
From ‘Gift’ to gift: producing organic solvents from CO2
Zhengkai
Chen
 a,
Shiying
Du
a,
Jiajun
Zhang
a and
Xiao-Feng
Wu
*abc
a,
Shiying
Du
a,
Jiajun
Zhang
a and
Xiao-Feng
Wu
*abc
aDepartment of Chemistry, Zhejiang Sci-Tech University, Hangzhou 310018, People's Republic of China. E-mail: xiao-feng.wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Science, 116023, Dalian, Liaoning, China
cLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Straβe 29a, 18059 Rostock, Germany
First published on 2nd November 2020
Abstract
‘Gift’ means ‘poison’ in German, which fits the situation of CO2 in our atmosphere from some points of view. Hence, the utilization and transformation of CO2 into highly value-added chemicals are like going from ‘Gift’ to gift and have attracted considerable attention from the chemical community. In this review, the latest advances in the field of production of commonly used organic solvents from CO2 are summarized and discussed.
 Zhengkai Chen | Zhengkai Chen was born in 1988 in Henan, China. He obtained his B.S. degree from Zhengzhou University in 2010. He received a Ph.D. degree from Zhejiang University under the supervision of Prof. Yuhong Zhang in 2015. Then he joined the Department of Chemistry, Zhejiang Sci-Tech University, and was promoted to associate professor in 2019. His current research interests focus on transition-metal-catalyzed carbonylative transformations and new methods for the synthesis of functionalized nitrogen-containing heterocycles. |
 Shiying Du | Shiying Du was born in 1994 in Jilin, China. He obtained his B.S. degree from Qingdao University of Science and Technology in 2016. He is currently studying towards an M.S. degree at Zhejiang Sci-Tech University under the supervision of Dr Zhengkai Chen and Prof. Xiao-Feng Wu. His current research interests focus on the transition-metal-catalyzed synthesis of functionalized nitrogen-containing heterocycles. |
 Jiajun Zhang | Jiajun Zhang was born in 1996 in Hebei, China. He obtained his B.S. degree from Dezhou University in 2018. He is currently studying towards an M.S. degree at Zhejiang Sci-Tech University under the supervision of Dr Zhengkai Chen and Prof. Xiao-Feng Wu. His current research interests focus on transition-metal-catalyzed carbonylative transformations and metal-free-mediated synthesis of nitrogen-containing heterocycles. |
 Xiao-Feng Wu | Xiao-Feng Wu was born in China. He studied chemistry at Zhejiang Sci-Tech University (China), where he got his bachelor's degree in science (2007). In the same year, he went to Rennes 1 University (France) and earned his master's degree in 2009. Then he joined Matthias Beller's group at the Leibniz Institute for Catalysis (Germany), where he completed his PhD defense in January 2012. Subsequently he started his independent research at LIKAT and ZSTU where he was promoted to professor in 2013. In March 2017, Xiao-Feng defended his Habilitation successfully from Rennes 1 University (France). In 2020, he joined the Dalian Institute of Chemical Physics (DICP) and is leading a research group on practical synthesis. Xiao-Feng has authored >350 publications in international journals; meanwhile, he is also the editor or author of >10 books. In 2019, Xiao-Feng was invited to publish his Author Profile in Angew. Chem., Int. Ed. (Angew. Chem., Int. Ed., 2019, 58, 8624). |
1 Introduction
As one of the main components of greenhouse gases, the steady increase of CO2 concentration has become the chief culprit of global climate change, which has caused serious social concerns. Contemporary chemists are obliged to face this severe challenge and seek suitable and sustainable methods to solve this problem. On the other hand, as an ideal and versatile C1 building block, much attention on the valorization of CO2 has been attracted among chemists due to its abundance, easy availability, nontoxicity and renewability.1–8 Recent years have witnessed the rapid development of chemical transformations of CO2 into a variety of high value-added chemicals.9–18 Among these transformations, the production of various organic solvents from CO2 is of prime significance, as organic solvents are widely used in academic research and are usually employed as raw materials in the chemical industry. In addition, the utilization of supercritical CO2 (scCO2) as a solvent in various chemical transformations has been flourishing in the past decades; these have found wide applications in a range of research fields due to the nontoxic and nonflammable properties of CO2.19–23Considering the huge demand for organic solvents, the transformation of CO2 into organic solvents has emerged as an attractive and promising research area, especially for industrial processes. However, CO2 is a highly oxidized form of carbon and thermodynamically stable and/or kinetically inert; hence its activation and utilization typically require the involvement of reactive substances with high energy or severe reaction conditions,24,25 limiting the wide utility of CO2 transformations at some extent. With respect to solvent synthesis from CO2, relevant research progress mainly focuses on methanol, N,N-dimethylformamide (DMF), dimethyl carbonate (DMC), dimethyl ether (DME), and so on. Virtually, there are a large number of publications about methanol synthesis from CO2via diverse catalytic systems and there have been several excellent reviews on this subject during the past decades.4,7,26–28 Therefore, this review will emphatically introduce the preparation of DMF and DMC from CO2 and select some of the mainstream studies about methanol synthesis, as well as the synthesis of some other useful solvents (Scheme 1).
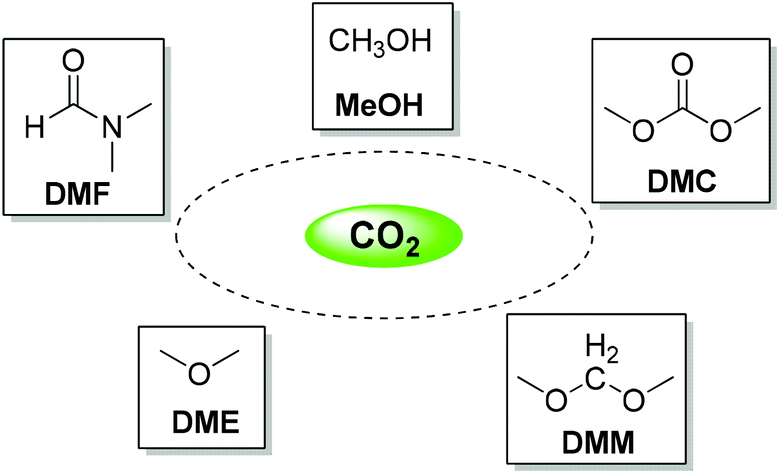 | ||
| Scheme 1 Producing organic solvents from CO2. | ||
2 Synthesis of DMF from CO2
N,N-Dimethylformamide (DMF) is a frequently used polar solvent and extremely versatile chemical reagent in organic synthesis.29,30 The conventional method for the construction of DMF is the reaction of dimethylamine with carbon monoxide in methanol, which was first reported by Haynes and co-workers in 1970.31 Afterwards, Kudo and co-workers developed a PdCl2-catalyzed protocol for the synthesis of DMF with 40 bar of CO2 and 80 bar of H2 at 170 °C.32 Vaska's group optimized the reaction by testing diverse metal complexes and DMF could be produced in the presence of an active platinum-based catalyst ([Pt2(μ-dppm)3]).33,34 Later, Noyori and co-workers achieved Ru-catalyzed production of DMF in supercritical CO2 (130 bar) with a higher turnover number (TON) of up to 370![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.35,36 A well-defined homogeneous RuCl2[P(CH3)3]4 catalyst was the key factor for the reaction and scCO2 was regarded as both reaction medium and reactant. In the reaction, ammonium formate salt was generated.
000.35,36 A well-defined homogeneous RuCl2[P(CH3)3]4 catalyst was the key factor for the reaction and scCO2 was regarded as both reaction medium and reactant. In the reaction, ammonium formate salt was generated.
An improved protocol for the synthesis of DMF was demonstrated by Baiker and co-workers (Scheme 2).37 A bidentate Ru/phosphine complex [RuCl2(dppe)2] was applied as a highly efficient catalyst to realize a TON of up to 740000 and the reaction was conducted at 100 °C under 130 bar of CO2 and 85 bar of H2. The reaction could be scaled up to produce 530 kg of DMF within 2 hours by using 1 gram of ruthenium catalyst. Notably, the solvent methyl formate was formed under a similar catalytic system in the presence of methanol and trimethylamine. Then, a sol–gel-derived heterogeneous catalyst containing ruthenium complexes was synthesized and utilized for DMF synthesis with high turnover frequencies (up to 18400 h−1), which was completed by the group of Baiker.38–40 The serial positive results for DMF synthesis under ruthenium catalysis provide many insights for further intensive investigation upon industrialized application, such as developing soluble catalytic complexes in the supercritical system and suitable ligands.
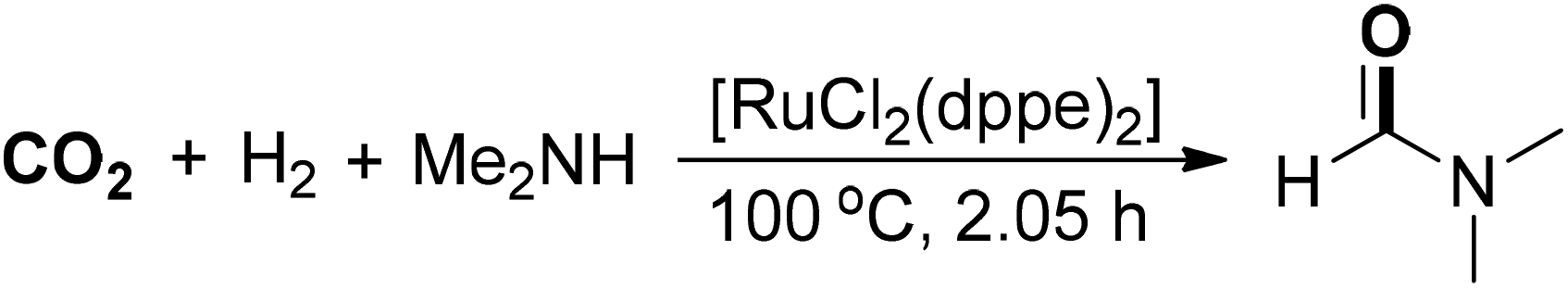 | ||
| Scheme 2 Ru-catalyzed DMF synthesis from CO2. | ||
Ito and co-workers designed a molybdenum complex, MoH3[Si(Ph)[Ph2PCH2CH2P(Ph)C6H4-o]2], and first applied it to catalytic DMF synthesis from CO2.41 The insertion of CO2 into the Mo–H bond produced another formal molybdenum complex, which was identified as an active catalyst and the key intermediate in DMF synthesis. A cheap heterogeneous Cu–ZnO catalyst mediated synthesis of DMF from CO2, H2, and dimethylamine with high efficiency was disclosed by Han and co-workers.42 Dimethylammonium dimethylcarbamate (DIMCARB) was used as a safe and convenient source of dimethylamine. The low-toxic Cu and ZnO catalyst showed good synergistic effects and great potential for application in the solvent-free conditions for producing DMF. After four years, another heterogeneous trans-bis(glycinato)copper(II) complex was developed to catalyze DMF synthesis in a highly efficient manner, which was disclosed by Jain's group.43 In 2018, Sadeghzadeh and co-workers reported a new class of copper(II) complex based on FeNi3/KCC-1 for the N-formylation of amines via CO2 reductive hydrogenation and DMF could be delivered in 91% yield.44
Beller, Laurenczy and co-workers developed a series of nonprecious metals to catalyze DMF synthesis from CO2. An active iron catalyst system was in situ generated from Fe(BF4)2·6H2O and the tetradentate ligand P(CH2CH2PPh2)3 for the hydrogenation of carbon dioxide (Scheme 3a).45 Under the established catalytic system, DMF was produced in 75% yield with a TON of 727 with 30 bar of CO2 and 60 bar of H2 at 100 °C. Soon after, a novel cobalt dihydrogen complex (Co(BF4)2·6H2O/PP3) was applied to DMF synthesis under similar conditions, showing an improved catalytic activity with a TON of 1308 (Scheme 3b).46 Another efficient iron-based catalyst with a tetradentate phosphorus ligand [tris(2-(diphenylphosphino)phenyl)phosphine] could enable the production of DMF in good yield with higher TONs (5104) (Scheme 3c).47 Similarly, diethylformamide could be obtained in moderate yield with a TON of 2114, which was substantially higher than that of a previously reported iron-catalyzed system. These well-defined iron- or cobalt-based catalyst systems achieved significantly higher yields and TONs for CO2 hydrogenation than those of other non-precious-metal-based catalysts and even superior to many known precious-metal catalytic systems.
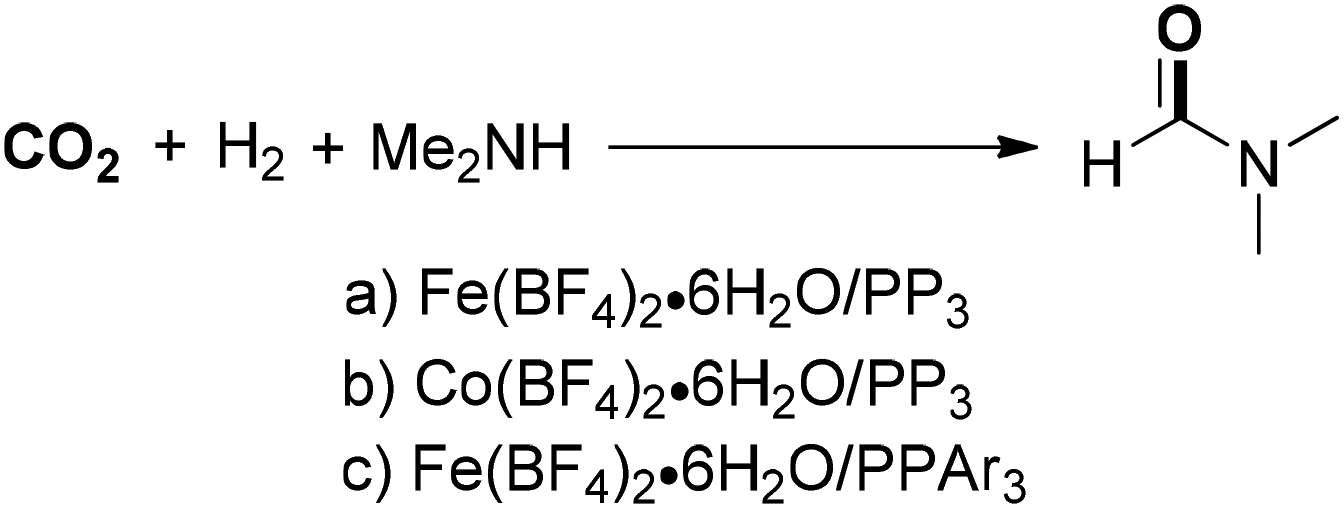 | ||
| Scheme 3 Fe- or Co-catalyzed DMF synthesis from CO2. | ||
Except for commonly used dihydrogen (H2), other reductants with high activity were also explored for the hydrogenation of CO2, such as hydrosilanes (R3Si–H) and hydroboranes (R2B–H), due to active Si–H and B–H bonds. In 2011, Cantat and co-workers reported a diagonal transformation for the synthesis of formamides from CO2, in which organosilanes were chosen as cheap and nontoxic reducing agents (Scheme 4).48 In the organocatalytic process, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) was an effective catalyst to promote the insertion of CO2 into N–H bonds to form the CO2 adduct A. The carbamate [Me2NCO2][H2NMe2] B reacted with A to give salt C, which was reduced by phenylsilane to lead to the DMF product and silanol by-product. The protocol exhibited several advantages over the previously established amine/CO2/H2 formylation system, including no use of metal catalyst, a lower pressure, solvent-free conditions and wide scope of amines. Considering the use of cheap and nontoxic silanes as reductants, the organocatalytic protocols for the formylation of amines with CO2 perhaps have broader prospects on the way to industrialization.
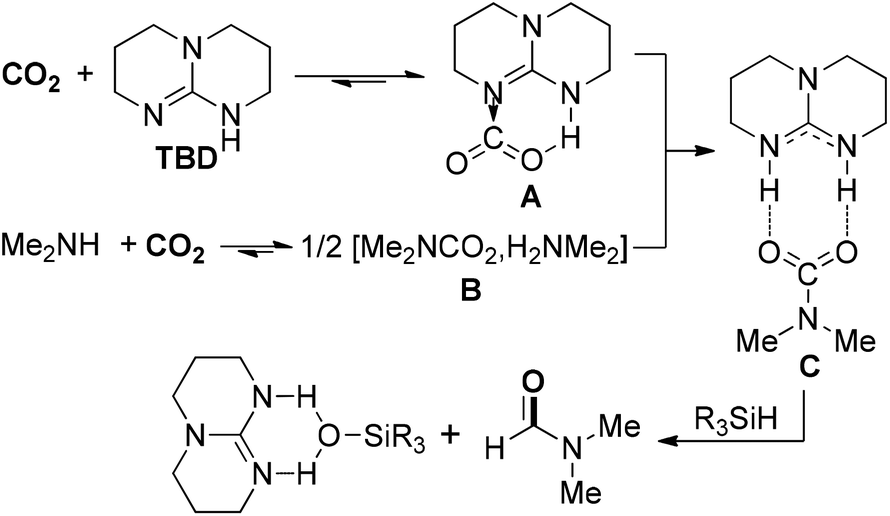 | ||
| Scheme 4 TBD-catalyzed synthesis of DMF from CO2. | ||
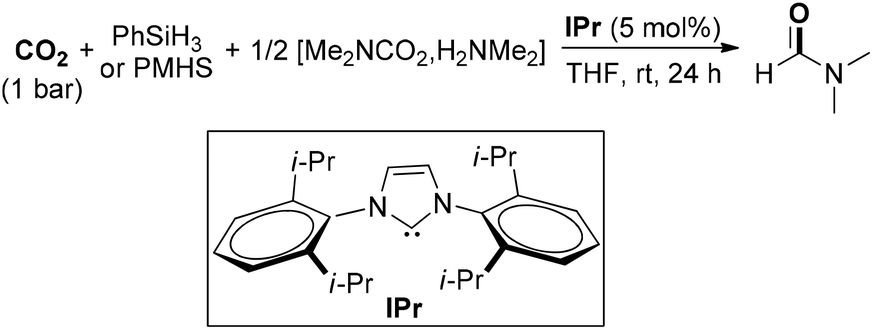
Cantat and co-workers further explored the organocatalytic formylation of N–H bonds by using N-heterocyclic carbenes, which featured mild reaction conditions and the utilization of two abundant and nontoxic chemical wastes of CO2 and polymethylhydrosiloxane (PMHS) (Scheme 5).49 The combination of NHC and silane enabled the formation of DMF in high yield at room temperature under 1 bar of CO2 starting from carbamate [Me2NCO2][H2NMe2]. The highly active organocatalytic system was also amendable for the formylation of various N–H bonds, including amines, anilines, imines, hydrazines, hydrazones and N-heterocycles. Under this efficient organocatalytic system, a large-scale CO2 recycling process is expected to be achieved. Another reaction for the formylation of amines using CO2 and PMHS was reported by Bhanage and Nale, who applied K2CO3 as the catalyst.50
 | ||
| Scheme 5 N-Heterocyclic carbene (NHC)-catalyzed synthesis of DMF from CO2. | ||
In 2014, Shi and co-workers prepared a heterogeneous Pd/Al2O3-NR-RD catalyst and utilized it for the catalytic formylation of amines with CO2–H2 under mild conditions.51 High catalytic activity was observed for the production of DMF in 84% yield. In the same year, another heterogeneous catalytic reaction for exclusive DMF synthesis was described by Cao and co-workers.52 A bifunctional catalyst based on partially reduced iridium oxide supported on TiO2 was created and found to show excellent activity for DMF synthesis through reductive activation of CO2 under a H2 atmosphere. The method directly employed available aqueous NHMe2 as the amine source instead of previously reported expensive dimethylammonium dimethylcarbamate.
Tanaka and co-workers developed a photocatalytic CO2 reduction reaction for the selective synthesis of DMF (Scheme 6).53 In the transformation, a [Ru(bpy)2(CO)2]2+/[Ru(bpy)3]2+/Me2NH/Me2NH2+ system was established to enable photocatalytic CO2 reduction. The nucleophilic attack of Me2NH on [Ru(bpy)2(CO)2]2+ produced [Ru(bpy)2(CO)(CONMe2)]+, which acted as the precursor of DMF.54 Me2NH and Me2NH2+ were used as electron donor and proton source, respectively. It was also found that the presence of the Li+ ion could greatly inhibit DMF production because Li+ blocked the formation of the RuCO complex through its strong stabilization of the Ru–CO2 scaffold.
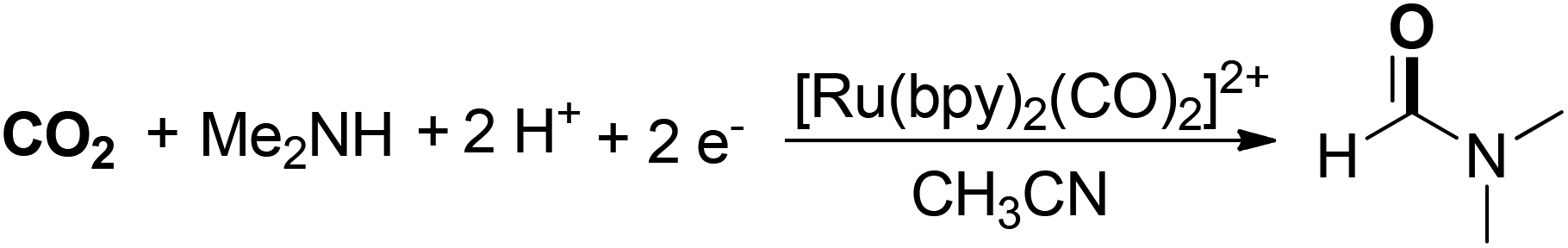 | ||
| Scheme 6 Photocatalytic CO2 reduction for the synthesis of DMF. | ||
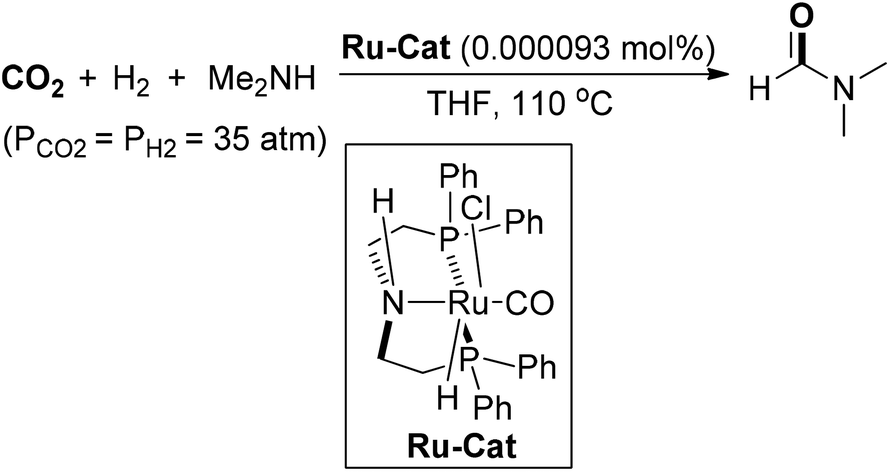
In 2015, Ding and co-workers demonstrated a highly efficient catalytic system for N-formylation of amines with CO2 and H2, which utilized ruthenium-pincer-type complexes as catalysts to produce diverse formamides with high efficiency and selectivity (Scheme 7).55 With regard to DMF synthesis, the N-formylation of dimethylamine was achieved with the ruthenium catalyst at a loading of 0.000093 mol%, producing DMF in 56% yield with a TON value of up to 599000. Noteworthy was that the ruthenium catalyst could be recycled for 12 runs for the preparation of DMF without significant loss of activity, which showed huge potential of practical utilization of this protocol. Gratifyingly, the industrial application of this cost-effective reaction of CO2 hydrogenation for DMF production was successfully realized in 2019. It is the first kiloton pilot plant of DMF production in the world, which provides a useful and practical pathway for CO2 utilization.
 | ||
| Scheme 7 Ruthenium-catalyzed DMF synthesis from CO2. | ||
Fu, Lin and co-workers reported readily available alkali-metal carbonates especially a cesium carbonate catalyzed reaction for the formylation of amines with CO2 and hydrosilanes (Scheme 8).56 Under mild conditions (1 bar of CO2 at room temperature), DMF could be delivered in 95% yield in the presence of PhSiH3 as a reductant. In addition, methylation of amines could also be attained and the selectivity was readily controlled by tuning the reaction temperature and silane. An obvious “cesium effect” upon the catalytic activity of alkali-metal carbonates was observed in the reaction, which might originate from the increased solubility of the carbonate salt.
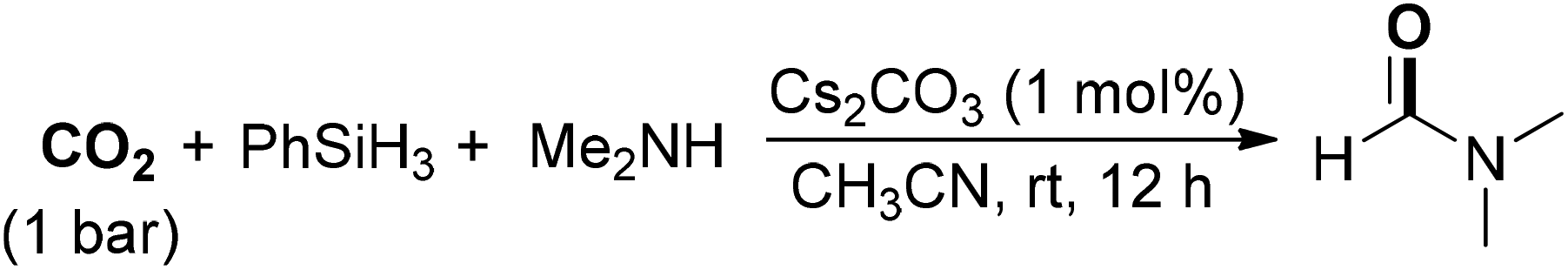 | ||
| Scheme 8 Cesium carbonate catalyzed DMF synthesis from CO2. | ||
Jain and co-workers synthesized a graphene oxide (GO)-immobilized heteroleptic iridium complex and used it as the first heterogenized homogeneous catalyst for DMF preparation from CO2, H2 and dimethylamine under solvent-free conditions.57 The catalyst could be easily recovered and recycled six times without the loss of catalytic efficiency.
An amine-modified meso-Al2O3@MCM-41, developed by Bhanage and co-workers, was applied as a catalyst for the formylation of amines with CO2.58 Under the developed heterogeneous catalytic system, DMF could be obtained in excellent 98% yield with dimethylamine borane (DMAB) as a green reducing source. Then, DMAB was applied to another heterogeneous catalytic N-formylation of amines for the synthesis of N-formamides and benzimidazole,59 which employed ruthenium nanoparticles (Ru-NPs) supported on polymeric ionic liquids (PILs) as effective catalysts to give DMF in 89% yield with a TON of 296. Subsequently, the same authors discovered an N-heterocyclic olefin (NHO) organocatalyst promoted chemical fixation of CO2 through the N-formylation of amines by using polymethylhydrosiloxane (PMHS) or 9-borabicyclo[3.3.1]nonane (9-BBN) as the reducing agent.60 In the reaction, DMF could be produced in good yield under mild conditions. The in situ formed zwitterionic NHO–carboxylate (NHO–CO2) adducts were the key factor for the activation of CO2. By the use of B(C6F5)3 as an efficient organocatalyst and dimethylamine borane (Me2NHBH3) as a green hydrogen transfer source, N-formylation of amines via hydrogenation of CO2 led to DMF in 89% yield with a TON of 1112.61 The interaction of bulky boron catalyst with amines could effectively activate CO2 and Me2NHBH3 molecules. The abovementioned transformations all enable the production of DMF in high yields but with moderate TONs, which is seemingly far from industrial application.
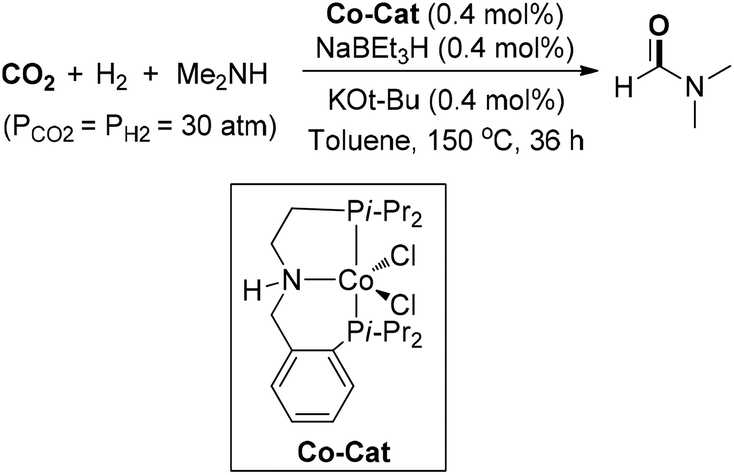
An Earth-abundant cobalt-catalyzed N-formylation of various amines under CO2 and H2 pressure (30 bar each) was disclosed by Milstein and co-workers (Scheme 9).62 A Co-PNP pincer complex was synthesized and could be transformed into active species in the presence of catalytic NaHBEt3 and t-BuOK. For large-scale DMF synthesis, the lower catalyst loading (0.4 mol%) rendered the formation of DMF in 54% yield with a TON of 130 with the addition of 4 Å molecular sieves. Later, Tu and co-workers explored a series of NHC–Ir coordination assemblies as solid molecular catalysts for N-formylation of diverse amines.63 Moderate yield of DMF was attained, even at 0.1 mol% catalyst loading, and the solid catalyst could be reused more than 10 runs.
 | ||
| Scheme 9 Ruthenium-catalyzed DMF synthesis from CO2. | ||
Jessop and co-workers also developed an abundant-metal-catalyzed formylation of amines by catalytic hydrogenation of CO2 to prepare formamides (Scheme 10).64 Dimethylammonium dimethylcarbamate was adopted under the catalysis of Ni(II)–phosphine complexes for the synthesis of DMF with a high TON value. Several metal complexes showed catalytic activity and Ni(II) salts were the superior choices. Another homogeneous catalyzed hydrogenation of carbon dioxide to DMF was achieved by using an in situ generated ruthenium catalyst from RuCl3·H2O and the phosphine ligand 2,2′-bis(diphenylphosphinomethyl)-1,1′-biphenyl (BISBI).65 An aqueous biphasic solvent system was utilized, where the catalyst could be recycled by immobilization in a nonpolar alcoholic solvent and the product could be extracted in situ into the aqueous phase. Then, the developed reaction system of DMF production was successfully transferred from lab-scale to a continuously operated miniplant with a low concentration of carbon monoxide as the only byproduct.66 The positive results highlighted long-term (over 95 h) stability and selectivity of the catalytic system. The continuous addition of the ternary amine could result in the increase of the yield by tuning the basicity of the reaction system. The obtained product could be easily isolated through distillation without decomposition, exhibiting good prospects for scaled-up production.
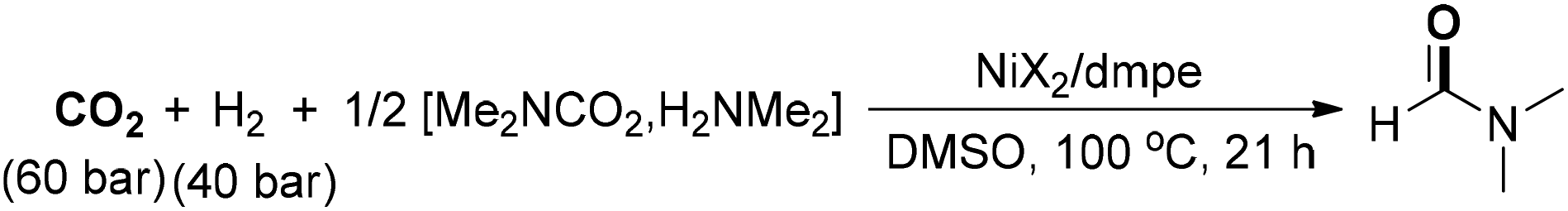 | ||
| Scheme 10 Ni(II)-catalyzed DMF synthesis from CO2. | ||
Recently, Shi and co-workers developed a method for the integration of nano- and molecular catalysis through the synthesis of N-doped carbon layers on AlOx-supported nano-Cu, which could be employed for the preparation of DMF in a controllable manner from dimethylamine and CO2/H2 (Scheme 11).67 The catalytic material was in situ formed by the reaction of CuAlOx and 1,10-phen under a H2 atmosphere. The active catalyst could be easily recycled in three runs for the production of DMF with high yield and selectivity. In addition, the CuAlOx catalyst also exhibited good performance for the catalytic hydrogenation of DMF to N(CH3)3. Later, the same authors demonstrated a heterogeneous Pd supported on natural palygorskite catalyst for amine formylation with CO2 and H2.68 In the transformation, DMF could be produced in 86% yield by using dimethylamine aqueous solution as the starting reagent. Another heterogeneous Pd-catalyzed CO2 fixation of amine to prepare DMF was described by Islam and co-workers, which utilized Merrifield resin supported palladium nanomaterial (Pd-PS-amtp catalyst) as a catalyst and poly(methylhydrosiloxane) (PMHS) as a hydride transferring agent.69
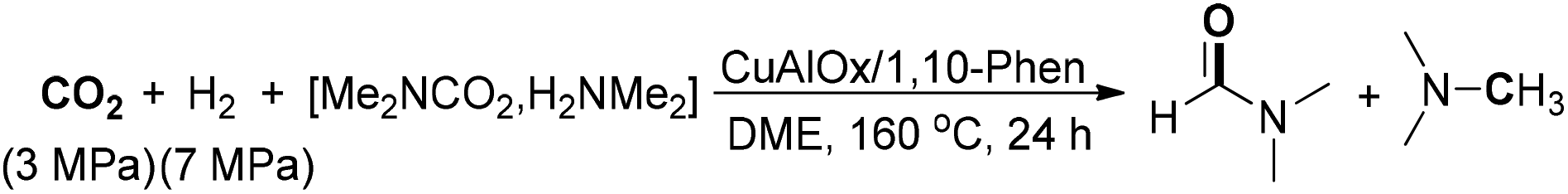 | ||
| Scheme 11 CuAlOx-catalyzed DMF synthesis from CO2. | ||
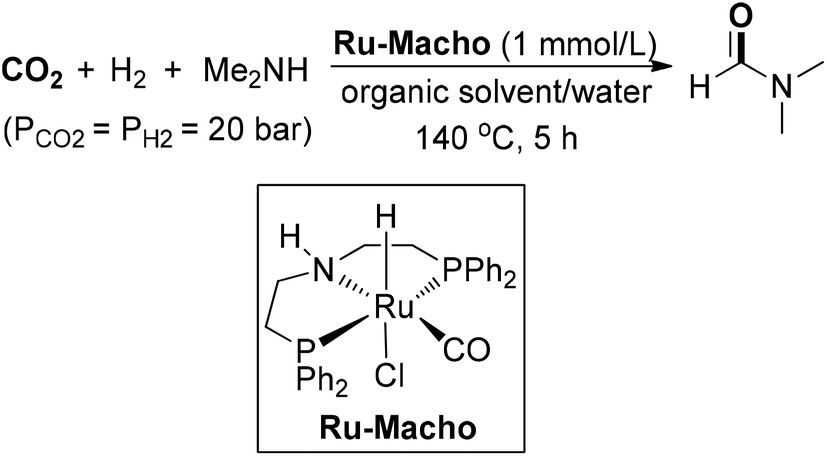
Vorholt and co-workers demonstrated a ruthenium-catalyzed synthesis of DMF from CO2 in a biphasic solvent system (Scheme 12).70 The PNP-pincer complex Ru-Macho was used as an active catalyst, which was identical to the catalyst in Ding and co-workers work.55 Several key factors of the reaction were investigated, including organic solvent, reaction pathways, the formate route and the concentration of CO2/amine. Adding carbon dioxide via reactive absorption to aqueous dimethylaminoethanol could enable DMF production in up to 81% yield, presumably due to the higher activities resulting from high basicity. Recently, this homogeneous biphasic protocol has been implemented on a miniplant scale by the same authors.71 The catalyst Ru-Macho complex could be recycled through immobilization in an alcohol phase and maintained high stability over 230 h, providing DMF in an average yield of 48%. A two-step process was designed to be applied for the synthesis of other formamides.
 | ||
| Scheme 12 Ru-Macho-catalyzed DMF synthesis from CO2. | ||
Very recently, an efficient heterogeneous catalytic system for DMF synthesis by hydrogenation of CO2 was established by Yoon and co-workers,72 which employed ruthenium-grafted bisphosphine-based porous organic polymer (Ru@PP-POP) as a recyclable catalyst. In a batch process, a TON of up to 160000 was obtained and excellent productivity and durability were observed in a continuous-flow process, thus offering great potential for industrial application of CO2 hydrogenation for DMF production.
A bifunctional heterogeneous Ru catalyst was developed by Ding, Yan and co-workers for the N-formylation of amine and CO2.73 The Ru-PPh3-SO3Na@POP catalyst, generated from the copolymerization of 3v-PPh3 and sodium p-styrenesulfonate, could immobilize metals and alkali on porous organic polymers, enabling the formation of DMF from dimethylamine in 62% yield with 3 MPa of CO2 and H2.
3 Synthesis of MeOH from CO2
As the simplest aliphatic alcohol and a kind of important industrial raw material, the annual production of methanol exceeds 100 million tons and still increases year by year.74 Methanol is currently produced from coal, biomass, especially natural gas and syngas,75–77 which is usually utilized as the precursor of many important industrial products, alternative fuels and hydrogen storage materials (12.5 wt% H2).78,79The industrial-scale synthesis of methanol lies in the catalytic reactions of syngas in the presence of Cu/ZnO/Al2O3-type heterogeneous catalysts under high pressure and elevated temperature.80,81 The production of MeOH solely from the hydrogenation of CO2 with heterogeneous catalysts has been intensively investigated and several excellent relevant reviews have been published.4,82–87 Very recently, Zhang and co-workers have summarized the significant advances in heterogeneous catalysis of CO2 hydrogenation to methanol in detail.88 In addition, the catalytic reduction of CO2 to methanol could be realized in the presence of stoichiometric amounts of reducing agents, such as boranes or silanes.89–95 In this section, we will selectively introduce some seminal work regarding MeOH synthesis from the hydrogenation of CO2 with homogeneous catalysts.
In 2011, Milstein and co-workers reported the first homogeneous catalytic synthesis of MeOH from the hydrogenation of carbonic acid derivatives and formates using pincer-type RuII catalysts, which constituted indirect routes from CO2 to methanol.96,97 Although the hydrogenation of dimethyl carbonate could generate methanol, the dimethyl carbonate was more expensive than methanol, which made it not an economical choice. After one year, Ding and co-workers described the catalytic hydrogenation of cyclic carbonates from CO2 and epoxides for the preparation of methanol and diols with readily available (PNP) RuII pincer complexes as catalysts.98 Starting from ethylene carbonate, two important bulk chemicals, methanol and ethylene glycol (EG), were readily produced with high TON values under relatively mild conditions. It should be noted that ethylene carbonate could be industrially obtained by reacting ethylene oxide with CO2. Poly(propylene carbonate) could also be hydrogenated to deliver 1,2-propylene diol and methanol. Another indirect methanol production from CO2 through a formic acid disproportionation strategy with a homogeneous iridium catalyst was reported by Laurenczy and co-workers.99
Sanford and Huff developed a cascade reaction with three different homogeneous catalysts to complete the hydrogenation of CO2 to methanol via formic acid and methyl formate intermediates with a maximum TON of 21.100 Then, they achieved the homogeneous Ru-pincer complex catalyzed reduction of CO2 to methanol in the presence of NHMe2 (Scheme 13).101 Under basic reaction conditions, MeOH could be produced with a TON of up to 550. The additional amine reacted with CO2 to generate dimethylammonium dimethylcarbamate, which coupled with formic acid to deliver DMF. In the same year, Ding and co-workers also accomplished a one-pot sequential N-formylation and hydrogenation process for the conversion of CO2 to MeOH via a formamide intermediate by using Ru-pincer complex as catalyst.55
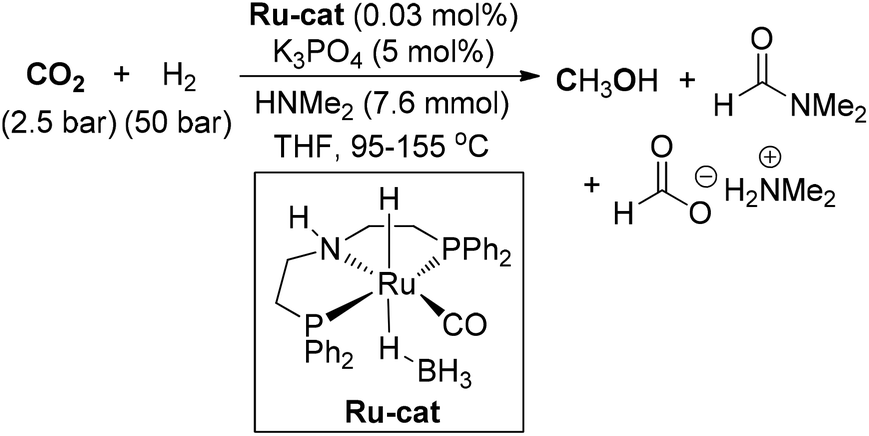 | ||
| Scheme 13 Ru-catalyzed hydrogenation of CO2 to MeOH. | ||
In 2012, a homogeneous catalytic hydrogenation of CO2 to methanol with a single ruthenium phosphine complex was investigated by Leitner, Klankermayer and co-workers (Scheme 14).102 By adopting (Triphos)Ru-(TMM) (TMM = trimethylenemethane) as catalyst and bis(trifluoromethane)sulfonimide (HNTf2) as acidic additive, a TON of up to 221 for MeOH synthesis was attained under 20/60 bar of CO2/H2. The counterion introduced from the acid exerted a great impact upon the catalyst's performance. The reaction was further explored through a detailed mechanistic study, which demonstrated that the hydrogenation of CO2 to methanol could occur under a single cationic Triphos–Ru catalytic system without the need for an alcohol additive.103 Furthermore, an aqueous biphasic system of 2-MTHF–water was developed for MeOH synthesis and catalyst recycling.
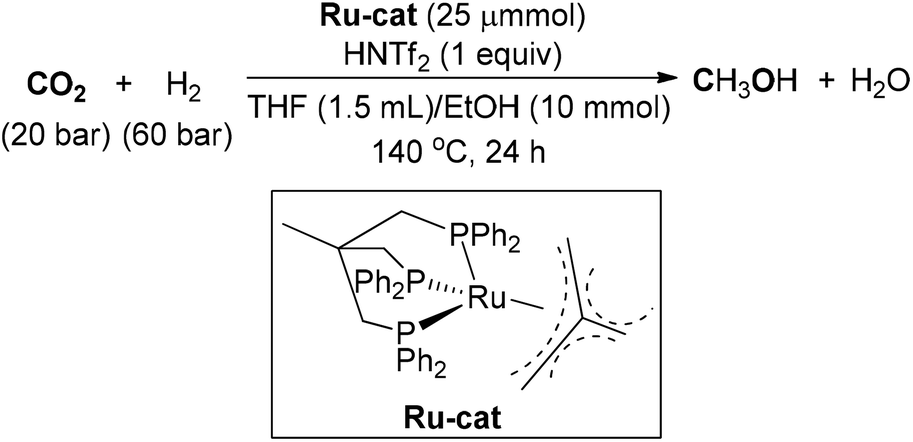 | ||
| Scheme 14 Ru-catalyzed synthesis of MeOH from CO2. | ||
Milstein and co-workers described a CO2 hydrogenation reaction by using PNN pincer Ru catalysts, in which CO2 was captured by aminoethanols combined with hydrogenation of the captured oxazolidinone product for the formation of MeOH.104 The above two procedures could be performed in the same reaction mixture without additional isolation or purification steps. Valinol was also used to capture CO2 at 1 bar in the presence of catalytic Cs2CO3. The approach features a low pressure of CO2 and high energy utilization rate, promoting the development of CO2 conversion into other value-added chemicals.
The Ru-pincer complex was applied for the synthesis of MeOH from CO2 with pentaethylenehexamine (PEHA) in an ethereal solvent, which was presented by Olah, Prakash and co-workers.105 It was the first example of reductive conversion of CO2 from air to MeOH with a homogeneous catalyst, despite the low CO2 concentration (400 ppm). Then, the group of Prakash developed a tandem system for CO2 capture in aqueous amine solution and hydrogenation to methanol in a biphasic 2-MTHF/water system.106 The catalytic combination of Ru-MACHO-BH complex with high-boiling polyamine PEHA exhibited the best activity for MeOH synthesis. Further investigation towards mechanistic insights into Ru pincer/amine mediated hydrogenation of CO2 to MeOH was performed by the same authors.107 The observation revealed that Ru-Macho showed the highest catalytic activity in both amine formylation and formamide hydrogenation. In addition, (di/poly)amines were also effective for MeOH formation.
In 2017, Beller and co-workers disclosed the first homogeneous non-noble-metal catalyst for the synthesis of MeOH from hydrogenation of CO2 (Scheme 15).108 The reaction could be operated at 100 °C with high reactivity in the presence of in situ generated catalyst from [Co(acac)3], Triphos, and HNTf2. Mechanistic studies revealed that a cationic cobalt/Triphos complex was regarded as an active catalyst, which was produced after slow removal of the acetylacetonate ligands. The study will stimulate the development of other non-noble-metal-catalyzed homogeneous hydrogenation of CO2 to methanol. Other studies involving Earth-abundant metal-based complexes as catalysts for hydrogenation of CO2 to methanol were consecutively reported in the same year.109,110
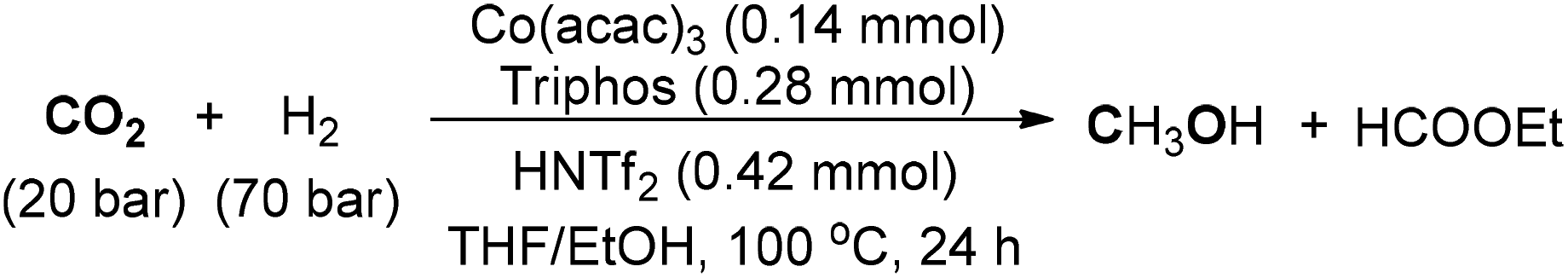 | ||
| Scheme 15 Co-catalyzed synthesis of MeOH from CO2. | ||
Zhou and co-workers designed a ruthenium complex with a tetradentate bipyridine ligand and applied it for the conversion of CO2 to achieve MeOH synthesis (Scheme 16).111 Under 2.5 atm of CO2 and 40 atm of H2, catalytic hydrogenation of CO2 could enable the production of MeOH with a TON of 2100 in the presence of HNMe2. The tetradentate bipyridine ligand proved to be the key factor for the stability of the ruthenium catalyst. In addition, hydrogenation of CO2 to formamides and hydrogenation of formamides to methanol and amines was accomplished with a high TON value under the catalytic system.
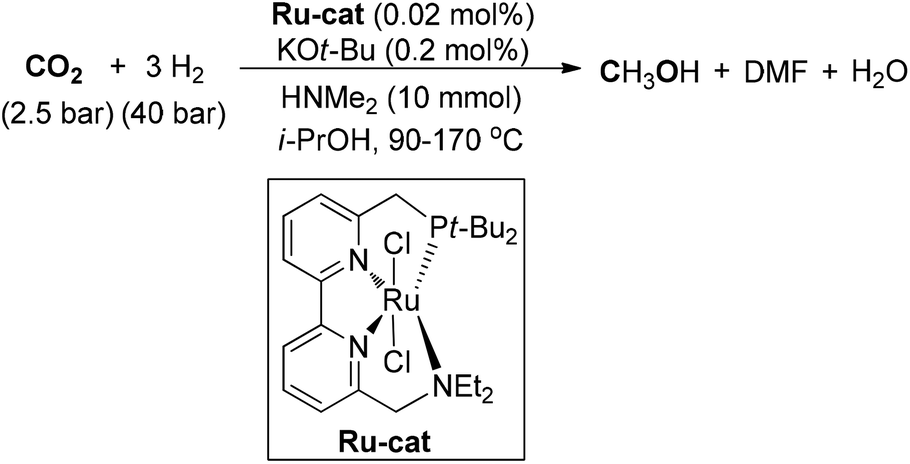 | ||
| Scheme 16 Ru-catalyzed synthesis of MeOH from CO2. | ||
In comparison with the three-catalyst cascade strategy established by Sanford and co-workers,100 an improved homogeneous catalytic cascade system for CO2 hydrogenation to MeOH was demonstrated by Goldberg and co-workers.112 The combination of Ru(H)2[P(CH2CH2PPh2)3]/Sc(OTf)3/Ir(tBuPCP)(CO) was identified as an active catalyst for the synthesis of MeOH with a TON of 428 via formic acid and formate ester intermediates. Sc(OTf)3 in the transformation served as both a Lewis and Brønsted acid donor.
Very recently, Leitner, Werlé and co-workers established a cobalt-based catalytic system that could achieve selective transformation of CO2 individually to the formic acid, the formaldehyde, or the methanol level.113 The catalyst was prepared based on the 3d transition-metal cobalt with a PNP pincer-type triazine ligand. For methanol synthesis, the catalytic hydrosilylation of CO2 was conducted in [D6]-DMSO at 80 °C under a constant flow of CO2 at 1 bar for 4 h with phenylsilane, producing the methoxysilane species with 99% selectivity.
Prakash and co-workers reported a Ru-catalyzed system for CO2 capture and conversion to MeOH by using alkali-metal hydroxide as a CO2 capturing agent and ethylene glycol as a formate ester promoter (Scheme 17).114 In the one-pot transformation, CO2 could be readily captured by ethylene glycol in the presence of KOH to form potassium glycolate (PG), which underwent in situ hydrogenation using Ru-PNP catalyst under 70 bar of H2 at 100–140 °C to realize MeOH production with high activity. The high CO2 capture efficiency of this protocol and regeneration of the hydroxide base rendered it superior to the previously established amine-based methods for the conversion of CO2 from ambient air to methanol in a scalable process. The novel protocol is of great significance for the development of CO2 capture and conversion in a highly efficient manner.
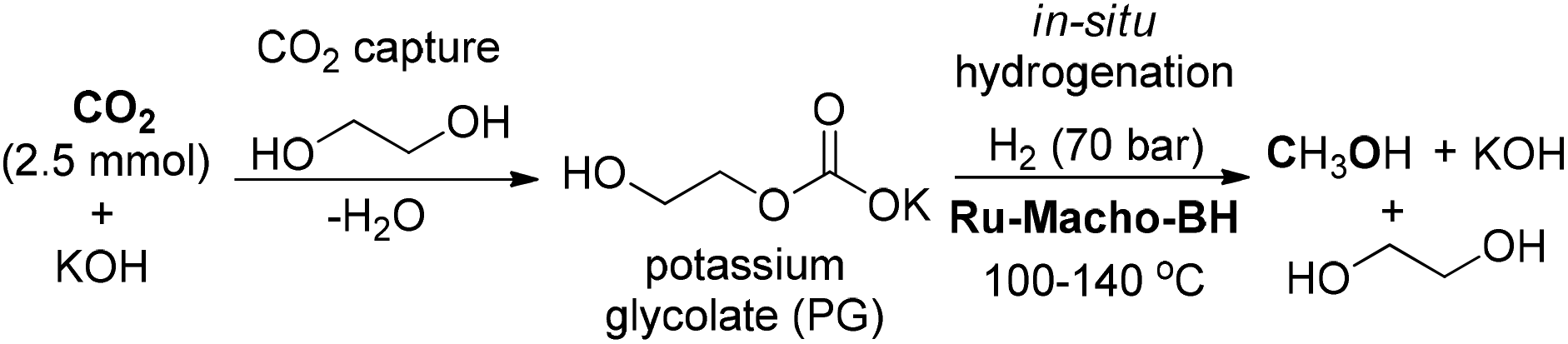 | ||
| Scheme 17 Ru-catalyzed hydrogenation of CO2 captured in ethylene glycol for MeOH synthesis. | ||
4 Synthesis of DMC from CO2
Dimethyl carbonate (DMC) is regarded as an environmentally benign solvent and versatile reagent for organic synthesis owing to its low toxicity, low viscosity and high biodegradability.115–119 DMC also act as an alternative for several toxic organic compounds and as non-aqueous electrolyte for lithium batteries.120,121 Conventional approaches for the synthesis of DMC lie in phosgenation and carbonylation of methanol, and transesterification of ethylene carbonate (EC) and methanol, which suffer from considerable issues regarding operational safety and environmental protection. Thus, direct DMC synthesis from abundant CO2 and methanol has emerged as an attractive and powerful pathway from the viewpoint of sustainable development and green chemistry.Although the direct synthesis of DMC from carbon dioxide and methanol constitutes a state-of-the-art process, some drawbacks still exist, which include the low yield and slow reaction rate, resulting from highly stable CO2 and thermodynamic limitations of the reaction. Key solutions to overcome these difficulties focus on the development of novel catalysts and diverse dehydrating agents. In this regard, tremendous effort has been devoted to this aspect and several excellent reviews summarizing relevant achievements have been thereby presented.122–124 Therefore, in this section we will emphatically describe the latest developments on DMC synthesis from CO2 in the last five years.
Due to their excellent catalytic activity, selectivity, oxidation–reduction properties and larger specific surface area, ceria-based oxide catalysts have attracted considerable attention for the direct synthesis of DMC from CO2 (Scheme 18).125–129 Ceria could provide selective catalytic sites to activate CO2 for the enhancement of the reaction rate of CO2 and alcohols to organic carbonates.130 Considering the unfavourable nature of bare CeO2, including small surface areas, poor durability and quick deactivation, doping CeO2 with transition metals (M) to form MxCe1−xO2 composites is usually employed in the catalytic reaction.
 | ||
| Scheme 18 Ceria-based oxide catalyzed synthesis of DMC from CO2. | ||
Wang and co-workers investigated the direct synthesis of DMC from CO2 by using a series of CeO2 nanocrystals of various structures (spindle, cube, and octahedron).131 With the addition of 2,2-dimethoxypropane (DMP) as an effective dehydrating agent, spindle-CeO2 exhibited a more favourable catalytic activity than other CeO2 catalysts. The hydrolysis of DMP could in situ remove water from the reaction system, thereby overcoming the thermodynamic limitation of the reaction and increasing the DMC yield. In 2017, Kumar and co-workers prepared several calcium–cerium mixed-metal oxide catalysts (CeO2–CaO) with various Ce/Ca molar ratios and applied them with 3 Å molecular sieves to the direct conversion of CO2 to DMC.132 The acidity and basicity of catalysts exerted a fundamental effect upon the DMC yield and the ceria–calcium oxide catalyst could be reused in five runs.
Owing to the surface properties and acid/base sites related to catalytic performance, the ceria/zirconia (CexZr1−xO2) solid solutions have attracted considerable attention in the field of the catalytic synthesis of DMC from CO2.133–135 Recently, Kim and co-workers synthesized a series of CexZr1−xO2 solid solutions (x = 0.25, 0.5, 0.75) with distinct morphology, acid/base sites and high surface area.136 The prepared CexZr1−xO2 solid solutions with modified physicochemical properties possessed excellent catalytic activity and stability in DMC synthesis from CO2 under mild conditions.
Diverse Zr-doped CeO2 nanorods with different ratios of Zr/Ce were prepared and used for the catalytic synthesis of DMC from CO2 and methanol at 6.8 MPa and 140 °C.137 Doping of Zr atoms into the ceria lattice resulted in the formation of oxygen vacancy sites, which could promote the adsorption and activation of CO2 and stabilize the final products by filling one oxygen atom of the CO2 molecule.138 It had been reported that CeO2 nanorods with high density of defect sites and acid–base sites showed higher activity than nanocubes and nano-octahedra.139 Among the developed Zr-doped CeO2 nanorods, Zr0.1Ce nanorods with the highest amount of oxygen vacancies showed the highest activity for DMC synthesis, implying a linear relationship between the concentration of surface oxygen vacancies and catalytic activity.
TiO2-doped CeO2 catalysts have also attracted considerable interest in the application of DMC synthesis because of the enhancement of the oxygen deficiency and acidity of the surface by doping TiO2 into CeO2.140 Meng and c-workers synthesized a number of TiO2-doped CeO2 nanorods and applied them to the direct synthesis of DMC from CO2 and CH3OH in a fixed-bed reactor.141 Better catalytic activities of ceria nanorods were observed with the introduction of TiO2 and the highest catalytic performance was achieved on Ti0.04Ce0.96O2 nanorod catalyst with a DMC selectivity of 83.1%. Kinetic and mechanistic studies revealed CO2 adsorption and activation were rate-determining steps.
Ionic liquids have many excellent properties, such as thermal stability, negligible vapor pressure, reusability, high solubility in various solvents, and high CO2 adsorption capacity under mild conditions.142 The combination of porous metal oxide with ionic liquids (ILs) could be used as an effective method to overcome the limitations of the direct conversion of CO2 to DMC. Very recently, Kim and co-workers prepared a series of ionic liquids with electrospun MgO–CeO2 metal oxide nanofiber sponge-500 (EMCMONS-500).143 The synergism between the ILs and EMCMONS-500 enabled a high catalytic performance towards DMC synthesis, as demonstrated that around 73.1 mmol g−1 cat DMC yield and 98.9% selectivity were obtained under 3 MPa CO2 pressure. The ILs-EMCMONS-500 catalyst exhibited superior catalytic activity for DMC production to that of other ceria or ILs-based catalysts.132,144–147
Except for ceria-based oxide catalysts, a variety of other catalysts, namely, zirconia,148,149 heteropoly compounds,150,151 ionic liquids,152,153 potassium methoxide,154 tin(IV)oxide,155–157 H3PO4–V2O5,158 and Cu–Ni or Cu–Fe bimetallic species supported on different carriers,159–162 have been extensively studied for DMC preparation through the conversion of CO2 with methanol.
In 2017, Wu, Hu and co-workers employed an imidazolium hydrogen carbonate ionic liquid ([CnCmIm][HCO3]) for the direct synthesis of DMC from CO2 and methanol at room temperature.163 Bifunctional [C1C4Im][HCO3] was used as an effective and recyclable catalyst and dehydrant, producing DMC with 74% CH3OH conversion (Scheme 19). The two roles of the imidazolium hydrogen carbonate ionic liquid system have many advantages and remarkably simplify the whole chemical process, offering the possibility of the further scaled application of this method.
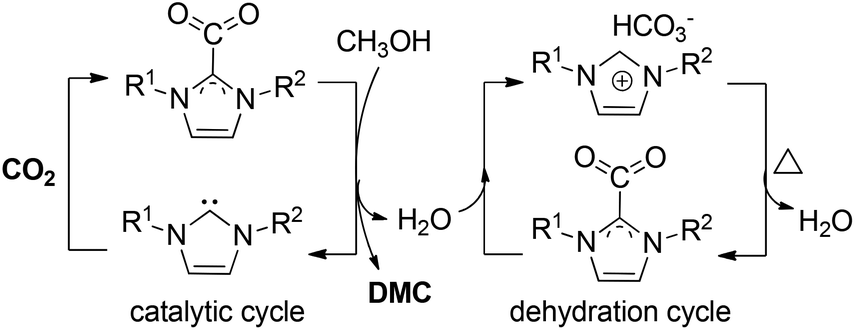 | ||
| Scheme 19 The catalytic and dehydration cycle of [CnCmIm][HCO3]. | ||
The utilization of zirconium oxide (ZrO2) as catalyst for the synthesis of DMC from CO2 was previously reported by Tomishige and co-workers.133,134,148,164 Bell and Jung performed a relevant mechanistic investigation in detail using in situ infrared (IR) spectroscopy.165,166 Inumaru and co-workers prepared diverse ZrO2 nanocrystals by hydrothermal synthesis at different temperatures and tested the catalytic activity in DMC formation.167 Due to their high surface area and high reaction rate per unit surface area, a high catalytic activity of ZrO2 nanocrystals was observed and the surface sites had a positive impact on the rate-determining step of the reaction.
Meng, Xiao and co-workers designed and prepared a series of alkali-adopted Cu–Ni/diatomite catalysts for application in DMC synthesis from CO2 and methanol.168 The results revealed that alkali adoption could promote polarization of the Cu–Ni lattice and increase electron transformation from Cu–Ni to CO2, thereby greatly promoting the catalytic activity of Cu–Ni bimetallic catalysts. DMC could be obtained with 85.9% selectivity and 9.22% CH3OH conversion under the reaction conditions. The incorporation of an alkali into the Cu–Ni bimetallic catalyst could decrease the decomposition and reduction temperatures, which contributed to the formation of a nano-scale dispersion of bimetallic particles. The authors further investigated the highly active K2O-promoted Cu–Ni catalyzed direct DMC synthesis with 3 Å molecular sieves (MS) as a dehydrating agent.169 The developed in situ dehydrating system enabled higher methanol conversion and selectivity by shifting the reaction equilibrium to a high DMC yield. Recently, a novel CuxNiy@POP-PPh3 catalyst was synthesized and used in direct DMC synthesis by Chen, Ye and co-workers.170 The CuNi alloy nanoparticles were encapsulated in thermally stable triphenylphosphine porous organic polymers, which exhibited high catalytic performance to achieve DMC production with 10.5% CH3OH conversion and 80% selectivity.
Wang and co-workers prepared a series of Y2O3-T catalysts through a one-pot calcination method from Y(NO3)3·6H2O and applied them to study the thermodynamics of the direct synthesis of DMC from CO2.171 The efficiency of DMC formation was closely related to the moderate acidity–basicity amounts of the catalysts. Y2O3-750 showed the best catalytic performance to give the highest yield of DMC at 90 °C under 8 MPa of CO2. It is the first example of utilizing yttrium oxide as a single metal oxide catalyst to catalyze DMC synthesis from CO2 and methanol.
5 Synthesis of DME/DMM from CO2
Dimethyl ether (DME) and dimethoxymethane (DMM) are known to be excellent fuel additives and green solvents, which have attracted considerable interest in recent years.172–174 DME can be synthesized by acid-catalyzed dehydration of methanol or from syngas with bifunctional catalysts.175 DMM is industrially produced through direct oxidation of methanol and a condensation sequence in the presence of suitable catalysts.176–178 On the basis of extensive studies on CO2 hydrogenation transformation, the direct conversion of CO2 has emerged as an appealing alternative route to the generation of DME/DMM.DME is prepared through CO2 hydrogenation in a one-pot reaction comprising a methanol synthesis and methanol dehydration sequence. The latter process can facilitate CO2 hydrogenation to methanol by shifting the thermodynamic equilibrium,179 and an effective catalyst for promoting methanol production is also significant. The Cu–Zn bimetallic system has been considered as an excellent catalyst for methanol synthesis from CO2 hydrogenation.180 In 2015, Zha and co-workers developed HZSM-5 packed CuO–ZnO–Al2O3 nanoparticles for the catalysis of CO2 hydrogenation with methanol to produce DME. Under the conditions of 3 MPa of CO2/H2 at 270 °C, 23.4% yield of DME was obtained with 48.5% selectivity.181 The combination of sulfated zirconia catalysts with a Cu/ZnO/ZrO2 catalyst for the synthesis of DME was developed by Chareonpanich and co-workers, in which the prepared hybrid catalyst with 20 wt% sulfur loading could lead to a superior DME production yield.182
Later, Mota and co-workers prepared several Cu·ZnO catalysts supported on Al2O3/Nb2O5 and employed them to study the conversion of CO2 to methanol and dimethyl ether (Scheme 20).183 The preparation procedure of the catalyst had a crucial influence on DME production, as demonstrated that the impregnated Cu·ZnO/Al2O3 catalyst with better reducibility exhibited the highest activity and selectivity for the formation of DME. The acid function was not the key factor in the process of DME synthesis. Pd/HZSM-5 bifunctional catalysts modified with CeO2 and CaO promotors were applied for the synthesis of DME through sulfur-containing CO2 hydrogenation.184 The addition of CaO reduced the acidity and weakened the CO2 adsorption of the catalyst, whereas the promotor CeO2 could increase the acidity and the quantity of adsorbed CO2, thereby greatly improving the catalytic activity. Optimal amounts of the promotors were 3 wt% CeO2 and 1 wt% CaO.
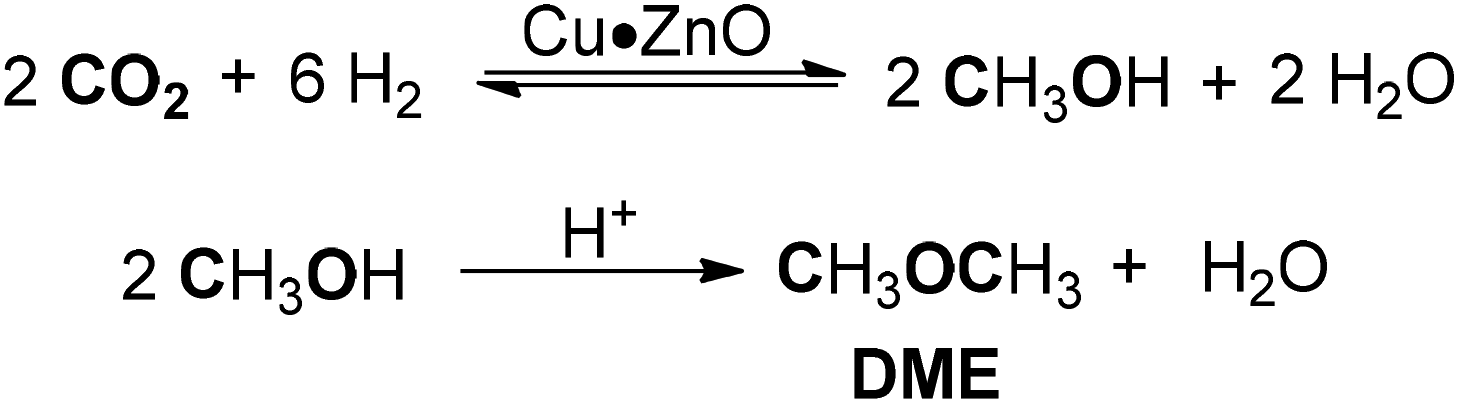 | ||
| Scheme 20 Cu·ZnO-catalyzed synthesis of DME from CO2 hydrogenation. | ||
Klankermayer and co-workers reported a ruthenium-catalyzed multistep reaction for the synthesis of dimethoxymethane (DMM) from CO2/H2 and methanol (Scheme 21).185 Ru(Triphos)(tmm) was chosen as a catalyst and the Lewis acid Al(OTf)3 was employed as an effective additive. Mechanistic studies indicated that methyl formate (MF) and methoxymethanol (MM) were formed as possible intermediates during the reaction. The reaction presumably involved the hydrogenation of CO2 to form the formic acid and subsequent esterification for producing MF. Then, MF was hydrogenated to MM in the presence of the Ru-Triphos-H2 catalyst system. Finally, transacetalization of MM with the solvent methanol could give the DMM product. Furthermore, the versatility of this reaction was further investigated by using several other alcohols to successfully produce various dialkoxymethanes with slightly lower TONs. The present catalytic reaction constitutes the first example of the selective conversion of CO2 and H2 to formaldehyde.
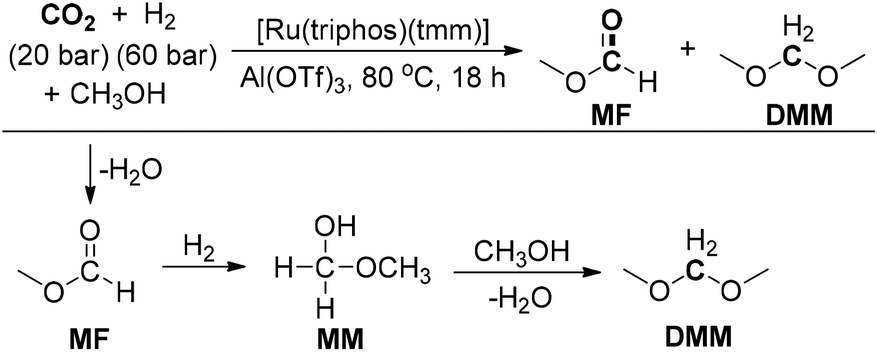 | ||
| Scheme 21 Ruthenium-catalyzed synthesis of DMM from CO2/H2. | ||
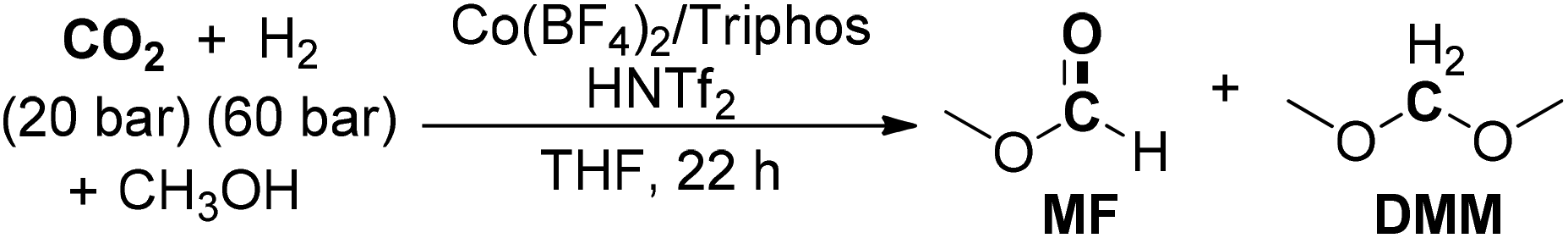
After one year, the group of Klankermayer achieved the selective transformation of CO2 to dialkoxymethane ethers using a non-precious transition-metal catalyst system.186 A tailored Co(BF4)2/Triphos/HNTf2 catalytic system was utilized for the production of DMM with a TON of up to 92 at 100 °C (Scheme 22). The developed protocol was also amenable for the formation of dialkoxymethane ethers (DAM) from other selected alcohol substrates. When isopropanol and more acidic hexafluoroisopropanol were used as substrates, methanol was selectively generated as the major product, implying switching of the catalyst system for methanol formation by variation of the alcohol additive or solvent. Moreover, the alteration of substituents on the phenyl groups of the Triphos ligand also affected the catalytic activity, as verified that sterically more demanding and electron-richer TriphosXyl and TriphosTol could enable the production of DMM with higher TONs of 120 and 157, respectively.
 | ||
| Scheme 22 Cobalt-catalyzed synthesis of DMM from CO2/H2. | ||
The conversion of methanol and CO2 to produce DMM at 3 MPa and 150 °C in the basic functionalized ionic liquid BmimOH was developed by Cai and co-workers.187 It was the first example of employing an ionic liquid as catalyst for the synthesis of DMM from CO2 and methanol.
On the basis of Klankermayer's work on the ruthenium-Triphos-aluminum triflate catalytic system, Trapp and co-workers modified the structure of Triphos ligands and ruthenium catalysts to tune the steric and electronic properties, as well as the coordinating units, thereby increasing the catalytic activity for the production of DMM with higher TONs.188 Under 90 bar of H2 and 20 bar CO2 in the presence of methanol, DMM and MF were produced using a ruthenium catalyst with an N-TriphosPh ligand with maximum turnover numbers of 786 and 1290, in which MF was transformed into DMM with high selectivity. Formaldehyde and methanol could be readily obtained by distillation from the hydrolysis of DMM. Later, the same authors further investigated steric and electronic modifications of the Triphos ligand structure and varied the substitution of the apical atom in the backbone with silicon and phosphorus to explore the catalytic activity.189 Under the optimized conditions, DMM and MF could be formed with maximum turnover numbers of 685 and 1370, which were comparable with the reactivity of their previous work.188 For ruthenium-catalyzed DMM synthesis, the employment of PhSi-TriphosPh and MeSi-TriphosPh as ligands was also a good choice to attain high reactivity.
6 Conclusions
Considering the increased global emission of CO2 and from the viewpoint of sustainability, the development of efficient transformations of CO2 into useful organic solvents constitutes an attractive and long-term goal for the chemical community. A wide range of strategies of CO2 conversion have been exploited to produce diverse solvents in the past few decades. Apart from transformations involving precious transition-metal catalysts, reactions using inexpensive Earth-abundant metals have also developed rapidly. However, the wide industrialization of the developed protocol for the transformation of CO2 to organic solvents is far from realized. Obvious limitations of the developed methodologies still remain, such as the necessity for precious-metal catalysts, harsh reaction conditions, low conversion rate and relatively poor chemo- and regioselectivity. Tremendous efforts should be devoted to solving these troublesome problems in the future. It will be of great significance for developing the efficient industrial-scale production of organic solvents from CO2 due to the huge demand for solvents in academic research and the chemical industry. It is a very promising and fascinating, but also challenging, field for chemists working on organic synthesis or industrial catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21772177), the Natural Science Foundation of Zhejiang Province (LY19B020016) and the Fundamental Research Funds of Zhejiang Sci-Tech University (2019Q065).Notes and references
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- K. Huang, C. L. Sun and Z. J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- K. Dong, R. Razzaq, Y. Hu and K. Ding, Top. Curr. Chem., 2017, 375, 23 CrossRef.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS.
- W. Zhang and X. Lv, Chin. J. Catal., 2012, 33, 745–756 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef.
- A. Tortajada, F. Julia-Hernandez, M. Borjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS.
- S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439–463 CrossRef CAS.
- S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382–404 RSC.
- C. S. Yeung, Angew. Chem., Int. Ed., 2019, 58, 5492–5502 CrossRef CAS.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- X. B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS.
- A. Baiker, Chem. Rev., 1999, 99, 453–474 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475–494 CrossRef CAS.
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121–191 CrossRef CAS.
- H.-F. Jiang, Curr. Org. Chem., 2005, 9, 289–297 CrossRef CAS.
- A. M. López-Periago, N. Portoles-Gil, P. López-Domínguez, J. Fraile, J. Saurina, N. Aliaga-Alcalde, G. Tobias, J. A. Ayllón and C. Domingo, Cryst. Growth Des., 2017, 17, 2864–2872 CrossRef.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797 CrossRef CAS.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221–231 CrossRef CAS.
- Y.-N. Li, R. Ma, L.-N. He and Z.-F. Diao, Catal. Sci. Technol., 2014, 4, 1498–1512 RSC.
- E. Alberico and M. Nielsen, Chem. Commun., 2015, 51, 6714–6725 RSC.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS.
- J. Muzart, Tetrahedron, 2009, 65, 8313–8323 CrossRef CAS.
- S. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 9226–9237 CrossRef CAS.
- P. Haynes, L. H. Slaugh and J. F. Kohnle, Tetrahedron Lett., 1970, 11, 365–368 CrossRef.
- K. Kudo, H. Phala, N. Sugita and Y. Takezaki, Chem. Lett., 1977, 6, 1495–1496 CrossRef.
- S. Schreiner, J. Y. Yu and L. Vaska, Inorg. Chim. Acta, 1988, 147, 139–141 CrossRef CAS.
- S. Schreiner, J. Y. Yu and L. Vaska, J. Chem. Soc., Chem. Commun., 1988, 602–603 RSC.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1994, 116, 8851–8852 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS.
- O. Kröcher, R. A. Köppel and A. Baiker, Chem. Commun., 1997, 453–454 RSC.
- O. Kröcher, R. A. Köppel and A. Baiker, J. Mol. Catal. A: Chem., 1999, 140, 185–193 CrossRef.
- O. Kröcher, R. A. Köppel, M. Fröba and A. Baiker, J. Catal., 1998, 178, 284–298 CrossRef.
- L. Schmid, M. Rohr and A. Baiker, Chem. Commun., 1999, 2303–2304 RSC.
- M. Minato, D.-Y. Zhou, K.-i. Sumiura, R. Hirabayashi, Y. Yamaguchi and T. Ito, Chem. Commun., 2001, 2654–2655 RSC.
- J. Liu, C. Guo, Z. Zhang, T. Jiang, H. Liu, J. Song, H. Fan and B. Han, Chem. Commun., 2010, 46, 5770–5772 RSC.
- S. Kumar and S. L. Jain, RSC Adv., 2014, 4, 64277–64279 RSC.
- R. Zhiani, S. M. Saadati, M. Zahedifar and S. M. Sadeghzadeh, Catal. Lett., 2018, 148, 2487–2500 CrossRef CAS.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS.
- C. Federsel, C. Ziebart, R. Jackstell, W. Baumann and M. Beller, Chem. – Eur. J, 2012, 18, 72–75 CrossRef CAS.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS.
- O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934–2937 CrossRef CAS.
- B. Bhanage and D. Nale, Synlett, 2016, 27, 1413–1417 CrossRef.
- X. Cui, Y. Zhang, Y. Deng and F. Shi, Chem. Commun., 2014, 50, 189–191 RSC.
- Q. Y. Bi, J. D. Lin, Y. M. Liu, S. H. Xie, H. Y. He and Y. Cao, Chem. Commun., 2014, 50, 9138–9140 RSC.
- K. Kobayashi, T. Kikuchi, S. Kitagawa and K. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 11813–11817 CrossRef CAS.
- H. Ishida, H. Tanaka, K. Tanaka and T. Tanaka, Chem. Lett., 1987, 16, 597–600 CrossRef.
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS.
- C. Fang, C. Lu, M. Liu, Y. Zhu, Y. Fu and B.-L. Lin, ACS Catal., 2016, 6, 7876–7881 CrossRef CAS.
- S. Kumar, P. Kumar, A. Deb, D. Maiti and S. L. Jain, Carbon, 2016, 100, 632–640 CrossRef CAS.
- D. B. Nale, D. Rath, K. M. Parida, A. Gajengi and B. M. Bhanage, Catal. Sci. Technol., 2016, 6, 4872–4881 RSC.
- V. B. Saptal, T. Sasaki and B. M. Bhanage, ChemCatChem, 2018, 10, 2593–2600 CrossRef CAS.
- V. B. Saptal and B. M. Bhanage, ChemSusChem, 2016, 9, 1980–1985 CrossRef CAS.
- V. B. Saptal, G. Juneja and B. M. Bhanage, New J. Chem., 2018, 42, 15847–15851 RSC.
- P. Daw, S. Chakraborty, G. Leitus, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 2500–2504 CrossRef CAS.
- Y. Zhang, J. Wang, H. Zhu and T. Tu, Chem. – Asian J., 2018, 13, 3018–3021 CrossRef CAS.
- M. A. Affan and P. G. Jessop, Inorg. Chem., 2017, 56, 7301–7305 CrossRef CAS.
- R. Kuhlmann, S. Schmitz, K. Haßmann, A. Prüllage and A. Behr, Appl. Catal., A, 2017, 539, 90–96 CrossRef CAS.
- R. Kuhlmann, A. Prüllage, K. Künnemann, A. Behr and A. J. Vorholt, J. CO2 Util., 2017, 22, 184–190 CrossRef CAS.
- Y. Wu, T. Wang, H. Wang, X. Wang, X. Dai and F. Shi, Nat. Commun., 2019, 10, 2599 CrossRef.
- X. Dai, B. Wang, A. Wang and F. Shi, Chin. J. Catal., 2019, 40, 1141–1146 CrossRef CAS.
- S. Ghosh, A. Ghosh, S. Biswas, M. Sengupta, D. Roy and S. M. Islam, ChemistrySelect, 2019, 4, 3961–3972 CrossRef CAS.
- R. Kuhlmann, M. Nowotny, K. U. Künnemann, A. Behr and A. J. Vorholt, J. Catal., 2018, 361, 45–50 CrossRef CAS.
- R. Kuhlmann, K. U. Künnemann, L. Hinderink, A. Behr and A. J. Vorholt, ACS Sustainable Chem. Eng., 2019, 7, 4924–4931 CrossRef CAS.
- G. H. Gunasekar, S. Padmanaban, K. Park, K. D. Jung and S. Yoon, ChemSusChem, 2020, 13, 1735–1739 CrossRef CAS.
- G. Wang, M. Jiang, G. Ji, Z. Sun, C. Li, L. Yan and Y. Ding, ACS Sustainable Chem. Eng., 2020, 8, 5576–5583 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS.
- J.-P. Lange, Catal. Today, 2001, 64, 3–8 CrossRef CAS.
- C. N. Hamelinck and A. P. C. Faaij, J. Power Sources, 2002, 111, 1–22 CrossRef CAS.
- T. Chmielniak and M. Sciazko, Appl. Energy, 2003, 74, 393–403 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS.
- M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893 CrossRef CAS.
- F. Liao, Y. Huang, J. Ge, W. Zheng, K. Tedsree, P. Collier, X. Hong and S. C. Tsang, Angew. Chem., Int. Ed., 2011, 50, 2162–2165 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296 CrossRef CAS.
- S. Dang, H. Yang, P. Gao, H. Wang, X. Li, W. Wei and Y. Sun, Catal. Today, 2019, 330, 61–75 CrossRef CAS.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS.
- F. Huang, C. Zhang, J. Jiang, Z.-X. Wang and H. Guan, Inorg. Chem., 2011, 50, 3816–3825 CrossRef CAS.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS.
- F. Huang, G. Lu, L. Zhao, H. Li and Z.-X. Wang, J. Am. Chem. Soc., 2010, 132, 12388–12396 CrossRef CAS.
- Z. Lu and T. J. Williams, ACS Catal., 2016, 6, 6670–6673 CrossRef CAS.
- C. Erken, A. Kaithal, S. Sen, T. Weyhermuller, M. Holscher, C. Werle and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef.
- H. Li, T. P. Goncalves, Q. Zhao, D. Gong, Z. Lai, Z. Wang, J. Zheng and K. W. Huang, Chem. Commun., 2018, 54, 11395–11398 RSC.
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS.
- E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 11702–11705 CrossRef CAS.
- Z. Han, L. Rong, J. Wu, L. Zhang, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041–13045 CrossRef CAS.
- K. Sordakis, A. Tsurusaki, M. Iguchi, H. Kawanami, Y. Himeda and G. Laurenczy, Chem. – Eur. J., 2016, 22, 15605–15608 CrossRef.
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS.
- S. Wesselbaum, T. Vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS.
- S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. V. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC.
- J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS.
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS.
- S. Kar, R. Sen, J. Kothandaraman, A. Goeppert, R. Chowdhury, S. B. Munoz, R. Haiges and G. K. S. Prakash, J. Am. Chem. Soc., 2019, 141, 3160–3170 CrossRef CAS.
- J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS.
- S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- A. P. C. Ribeiro, L. M. D. R. S. Martins and A. J. L. Pombeiro, Green Chem., 2017, 19, 4811–4815 RSC.
- F.-H. Zhang, C. Liu, W. Li, G.-L. Tian, J.-H. Xie and Q.-L. Zhou, Chin. J. Chem., 2018, 36, 1000–1002 CrossRef CAS.
- W.-Y. Chu, Z. Culakova, B. T. Wang and K. I. Goldberg, ACS Catal., 2019, 9, 9317–9326 CrossRef CAS.
- H. H. Cramer, B. Chatterjee, T. Weyhermuller, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2020, 59, 15674–15681 CrossRef CAS.
- R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS.
- P. Tundo, M. Musolino and F. Aricò, Green Chem., 2018, 20, 28–85 RSC.
- G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef.
- F. Aricò and P. Tundo, Russ. Chem. Rev., 2010, 79, 479–489 CrossRef.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS.
- S. Hess, M. Wohlfahrt-Mehrens and M. Wachtler, J. Electrochem. Soc., 2015, 162, A3084–A3097 CrossRef CAS.
- B. A. V. Santos, V. M. T. M. Silva, J. M. Loureiro and A. E. Rodrigues, ChemBioEng Rev., 2014, 1, 214–229 CrossRef CAS.
- A. A. Chaugule, A. H. Tamboli and H. Kim, Fuel, 2017, 200, 316–332 CrossRef CAS.
- A. H. Tamboli, A. A. Chaugule and H. Kim, Chem. Eng. J., 2017, 323, 530–544 CrossRef CAS.
- T. Baidya, A. Gayen, M. S. Hegde, N. Ravishankar and L. Dupont, J. Phys. Chem. B, 2006, 110, 5262–5272 CrossRef CAS.
- G. Dutta, A. Gupta, U. V. Waghmare and M. S. Hegde, J. Chem. Sci., 2011, 123, 509–516 CrossRef CAS.
- T. Vinodkumar, D. N. Durgasri, S. Maloth and B. M. Reddy, J. Chem. Sci., 2015, 127, 1145–1153 CrossRef CAS.
- S. Wang, L. Zhao, W. Wang, Y. Zhao, G. Zhang, X. Ma and J. Gong, Nanoscale, 2013, 5, 5582–5588 RSC.
- Z. Li, W. Li, H. Abroshan, Q. Ge, G. Li and R. Jin, Nanoscale, 2018, 10, 6558–6565 RSC.
- X. Tong, T. Luo, X. Meng, H. Wu, J. Li, X. Liu, X. Ji, J. Wang, C. Chen and Z. Zhan, Small, 2015, 11, 5581–5588 CrossRef CAS.
- S. Wang, J. Zhou, S. Zhao, Y. Zhao and X. Ma, Chem. Eng. Technol., 2016, 39, 723–729 CrossRef CAS.
- P. Kumar, V. C. Srivastava, R. Gläser, P. With and I. M. Mishra, Powder Technol., 2017, 309, 13–21 CrossRef CAS.
- K. Tomishige, Y. Furusawa, Y. Ikeda, M. Asadullah and K. Fujimoto, Catal. Lett., 2001, 76, 71–74 CrossRef CAS.
- K. Tomishige and K. Kunimori, Appl. Catal., A, 2002, 237, 103–109 CrossRef CAS.
- X. Deng, M. Li, J. Zhang, X. Hu, J. Zheng, N. Zhang and B. H. Chen, Chem. Eng. J., 2017, 313, 544–555 CrossRef CAS.
- A. H. Tamboli, A. A. Chaugule, S. W. Gosavi and H. Kim, Fuel, 2018, 216, 245–254 CrossRef CAS.
- B. Liu, C. Li, G. Zhang, X. Yao, S. S. C. Chuang and Z. Li, ACS Catal., 2018, 8, 10446–10456 CrossRef CAS.
- S. Huygh, A. Bogaerts and E. C. Neyts, J. Phys. Chem. C, 2016, 120, 21659–21669 CrossRef CAS.
- S.-P. Wang, J.-J. Zhou, S.-Y. Zhao, Y.-J. Zhao and X.-B. Ma, Chin. Chem. Lett., 2015, 26, 1096–1100 CrossRef CAS.
- S. Watanabe, X. Ma and C. Song, J. Phys. Chem. C, 2009, 113, 14249–14257 CrossRef CAS.
- Z. Fu, Y. Zhong, Y. Yu, L. Long, M. Xiao, D. Han, S. Wang and Y. Meng, ACS Omega, 2018, 3, 198–207 CrossRef CAS.
- K. V. Wagh and B. M. Bhanage, Green Chem., 2015, 17, 4446–4451 RSC.
- A. A. Pawar, D. Lee, W.-J. Chung and H. Kim, Chem. Eng. J., 2020, 395, 124970 CrossRef CAS.
- U. P and S. Darbha, J. Chem. Sci., 2016, 128, 957–965 CrossRef CAS.
- Y. Pu, K. Xuan, F. Wang, A. Li, N. Zhao and F. Xiao, RSC Adv., 2018, 8, 27216–27226 RSC.
- Z.-F. Zhang, Z.-W. Liu, J. Lu and Z.-T. Liu, Ind. Eng. Chem. Res., 2011, 50, 1981–1988 CrossRef CAS.
- A. A. Chaugule, H. A. Bandhal, A. H. Tamboli, W.-J. Chung and H. Kim, Catal. Commun., 2016, 75, 87–91 CrossRef CAS.
- K. Tomishige, T. Sakaihori, Y. Ikeda and K. Fujimoto, Catal. Lett., 1999, 58, 225–229 CrossRef CAS.
- B. Peng, H. Dou, H. Shi, E. E. Ember and J. A. Lercher, Catal. Lett., 2018, 148, 1914–1919 CrossRef CAS.
- K. W. La, M. H. Youn, J. S. Chung, S. H. Baeck and I. K. Song, Solid State Phenom., 2007, 119, 287–290 CAS.
- K. W. La, J. C. Jung, H. Kim, S.-H. Baeck and I. K. Song, J. Mol. Catal. A: Chem., 2007, 269, 41–45 CrossRef CAS.
- H. Zhao, B. Lu, X. Li, W. Zhang, J. Zhao and Q. Cai, J. CO2 Util., 2015, 12, 49–53 CrossRef CAS.
- M. O. Vieira, A. S. Aquino, M. K. Schütz, F. D. Vecchia, R. Ligabue, M. Seferin and S. Einloft, Energy Procedia, 2017, 114, 7141–7149 CrossRef CAS.
- Q. Cai, B. Lu, L. Guo and Y. Shan, Catal. Commun., 2009, 10, 605–609 CrossRef CAS.
- D. Aymes, D. Ballivet-Tkatchenko, K. Jeyalakshmi, L. Saviot and S. Vasireddy, Catal. Today, 2009, 147, 62–67 CrossRef CAS.
- P. Švec, H. Cattey, Z. Růžičková, J. Holub, A. Růžička and L. Plasseraud, New J. Chem., 2018, 42, 8253–8260 RSC.
- S. R. Sanapureddy and L. Plasseraud, Appl. Organomet. Chem., 2017, 31, e3807 CrossRef.
- X. L. Wu, M. Xiao, Y. Z. Meng and Y. X. Lu, J. Mol. Catal. A: Chem., 2005, 238, 158–162 CrossRef CAS.
- J. Bian, M. Xiao, S.-J. Wang, Y.-X. Lu and Y.-Z. Meng, Appl. Surf. Sci., 2009, 255, 7188–7196 CrossRef CAS.
- J. Bian, M. Xiao, S. J. Wang, Y. X. Lu and Y. Z. Meng, Catal. Commun., 2009, 10, 1529–1533 CrossRef CAS.
- Y. Zhou, S. Wang, M. Xiao, D. Han, Y. Lu and Y. Meng, RSC Adv., 2012, 2, 6831–6837 RSC.
- Y.-J. Zhou, M. Xiao, S.-J. Wang, D.-M. Han, Y.-X. Lu and Y.-Z. Meng, Chin. Chem. Lett., 2013, 24, 307–310 CrossRef CAS.
- T. Zhao, X. Hu, D. Wu, R. Li, G. Yang and Y. Wu, ChemSusChem, 2017, 10, 2046–2052 CrossRef CAS.
- K. Tomishige, Y. Ikeda, T. Sakaihori and K. Fujimoto, J. Catal., 2000, 192, 355–362 CrossRef CAS.
- K. T. Jung and A. T. Bell, J. Catal., 2001, 204, 339–347 CrossRef CAS.
- K. T. Jung and A. T. Bell, Top. Catal., 2002, 20, 97–105 CrossRef CAS.
- T. Akune, Y. Morita, S. Shirakawa, K. Katagiri and K. Inumaru, Langmuir, 2018, 34, 23–29 CrossRef CAS.
- D. Han, Y. Chen, S. Wang, M. Xiao, Y. Lu and Y. Meng, Catalysts, 2018, 8, 302 CrossRef.
- D. Han, Y. Chen, S. Wang, M. Xiao, Y. Lu and Y. Meng, Catalysts, 2018, 8, 343 CrossRef.
- Y. Chen, Y. Yang, S. Tian, Z. Ye, Q. Tang, L. Ye and G. Li, Appl. Catal., A, 2019, 587, 117275 CrossRef.
- W. Sun, L. Zheng, Y. Wang, D. Li, Z. Liu, L. Wu, T. Fang and J. Wu, Ind. Eng. Chem. Res., 2020, 59, 4281–4290 CrossRef CAS.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- J. Burger, E. Ströfer and H. Hasse, Chem. Eng. Res. Des., 2013, 91, 2648–2662 CrossRef CAS.
- S. P. Naik, T. Ryu, V. Bui, J. D. Miller, N. B. Drinnan and W. Zmierczak, Chem. Eng. J., 2011, 167, 362–368 CrossRef CAS.
- Y. Fu and J. Shen, Chem. Commun., 2007, 2172–2174 RSC.
- N. T. Prado, F. G. E. Nogueria, A. E. Nogueira, C. A. Nunes, R. Diniz and L. C. A. Oliveira, Energy Fuels, 2010, 24, 4793–4796 CrossRef CAS.
- M. Li, Y. Long, Z. Deng, H. Zhang, X. Yang and G. Wang, Catal. Commun., 2015, 68, 46–48 CrossRef CAS.
- W.-J. Shen, K.-W. Jun, H.-S. Choi and K.-W. Lee, Korean J. Chem. Eng., 2000, 17, 210–216 CrossRef CAS.
- M. V. Twigg and M. S. Spencer, Appl. Catal., A, 2001, 212, 161–174 CrossRef CAS.
- R. Liu, H. Tian, A. Yang, F. Zha, J. Ding and Y. Chang, Appl. Surf. Sci., 2015, 345, 1–9 CrossRef CAS.
- T. Witoon, T. Permsirivanich, N. Kanjanasoontorn, C. Akkaraphataworn, A. Seubsai, K. Faungnawakij, C. Warakulwit, M. Chareonpanich and J. Limtrakul, Catal. Sci. Technol., 2015, 5, 2347–2357 RSC.
- R. J. da Silva, A. F. Pimentel, R. S. Monteiro and C. J. A. Mota, J. CO2 Util., 2016, 15, 83–88 CrossRef CAS.
- R. Chu, C. Song, W. Hou, X. Meng, Z. Miao, X. Li, G. Wu, Y. Wan and L. Bai, J. Taiwan Inst. Chem. Eng., 2017, 80, 1041–1047 CrossRef CAS.
- K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS.
- B. G. Schieweck and J. Klankermayer, Angew. Chem., Int. Ed., 2017, 56, 10854–10857 CrossRef CAS.
- Q. Zhang, H. Zhao, B. Lu, J. Zhao and Q. Cai, J. Mol. Catal. A: Chem., 2016, 421, 117–121 CrossRef CAS.
- M. Siebert, M. Seibicke, A. F. Siegle, S. Krah and O. Trapp, J. Am. Chem. Soc., 2019, 141, 334–341 CrossRef CAS.
- M. Seibicke, M. Siebert, A. F. Siegle, S. M. Gutenthaler and O. Trapp, Organometallics, 2019, 38, 1809–1814 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |