
Deep-blue emission and thermally activated delayed fluorescence via Dimroth rearrangement of tris(triazolo)triazines†
Ryoga
Hojo
 *,
Don M.
Mayder
and
Zachary M.
Hudson
*
*,
Don M.
Mayder
and
Zachary M.
Hudson
*
Department of Chemistry, The University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: zhudson@chem.ubc.ca; Fax: +1-604-822-2847; Tel: +1-604-822-3266
First published on 12th May 2022
Abstract
Three luminescent donor–acceptor compounds were prepared based on the Dimroth rearrangement of tris(triazolo)triazines (TTT). In comparison to the non-rearranged TTT isomers, the Dimroth isomer (TTTD) exhibits a substantial blue-shift in emission while maintaining thermally activated delayed fluorescence (TADF) properties. Out of the series of emitters, TTTD-3HMAT exhibits deepest blue emission with CIE(x, y) < (0.16, 0.03) and unity quantum yield in toluene, a narrower emission band, and highest two-photon cross-section of 1001 GM owing to the nature of the planarized HMAT donor. TTTD-3tBu also exhibits unity quantum yield and deep-blue emission with CIE(x, y) < (0.16, 0.05) in toluene. Finally, TTTD-3ACR exhibits TADF with blue-shifted emission and prolonged delayed lifetime due to the slightly larger ΔEST of 0.26 eV in comparison to the non-rearranged isomer. Overall, this work demonstrates a practical strategy to convert TTT-based donor–acceptor materials to their Dimroth isomers, opening the door to deeper blue-emitting TADF materials with TTT-type acceptors.
 Zachary M. Hudson | Dr Zachary M. Hudson is an Associate Professor and Canada Research Chair in Sustainable Chemistry at the University of British Columbia. Zac completed his BSc and PhD in Chemistry at Queen's University, developing luminescent materials for organic electronics. During his PhD he also held graduate fellowships at Jilin University (China) as well as Nagoya University (Japan). He then completed postdoctoral fellowships in polymer science at the University of Bristol (UK) and the University of California, Santa Barbara (USA). He currently leads a research program at UBC developing luminescent materials for optoelectronics, bioimaging, and photocatalysis. |
Introduction
Since the seminal work by Adachi and co-workers revealing the phenomenon of thermally activated delayed fluorescence (TADF) in purely organic donor–acceptor materials,1 substantial progress has been made in realizing their use in the organic light-emitting diode (OLED) display industry.2–6 TADF materials are of interest as they can harvest energy from both singlet and triplet excitons through reverse intersystem crossing (RISC).1,3,5,7 Therefore, with TADF materials, 100% internal quantum efficiencies can be achieved in OLED devices without the need for rare-earth metals.1,8 In recent years, the applications of TADF materials have grown beyond display technology, most notably to encompass time-gated biological imaging9–14 and organo-photocatalysis.15–19 To further encourage the mainstream adoption of TADF materials in both biology and chemistry, new donor–acceptor motifs displaying TADF remain critical to furthering emitter design and practical applications.TADF behaviour is achieved by reducing the energy gap between a molecule's lowest excited singlet and triplet states (ΔEST), which can be achieved by lowering the electron exchange energy through the spatial separation of the HOMO and LUMO.1,20 In terms of donor–acceptor design, this process of minimizing ΔEST can be achieved by inducing a large dihedral angle between the donor–acceptor moieties.1,21 If ΔEST is sufficiently small (<0.3 eV), thermal energy enables upconversion from the triplet to singlet excited state, allowing for RISC. Despite this seemingly simple design strategy, minimizing ΔEST is often done at the cost of broad and red-shifted emission, due to the strong charge-transfer (CT) character of this donor–acceptor design.22,23 For this reason, designing efficient deep-blue emitting TADF materials remains one of the biggest challenges in this field.2,24,25
In recent years [1,2,4]-triazolo-[1,3,5]-triazine (TTT) has attracted significant attention in the realm of blue-emitting TADF materials due to their weak electron-withdrawing ability.26–30 The synthesis for this unique heterocyclic motif features cyclization of aryl tetrazoles onto cyanuric chloride under basic conditions.31,32 To date, several derivatives of TTT-based TADF materials have been successfully synthesized with respectable device performance.26–30 Our group has also revealed the remarkable two-photon cross-section of TTT-based emitters using hexamethylazatriangulene (HMAT) donors, which has great potential for biological imaging using two-photon excited fluorescence (2PEF).33
Following the synthesis of TTT reported in the original paper by Hoffman et al.,34 Tartakovsky and co-workers31 reported the thermal isomerization process of TTT derivatives, uncovering the Dimroth isomer, TTTD. Similarity in 2018, Detert explored the TTTD isomer in the design of discotic liquid crystals.35 The Dimroth rearrangement involves thermal ring-transformation of 1,2,3 triazoles,36,37 whereas for TTT derivatives, the process involves a threefold ring-opening and ring-closing mechanism (Scheme 1).31,35 More recently, the You group demonstrated a mono-fused (triazolo)triazine derivative undergoing the Dimroth rearrangement to produce efficient TADF emitters with minimal ΔEST (Scheme S1A, ESI†).38 Inspired by this work, we hypothesized that the Dimroth rearrangement would provide a unique opportunity to observe the effect of thermal isomerization on TTT compounds; and precisely observe the change in TADF behavior.
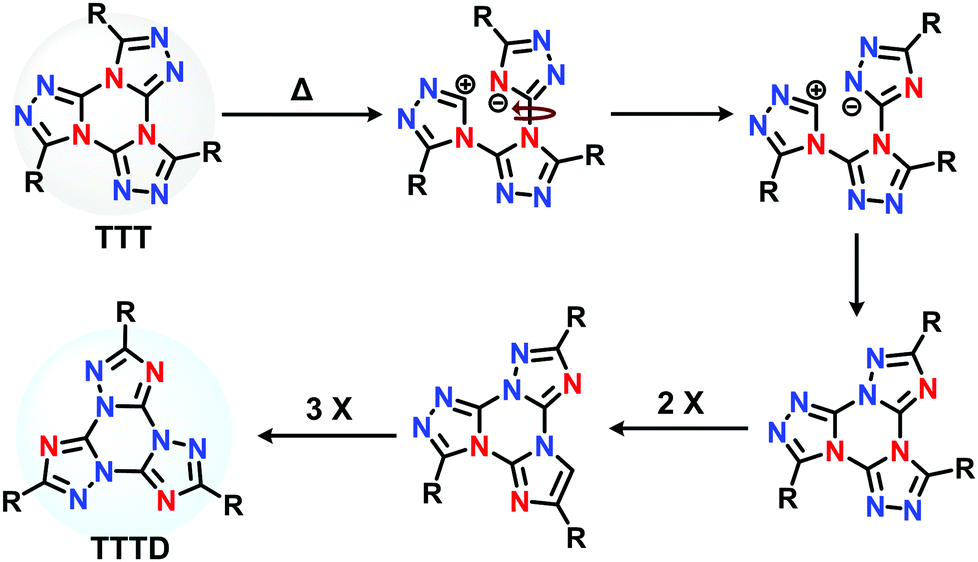 | ||
| Scheme 1 The isomerization of TTT to TTTD.35 | ||
Herein we report the synthesis of three compounds, TTTD-3HMAT, TTTD-3tBu, and TTTD-3ACR, based on the Dimroth rearrangement of donor-acceptor materials bearing a TTT core. TTTD-3HMAT and TTTD-3tBu exhibit unity photoluminescence quantum yields (PLQYs) in both solution and the solid-state, with TTTD-3HMAT exhibiting deeper-blue emission and a higher two-photon cross-section (σ2) in toluene relative to TTTD-3tBu. Finally, TTTD-3ACR retains TADF character with blue-shifted emission compared to the non-rearranged TTT emitter. Indeed, TTTD-3ACR represents the deepest blue emitter displaying TADF behaviour to date based on the TTT acceptor motif.
Results and discussion
The pre-rearranged TTT compounds were synthesized according to previous reports, involving reaction of donor-functionalized tetrazoles with cyanuric chloride (Scheme S1B, ESI†).33 The procedure for the Dimroth rearrangement was adapted from the procedure of You and coworkers (Scheme S1A, ESI†),38 by heating the TTT compounds to 200 °C under basic and inert conditions. Reaction times varied from 3–7 days, as monitored by thin layer chromatography (Scheme 2). Specifically, the thermal conversion to synthesize TTTD-3tBu and TTTD-3ACR was completed after three days, while synthesis of TTTD-3HMAT required seven days to achieve full conversion. The characteristic downfield shift in the donor aryl-hydrogen signals at ∼8 ppm in the 1H NMR spectra relative to the starting material TTT isomer, and the retained symmetry in both 1H and 13C{1H} NMR spectra (Fig. S1–S7, ESI†) agree with previous reports suggesting a symmetric Dimroth-rearranged tris(triazolo)triazine.35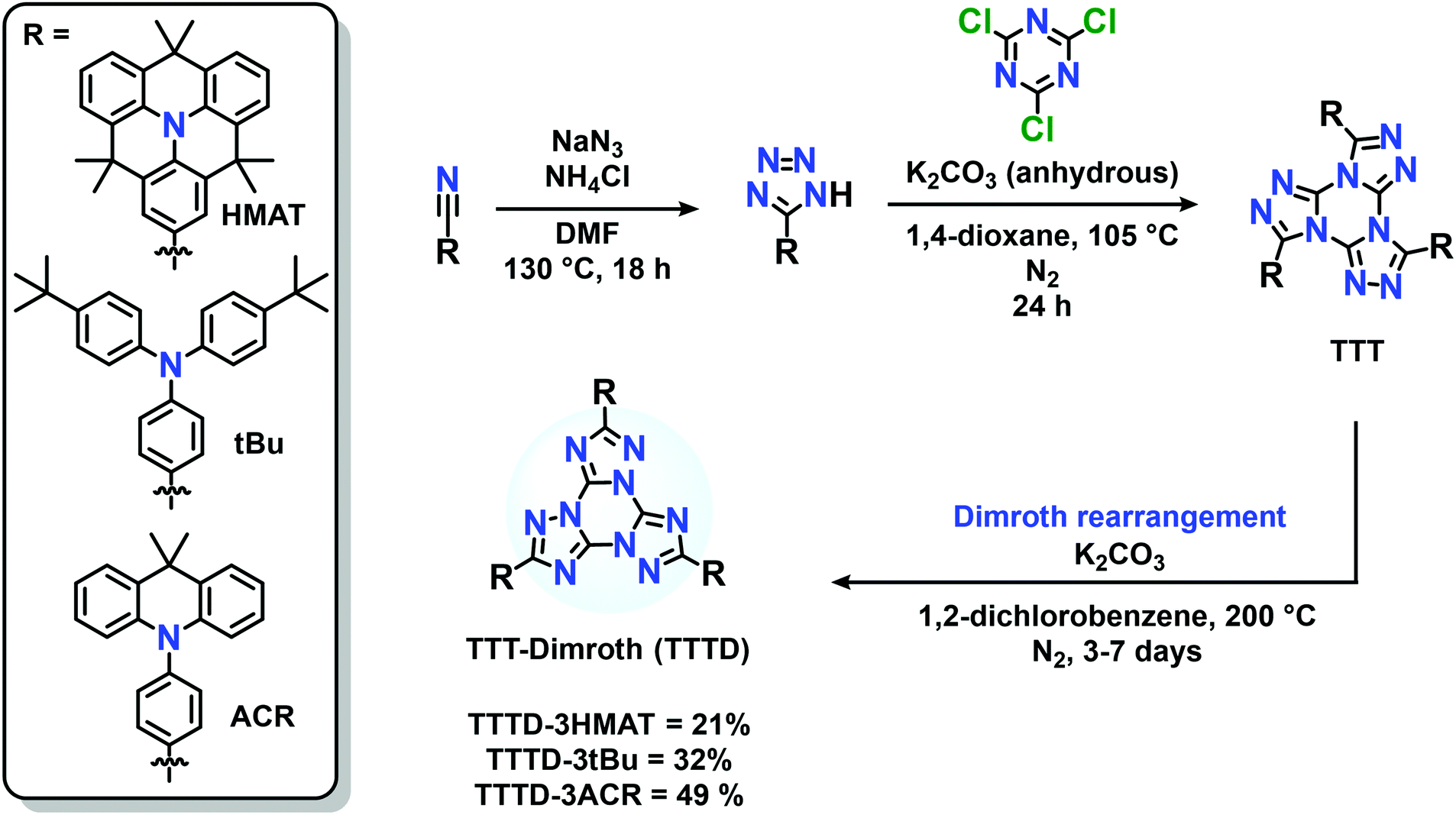 | ||
| Scheme 2 Synthetic route to donor–acceptor Dimroth-rearranged TTTD compounds. | ||
Our method contrasts with the synthetic route reported by You and co-workers involving a Pd-catalyzed cross-coupling on the pre-assembled and Dimroth-rearranged (triazolo)triazine (Scheme S1A, ESI†). Our results demonstrate that thermal rearrangement can reliably occur after the donors are appended to the acceptor, providing a direct synthetic route without the use of palladium. Moreover, we also attempted a Pd-catalyzed coupling using a triiodide-functionalized Dimroth-rearranged TTT acceptor, for cross-coupling with amine-based donors; the insolubility of tris(p-iodophenyl)-TTTD made this method unfeasible and challenging to characterize (Scheme S1C, ESI†). We also attempted to synthesize and characterize TTTD derivatives bearing 10H-phenoxazine (TTTD-3POX) or 10H-phenothiazine donors (TTTD-3PTZ) by thermal rearrangement. In these cases, however, the products were highly insoluble in common organic solvents, likely due to extensive π-stacking (Scheme S1C, ESI†). No glass transitions could be observed for the three isolated compounds between 30–200 °C by differential scanning calorimetry (Fig. S16, ESI†).
The UV-visible absorption and photoluminescence (PL) spectra of TTTD emitters in toluene are given in Fig. 1A. The absorption maxima ranged from 298 nm to 391 nm, with TTTD-3HMAT and TTTD-3tBu exhibiting small Stokes shifts of 25 nm and 58 nm, respectively (Table 1). PLQYs of 1.0 were observed for both TTTD-3HMAT and TTTD-3tBu, while TTTD-3HMAT exhibited deeper blue emission (416 nm) and smaller FWHM (42 nm) in toluene, attributed to the restricted intermolecular motion of the planarized donors. In contrast, a weak charge-transfer absorption band is observed for TTTD-3ACR, with a broader emission profile and an emission maximum at 461 nm in toluene. In addition, an increase in PLQY from 0.26 under air to 0.41 in N2-sparged solution is observed for TTTD-3ACR in toluene, suggesting the likely involvement of triplet states in the emission mechanism. The prompt fluorescence PL decays in toluene (Fig. S7A, ESI†) were also measured using time-correlated single-photon counting (TCSPC): the lifetime for TTTD-3HMAT and TTTD-3tBu ranged from 2.1 to 2.6 ns, while the lifetime for TTTD-3ACR is slightly longer at 7.6 ns (Table 1). Multi-channel scaling (MCS) was then used to analyze the longer-lived PL decays in solution (Fig. S8A, ESI†). While TTTD-3HMAT and TTTD-3tBu show no observable delayed lifetime in both ambient and inert conditions, TTTD-3ACR exhibits a delayed lifetime of 19 μs in oxygen-free conditions, likely arising from delayed fluorescence.
 | ||
| Fig. 1 (A) Solution-state absorption (dashed) and fluorescent emission under air (solid) measured in toluene at optical densities of 0.05 (λexc = 350 nm). (B) Two-photon absorption of TTTD emitters (C) CIE diagram showing the chromaticity of the emitters in toluene measured under air. | ||
| Entry | Solution | 3 wt% in PMMA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ abs (nm) ε (104 cm−1 M−1) | λ em/FWHM (nm) | σ 740 2 (GM) | Φ air/ΦN2b/c |
τ
air![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
τ
N2
|
λ em/FWHM (nm) | Φ air |
τ
aird |
τ vac | |
| a Measured in toluene under air at 0.01 mg mL−1. b Absolute photoluminescence quantum yields determined using an integrating sphere. c Measured in toluene solutions sparged with N2. d Measured under air at 298 K. e Measured under vacuum at 298 K. f Amplitude-weighted lifetime measurement via multiexponential tail fitting. | ||||||||||
| TTTD-3HMAT | 298 (11), 391 (11) | 416/42 | 1001 | 1.00/1.00 | 2.1 ns | 2.3 ns | 425/56 | 1.00 | 2.7 ns | 2.7 ns |
| TTTD-3tBu | 298 (4.1), 370 (10) | 428/58 | 845 | 1.00/1.00 | 2.6 ns | 2.6 ns | 432/60 | 1.00 | 3.5 ns | 3.5 ns |
| TTTD-3ACR | 363 (0.5) | 461/74 | 83 | 0.26/0.41 | 7.6 ns | 11 ns/19 μs | 453/130 | 0.14 | 9.2 ns/107 μsf | 9.2 ns/13 msf |
Aggregation-induced emission (AIE) and solvatochromism experiments were conducted to investigate the nature of the CT properties of these three materials. AIE experiments were conducted using water/THF mixtures, with water fractions (fw) ranging from 0% to 99%. The emission intensity increased as the percentage of water increased for TTTD-3ACR, with an overall 170% enhancement at fw = 70% Fig. S9, ESI†). In contrast, TTTD-3HMAT and TTT-3tBu lack AIE character. A positive solvatochromic shift is observed for all emitters, from least to most polar solvents (Fig. S10, ESI†). However, a minimal solvatochromic shift is observed with TTTD-3HMAT and TTT-3tBu. In contrast, the significant solvatochromic shift observed for TTTD-3ACR is evidence of strong CT character, consistent with its TADF behaviour (Fig. S10, ESI†).21,39,40
The two-photon cross-sections (σ2) for TTTD compounds were examined over the range of 710–850 nm using 2PEF measurements (Fig. 1B). Using (E)-4,4′-(ethene-1,2-diyl)bis(N,N-diphenylaniline) in dichloromethane as a blue-emitting reference dye standard, all measurements were periodically checked to verify a square dependence of signal intensity with excitation power. The highest σ2 was observed with TTTD-3HMAT (σ7402 = 1001 GM), followed by TTTD-3tBu (σ7402 = 845 GM) and TTTD-3ACR (σ7402 = 83 GM). In agreement with previous reports, the structural constraints and planar nature of HMAT donors allow for superior two-photon cross-sections, where a high σ2 value are enhanced with higher planarity and symmetry of the molecule (Table 1).33,41–43
Photophysical properties of the emitters in the solid-state were analyzed by preparing poly(methyl methacrylate) (PMMA) films doped with 3 wt% of emitter. Blue-shifted emission is observed with TTTD-3ACR (453 nm) relative to the emission maximum in toluene (461 nm). Red-shifted emission is observed with TTTD-3HMAT and TTTD-3tBu, which can arise from aggregation due to greater π-stacking in these less-twisted materials.26 Moreover, PLQY values of unity were observed for TTTD-3HMAT and TTTD-3tBu in the solid state, while that of TTTD-3ACR reached 0.14. Under vacuum, the emission intensity of TTTD-3ACR rises 2.0-fold compared to an air atmosphere, while intensities for TTTD-3HMAT and TTTD-3tBu remain essentially unchanged (Fig. S13, ESI†).
PL decays were measured for PMMA-doped films using TSCPC to measure prompt fluorescence (Fig. S1B, ESI†), and MCS was used to analyze the longer-lived PL decays (Fig. S2B, ESI†). Lifetimes of TTTD-3HMAT and TTTD-3tBu were consistent under air or vacuum at 298 K, only exhibiting prompt fluorescence in each case. In contrast, TTTD-3ACR has a long-lived PL lifetime of 107 μs in air, further lengthened to 13 ms under vacuum (Table 1). To elucidate if the long-lived emission of TTTD-3ACR was due to TADF, temperature-dependent lifetime measurements were conducted in the range of 77 to 298 K. As predicted, suppression of delayed fluorescence was observed upon cooling from 298 K to 77 K (Fig. 2). However, the emergence of long-lived phosphorescence is also observed below 200 K, which can be attributed to the restriction of roto-vibrational motion and dominance of phosphorescence emission when reaching cryogenic temperatures. Time-gated and steady-state emission measurements at 77 K of TTTD-3ACR in 2-methyltetrahydrofuran further support TADF, with a measured value for ΔEST of 0.26 eV (Fig. S14, ESI†). Conversely, the larger values of ΔEST measured for TTTD-3HMAT (0.51 eV) and TTTD-3tBu (0.35 eV) likely preclude RISC.
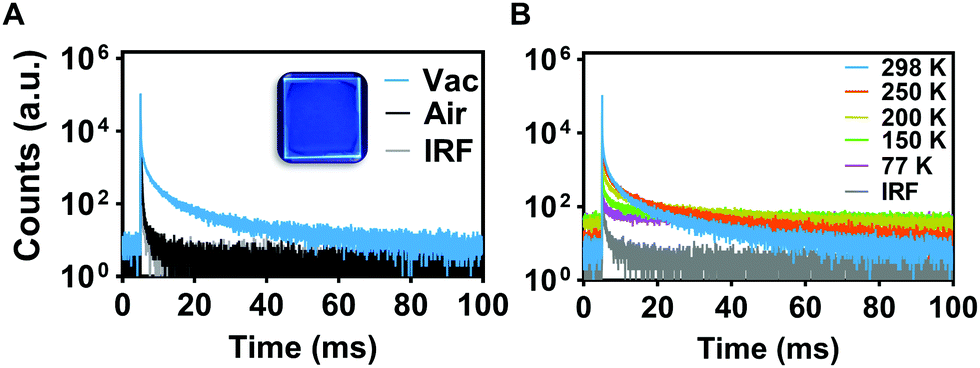 | ||
| Fig. 2 (A) PL decays for 3 wt% doped PMMA films under air (black) vs. vacuum (colored) of TTTD-3ACR. (B) PL decays for TTTD-3ACR (3 wt% doped PMMA film) at various temperatures between 77–298 K. λex = 350 nm. | ||
The determination of EHOMO and ELUMO were identified using cyclic voltammetry (CV) and Tauc plots (Fig. S11, ESI†). For all emitters, quasi-reversible one-electron oxidation is observed, which is attributed to each donor moiety. An irreversible reduction event is shown for TTTD acceptors within the solvent window of o-difluorobenzene. Therefore, Tauc plots were necessary to obtain optical gaps (Egap) from UV-Vis absorption measurements, where the ELUMO ranged from −1.85 eV to −1.92 eV. The trend for Egap is in slight disagreement with the calculated values using DFT (Fig. S12, ESI†). Nevertheless, all emitters display a large Egap around 3.1 eV, expected for deep-blue emitting chromophores (Table 2). Finally, we provide a comprehensive comparison of the photophysical and electrochemical properties of these TTTD compounds and their TTT isomers in Table S8 (ESI†).
| Entry | HOMO/LUMOab (eV) |
E
DFTgapc (eV) |
E
optgapd (eV) |
ΔEST (eV) | |
|---|---|---|---|---|---|
| Calc.c | Exp.e | ||||
| a HOMO = −(Eox1/2 + 4.8 eV). b LUMO = (Eoptgap + HOMO). c Theoretical calculations from TDA-DFT at the B3LYP/6-31g(d) level. d Calculated using a Tauc plot of the UV-Vis spectrum in toluene. e Determined from onsets of time-gated phosphorescence and fluorescence spectra measured in 2-MeTHF at 77 K. | |||||
| TTTD-3HMAT | −4.97/−1.91 | 3.42 | 3.06 | 0.38 | 0.51 |
| TTTD-3tBu | −5.01/−1.85 | 3.03 | 3.16 | 0.46 | 0.35 |
| TTTD-3ACR | −4.98/−1.92 | 3.01 | 3.06 | 0.006 | 0.26 |
Density functional theory (DFT) at the B3LYP/6-31g+(d) level was used further to analyze the photophysical and electronic properties of the emitters (Fig. 3). Structural optimization was performed at the B3LYP/6-31g+(d) level, followed by the use of the Tamm–Dancoff approximation (TDA) for time-dependent calculations at the B3LYP/6-31g(d) level. TDA-DFT was selected for theoretical calculations, as it has been shown to be suitable for larger molecules that suffer from the triplet instability problem.44 While diffuse orbitals were used during structural optimization at the B3LYP/6-31g+(d) level, they were not included for TDA-DFT due to the higher computational demand for the large molecules presented here.
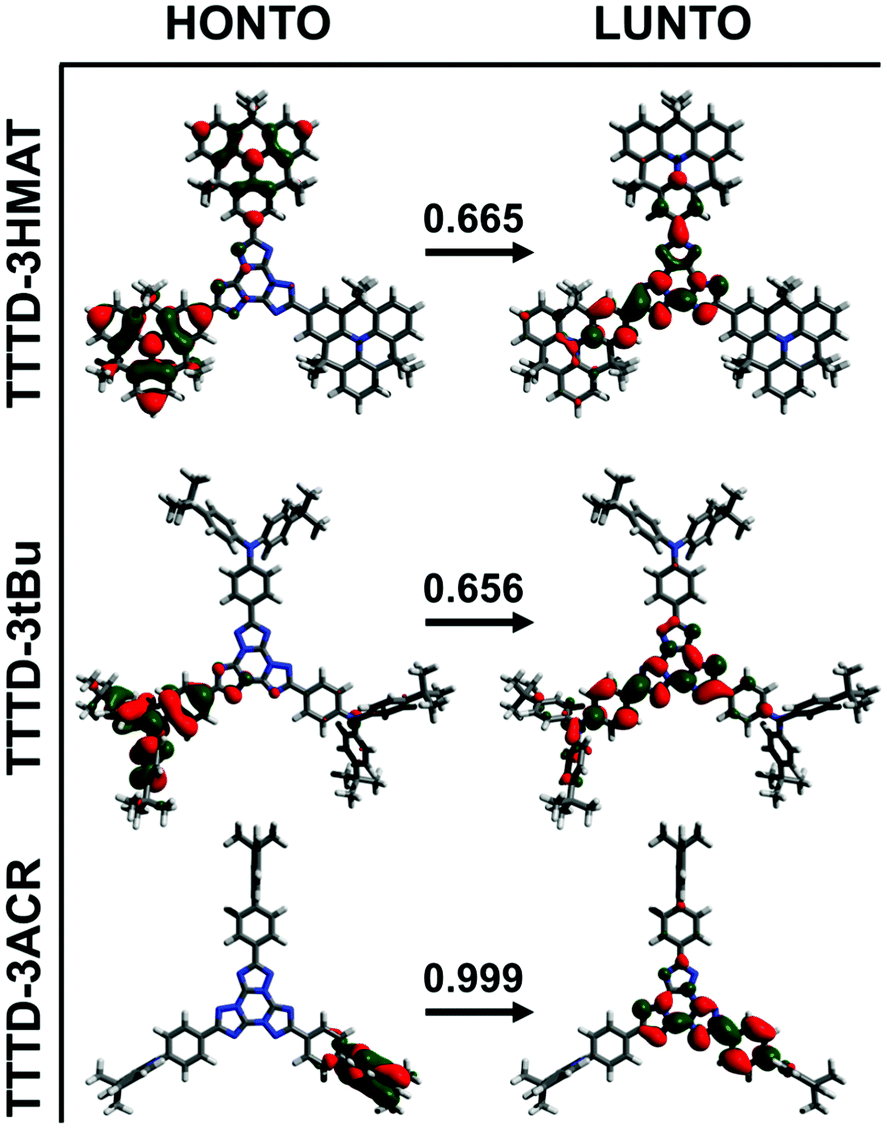 | ||
| Fig. 3 Pictorial representation of optimized S0 geometries for TTTD emitters. The weight of the transition between HONTO and LUNTO is indicated above the arrow. | ||
In general, theoretical calculations yielded trends in agreement with experimental observations. The calculated ΔEST for TTTD-3ACR is the smallest at 0.006 eV, while both TTTD-3HMAT and TTTD-3tBu have larger calculated ΔEST values above 0.38 eV. Molecular orbital visualization through natural transition orbitals (NTOs) indicates poor spatial separation of frontier molecular orbitals of the donor and acceptor in TTTD-3HMAT and TTTD-3tBu. Consequently, the higher calculated oscillator strengths for the S0 to S1 transition support the stronger observed CT absorption, and subsequently the high PLQY for these materials as well (Table S1, ESI†). In contrast, the near-orthogonal predicted dihedral angle between the ACR donor and TTTD acceptor in TTTD-3ACR induces strong spatial separation between the highest-occupied NTO (HONTO) and lowest-unoccupied NTO (LUNTO). This likely explains the TADF behaviour of TTTD-3ACR, yet also limits strong absorption from S0 to S1 as indicated by the low oscillator strength of this transition (Fig. 3 and Table S1, ESI†).
Interestingly, a general comparison between TTTD and TTT isomers indicates that each isomer is of similar total energy, with a minimal predicted change in the energy of the S1 state between each isomer pair (Fig. S15 and Table S1, ESI†). In all three cases, the predicted energies of the S1 states of the TTT isomers were within 50 meV of the TTTD isomers, when simulated in toluene using a polarized continuum model (PCM).
Conclusion
Here, we have employed the Dimroth rearrangement for a series of donor–acceptor compounds containing the TTT acceptor, revealing the photophysical properties of the TTTD isomer. All three emitters, TTTD-3HMAT, TTTD-3tBu, and TTTD-3ACR, exhibit a deeper blue emission than the TTT isomer. TTTD-3HMAT and TTTD-3tBu exhibit deep blue emission at λmax = 416 nm and 428 nm, respectively in toluene, while TTTD-3HMAT displays a narrow FWHM (42 nm) and high 2PA (σ7402 = 1001 GM) owing to the nature of the planarized donor groups (Table 1). In contrast, TTTD-3ACR retains its TADF character while exhibiting a deeper blue emission (454 nm) in comparison to its TTT isomer. Overall, this work demonstrates the effect of the Dimroth rearrangement on the photophysical and electronic properties of TTT-based donor–acceptor materials. In doing so, we have provided the path toward deeper blue materials based on the TTT framework. Finally, these novel TTTD-based materials may find their use in diverse applications for deep-blue emitters, with potential as high energy sensitizers in photocatalysis, two-photon bioimaging dyes, or as emitters in OLEDs.Experimental details
General considerations
All reagents were purchased from Oakwood Chemical, Sigma-Aldrich, or Alfa Aesar and used without purification. HMAT was synthesized according to previously reported procedures (Scheme S1, ESI†).45 All yields correspond to the isolated yield. The 1H and 13C{1H} nuclear magnetic resonance (NMR) spectra were measured on a Bruker Advance 300 Spectrometer or Bruker AV III HD 400 MHz spectrometer with methylene chloride-d2 (CD2Cl2) or chloroform-d (CDCl3) as the solvent. High resolution mass spectra (HRMS) were obtained with atmospheric pressure chemical ionization (ACPI) or field desorption (FC) using a Waters/Micromass LCT spectrometer. Absorbance measurements were taken using a Cary 60 spectrometer, while fluorescence measurements were obtained from Edinburgh Instruments FS5 spectrofluorometer and Edinburgh Instruments FLS1000 spectrofluorometer was specifically used for time-gated emission measurements. Absolute photoluminescence quantum yields were determined using an Edinburgh Instruments SC-30 Integrating Sphere Module, and samples were prepared at concentrations with optical densities of 0.10 at the excitation wavelength.Electrochemical methods
Cyclic voltammograms were measured using a BASi Epsilon Eclipse potentiostat at room temperature using a three-electrode configuration (working electrode: 3 mm diameter glassy carbon; reference electrode: RE-5B Ag/AgCl electrode in saturated aqueous KCl (BASi Inc.), referenced externally to ferrocene/ferrocenium; counter electrode: Pt wire) in 0.2 M tetrabutylammonium hexafluorophosphate in o-difluorobenzene. Experiments were conducted at a scan rate of 20 mV s−1 in N2-sparged electrolyte solution with 2 mg mL−1 of the analyte.Density functional theory
Quantum chemical calculations were conducted using the Gaussian 16 Rev. B.01 package with default settings unless otherwise stated. Ground state energies and corresponding geometries were calculated at the B3LYP/6-31g+(d) level of theory. Tamm–Dancoff approximation (TDA)-DFT was performed from the optimized frequencies at the B3LYP/6-31g(d) level of theory. Molecular orbitals were visualized using Avogadro version 1.20 and rendered using POV-Ray version 3.7.0.General procedure for the Dimroth rearrangement from TTT to TTTD
The Dimroth rearrangements of tris(triazolo)triazines were conducted according to a modified literature procedure.38 To a 100 mL Schlenk flask equipped with a magnetic stir bar was added tris(triazolo)triazines (0.493 mmol, 1 equiv.), K2CO3 (1.97 mmol, 4 equiv.), and anhydrous 1,2-dichlorobenzene (50 mL). The reaction mixture was sparged with N2 for 15 min before being heated to 200 °C for 3–7 days (3 days for TTTD-3tBu and TTTD-3ACR, 7 days for TTTD-3HMAT) using a preheated oil bath under N2 atmosphere. Upon completion, the reaction was cooled to room temperature. Once cooled, the crude reaction was filtered to remove K2CO3, and the filtrate was concentrated in vacuo. The crude residue was purified by column chromatography over silica gel to yield the pure tris(triazolo)triazine compound.:1 hexanes:EtOAc) resulting in yellow solid. Yield: 21 mg (21%). 1H NMR (300 MHz, CD2Cl2) δ 8.42 (s, 6H), 7.51–7.45 (m, 12H), 7.20 (dd, J = 7.6 Hz, 6H), 1.79 (s, 36H), 1.68 (s, 18H) ppm. 13C{1H} NMR (101 MHz, CD2Cl2): 13C NMR (101 MHz, CD2Cl2) δ 165.1, 144.7, 135.1, 131.3, 130.4, 130.3, 130.1, 124.0, 123.6, 123.5, 122.8, 122.5, 35.7, 35.4, 33.4, 32.7 ppm. HRMS (APCI) m/z: [M]+ calc’d for [C87H78N12]+ 1290.6466; found 1290.6472; 0.49 difference ppm.
:2 hexanes:EtOAc) resulting in yellow solid. Yield: 198 mg (32%). 1H NMR (300 MHz, CD2Cl2) δ 8.14 (ddd, J = 8.2 Hz, 6H), 7.38–7.35 (m, 12H), 7.14–7.11 (m, 12H), 7.07 (ddd, J = 8.8 Hz, 6H) 1.34 (s, 54H) ppm. 13C{1H} NMR (101 MHz, CD2Cl2): 13C NMR (101 MHz, CDCl3) δ 165.1, 151.1, 147.1, 144.4, 144.1, 128.8, 126.3, 125.3, 120.2, 119.8, 34.4, 31.5 ppm. HRMS (APCI) m/z: [M]+ calc’d for [C83H87N12]+ 1266.7399; found 1266.7411; difference 0.99 ppm.
:1 hexanes:CHCl3) resulting in yellow solid. Yield: 295 mg (49%). 1H NMR (300 MHz, CDCl3) δ 8.74 (ddd, J = 8.4 Hz, 6H), 7.59 (ddd, J = 8.4 Hz, 6H), 7.49 (ddd, J = 5.9 Hz, 6H), 7.04–6.93 (m, 12H), 6.38 (ddd, J = 6.5 Hz, 6H), 1.72 (s, 18H) ppm. 13C{1H} NMR (101 MHz, CDCl3): δ 164.8, 144.8, 144.7, 140.5, 132.0, 130.5, 130.4, 127.8, 126.4, 125.4, 121.0, 114.1, 36.1, 31.2 ppm. HRMS (FD) m/z: [M]+ calc’d for [C69H54N12]+ 1050.4536; found 1050.4545; difference 0.92 ppm.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for support of their research program. RH is grateful for the David W. Strangway Fellowship from UBC and DMM thanks NSERC for a Canada Graduate Scholarship. ZMH is also grateful for support from the Canada Research Chairs program.References
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- A. Monkman, ACS Appl. Mater. Interfaces DOI:10.1021/acsami.1c09189.
- X. Cai and S. J. Su, Adv. Funct. Mater., 2018, 28, 1–33 Search PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- H. Lim, H. J. Cheon, S. J. Woo, S. K. Kwon, Y. H. Kim and J. J. Kim, Adv. Mater., 2020, 32, 1–8 Search PubMed.
- Y. Tang, Y. Liu, W. Ning, L. Zhan, J. Ding, M. Yu, H. Liu, Y. Gao, G. Xie and C. Yang, J. Mater. Chem. C, 2022, 10, 4637–4645 RSC.
- D. Hu, L. Yao, B. Yang and Y. Ma, Philos. Trans. R. Soc., A, 2015, 373, 20140318 CrossRef PubMed.
- Y. C. Duan, L. L. Wen, Y. Gao, Y. Wu, L. Zhao, Y. Geng, G. G. Shan, M. Zhang and Z. M. Su, J. Phys. Chem. C, 2018, 122, 23091–23101 CrossRef CAS.
- N. R. Paisley, S. V. Halldorson, M. V. Tran, R. Gupta, S. Kamal, W. R. Algar and Z. M. Hudson, Angew. Chem., Int. Ed., 2021, 60, 18630–18638 CrossRef CAS PubMed.
- C. J. Christopherson, N. R. Paisley, Z. Xiao, W. R. Algar and Z. M. Hudson, J. Am. Chem. Soc., 2021, 143, 13342–13349 CrossRef CAS PubMed.
- A. M. Polgar and Z. M. Hudson, Chem. Commun., 2021, 57, 10675–10688 RSC.
- Q. Zhang, S. Xu, M. Li, Y. Wang, N. Zhang, Y. Guan, M. Chen, C. F. Chen and H. Y. Hu, Chem. Commun., 2019, 55, 5639–5642 RSC.
- V. N. Nguyen, A. Kumar, M. H. Lee and J. Yoon, Coord. Chem. Rev., 2020, 425, 213545 CrossRef CAS.
- S. Qi, S. Kim, V. N. Nguyen, Y. Kim, G. Niu, G. Kim, S. J. Kim, S. Park and J. Yoon, ACS Appl. Mater. Interfaces, 2020, 12, 51293–51301 CrossRef CAS PubMed.
- E. R. Sauvé, C. M. Tonge and Z. M. Hudson, J. Mater. Chem. C, 2021, 9, 4164–4172 RSC.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- Z. Mao, A. Huang, L. Ma and M. Zhang, RSC Adv., 2021, 11, 38235–38238 RSC.
- C.-L. Dong, L.-Q. Huang, Z. Guan, C.-S. Huang and Y.-H. He, Adv. Synth. Catal., 2021, 363, 3803–3811 CrossRef CAS.
- D.-F. Chen, C. H. Chrisman and G. M. Miyake, ACS Catal., 2020, 10, 2609–2614 CrossRef CAS PubMed.
- C. M. Marian, Annu. Rev. Phys. Chem., 2020, 72, 617–640 CrossRef PubMed.
- S. Weissenseel, N. A. Drigo, L. G. Kudriashova, M. Schmid, T. Morgenstern, K. H. Lin, A. Prlj, C. Corminboeuf, A. Sperlich, W. Brütting, M. K. Nazeeruddin and V. Dyakonov, J. Phys. Chem. C, 2019, 123, 27778–27784 CrossRef CAS.
- X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- R. Huang, N. A. Kukhta, J. S. Ward, A. Danos, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2019, 7, 13224–13234 RSC.
- D. Zhang and L. Duan, Nat. Photonics, 2021, 15, 173–174 CrossRef CAS.
- J. H. Lee, C. H. Chen, P. H. Lee, H. Y. Lin, M. K. Leung, T. L. Chiu and C. F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- S. K. Pathak, Y. Xiang, M. Huang, T. Huang, X. Cao, H. Liu, G. Xie and C. Yang, RSC Adv., 2020, 10, 15523–15529 RSC.
- F. Hundemer, E. Crovini, Y. Wada, H. Kaji, S. Bräse and E. Zysman-Colman, Mater. Adv., 2020, 1, 2862–2871 RSC.
- S. Wang, X. Wang, K. H. Lee, S. Liu, J. Y. Lee, W. Zhu and Y. Wang, Dyes Pigm., 2020, 182, 108589 CrossRef CAS.
- Z. Fang, S. Wang, J. Liao, X. Chen, Y. Zhu, W. Zhu and Y. Wang, J. Mater. Chem. C, 2022, 38–43 Search PubMed.
- S. Zeng, C. Xiao, J. Zhou, Q. Dong, Q. Li, J. Lim, H. Ma, J. Y. Lee, W. Zhu and Y. Wang, Adv. Funct. Mater., 2022, 2113183 CrossRef.
- V. A. Tartakovsky, A. E. Frumkin, A. M. Churakov and Y. A. Strelenko, Russ. Chem. Bull., 2005, 54, 719–725 CrossRef CAS.
- S. Glang, T. Rieth, D. Borchmann, I. Fortunati, R. Signorini and H. Detert, Eur. J. Org. Chem., 2014, 3116–3126 CrossRef CAS.
- R. Hojo, D. M. Mayder and Z. M. Hudson, J. Mater. Chem. C, 2021, 9, 14342–14350 RSC.
- O. E.-K. A. Hofmann, Ber. Dtsch. Chem. Ges., 1911, 44, 2713–2717 CrossRef.
- H. Detert, Eur. J. Org. Chem., 2018, 4501–4507 CrossRef CAS.
- C. Wentrup, M. S. Mirzaei, D. Kvaskoff and A. A. Taherpour, J. Org. Chem., 2021, 86, 8286–8294 CrossRef CAS PubMed.
- A. Champiré, C. Vala, A. Laabid, A. Benharref, M. Marchivie, K. Plé and S. Routier, J. Org. Chem., 2016, 81, 12506–12513 CrossRef PubMed.
- R. Su, Y. Zhao, F. Yang, L. Duan, J. Lan, Z. Bin and J. You, Sci. Bull., 2021, 66, 441–448 CrossRef CAS.
- R. Ansari, W. Shao, S.-J. Yoon, J. Kim and J. Kieffer, ACS Appl. Mater. Interfaces, 2021, 13, 28529–28537 CrossRef CAS PubMed.
- M. Wang, H. S. Shin, F. Zhou, H. Xu, P. Prabhakaran, B. Dryzhakov, H. Su, K. S. Lee and B. Hu, J. Phys. Chem. C, 2020, 124, 14832–14837 CrossRef CAS.
- D. M. Mayder, C. M. Tonge, G. D. Nguyen, R. Hojo, N. R. Paisley, J. Yu, G. Tom, S. A. Burke and Z. M. Hudson, Chem. Mater., 2022, 34(6), 2624–2635 CrossRef CAS.
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed.
- Z. Fang, T. L. Teo, L. Cai, Y. H. Lal, A. Samoc and M. Samoc, Org. Lett., 2009, 11, 1–4 CrossRef CAS PubMed.
- F. J.-A. Ferrer, J. Cerezo, E. Stendardo, R. Improta and F. Santoro, J. Chem. Theory Comput., 2013, 9, 2072–2082 CrossRef PubMed.
- X. K. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J. L. Brédas, Adv. Mater., 2017, 29, 1–8 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: A synthetic scheme towards aryl tetrazoles; 1H and 13C{1H} NMR spectra, solvatochromic and AIE properties, nanosecond PL decay curves, cyclic voltammetry data, Tauc plots, time-resolved emission spectra and computational results for TTTD-3HMAT, TTTD-3tBu, TTTD-3ACR. See DOI: https://doi.org/10.1039/d2tc01153k |
| This journal is © The Royal Society of Chemistry 2022 |