
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZr-containing UiO-66 metal–organic frameworks as efficient heterogeneous catalysts for glycerol valorization: synthesis of hyacinth and other glyceryl acetal fragrances†
A.
Rapeyko
 *,
J. C.
Díaz Infante
and
F. X.
Llabrés i Xamena
*
*,
J. C.
Díaz Infante
and
F. X.
Llabrés i Xamena
*
Instituto de Tecnología Química UPV-CSIC, Universitat Politècnica de València, Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos, s/n, Valencia 46022, Spain. E-mail: arapeyko@itq.upv.es; fllabres@itq.upv.es
First published on 8th February 2023
Abstract
Zr-containing UiO-66 and UiO-66-NH2 are good heterogeneous catalysts for the acetalization of phenylacetaldehyde with glycerol, producing the corresponding hyacinth fragrance in high yields after short (2 h) reaction times. Mixtures of 1,3-dioxolanes and 1,3-dioxanes are obtained, whose ratios can be modified between 2.8 and 4.6 depending on the catalyst used, the amount of missing linker defects of the solid, and the reaction time. The catalysts are stable under the reaction conditions used, and they can be reused without loss of activity or selectivity. The scope of UiO-66 materials is demonstrated for the formation of other glyceryl acetals of interest for the flavoring industry, which represents an interesting route for glycerol valorization.
Design, System, ApplicationHerein, zirconium-containing UiO-66 metal–organic frameworks are used as heterogeneous catalysts for the acetalization of phenylacetaldehyde with glycerol to produce hyacinth fragrances. This solid contains the necessary Brønsted acid sites for this reaction, arising from the presence of strongly polarized H2O molecules adsorbed onto coordinatively unsaturated Zr4+ associated with missing linker defects. We observe a direct correlation between the number of such defective sites in the solid and its catalytic activity for this reaction. Meanwhile, introduction of –NH2 groups in the organic linkers, together with the concentration of defects in the solid, allows us to modify the dioxolane/dioxane ratio in the reaction product between 2.8 and 4.6. This affords a means to fine-tune the organoleptic characteristics of the final hyacinth fragrance mixture. Furthermore, a fair scope of UiO-66 has been demonstrated for the formation of other glyceryl acetals with other aldehydes and ketones of interest in the flavoring industry. Overall, our work represents an interesting route for glycerol valorization and the production of new molecules for the flavoring industry, since the production of acetals can drastically change the viscosity, solubility and aroma properties of existing aldehyde and ketone fragrances. |
Introduction
The increasing demand for environmentally friendly renewable energy sources has resulted in a fast growth of biofuel production. Biofuels produced from sugar, starch, vegetable oil, algae or lignocellulosic biomass are considered as promising green alternatives to non-renewable fossil fuels.1 Among different kinds of biofuels, the production of biodiesel by transesterification of triglycerides has increased considerably in the last decade. As a result, a large amount of glycerol is produced as a main by-product of the biodiesel process. It is estimated that 1 kg of glycerol is formed for every 9 kg of biodiesel during the transesterification of triglycerides.2 Due to increasing oversupply of glycerol in the market and its biomass nature, the development of new processes for the transformation of glycerol into value added products (glycerol valorization) is now a subject of interest for many researchers and industries.Glycerol is considered as one of the main biomass platform molecules3 that can be easily transformed into many valuable chemical products via hydrogenation,4 oxidation,5 dehydration,6 esterification,7 etherification8 or acetalization,9,10 among other chemical reactions (Fig. 1).
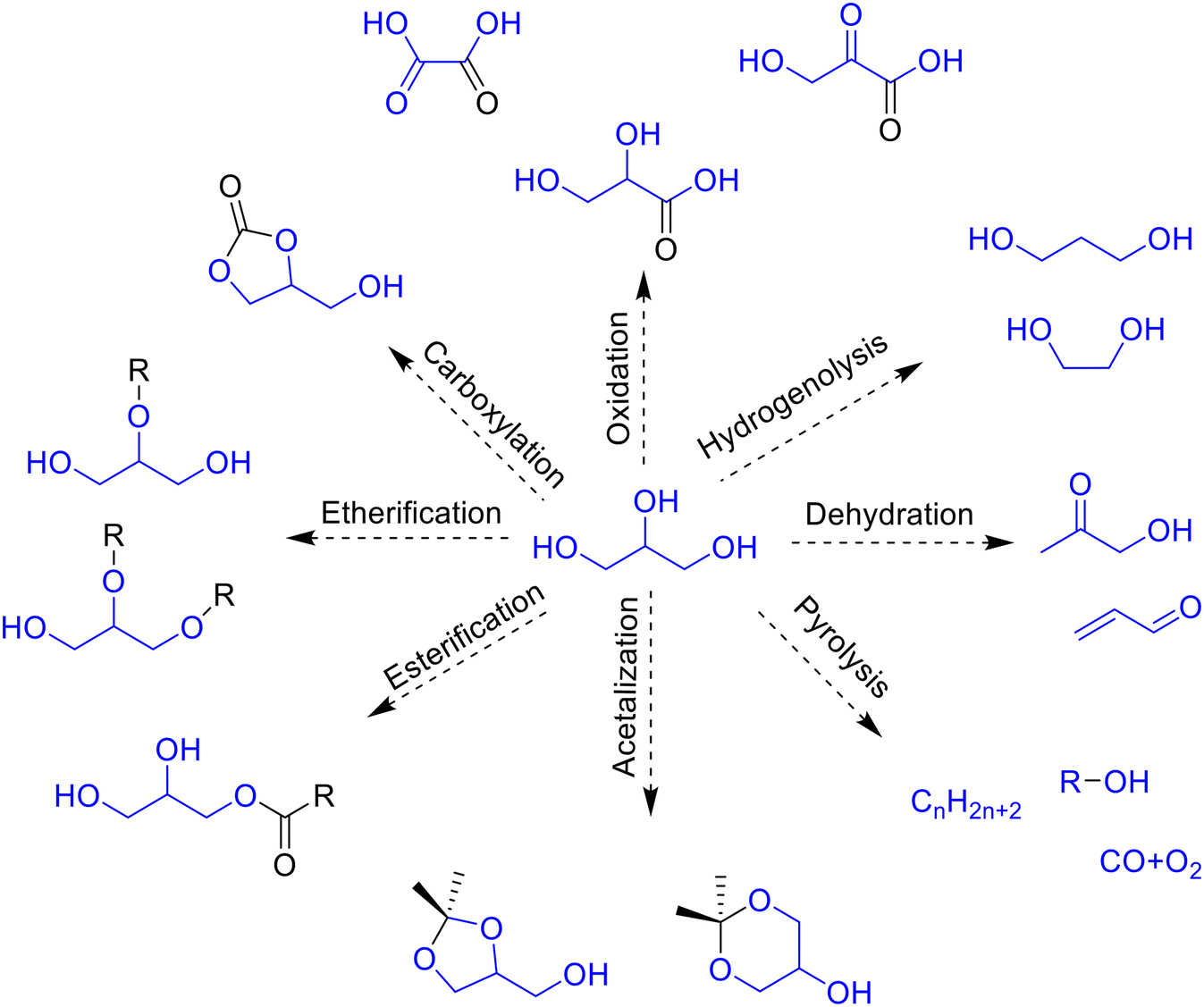 | ||
| Fig. 1 Main catalytic routes for glycerol valorization. | ||
One promising route for glycerol valorization is through the acetalization reaction with carbonyl compounds to obtain cyclic ketals (dioxanes and dioxolanes) with a wide range of applications as fragrances,11,12 pharmaceuticals,13 surfactants,14 or fuel additives.15–18 In particular, it is well known that conversion of flavor carbonyl compounds into acetals can drastically change their viscosity, solubility and aroma properties.19 For example, glycerol acetals with orange blossom, hyacinth, menthone or vanilla scents20,21 are widely used in the fragrance industry.
Acetalization of glycerol with aldehydes and ketones is an acid catalyzed reaction that can be promoted by homogeneous or heterogeneous catalysts. The reaction produces cyclic acetals, 1,3-dioxolanes and 1,3-dioxanes, as main products (see Fig. 2). Homogeneous Brønsted (e.g., H2SO4, HCl, p-TSOH22,23) and Lewis (e.g., AuCl3, AgBF4, FeCl4·H2O (ref. 24 and 25) acid catalysts have been previously used for acetalization of glycerol. However, the use of homogeneous catalysts presents several economic and environmental drawbacks, such as difficult recovery and reuse, generation of toxic solid waste, and multiple steps for product purification. Therefore, the development of new heterogeneous sustainable catalytic systems is a main subject of recent research. Among them, zeolites,26,27 mesoporous silicas,17,28 carbons,21,29 heteropolyacids30 and metal–organic frameworks15,31 have been recently reported as promising heterogeneous acid catalysts for acetalization reactions with glycerol. In this sense, metal–organic frameworks are widely used in acid catalysis32–36 due to the presence of Lewis (unsaturated metal sites) or Brønsted acid groups in their structure. In particular, UiO-66 (ref. 15) and isostructural MIL-100 and MIL-53 (ref. 37) metal–organic frameworks have been studied for the acetalization of acetone and levulinic acid with glycerol. Additionally, the well-defined porous structure and high surface area of MOFs, as well as their good thermal and chemical stability, make them feasible green alternatives to conventional homogeneous catalysts.
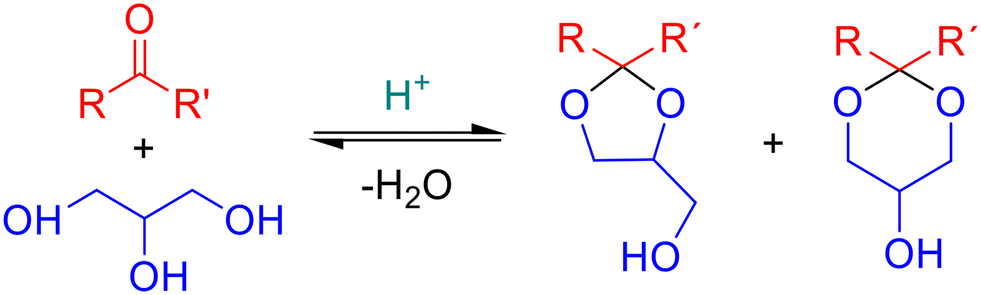 | ||
| Fig. 2 General scheme of acetalization with glycerol. | ||
Continuing with our exploration on the catalytic potential of zirconium MOFs in acid catalyzed reactions for biomass revalorization, herein we have applied UiO-66 in the acetalization of phenylacetaldehyde with glycerol to produce cyclic acetals with hyacinth fragrance. To date, only a couple of examples of heterogeneous catalysts have been reported for this reaction. Climent et al.20 have studied the catalytic performance of zeolites with different structures, pore sizes and hydrophobic properties for the synthesis of hyacinth, vanilla, and blossom orange fragrances. High yields of hyacinth acetals (as mixtures of dioxanes/dioxolanes) were achieved using large pore USY and beta zeolites. More recently, Silva et al.21 have used oxidized and sulfonated-activated carbons, attaining 95% conversion to the corresponding acetals. However, to the best of our knowledge, MOFs have not been employed so far for this reaction. Recently, our group have reported high catalytic activities of Zr-containing UiO-66 catalysts for the selective ketalization and esterification of levulinic acid.38–40 We have observed that strongly polarized water molecules adsorbed onto Zr4+ sites associated with linker defects introduce relatively strong Brønsted acid sites on hydrated UiO-66, which are responsible for a high catalytic activity. However, when the material is dehydrated and these polarized water molecules are removed, the Brønsted acidity is lost, leaving coordinately unsaturated Zr4+ sites at the linker defects that can act as Lewis acid centers. The defect-dependent reactivity and tunable Brønsted/Lewis acid character of UiO-66 MOFs, along with high porosity and thermal stability, provide them with unique properties that can be exploited to design highly selective catalytic processes.
Therefore, the aim of this work is to continue exploring the possibilities of UiO-66 MOFs as heterogeneous catalysts in industrially and environmentally relevant organic reactions. Particularly, we have used UiO-66 as a heterogeneous catalyst for the valorization of glycerol into industrially important hyacinth fragrance molecules. We have paid special attention to the analysis of the influence of the number of linker defects on the reactivity and selectivity of the reaction.
Experimental section
Materials and reagents
Glycerol, phenylacetaldehyde, toluene, ZrCl4, ZrOCl2·8H2O, N,N-dimethylformamide, benzene-1,3,5-tricarboxylic acid (BTC), 1,4-dioxane, hydrochloric acid, formic acid, and benzene-1,4-dicarboxylic acid (BDC) were supplied by Sigma-Aldrich and used as received.Synthesis of catalysts
Zr containing UiO-66-X samples with different percentages of linker defects X (%) were synthesized in the absence of a modulator following the procedure reported previously.41 Briefly, 375 mg of ZrCl4 and 370 mg of 1,4-benzenedicarboxylic acid (BDC) were dissolved in 20 mL and 25 mL of N,N-dimethylformamide (DMF), respectively. Both solutions were sonicated for 15 min and mixed in a screw-capped glass bottle. The resulting mixture was placed in an oven at 80 °C for 24 h and then the temperature was raised to 100 °C and kept for another 24 h. The resulting white precipitate was recovered by vacuum filtration, washed with DMF and CH2Cl2 and dried under vacuum at ambient temperature. Three samples of UiO-66-X% with different amounts of linker defects (calculated using TGA curves, Fig. S3 in the ESI†) were prepared. It is important to note that these missing linker defects have a stochastic origin, due to very small local variations in nucleation and crystal growth from batch to batch; therefore, it is not possible to know a priori what will be the final concentration of defects in each sample.Zr containing UiO-66-NH2-X samples were prepared following the same procedure as described above for the UiO-66-X samples but using 2-aminoterephthalic acid (400 mg) instead of BDC.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ratio. Then, 158 mg of trimesic acid (BTC) was dissolved in 11.2 mL of DMF. Both solutions were mixed and placed in a Teflon lined autoclave. Then, the autoclave was introduced into an oven at 130 °C for 48 h. After cooling down to room temperature, the material was recovered by centrifugation. Finally, the obtained solid was washed 3 times with DMF (changing the solvent every 15 min) and 3 times with ethanol (changing the solvent every 30 min). After removing the solvent by centrifugation, it was dried in air at ambient temperature.
:1 (v/v) ratio. Then, 158 mg of trimesic acid (BTC) was dissolved in 11.2 mL of DMF. Both solutions were mixed and placed in a Teflon lined autoclave. Then, the autoclave was introduced into an oven at 130 °C for 48 h. After cooling down to room temperature, the material was recovered by centrifugation. Finally, the obtained solid was washed 3 times with DMF (changing the solvent every 15 min) and 3 times with ethanol (changing the solvent every 30 min). After removing the solvent by centrifugation, it was dried in air at ambient temperature.
Characterization of catalysts
All the synthesized catalyst samples were characterized by powder X-ray diffraction (Phillips X'Pert, Cu Kα radiation) to confirm the expected structure and crystallinity of the materials. XRD diffraction patterns of UiO-66-X, UiO-66-HN2-X, MOF-808 and MOF-808-HCl samples are shown in Fig. S1 and S2 (ESI†).Thermogravimetric analysis (TGA) of the synthesized MOFs was performed under an air flow and a heating ramp of 10 °C min−1 using a NETZSCH STA 449 F3 Jupiter analyzer. The TG curves obtained are presented in Fig. S3 (see the ESI†). From these TG curves, the amount of linker defects in each UiO-66 sample was determined following the method reported by Valenzano et al.46 as described in the ESI.†
Textural properties of the UiO-66 samples were determined from the corresponding N2 adsorption isotherms (at 77 K) and the results are summarized in Table S1 (ESI†). The values obtained for the specific surface area (SBET) and pore volume are in line with other data reported in the literature for UiO-66 materials.
General procedure for the acetalization of phenylacetaldehyde with glycerol
Acetalization of phenylacetaldehyde (PhA) with glycerol was carried out with the following procedure: 2 mmol of PhA, 4 mmol of glycerol, 6 mL of toluene and 10 mg of catalyst (ca. 1.5 mol% Zr with respect to PhA) were placed into a round bottom two-necked flask connected to Dean–Stark apparatus to remove water formed during the reaction. The mixture was stirred at 400 rpm (which is enough to avoid external diffusion control of the reaction) under reflux. The reaction was followed by taking sample aliquots at regular time intervals and analyzing them by Gas Chromatography (GC) using a Shimadzu 2010 instrument equipped with a capillary column (HP5, 30 m × 0.25 mm × 0.25 μm) and a flame ionization detector (FID), and using dodecane as an internal standard. All the products were identified by combined gas chromatography-mass spectrometry analysis (GC-MS) and nuclear magnetic resonance (NMR). 1H NMR spectra were recorded at 400 MHz on a Bruker 300 Ultrashield instrument, using chloroform-d (CDCl3) as a solvent. Conversion, yield and selectivity were determined from the areas of the corresponding GC peaks upon correction by the response factor of each product determined for the pure compounds with respect to the internal standard (dodecane).Regeneration and reuse of the catalyst
After each reaction cycle, the UiO-66 catalyst was separated from the reaction mixture by filtration, thoroughly washed with ethanol and dried overnight at room temperature before next use.Results and discussion
Acetalization of phenylacetaldehyde with glycerol over UiO-66-X
The acetalization of phenylacetaldehyde (PhA) with glycerol in the presence of UiO-66 was carried out under reflux of toluene and using Dean–Stark apparatus to remove water formed during the reaction. In all cases, four main products were detected by GC-MS, corresponding to the E and Z isomers of 1,3-dioxolane (2-benzyl-4-hydroxymethyl-1,3-dioxolane) and 1,3-dioxane (2-benzyl-5-hydroxy-1,3-dioxane), as shown in Fig. 3.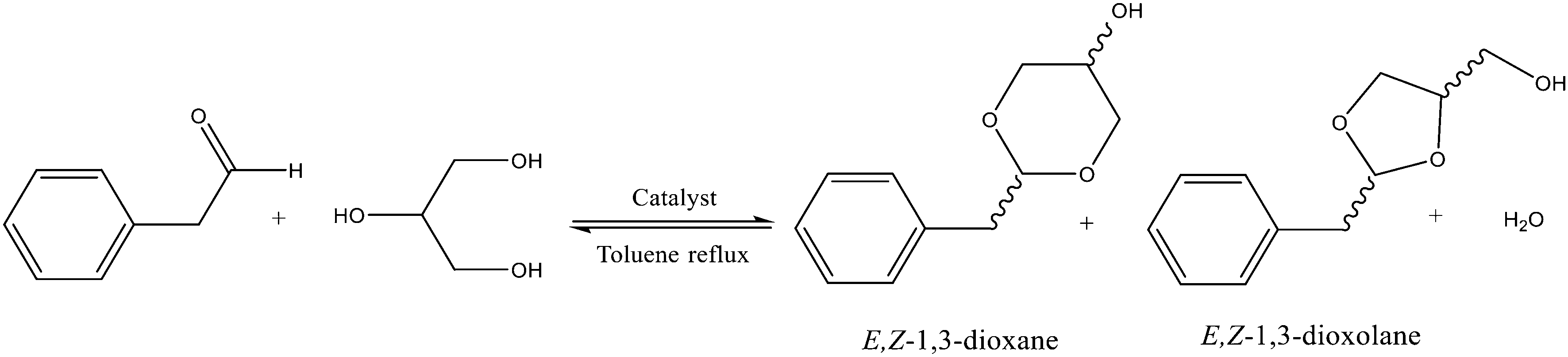 | ||
| Fig. 3 General reaction scheme of the formation of cyclic acetals, 1,3-dioxane and 1,3-dioxolane. | ||
The MS spectra of all four products (see Fig. S4†) showed a strong m/z 103 ion fragment characteristic of glyceryl acetals.47,48 However, the mass spectra of the four detected isomers were almost identical (see Fig. S4†); so, an accurate assignment of E and Z dioxane/dioxolane structures was done by NMR analysis. Thus, the purified crude obtained at the end of the reaction (24 h) was analyzed by 1H NMR spectroscopy. The spectra containing a mixture of the four acetals showed four triplets (Fig. S5†), which can be attributed to the hydrogen attached to the carbonyl carbon in each acetal structure. The shifts at δH = 5.16 and 5.08 ppm were assigned to E and Z dioxolanes, while the shifts at δH = 4.68 and 4.53 ppm were attributed to E and Z dioxanes, in accordance with the data reported previously by Silva21 and by Pawar.49 Quantitative analysis of these 1H-NMR signals was compared with the analysis obtained by GC using dodecane as the internal standard, which allowed a straightforward assignment of the four chromatographic peaks to their corresponding isomers (see Table S2†).
Concerning the time-evolution of products, the acetalization of PA with glycerol in the presence of UiO-66-15 (a sample containing 15% missing linker defects) resulted in high conversion (98.5%) and 100% selectivity to cyclic acetals (mixture of dioxanes and dioxolanes) after 24 h of reaction (Fig. 4). 1,3-Dioxolanes with an overall yield of 77.7% were detected as major products, while 1,3-dioxanes were obtained as minor products with 20.8% yield. In order to confirm the catalytic contribution of UiO-66, the reaction was performed in the absence of any catalyst. The results of the blank experiment (see entry 1, Table 1) showed that only 34.7% conversion was achieved after 24 h of reaction.
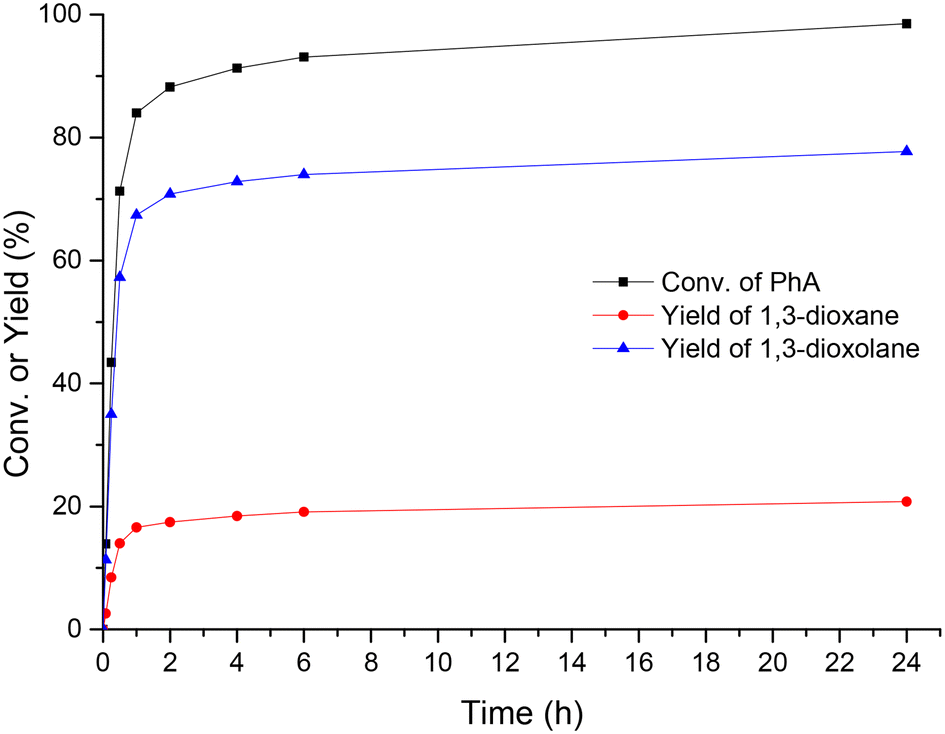 | ||
| Fig. 4 Yield/conversion & time plots for the acetalization reaction of phenylacetaldehyde with glycerol using UiO-66-15 as a catalyst. Reaction conditions: PhA – 2 mmol; glycerol – 4 mmol; catalyst – 10 mg (1.5 mol% Zr); reflux of toluene (6 ml), Dean–Stark. | ||
| Entry | Catalyst | Conv. (%) PhA | Yield (%) dioxanes (1) | Yield (%) dioxolanes (2) | Ratio 2:1 |
Zrtotal TOF (h−1) | Zrdefect TOF (h−1) |
|---|---|---|---|---|---|---|---|
| Reaction conditions: PhA – 2 mmol; glycerol – 4 mmol; catalyst – 10 mg (ca. 1.5 mol% Zr for the UiO-66 materials); reflux of toluene (6 ml), time 24 h. Dean–Stark.a Turnover frequency (TOF) is calculated as moles of PhA converted per mol of Zrtotal/Zrdefect.b Turnover frequency (TOF) is calculated as moles of PhA converted per hour at a short reaction time and low PhA conversion.c Dehydrated UiO-66-15, obtained by pretreating the catalyst at 150 °C for 2 h under a vacuum. | |||||||
| 1 | — | 34.7 | 5.0 | 29.8 | 6.0 | — | — |
| 2 | UiO-66-15 | 98.5 | 20.8 | 77.7 | 3.7 | 97 | 325 |
| 3 | UiO-66-15c | 96.0 | 17.2 | 78.8 | 4.5 | 44 | 84 |
| 4 | MOF-808 | 29.2 | 4.3 | 24.9 | 5.8 | 0.7 | — |
| 5 | MOF-808-HCl | 97.9 | 24.7 | 73.2 | 3.0 | n.d. | |
| 6 | Zr-MCM-41 | 97.6 | 22.7 | 74.9 | 3.3 | 59 | — |
The high catalytic activity of UiO-66 in the acetalization reaction is in line with previous results reported by our group for the esterification and ketalization of levulinic acid.40,50,51 According to the previous studies, the high catalytic performance of UiO-66 in acid catalyzed acetalization and esterification reactions is related to the presence of Brønsted-induced acid sites in its structure arising from strongly polarized water molecules adsorbed on defective Zr4+. When the Brønsted-induced acid sites present in hydrated UiO-66 were removed upon dehydration, the activity of the catalyst for esterification and ketalization reactions of levulinic acid was considerably decreased. In order to determine if the Brønsted-induced acid sites in hydrated UiO-66 are also responsible for its catalytic activity in the PhA acetalization reaction, an additional experiment was performed using dehydrated UiO-66-15 as a catalyst. According to our results (entry 3, Table 1), dehydrated UiO-66 also exhibits a very high PhA conversion (96% after 24 h). However, the initial reaction rate per mmol of Zrtotal or Zrdefect (see the TOF values in Table 1 and Fig. S6†) is significantly lower than that for dehydrated UiO-66. Note that Zrtotal is related to the overall content of zirconium present in the sample, whereas Zrdefect is related to accessible Zr4+ active sites only, which are associated to missing linker defects. As it was mentioned before,40,50,51 each missing ligand creates two coordinatively unsaturated Zr4+ active sites. Therefore, the number of active sites in each UiO-66 sample will correspond to twice the number of linker defects. Note that when UiO-66 is dehydrated, coordinated water molecules are removed, leaving coordinatively unsaturated (cus) Zr4+ Lewis acid sites. During the course of the acetalization reaction, glycerol can adsorb onto these cus sites, thus generating new Brønsted-induced acid sites (see Fig. 5), which are weaker than those formerly present in hydrated UiO-66.52 This explains the observed decrease of the catalytic activity of dehydrated versus hydrated UiO-66.
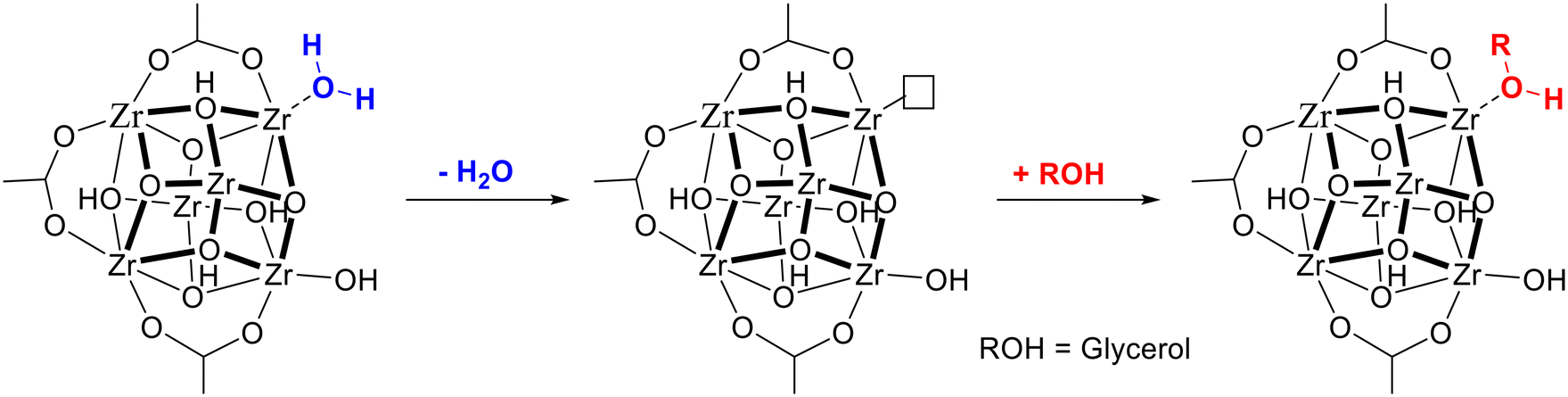 | ||
| Fig. 5 Removal of polarized H2O molecules during UiO-66 dehydration and formation of novel Brønsted-induced acid sites upon glycerol adsorption. | ||
For the sake of comparison with other Zr-containing catalysts, PhA acetalization was performed in the presence of MOF-808. This MOF contains hexanuclear Zr6 clusters similar to those in UiO-66, but with a different (wider) pore structure and nature of acid sites. Indeed, hydrated MOF-808 contains much weaker Brønsted-induced acid sites than UiO-66,51 which is due to the presence of geminal OH groups, which decreases the polarizing power of the Zr4+ sites over the adsorbed H2O molecules.50 As a result, the catalytic activity of MOF-808 for PhA acetalization is considerably lower compared to that of UiO-66: only 21.7% PhA conversion was achieved after 24 h (see entry 4 in Table 1).
In order to increase the acidity of MOF-808 (and, hopefully, also its catalytic activity), the solid was subjected to a chlorination process analogous to that recently described by Liu et al. for Zr-containing NU-1000 (ref. 53) (see the Experimental section for details). This chlorination process clearly increased the Brønsted acid strength of MOF-808, as confirmed by direct pH measurements of methanol suspensions, following the procedure described before:50,54 the pH decreased from 5.8 for pristine MOF-808, down to 3.2 for the HCl-treated solid. Thus, the resulting MOF-808-HCl sample was used as a catalyst for PhA acetalization, affording a considerably better activity than non-treated MOF-808 (compare entries 4 and 5 in Table 1). This confirms the acetalization reaction of PhA catalyzed by Brønsted acid sites.
To complete the comparison of the catalytic activity of UiO-66 with other Zr heterogeneous catalysts, we prepared a Zr-grafted MCM-41 mesoporous silica material, as described in the Experimental section. This catalyst was found to be also active for the PhA acetalization with glycerol, affording 97.6% conversion after 24 h. However, the TOF calculated for this material was considerably lower than that obtained for hydrated UiO-66-15 (59 vs. 97 h−1, respectively).
Fig. S7† compares the time-conversion plots obtained for the catalysts considered in Table 1 with respect to hydrated UiO-66-15.
Effect of linker defects on the catalytic performance of UiO-66
In previous studies, we have observed that the number of linker defects can affect the catalytic activity of UiO-66 materials for levulinic acid esterification and acetalization reactions.39,51 In order to study how these defects influence the catalytic performance of UiO-66 for PhA acetalization, different catalysts were prepared by varying the type of organic linker (either terephthalic acid or 2-aminoterephthalic acid) and the amount of linker defects. These samples will be hereafter referred to as UiO-66-X and UiO-66-NH2-X, where X indicates the percentage of missing linkers in each material. The results obtained are summarized in Table 2. In all cases, all UiO-66-X and UiO-66-NH2-X samples tested exhibited high catalytic activities for this reaction, with PhA conversions in the range 88–92% after only 2 h of reaction. Mixtures of dioxane/dioxolane acetals in different ratios were detected as the only reaction products, being the dioxolanes the major products in all cases.| Catalyst | Time (h) | Conv. (%) PhA | Yield (%) dioxanes (1) | Yield (%) dioxolanes (2) | Ratio 2:1 |
TOF Zrtotal (h−1) | TOF Zrdefect (h−1) |
|---|---|---|---|---|---|---|---|
| Reaction conditions: PhA (2 mmol), Gly (4 mmol), MOF (1.5 mol% Zr) 10 mg, reflux of toluene (6 mL), Dean–Stark, 24 h.a Turnover frequency (TOF) is calculated as moles of PhA converted per mol of Zr total or Zrdefect and per hour at short reaction time. | |||||||
| UiO-66-12.4 | 2 | 89.6 | 18.9 | 70.7 | 3.7 | 94 | 377 |
| 24 | 97.9 | 22.3 | 75.6 | 3.4 | |||
| UiO-66-15.0 | 2 | 88.2 | 17.3 | 70.8 | 4.1 | 97 | 325 |
| 24 | 98.5 | 20.8 | 77.7 | 3.7 | |||
| UiO-66-25.6 | 2 | 84.5 | 15.2 | 69.3 | 4.6 | 201 | 392 |
| 24 | 96.6 | 18.7 | 77.9 | 4.2 | |||
| UiO-66-NH2-14.8 | 2 | 92.3 | 20.3 | 72.0 | 3.5 | 114 | 384 |
| 24 | 99.8 | 26.6 | 73.2 | 2.8 | |||
| UiO-66-NH2-15.2 | 2 | 90.4 | 18.6 | 71.9 | 3.9 | 102 | 335 |
| 24 | 97.5 | 21.7 | 75.8 | 3.5 | |||
| UiO-66-NH2-18.0 | 2 | 88.7 | 17.1 | 71.6 | 4.2 | 132 | 366 |
| 24 | 96.0 | 19.3 | 76.8 | 4.0 | |||
| 96 | 97.9 | 19.9 | 78.1 | 3.9 | |||
For both UiO-66-X and UiO-66-NH2-X series of samples, a progressive increase of the turnover frequency calculated per total zirconium of the catalyst, TOF (Zrtotal), was observed with the number of linker defects, irrespective of the ligand substituent of the MOF (Fig. 6a). However, the TOF calculated considering only the accessible Zr4+ sites associated to missing linker defects, TOF (Zrdefect), was maintained roughly constant (∼350 h−1) and independent of the number of linker defects (Fig. 6b). This relatively constant TOF (Zrdefect) value reflects that all the active centers of UiO-66 have basically the same intrinsic catalytic activity, so it can be considered that these active sites are isolated and independent from each other.
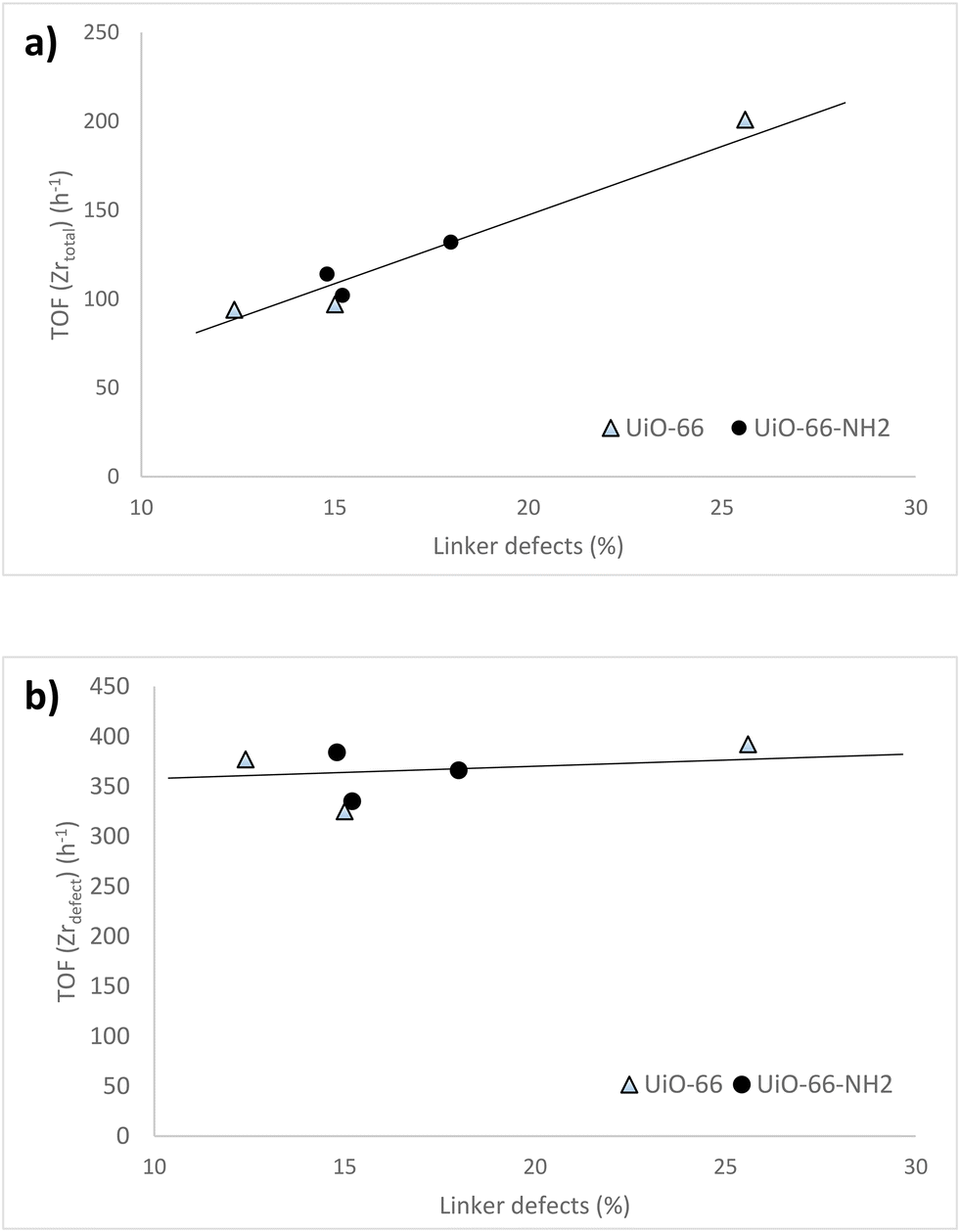 | ||
| Fig. 6 a) TOF (Zrtotal) & linker defect and b) TOF (Zrdefect) & linker defect plots for the acetalization reaction of phenylacetaldehyde with glycerol in the presence of UiO-66 and UiO-66-NH2 catalysts. Reaction conditions: PhA (2 mmol), glycerol (4 mmol), MOF 10 mg (ca. 1.5 mol% Zr), reflux of toluene (6 mL), Dean–Stark. | ||
Although mixtures of dioxanes/dioxolanes in different proportions are accepted as commercial hyacinth fragrances,20 it can be of practical interest to control the selectivity of both isomers and therefore the intensity of the final fragrance mixture. In this context, Climent et al.20 reported the influence of the pore size and structure of zeolites on their activity and relative yields of acetal isomers. They suggested that large pore USY and beta zeolites could promote the isomerization of dioxolanes to dioxanes, thus increasing the selectivity to dioxanes. In contrast, small pore ZSM-5 and unidirectional MOR zeolites not only showed lower conversions of PhA, but also afforded higher dioxolane/dioxane ratios. This fact was explained by the authors as being due to diffusional restrictions inside the zeolite pores, which prevented the formation of the bulky transition state of the isomerization reaction. In the case of the UiO-66-X and UiO-66-NH2-X catalysts, dioxolanes were always found to be the main reaction product, with dioxolane/dioxane ratios after 2 h of reaction ranging from 3.7 to 4.6 for UiO-66-X, and from 3.5 to 4.2 for UiO-66-NH2-X (2:1 ratio in Table 2). Although these variations are small, a progressive increase of the 2:1 ratio is observed with the amount of missing linker defects. These ratios are just slightly lower than those for the UiO-66-NH2-X series. At longer reaction times (24 h) and at practically full PhA conversion, a small decrease of the 2:1 ratio is observed in all cases, which reflects that the dioxolane-to-dioxane isomerization takes place only to a minor extent. To check whether the use of still longer reaction times could bring about further decrease of the 2:1 ratio, the acetalization reaction was carried out in the presence of UiO-66-NH2 for up to 96 h. However, we did not observe significant additional changes in the relative yields of dioxolanes and dioxanes (see last entry in Table 2).
In conclusion, although the observed variations of the 2:1 ratio were small, it was still possible to modify this value between 2.8 and 4.6, depending on the reaction conditions used. Short reaction times and the use of UiO-66 with a high content of defects favored the formation of dioxolanes (2:1 = 4.6), while longer reaction times and the use of UiO-66-NH2 with few ligand defects have the opposite effect (2:1 = 2.8).
Reaction mechanism
According to the generally accepted mechanism of acetalization reaction and the results presented above for the UiO-66 materials, the two-step reaction mechanism shown in Fig. 7 was proposed. In the first step, addition of the primary (1a) or the secondary (1b) hydroxyl group of glycerol to PhA leads to the formation of the corresponding hemiacetal (HAc1 or/and HAc2). In the second step, the intramolecular addition of a second hydroxyl group to the intermediate hemiacetal finally yields the corresponding five- or six-membered cyclic acetal. Specifically, HAc1 can evolve into dioxane or dioxolane depending on whether the OH addition comes from the primary (2a) or the secondary (2b) hydroxyl group of glycerol, while HAc2 can only form dioxolane (2c), since the two remaining OH groups are equivalent. Both the first and second reaction steps produce a H2O molecule as a by-product, which is rapidly removed from the reaction medium by the Dean–Stark trap. Therefore, all the reactions depicted in Fig. 7 can be considered irreversible. The catalytic role of the (Brønsted) acid sites of UiO-66 would then be assisting the OH addition to PhA by increasing the electrophilic character of the carbonyl carbon, through a general acid catalysis mechanism.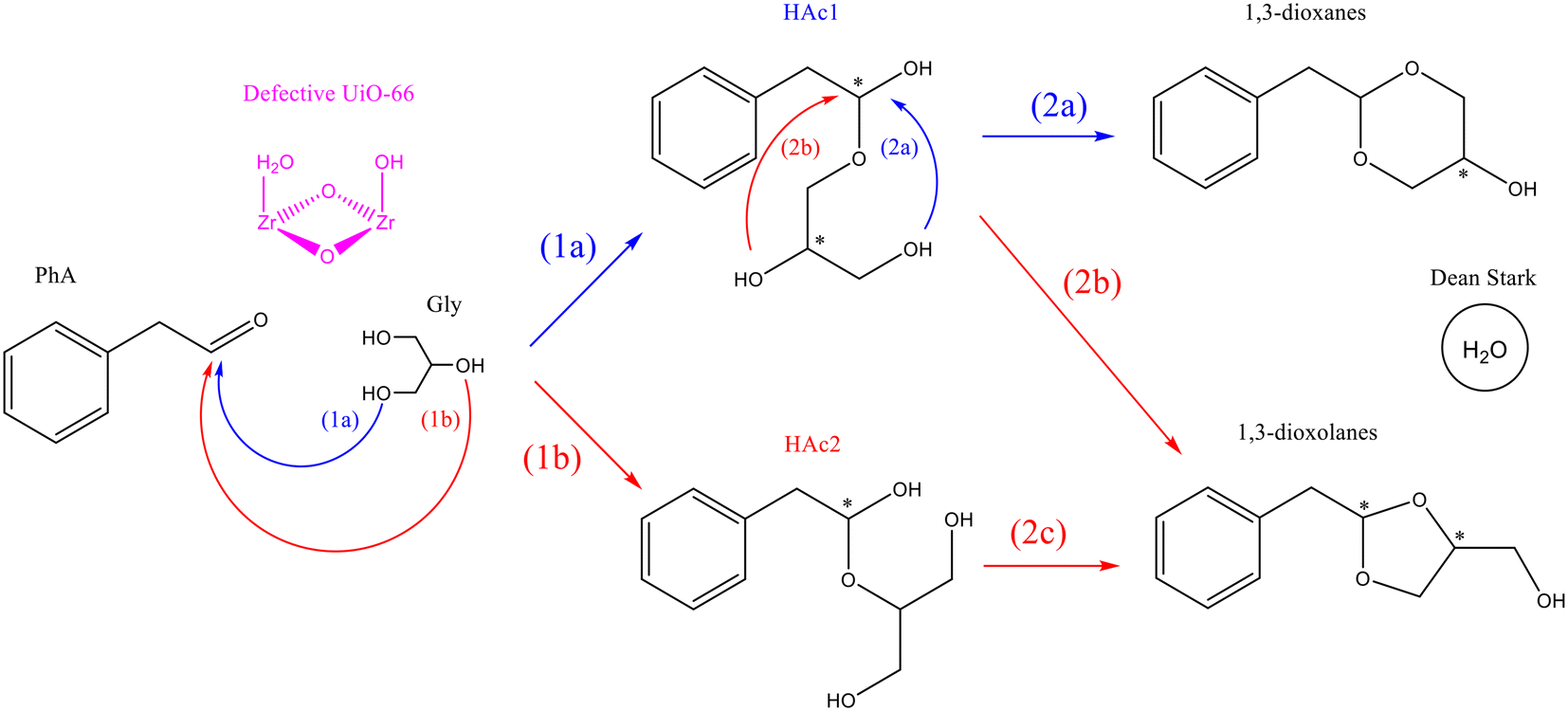 | ||
| Fig. 7 Proposed mechanism of acetalization of phenylacetaldehyde with glycerol in the presence of UiO-66. | ||
According to the catalytic results discussed above, the dioxolane/dioxane ratio is maintained quite constant during the reaction, even at very long reaction times, suggesting that the dioxolane-to-dioxane isomerization process does not take place within the UiO-66 pores, as was the case for zeolites. Therefore, the difference in selectivity of the two isomers in the final mixture should be determined by the relative reaction rate constants of the individual reactions leading to the formation of the hemiacetals (1a and 1b) and acetals (2a/2b and 2c).
Catalyst reusability and stability
To illustrate the reusability and stability of UiO-66 materials, Fig. 8 shows four consecutive reaction cycles carried out in the presence of the UiO-66-NH2-18 catalyst, though similar results were obtained for all the UiO-66 materials considered in this work (see Fig. S8a†). After each reaction cycle, the catalyst was separated from the reaction mixture by filtration, washed with toluene and ethanol and finally, dried at ambient temperature before next use. After four consecutive reuses, the conversion of PhA was maintained constant in the range of 93–95% after 6 h of reaction. The dioxolane/dioxane selectivity was also maintained after four consecutive reuses. Additionally, comparative XRD analysis revealed that the solid recovered after four catalytic runs still maintained the characteristic peaks of UiO-66 (see Fig. S8b†), confirming that the crystalline structure of UiO-66-NH2 was maintained even after several reuses.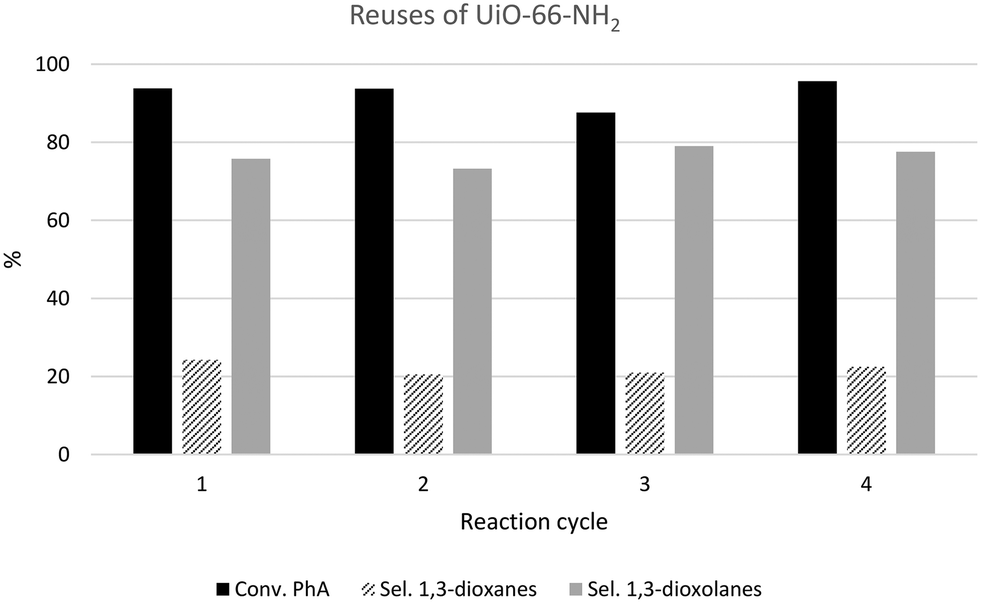 | ||
| Fig. 8 Results of the consecutive reuses of the UiO-66-NH2-18 sample in the acetalization reaction of phenylacetaldehyde with glycerol after 6 h. | ||
To confirm the heterogeneous character of the UiO-66-NH2 catalyst and the absence of leaching of active sites to the reaction medium, an additional hot filtration experiment was carried out. Briefly, after 15 min of reaction, the catalyst was removed by filtration at the reaction temperature, and the reaction was continued without a catalyst. No further conversion of PhA was observed after removing the catalyst (see Fig. S9†). Additionally, ICP-AES analysis of the reaction filtrate didn't detect the presence of Zr4+ species, thus confirming the true heterogeneous character of the UiO-66-NH2 catalyst.
Comparison of catalytic activity of UiO-66 and UiO-66-NH2 with previously reported catalysts for synthesis of hyacinth fragrance acetals
In order to put into context the catalytic results obtained herein with UiO-66 for the acetalization of PhA with glycerol, a comparison was made with other results previously reported in the literature. As it can be seen in Table 3, UiO-66 and UiO-66-NH2 exhibit high catalytic activity for this reaction, reaching conversions of 89–93% after a short reaction time. These results are similar to those obtained over large pore USY and beta zeolites, homogeneous p-toluenesulfonic acid (PTSA) and acid treated activated carbon (AC-NS), which are reported as the most promising state-of-the-art catalysts for acetalization reactions.| Catalyst | Reaction temp. (°C) | Cat (wt%) | Time | Conv. (%) | Select. 1,3-dioxolanes (%) | Ref. |
|---|---|---|---|---|---|---|
| General conditions: Gly/PhA mol ratio – 2; toluene as a solvent; Dean–Stark. | ||||||
| USY | 147 | 7.5 | 1 h | 93 | 62 | 20 |
| Beta | 147 | 7.5 | 1 h | 92 | 66 | |
| Mordenite | 147 | 7.5 | 1 h | 33 | 85 | |
| MCM-41 | 147 | 7.5 | 1 h | 36 | 72 | |
| ZSM-5 | 147 | 7.5 | 1 h | 54 | 85 | |
| PTSA | 147 | 1.7 | 1 h | 97 | 68 | |
| AC-NS | 110 | 7.5 | 90 min | 95 | 88 | 21 |
| Microwave | 140 | — | 15 min | 98 | 60 | 55 |
| UiO-66 (12.4) | 130 | 4.2 | 2 h | 89 | 71 | This work |
| UiO-66-NH2 (14.8) | 130 | 4.2 | 2 h | 92 | 72 | |
Synthesis of other glyceryl acetal fragrances over UiO-66 catalysts
As it was mentioned above, several acetals are widely used in the fragrance industry, since acetalization can profoundly change the vapor pressure, solubility and aroma characteristics of the corresponding aldehyde or ketone flavors. Therefore, in order to evaluate the scope and potential of UiO-66 as a heterogeneous catalyst, we extended the glycerol acetalization reaction to other aldehydes and ketones used as flavor products. The results obtained are summarized in Table 4.| Aldehyde | Time (h) | Conv. (%) | Select. 1,3-dioxane (%) | Select. 1,3-dioxolane (%) |
|---|---|---|---|---|
| Reaction conditions: aldehyde or ketone (1 mmol), glycerol (2 mmol), toluene (3 mL), UiO-66 (10 mg), Dean–Stark, 130 °C. | ||||
 Cinnamaldehyde Cinnamaldehyde |
3 | 100 | 57.8 | 42.2 |
 Benzaldehyde Benzaldehyde |
1 | 100 | 33.4 | 66.6 |
 Citral (mixture of geranial and neral) Citral (mixture of geranial and neral) |
3 | 100 | 60.9 | 39.1 |
 L-Menthone
L-Menthone |
24 | 18.7 | 17.7 | 82.3 |
 Isovaleraldehyde Isovaleraldehyde |
3 | 99 | 30.5 | 69.5 |
 2-Butanone 2-Butanone |
7 | 100 | 0 | 100 |
According to the results presented in Table 4, both aromatic and lineal aldehydes were successfully reacted with glycerol over UiO-66, obtaining high yields and total selectivity to the corresponding acetals (mixture of dioxanes and dioxolanes) after a relatively short reaction time. In all cases, the dioxane/dioxolane ratios were determined from the corresponding 1H NMR spectra (see Fig. S10-S14†). Additionally, the total conversion of 2-butanone to the corresponding dioxolanes (cis and trans isomers) was also achieved, confirming the high catalytic performance of UiO-66. However, a relatively low conversion of L-menthone was observed, which is probably caused by the steric hindrance of the large isopropyl group in the ortho position to the carbonyl group.
Conclusions
Herein, we have shown that UiO-66 and UiO-66-NH2 materials are highly active, selective and reusable heterogeneous catalysts for the acetalization of PhA with glycerol to yield hyacinth fragrance, which represents an interesting route for glycerol valorization. Mixtures of 5- and 6-membered cyclic ketals, 1,3-dioxolanes and 1,3-dioxanes, were obtained as the sole products in good yields after a short (2 h) reaction time. The catalytic activity of the solid depends on the amount of available Zr4+ sites, which are associated to missing linker defects. However, no significant differences in catalytic activity are observed between UiO-66 and UiO-66-NH2. The dioxolane–dioxane ratio can be modified to some extent (between 2.8 and 4.6), depending on the catalyst used (UiO-66 or UiO-66-NH2), the amount of missing linker defects of the solid and the reaction time. This could afford a means to fine-tune the organoleptic characteristics of the final hyacinth fragrance mixture. Furthermore, a fair scope of UiO-66 has been demonstrated for the formation of other glyceryl acetals with other aldehydes and ketones of interest in the flavoring industry.Author contributions
FXLiX: conceptualization, supervision, writing – original draft, writing – review & editing, and funding acquisition; AR: conceptualization, supervision, investigation, and writing – review & editing; JCDI: investigation and methodology.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors are grateful for grant PID2020-112590GB-C21 funded by MCIN/AEI/10.13039/501100011033References
- W. H. Liew, M. H. Hassim and D. K. S. Ng, J. Cleaner Prod., 2014, 71, 11–29 CrossRef CAS.
- C. Xu and Q. Xu, Ind. Eng. Chem. Res., 2018, 57, 16809–16816 CrossRef CAS.
- K. S. Arias, A. Garcia-Ortiz, M. J. Climent, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2018, 6, 4239–4245 CrossRef CAS.
- M. H. Haider, N. F. Dummer, D. W. Knight, R. L. Jenkins, M. Howard, J. Moulijn, S. H. Taylor and G. J. Hutchings, Nat. Chem., 2015, 7, 1028–1032 CrossRef CAS PubMed.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- B. Katryniok, S. Paul, V. Bellière-Baca, P. Rey and F. Dumeignil, Green Chem., 2010, 12, 2079–2098 RSC.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- Y. Gu, A. Azzouzi, Y. Pouilloux, F. Jérôme and J. Barrault, Green Chem., 2008, 10, 164–167 RSC.
- C. J. A. Mota, C. X. A. Da Silva, N. Rosenbach, J. Costa and F. Da Silva, Energy Fuels, 2010, 24, 2733–2736 CrossRef CAS.
- I. Fatimah, I. Sahroni, G. Fadillah and M. M. Musawwa, Energies, 2019, 12, 2872 CrossRef CAS.
- M. J. Climent, A. Corma and A. Velty, Appl. Catal., A, 2004, 263, 155–161 CrossRef CAS.
- C. A. C. Silva, F. C. A. Figueiredo, R. Rodrigues, M. I. Sairre, M. Gonçalves, I. Matos, I. M. Fonseca, D. Mandelli and W. A. Carvalho, Clean Technol. Environ. Policy, 2016, 18, 1551–1563 CrossRef CAS.
- P. Sari, M. Razzak and I. G. Tucker, Pharm. Dev. Technol., 2004, 9, 97–106 CrossRef CAS PubMed.
- A. Piasecki, A. Sokołowski, B. Burczyk and U. Kotlewska, J. Am. Oil Chem. Soc., 1997, 74, 33–37 CrossRef CAS.
- V. R. Bakuru, S. R. Churipard, S. P. Maradur and S. B. Kalidindi, Dalton Trans., 2019, 48, 843–847 RSC.
- P. H. R. Silva, V. L. C. Gonçalves and C. J. A. Mota, Bioresour. Technol., 2010, 101, 6225–6229 CrossRef CAS PubMed.
- I. Fatimah, I. Sahroni, G. Fadillah and M. M. Musawwa, Energies, 2019, 12, 2872 CrossRef CAS.
- C. J. A. Mota, C. X. A. Da Silva, N. Rosenbach, J. Costa and F. Da Silva, Energy Fuels, 2010, 24, 2733–2736 CrossRef CAS.
- K. Woelfel and T. G. Hartman, ACS Symp. Ser., 1998, 705, 193–210 CrossRef CAS.
- M. J. Climent, A. Corma and A. Velty, Appl. Catal., A, 2004, 263, 155–161 CrossRef CAS.
- C. A. C. Silva, F. C. A. Figueiredo, R. Rodrigues, M. I. Sairre, M. Gonçalves, I. Matos, I. M. Fonseca, D. Mandelli and W. A. Carvalho, Clean Technol. Environ. Policy, 2016, 18, 1551–1563 CrossRef CAS.
- T. Coleman and A. Blankenship, WO/2010/022263, 2010.
- E. V. Gromachevskaya, F. V. Kvitkovsky, E. B. Usova and V. G. Kulnevich, Chem. Heterocycl. Compd., 2004, 40, 979–985 CrossRef CAS.
- B. L. Wegenhart, S. Liu, M. Thom, D. Stanley and M. M. Abu-Omar, ACS Catal., 2012, 2, 2524–2530 CrossRef CAS.
- S. Zaher, L. Christ, M. Abd El Rahim, A. Kanj and I. Karamé, Mol. Catal., 2017, 438, 204–213 CrossRef CAS.
- P. Manjunathan, S. P. Maradur, A. B. Halgeri and G. V. Shanbhag, J. Mol. Catal. A: Chem., 2015, 396, 47–54 CrossRef CAS.
- J. Kowalska-Kus, A. Held, M. Frankowski and K. Nowinska, J. Mol. Catal. A: Chem., 2017, 426, 205–212 CrossRef CAS.
- K. S. Arias, A. Garcia-Ortiz, M. J. Climent, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2018, 6, 4239–4245 CrossRef CAS.
- N. Oger, Y. F. Lin, E. Le Grognec, F. Rataboul and F. X. Felpin, Green Chem., 2016, 18, 1531–1537 RSC.
- L. Chen, B. Nohair, D. Zhao and S. Kaliaguine, ChemCatChem, 2018, 10, 1918–1925 CrossRef CAS.
- M. N. Timofeeva, V. N. Panchenko, N. A. Khan, Z. Hasan, I. P. Prosvirin, S. V. Tsybulya and S. H. Jhung, Appl. Catal., A, 2017, 529, 167–174 CrossRef CAS.
- W.-G. Cui, T.-L. Hu, W.-G. Cui and T.-L. Hu, Small, 2021, 17, 2003971 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. Son Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- A. Corma, H. Garcia and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. v. Ramos Fernandez, F. X. Llabrés i Xamena, V. van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- M. N. Timofeeva, V. N. Panchenko, N. A. Khan, Z. Hasan, I. P. Prosvirin, S. V. Tsybulya and S. H. Jhung, Appl. Catal., A, 2017, 529, 167–174 CrossRef CAS.
- A. Rapeyko, M. Rodenas and F. X. Llabrés i Xamena, Adv. Sustainable Syst., 2022, 6, 1–9 Search PubMed.
- F. G. Cirujano, A. Corma and F. X. Llabrés i Xamena, Chem. Eng. Sci., 2015, 124, 52–60 CrossRef CAS.
- F. G. Cirujano and F. X. Llabrés i Xamena, J. Phys. Chem. Lett., 2020, 11, 4879–4890 CrossRef CAS PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- H. Furukawa, F. Gándara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- J. Liu, Z. Chen, R. Wang, S. Alayoglu, T. Islamoglu, S. J. Lee, T. R. Sheridan, H. Chen, R. Q. Snurr, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2021, 13, 22485–22494 CrossRef CAS PubMed.
- H. I. Meléndez-Ortiz, L. A. García-Cerda, Y. Olivares-Maldonado, G. Castruita, J. A. Mercado-Silva and Y. A. Perera-Mercado, Ceram. Int., 2012, 38, 6353–6358 CrossRef.
- W. H. Zhang, J. L. Shi, L. Z. Wang and D. S. Yan, Mater. Lett., 2000, 46, 35–38 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- I. Agirre, M. B. Güemez, A. Ugarte, J. Requies, V. L. Barrio, J. F. Cambra and P. L. Arias, Fuel Process. Technol., 2013, 116, 182–188 CrossRef CAS.
- K. Woelfel and T. G. Hartman, ACS Symp. Ser., 1998, 705, 193–210 CrossRef CAS.
- R. R. Pawar, S. V. Jadhav and H. C. Bajaj, Chem. Eng. J., 2014, 235, 61–66 CrossRef CAS.
- J. M. Guarinos, F. G. Cirujano, A. Rapeyko and F. X. Llabrés i Xamena, Mol. Catal., 2021, 515, 111925 CrossRef CAS.
- A. Rapeyko, M. Rodenas and F. X. Llabrés i Xamena, Adv. Sustainable Syst., 2022, 6, 1–9 Search PubMed.
- A. Vimont, H. Leclerc, F. Maugé, M. Daturi, J. C. Lavalley, S. Surblé, C. Serre and G. Férey, J. Phys. Chem. C, 2007, 111, 383–388 CrossRef CAS.
- J. Liu, Z. Chen, R. Wang, S. Alayoglu, T. Islamoglu, S. J. Lee, T. R. Sheridan, H. Chen, R. Q. Snurr, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2021, 13, 22485–22494 CrossRef CAS PubMed.
- A. Herbst, A. Khutia and C. Janiak, Inorg. Chem., 2014, 53, 7319–7333 CrossRef CAS PubMed.
- R. R. Pawar, S. v. Jadhav and H. C. Bajaj, Chem. Eng. J., 2014, 235, 61–66 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00255h |
| This journal is © The Royal Society of Chemistry 2023 |