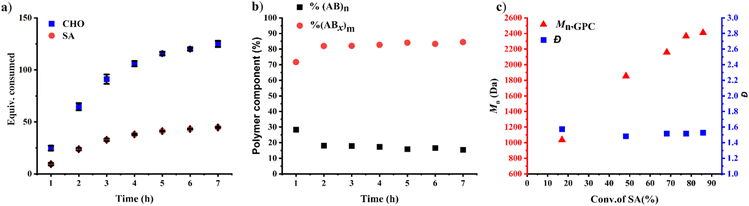 |
| | Fig. 3 ROCOP of CHO and SA using CHO:SA:SnOct2 = 300:50:1 with initial [CHO]0 = 2.0 M at 80 °C. (a) Plots of consumed monomers versus time, (b) plots of polymer composition versus time and (c) plots of Mn and dispersity versus conversion of SA. | |
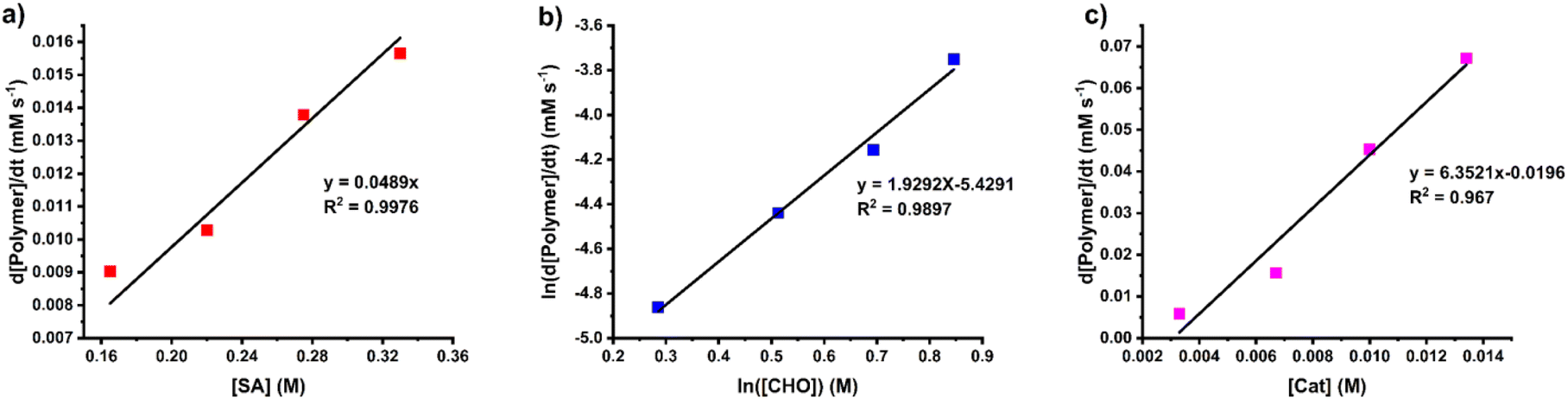
The rate law for the ROCOP of SA and CHO was determined using initial rate analysis within the first 20% conversion of SA using the relationship of rate = kp[SA]x[CHO]y[SnOct2]z. For SA dependence, the initial rates were determined at different initial SA concentrations ([SA]0 = 0.165, 0.220, 0.275, and 0.330 M) while [CHO]0 and [SnOct2]0 were kept constant at 2.0 M and 6.7 mM, respectively. The plot of initial rates versus the SA concentration demonstrated a linear correlation indicating that the reaction has a first-order dependence on SA concentration (Fig. 4a). The CHO dependence was determined similarly using different initial CHO concentrations ([CHO]0 = 1.33, 1.67, 2.00, and 2.33 M) while initial catalyst and SA concentrations were held constant ([SnOct2]0 = 6.7 mM and [SA]0 = 0.33 M). A plot of the initial polymerization rate (<20% SA conversion) and [CHO] was obtained but it deviated from a linear relationship. Therefore, the order of the reaction was determined by taking the natural logarithm of the above equation giving the relationship ln[rate] = yln[CHO]ini + ln(kobs) where kobs = kp[SA][SnOct2]z. As shown in Fig. 4b, a plot of ln[rate] vs. ln[CHO] gave a linear relationship with a slope of 1.93, which is approximated to 2. Therefore, the reaction is 2nd order with respect to the CHO concentration. Finally, the catalyst dependence was determined by varying the initial concentrations of [SnOct2]0 (3.3, 6.7, 10.0, and 13.4 mM) using [CHO]0 and [SA]0 of 2.0 M and 0.33 M, respectively. The initial rates of polymerization increased with increasing catalyst concentration in a linear fashion (Fig. 4c) also suggesting a first-order dependence on SnOct2. Therefore, the rate law of the polymerization is as follows:
| Rate = kp [SA][CHO]2[SnOct2] |
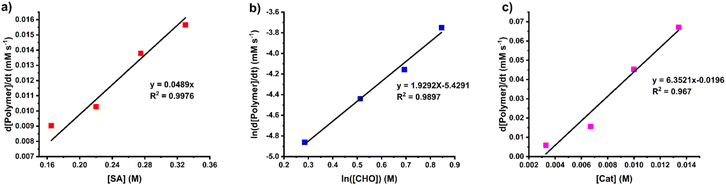 |
| | Fig. 4 Plots of initial rates versus (a) SA concentration, (b) CHO concentration and (c) SnOct2 concentration. | |
In other catalytic systems where alternating (AB)n polymers were obtained, the insertion of cyclic anhydride is relatively fast (normally zero-order with respect to cyclic anhydride concentrations).42,48 Since the reaction has first-order dependence with respect to the SA concentration in our system, the insertion rate of SA into the tin catalyst system is relatively slower. In addition, the 2nd-order dependence on the CHO concentration will also assist in multiple epoxide insertions to form ABx polymer sequences.
A general ROCOP mechanism involves metal alkoxide (after epoxide insertions) and metal carboxylate (after cyclic anhydride insertions) intermediates.5,6 Therefore, both alcohols and carboxylic acids are eligible as initiators or chain transfer agents allowing possible end-functionalization of the obtained polymers. This is a great advantage since carboxylic acids are not efficient initiators for common ROP of cyclic esters. We tested this hypothesis by adding benzyl alcohol and benzoic acid as initiators (Table 1, entries 8 and 9). The resulting polymers revealed a similar amount of the ABx polymer (77%) and xav values (3.2–3.3) compared to the polymerization without added initiators (entry 6). However, the obtained polymers have lower molecular weights as a result of more initiators being present in the polymerizations. Both 2-ethylhexanoate and benzyloxide (or benzoate) were observed as the end groups in the resulting polymers (Fig. S12 and S13†).
ROCOP using SnOct2 as the catalyst can be extended to other bulky cyclic anhydrides including phthalic anhydride (PA), 1,8-naphthalic anhydride (NA), terpene-based cyclic anhydride (TBA), maleic anhydride (MA) and diglycolic anhydride (DGA) using an [Anh]:[CHO]:[SnOct2] ratio of 50:300:1 (Table 1, entries 10–14). In all cases, copolymers having (ABx)m sequences were successfully obtained and (ABx)m percentages of 97% for PA, 72% for NA, 71% for MA and 26% for DGA were achieved. Although SnOct2 can catalyze ROCOP of CHO and TBA giving [CHO]:[TBA] = 2.8 in the copolymer, the accurate percentages of (AB)n and (ABx)m cannot be determined due to the broad and significantly overlapping signals around 4.5–4.8 ppm arising from the asymmetric nature of TBA (Fig. S20†). Because the polymer structure has a strong influence on thermal properties, such as glass transition temperature (Tg), the increasing amount of Bx units will play a significant role in this property. The Tg of poly(SA-co-CHO) (Table 1, entry 6) having 82% AB3 was found at 48 °C, which is in between the reported Tg of perfectly alternating poly(SA-alt-CHO) at 32 °C49 and poly(CHO) at 58–75 °C.50 A similar effect was also observed for MA (Table 1, entry 12) where the Tg of the polymer having 71% AB3.4 also demonstrated a higher value of 62 °C, which is higher than that of poly(MA-alt-CHO) reported at 51 °C.49 In contrast, for the bulkier cyclic anhydrides (PA, NA, and TBA listed in Table 1, entries 10, 11 and 14), the observed Tg values are lower than those previously reported for poly(PA-alt-CHO), poly(NA-alt-CHO), and poly(TBA-alt-CHO) of 133,49 159,51 and 162 °C,52 respectively. This is due to the greater flexibility of the cyclohexyl rings (in the ABx unit) compared to the more rigid aromatic and tricyclic units. In addition, the increase of CHO content in the ABx copolymer decreases the content of rigid cyclic anhydrides in the resulting copolymer. Therefore, the overall rigidity of the copolymer is decreased giving lower Tg values. The molecular weight of the polymer can be increased by changing the monomer:catalyst ratio as shown in Table 1 (compare entries 10 and 15) giving poly(PA-co-CHO) with a higher Mn value of 17580 Da (Đ = 1.25) and an impressive ABx content of 99% with xav of 3.4.
The preference for multiple insertions of epoxides is not limited to just CHO. When cyclopentene oxide (CPO) and propylene oxide (PO) were used instead of CHO at a ratio of [PA]:[CPO or PO]:[SnOct2] = 50:300:1, as shown in Table 1 entries 16 and 17, copolymers having high (ABx)m sequences of 77% for CPO and 88% for PO were successfully obtained. The Tg values of poly(PA-co-CPO) and poly(PA-co-PO) reported in entries 16 and 17 were found at 33 °C and −1 °C, respectively. These Tg values are significantly lower than the Tg values of alternate poly(PA-alt-CPO) and poly(PA-alt-PO) reported at 48 and 40 °C, respectively.53
Synthesis of poly(ABx-b-PLA) and ABx-based polyurethane
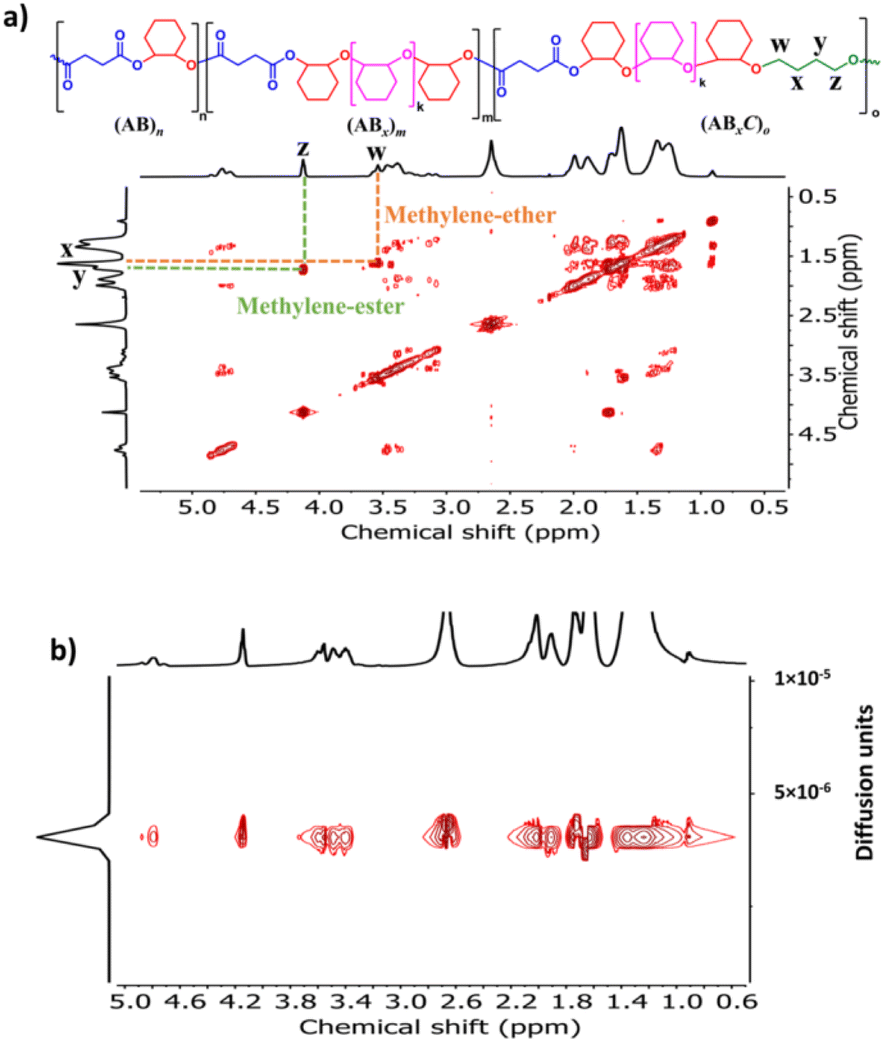
Because SnOct2 is a common catalyst for ROP of cyclic esters, such as lactides, it is therefore possible that SnOct2 can be used to combine ABx polymers in one pot with biodegradable polylactides widely used in food packaging and biomedical applications. MA was selected as a model cyclic anhydride because it contains a double bond capable of post-polymerization modifications if needed. To demonstrate this application, the first block of poly(ester-ether) was synthesized from ROCOP of MA and CHO using the [MA]:[CHO]:[SnOct2] ratio of 50:300:1 affording the polymer with 21% (AB)n and 79% (ABx)m; x = 3.2 (Mn = 2270 Da, Đ = 1.41). After MA was completely consumed, 50 equiv. of L-LA was subsequently added in one pot giving a diblock polymer having the (AB)n/(ABx)m/PLA ratio of 6:30:64 (Mn = 4880 Da, Đ = 1.35) as shown in Fig. 5a. A single diffusion coefficient was also observed in the DOSY-NMR spectrum confirming the success of polylactide addition to the existing poly(ester-ether) in one pot (Fig. 5b). The GPC trace of the final product was still observed as a monomodal narrow distribution (Fig. S50†). A single Tg value of 63 °C was observed for the block copolymer (Fig. S53†), which is indicative of polymer miscibility.
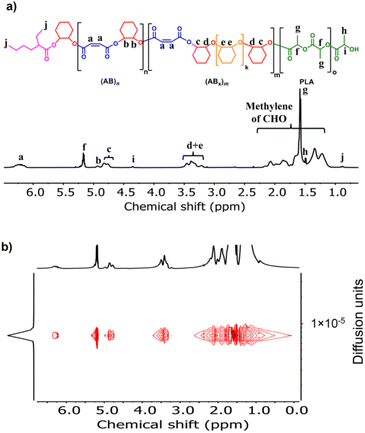 |
| | Fig. 5 (a) 1H NMR and (b) DOSY spectra (600 MHz, CDCl3, 30 °C) of poly(ester-ether)-b-PLA. | |
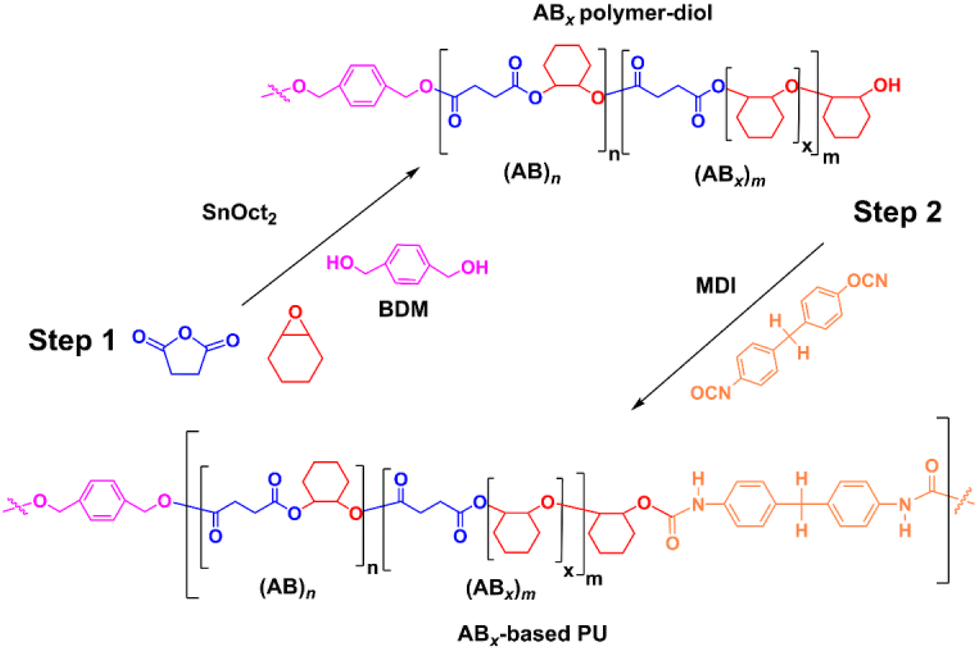
The versatility of SnOct2 can be further demonstrated in polyurethane synthesis as shown in Scheme 2 where tin-based complexes were commonly exploited as catalysts owing to their efficiency for reactions of isocyanates and alcohols.54 ROCOP of SA and CHO catalyzed by SnOct2 was first carried out using 1,4-benzendimethanol (BDM) as a bifunctional initiator using [CHO]:[SA]:[SnOct2]:[BDM] of 1200:200:1:20 in 42 h at 80 °C. A small polymer sample was taken and found to be poly(ester-ether)diol (Mn = 2450 g mol−1, Đ = 1.32) with 14% AB and 86% ABx; xav = 3.6. Subsequently in one pot, 20 equiv. of 4,4′-diphenylmethane diisocyanate (MDI) was added to the reaction mixture and stirred for 24 h affording a polyurethane with a higher molecular weight (Fig. S54†) of 5903 g mol−1 (Đ = 2.09). The 1H and DOSY NMR analyses also confirmed the existence of urethane linkages installed in the polymer chain (Fig. S55 and 56†). The FT-IR spectrum (Fig. S57†) of the poly(ester-ether)-based polyurethane showed the characteristic absorptions at 1526 cm−1 and 3351 cm−1 due to NH deformation and stretching vibrations, respectively.55
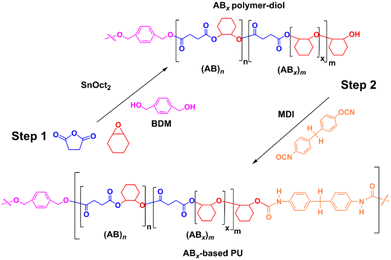 |
| | Scheme 2 Synthesis of the ABx-based polyurethane. | |
Synthesis of the novel ABxC poly(ester-ether) from additional cyclic ethers (C)
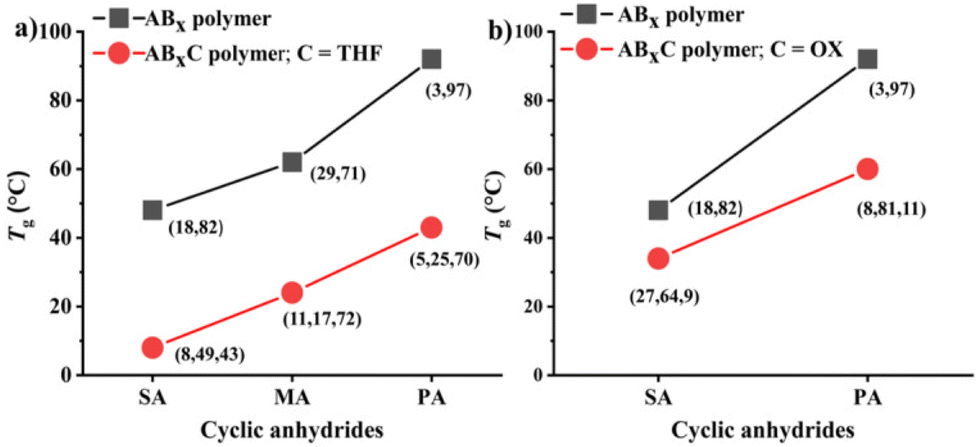
Recently, a zirconium complex was reported by Williams as a catalyst in the ROCOP of cyclic anhydride (A), epoxide (B), and THF (C), giving a unique copolymer having ABB and ABC repeating units possibly due to the large ionic radii of Zr(IV).31 Up to 29% selectivity for THF was reported. Inspired by this excellent work, we explored the possibility of THF incorporation into the copolymer using SnOct2 since Sn(II) has a larger ionic radius (1.36 Å)56 compared to that of Zr(IV) (0.72 Å).31 Therefore, novel ABxC polymer sequences having multiple B units could be synthesized in addition to the reported ABC sequence. ROCOP of only two monomers (SA and THF) using an [SA]:[THF]:[SnOct2] ratio of 50:150:1 was first carried out at 80 °C for 24 h in a closed reactor but resulted in no polymer formation (Table 2, entry 1). Polymerization using the [CHO]:[THF]:[SnOct2] ratio of 150:150:1 at 80 °C also showed no conversion after 44 h (Table 2, entry 2). Polymerization was then conducted with three monomers using an [SA]:[CHO]:[THF]:[SnOct2] ratio of 50:150:150:1 at 80 °C (Table 2, entry 3). As anticipated, THF could now be copolymerized as evidenced by the new distinctive 1H NMR peak of –CH2-C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2-OC(O)– at 4.10 ppm (shown in Fig. 6b as z protons and Fig. S59†). The COSY NMR spectrum revealed that the ring-opened THF contained methylene-ester on one end and methylene-ether at the other end strongly indicating CHO–THF–SA sequences (Fig. 7a). Therefore, only one THF unit was inserted after CHO insertions, followed immediately by the reaction with SA. This CHO–THF–SA sequence is highly possible because after the ring-opening of THF, the more reactive primary alkoxide (–O(CH2)4O−) is generated (compared to the secondary alkoxide after the CHO ring-opening) and can efficiently react with SA. The 1H NMR spectrum of the obtained polymer was carefully analyzed and found to contain 8% AB, 49% ABx, and 43% ABxC (see the ESI† for sequence analysis). A single diffusion coefficient was observed in the DOSY-NMR spectrum indicating that all monomers including THF were installed on the same polymer chains (Fig. 7b). The ring-opened THF is always connected to cyclic anhydride (SA) and will not exceed the amount of SA. Therefore, the selectivity for THF was defined and calculated relative to SA. A very high selectivity for THF incorporation of 43% was observed. The presence of THF in the polymer significantly reduced the Tg from 48 °C (Table 1, entry 6) to 8 °C (Table 2, entry 3) as shown in Fig. 8a due to the highly flexible –(CH2)4– backbone. The THF content could be increased impressively from 43% to 70%, and 74% by increasing the THF equivalents from 150 to 800, and 3000, respectively (Table 2, entries 3–5). This is the highest selectivity for THF ever reported for ROCOP. The major sequence of the polymer in entry 5, having THF content as high as 74% [AB2.2C]m, could also be rearranged to [CAB2.2]m or [(THF)(SA)(CHO)2.2]m. Therefore, this new polymer can be alternatively visualized as butylene succinate units separated by approximately two CHO units. This polymer has a structure resembling that of poly(butylene succinate), a very well-known biodegradable polymer. The polymerizations with THF could also be extended to PA and MA giving the terpolymers with high ABxC components of 70% and 72%, respectively with xav around 2.6 (Table 2, entries 6 and 7). The Tg values of these polymers containing high THF contents were significantly lower by about 40–50 °C (Fig. 8a) due to the highly flexible methylene units incorporated into the polymer chains.
2-OC(O)– at 4.10 ppm (shown in Fig. 6b as z protons and Fig. S59†). The COSY NMR spectrum revealed that the ring-opened THF contained methylene-ester on one end and methylene-ether at the other end strongly indicating CHO–THF–SA sequences (Fig. 7a). Therefore, only one THF unit was inserted after CHO insertions, followed immediately by the reaction with SA. This CHO–THF–SA sequence is highly possible because after the ring-opening of THF, the more reactive primary alkoxide (–O(CH2)4O−) is generated (compared to the secondary alkoxide after the CHO ring-opening) and can efficiently react with SA. The 1H NMR spectrum of the obtained polymer was carefully analyzed and found to contain 8% AB, 49% ABx, and 43% ABxC (see the ESI† for sequence analysis). A single diffusion coefficient was observed in the DOSY-NMR spectrum indicating that all monomers including THF were installed on the same polymer chains (Fig. 7b). The ring-opened THF is always connected to cyclic anhydride (SA) and will not exceed the amount of SA. Therefore, the selectivity for THF was defined and calculated relative to SA. A very high selectivity for THF incorporation of 43% was observed. The presence of THF in the polymer significantly reduced the Tg from 48 °C (Table 1, entry 6) to 8 °C (Table 2, entry 3) as shown in Fig. 8a due to the highly flexible –(CH2)4– backbone. The THF content could be increased impressively from 43% to 70%, and 74% by increasing the THF equivalents from 150 to 800, and 3000, respectively (Table 2, entries 3–5). This is the highest selectivity for THF ever reported for ROCOP. The major sequence of the polymer in entry 5, having THF content as high as 74% [AB2.2C]m, could also be rearranged to [CAB2.2]m or [(THF)(SA)(CHO)2.2]m. Therefore, this new polymer can be alternatively visualized as butylene succinate units separated by approximately two CHO units. This polymer has a structure resembling that of poly(butylene succinate), a very well-known biodegradable polymer. The polymerizations with THF could also be extended to PA and MA giving the terpolymers with high ABxC components of 70% and 72%, respectively with xav around 2.6 (Table 2, entries 6 and 7). The Tg values of these polymers containing high THF contents were significantly lower by about 40–50 °C (Fig. 8a) due to the highly flexible methylene units incorporated into the polymer chains.
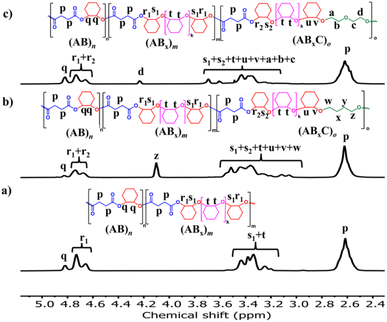 |
| | Fig. 6
1H NMR spectra (600 MHz, CDCl3, 30 °C) of (a) ABx copolymer (Table 1, entry 6, for comparison); (b) ABxC copolymer; C = THF (Table 2, entry 4); and (c) ABxC copolymer; C = OX (Table 2, entry 9). | |
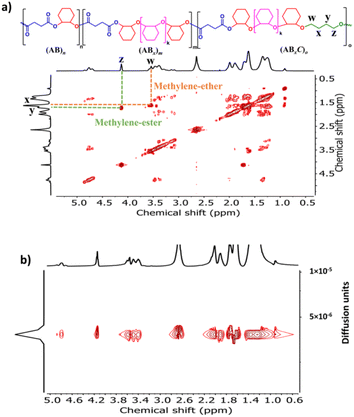 |
| | Fig. 7 COSY and DOSY-NMR spectra (600 MHz, CDCl3, 30 °C) of the copolymer from ROCOP of SA, CHO and THF using an SA/CHO/THF/SnOct2 molar ratio of 50:150:150:1 (Table 2, entry 3). | |
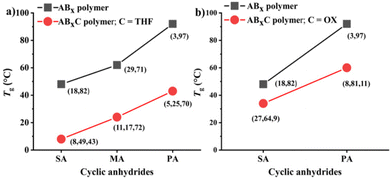 |
| | Fig. 8 Comparison of glass-transition temperatures (Tg) between ABx and ABxC polymers where (a) C = THF and (b) C = OX, given that (X,Y) = %AB and %ABx, respectively, and (X,Y,Z) = %AB, %ABx and %ABxC, respectively. | |
Table 2 ROCOP of cyclic anhydrides, CHO, and cyclic ethers catalyzed by 1 equiv. of SnOct2 at 80 °C
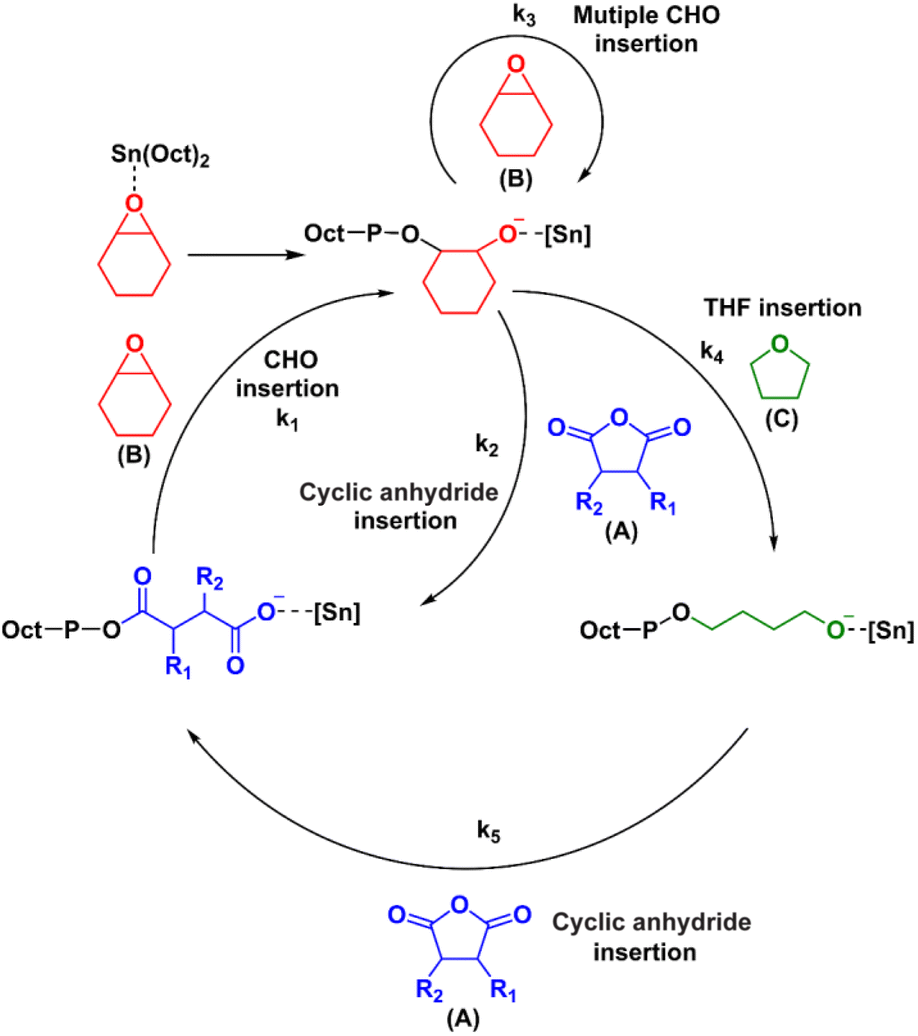
The mechanism for the terpolymerization leading to AB, ABx, and ABxC sequences is proposed and presented in Scheme 3. After CHO is initially ring-opened, there are three possible insertion reactions: (1) cyclic anhydride insertion (k2), (2) CHO insertion (or multiple insertions as k3), and (3) THF insertion (k4). The insertion of cyclic anhydride to metal alkoxides leads to alternating AB sequences as observed in many catalytic systems such as Al(III) and Cr(III) complexes where k2 > k3. However, in the SnOct2 system, k2 appeared to be slower than k3 leading to multiple insertions of CHO giving ABx sequences. This is supported by the results presented in Table 1 where xav = 2.5–3.4. It is evident that insertions of 2–4 CHO units were favored over the insertions of cyclic anhydrides. This could be a result of the large size of tin(II) metal. After multiple insertions of CHO, THF could be inserted giving ABxC sequences. From the results of the hydrolysis experiment (vide infra), multiple insertions of C units do not occur (Fig. S58†). Therefore, the generated [Sn]-O(CH2)4– species could react rapidly with the cyclic anhydride giving tin(II) carboxylates to complete the cycle. The factors that control monomer enchainment sequences are not certain at this point and will require further investigation.
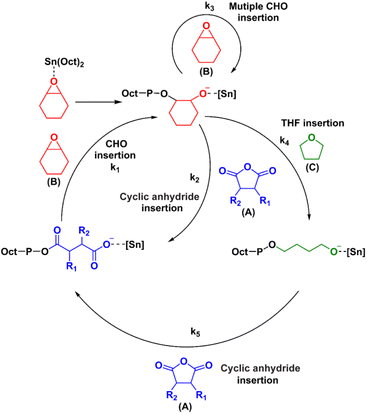 |
| | Scheme 3 Proposed mechanism for the formation of ABx and ABxC poly(ether-ester) from ROCOP of cyclic anhydrides (A), CHO (B) and THF (C). | |
In addition to THF, the more challenging highly stable 1,4-dioxane (OX) was investigated. Dioxane is commonly regarded as a stable solvent having inherently lower ring strain of 14 kJ mol−1 compared to that of THF at 25 kJ mol−1.57 Although ring-opening reactions of 1,4-dioxane have been reported, they are all quantitative reactions, not polymerizations.58,59 Therefore, there is no report of polymers obtained directly from 1,4-dioxane in the literature. Polymerization using a [PA]:[CHO]:[OX]:[SnOct2] ratio of 50:150:800:1 was carried out at 80 °C for 6 h (Table 2, entry 8). To our surprise, 1,4-dioxane could be incorporated into the polymer similar to THF having a dioxane selectivity of 11%. The obtained polymer contained 8% AB, 81% ABx, and 11% ABxC (see the ESI† for sequence analysis). The polymerization with OX could be extended to SA (Table 2, entry 9 and Fig. 6c) under the same conditions where 9% selectivity for dioxane was observed furnishing the polymer containing 27% AB, 64% ABx, and 9% ABxC. The presence of –CH2OC(O)– protons at 4.25 ppm (protons d in Fig. 6c) and a single diffusion coefficient in the DOSY NMR confirmed the dioxane incorporation in the copolymer (Fig. S79†). Significantly lower Tg values (15–30 °C) were also observed in ABxC polymers compared to ABx polymers for succinic anhydride and phthalic anhydride as shown in Fig. 8b.
Degradation of ABx and ABxC polymers
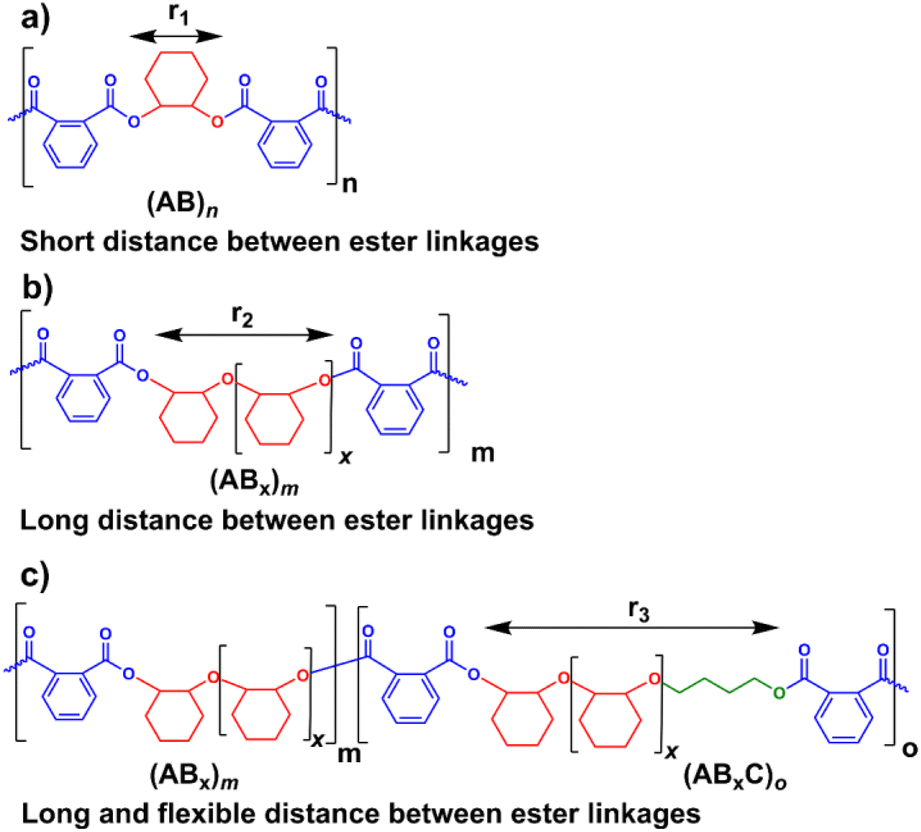
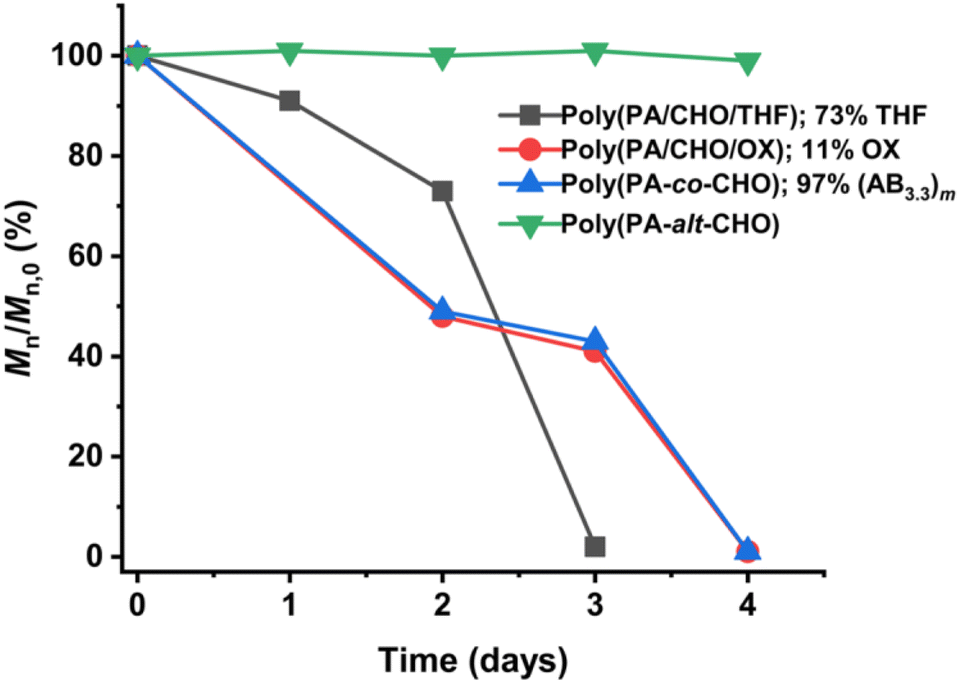
Several reports suggested that the increasing distance between ester linkages can accelerate the degradation rates of polyesters.31,60 Therefore, ABx and ABxC polymer sequences having long and flexible linkers between the ester linkages (Bx or BxC) might demonstrate higher hydrolysis rates compared to the shorter distance between the ester linkages of the alternating (AB)n polyester (Scheme 4, r1 < r2 < r3). Different polymer structures (ABx and ABxC) were evaluated for degradability compared to the conventional alternate (AB)n polyester targeted for hydrolysis at the ester bonds. The polymers having similar molecular weights in the range of 4.4–6.5 kDa were selected (Table S6†). The polymers were purified by precipitation in excess cold methanol 3 times and were subsequently subjected to the degradation test in 5 M NaOH at 70 °C for 4 days as shown in Fig. 9. Poly(PA-alt-CHO) (alternating (AB)n polymer) was intact as the molecular weight of poly(PA-alt-CHO) remained rather constant during the testing period. On the other hand, poly(PA/CHO) (3% AB, 97% AB3.3) and poly(PA/CHO/OX) (8% AB, 81% AB2.7, 11% AB2.7C) degraded rapidly at a similar rate within 4 days. This is possibly due to the longer distance between ester linkages found in (ABx)m sequences as shown in Scheme 4a and b where r2 > r1. In addition, the low amount of the OX content (11%) in the polymer may not yet cause substantial differences compared to the parent ABx polymer. Interestingly, poly(PA/CHO/THF) (4% AB, 23% AB2.5, 73% AB2.5C) degraded rapidly in 3 days. The APCI mass spectra of the degraded components were recorded and analyzed to be Bx and BxC moieties with x = 2,3 having B2C as the majority unit (Fig. S58†). This result indicated that only ester bonds (A–B and C–A) were hydrolyzed during the degradation. The degradation appeared to be slow at first to allow water diffusion into the polymer but subsequently accelerated as a result of enhanced hydrolysis at the –C(O)-O(CH2)4– ester bonds. The longer distances (r2 and r3) between ester linkages found in (ABx)m and (ABxC)o sequences in poly(PA/CHO/THF) also have a significant impact on the degradation rate of the polymer backbone allowing faster hydrolysis of the ester bonds. In addition, a large number of highly flexible and hydrolytically more accessible methylene units from THF (Scheme 4c) could also take part to accelerate polymer degradation.
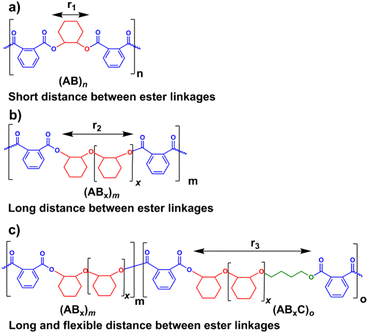 |
| | Scheme 4 Increasing distances between ester linkages in AB, ABx, and ABxC sequences. | |
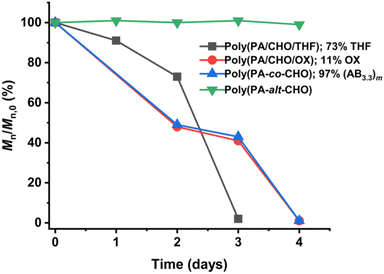 |
| | Fig. 9 Degradation profiles of AB, ABx, and ABxC (C = THF, OX) polymers. | |
Conclusions
Commercially available SnOct2 was demonstrated as a simple, inexpensive, versatile, and highly efficient catalyst for the ring-opening co(ter)polymerization of cyclic anhydrides (A), CHO (B), and THF or 1,4-dioxane (C) without the need for cocatalysts. In addition to the alternate (AB)n polymers commonly presented in the literature, the poly(ester-ether) of ABx type having an adjustable ether length (x) was synthesized with exceptionally high purity of up to 99%. The kinetic studies revealed a rate law of rate = kp[SA][CHO]2[SnOct2] supporting multiple epoxide insertions to form ABx polymer sequences. The versatility of SnOct2 was demonstrated and allowed the ABx polymer to seamlessly integrate with biodegradable polylactides and polyurethanes in one pot. In the presence of THF or 1,4-dioxane, a new poly(ester-ether) of ABxC sequences was synthesized with impressive selectivity of the third component of up to 74% for THF. This is also the first time that 1,4-dioxane has been directly incorporated into the polymer structures with 11% selectivity. The incorporation of the third component (C) was shown to significantly reduce the Tg value by 40–50 °C and increase the degradation rate of the polymers due to the flexible methylene units. The unique catalytic efficiency of SnOct2 has substantial implications for polymer engineering and synthesis because it allows new polymer architectures of ABx and ABxC sequences to effortlessly integrate with other existing ROP of cyclic esters widely used in biomedical and packaging applications through the use of a conventional air-stable SnOct2 catalyst.
Author contributions
Nattawat Jabprakon: investigation, visualization, formal analysis, and writing – original draft; Phongnarin Chumsaeng: visualization, formal analysis; Khamphee Phomphrai: conceptualization, validation, editing, and supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge financial support from the Vidyasirimedhi Institute of Science and Technology (M22KHP-VIS010) and the National Research Council of Thailand (NRCT) (No. N42A650196).
References
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- X. Yu, J. Jia, S. Xu, K. U. Lao, M. J. Sanford, R. K. Ramakrishnan, S. I. Nazarenko, T. R. Hoye, G. W. Coates and R. A. DiStasio Jr., Nat. Commun., 2018, 9, 2880–2888 CrossRef PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed.
- N. Laiwattanapaisarn, A. Virachotikul, P. Chumsaeng, T. Jaenjai and K. Phomphrai, Inorg. Chem., 2022, 61, 20616–20628 CrossRef CAS PubMed.
- F. D. Monica and A. W. Kleij, ACS Sustainable Chem. Eng., 2021, 9, 2619–2625 CrossRef CAS.
- F. Santulli, I. D'Auria, L. Boggioni, S. Losio, M. Proverbio, C. Costabile and M. Mazzeo, Organometallics, 2020, 39, 1213–1220 CrossRef CAS.
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS PubMed.
- X. Wang, Z. Huo, X. Xie, N. Shanaiah and R. Tong, Chem. – Asian J., 2023, 18, e202201147 CrossRef CAS PubMed.
- A. Takasu, M. Ito, Y. Inai, T. Herabayashi and Y. Nishimura, Polym. J., 1999, 31, 961–969 CrossRef CAS.
- Y. Maeda, A. Nakayama, L. Arvanityannis, N. Kawasaki, K. Hayashi, N. Yamamoto and S. Aiba, Polym. J., 2000, 32, 307–315 CrossRef CAS.
- Y. Maeda, A. Nakayama, N. Kawasaki, K. Hayashi, S. Aiba and N. Yamamoto, Polymer, 1997, 38, 4719–4725 CrossRef CAS.
- R. F. Fischer, J. Polym. Sci., 1960, 44, 155–172 CrossRef CAS.
- E. Fazekas, P. A. Lowy, M. A. Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252–3264 CrossRef CAS PubMed.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- W. T. Diment, W. Lindeboom, F. Fiorentini, A. C. Deacy and C. K. Williams, Acc. Chem. Res., 2022, 55, 1997–2010 CrossRef CAS PubMed.
- W. T. Diment and C. K. Williams, Chem. Sci., 2022, 13, 8543–8549 RSC.
- A. J. Plajer and C. K. Williams, ACS Catal., 2021, 11, 14819–14828 CrossRef CAS.
- N. V. Reis, A. C. Deacy, G. Rosetto, C. B. Durr and C. K. Williams, Chem. – Eur. J., 2022, 28, e202104198 CrossRef CAS PubMed.
- A. Thevenon, J. A. Garden, A. J. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS PubMed.
- T. Ungpittagul, T. Jaenjai, T. Roongcharoen, S. Namuangruk and K. Phomphrai, Macromolecules, 2020, 53, 9869–9877 CrossRef CAS.
- N. Yuntawattana, G. L. Gregory, L. P. Carrodeguas and C. K. Williams, ACS Macro Lett., 2021, 10, 774–779 CrossRef CAS PubMed.
- S. Yimthachote, P. Chumsaeng and K. Phomphrai, Dalton Trans., 2022, 51, 509–517 RSC.
- W. Thumrongpatanaraks, T. Pongpanit, P. Chumsaeng, T. Jaenjai, S. Yimthachote and K. Phomphrai, ChemistrySelect, 2022, 7, e202104450 CrossRef CAS.
- L. Cui, Y. Liu, B.-H. Ren and X.-B. Lu, Macromolecules, 2022, 55, 3541–3549 CrossRef CAS.
- R. W. F. Kerr and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 6882–6893 CrossRef CAS PubMed.
- M. A. Carnahan, C. Middleton, J. Kim, T. Kim and M. W. Grinstaff, J. Am. Chem. Soc., 2002, 124, 5291–5293 CrossRef CAS PubMed.
- C. Diaz and P. Mehrkhodavandi, Polym. Chem., 2021, 12, 783–806 RSC.
- R. Wang, H. Zhang, M. Jiang, Z. Wang and G. Zhou, Macromolecules, 2021, 55, 190–200 CrossRef.
- M. Hans, H. Keul and M. Moeller, Biomacromolecules, 2008, 9, 2954–2962 CrossRef CAS PubMed.
- F. Jia, X. Lu, D. Wang, X. Cao, X. Tan, H. Lu and K. Zhang, J. Am. Chem. Soc., 2017, 139, 10605–10608 CrossRef CAS PubMed.
- S. Theiler, S. E. Diamantouros, S. Jockenhoevel, H. Keul and M. Moeller, Polym. Chem., 2011, 2, 2273–2283 RSC.
- R. van Dijkhuizen-Radersma, S. Metairie, J. R. Roosma, K. de Groot and J. M. Bezemer, J. Controlled Release, 2005, 101, 175–186 CrossRef CAS PubMed.
- D. Merckle, O. King and A. C. Weems, ACS Sustainable Chem. Eng., 2023, 11, 2219–2228 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Polym. Chem., 2022, 13, 1618–1647 RSC.
- S. M. Weidner, A. Meyer, J. Falkenhagen and H. R. Kricheldorf, Eur. Polym. J., 2021, 153, 110508–110517 CrossRef CAS.
- C.-M. Chen, X. Xu, H.-Y. Ji, B. Wang, L. Pan, Y. Luo and Y.-S. Li, Macromolecules, 2021, 54, 713–724 CrossRef CAS.
- X. Xia, T. Gao, F. Li, R. Suzuki, T. Isono and T. Satoh, Macromolecules, 2022, 56, 92–103 CrossRef.
- X. Xia, T. Gao, F. Li, R. Suzuki, T. Isono and T. Satoh, J. Am. Chem. Soc., 2022, 144, 17905–17915 CrossRef CAS PubMed.
- X. Xia, R. Suzuki, K. Takojima, D.-H. Jiang, T. Isono and T. Satoh, ACS Catal., 2021, 11, 5999–6009 CrossRef CAS.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- H. Xie, L. Zheng, J. Feng, X. Wang, S. Kuang, L. Zhou, J. Jiang, Y. Xu, Y. Zhao and Z. Xu, Polym. Chem., 2023, 14, 1630–1638 RSC.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
- K. Ambrose, J. N. Murphy and C. M. Kozak, Macromolecules, 2019, 52, 7403–7412 CrossRef CAS.
- H. K. Ryu, D. Y. Bae, H. Lim, E. Lee and K.-s. Son, Polym. Chem., 2020, 11, 3756–3761 RSC.
- M. J. Sanford, L. P. Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- S. Ghosh, E. Glöckler, C. Wölper, J. Linders, N. Janoszka, A. H. Gröschel and S. Schulz, Eur. J. Inorg. Chem., 2022, e202101017 CAS.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS PubMed.
- T. Saito, K. Takojima, T. Oyama, S. Hatanaka, T. Konno, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, ACS Sustainable Chem. Eng., 2019, 7, 8868–8875 CrossRef CAS.
- S. Uma, J. Singh and V. Thakral, Inorg. Chem., 2009, 48, 11624–11630 CrossRef CAS PubMed.
- H. K. Eigenmann, D. M. Golden and S. W. Benson, J. Phys. Chem., 1973, 77, 1687–1691 CrossRef CAS.
- B. Birkmann, T. Voss, S. J. Geier, M. Ullrich, G. Kehr, G. Erker and D. W. Stephan, Organometallics, 2010, 29, 5310–5319 CrossRef CAS.
- Z. Zhang, W. Sun and Z. Cao, Chin. J. Org. Chem., 2018, 38, 1292–1318 CrossRef CAS.
- S. P. Zustiak and J. B. Leach, Biomacromolecules, 2010, 11, 1348–1357 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experiments and sequence calculations, NMR spectra, DSC data. See DOI: https://doi.org/10.1039/d3py00605k |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*