
Flower-like cobalt carbide for efficient carbon dioxide conversion†
Qing
Guo‡
ab,
Shu-Guang
Xia‡
ab,
Xu-Bing
Li
 *ab,
Yang
Wang
ab,
Fei
Liang
bc,
Zhe-Shuai
Lin
bc,
Chen-Ho
Tung
ab and
Li-Zhu
Wu
*ab
*ab,
Yang
Wang
ab,
Fei
Liang
bc,
Zhe-Shuai
Lin
bc,
Chen-Ho
Tung
ab and
Li-Zhu
Wu
*ab
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: lixubing@mail.ipc.ac.cn; lzwu@mail.ipc.ac.cn
bSchool of Future Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cKey Laboratory of Functional Crystals and Laser Technology, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 5th March 2020
Abstract
Catalytic conversion of carbon dioxide (CO2) to value-added chemicals under mild conditions is highly desired, albeit with significant challenges. Here, in terms of exposure of abundant active sites and excellent photo-to-thermal conversion properties, flower-like Co2C has been firstly used for effectively catalysing the cycloaddition of CO2 with epoxides to produce cyclic carbonates with yields of up to 95% under solar light. Density functional theory (DFT) calculations reveal that Lewis acid sites of the surface Co atoms can activate both CO2 and epoxide, thus opening up the possibility of a CO2-epoxide cycloaddition reaction.
The increasing atmospheric concentration of carbon dioxide (CO2), which is regarded as the main greenhouse gas, has caused serious environmental concerns (e.g., global warming, abnormal climate change and sea level rise).1–4 To address the anthropogenic CO2 emission issues,5–11 the approaches of physical CO2 capture and storage,12,13 and chemical conversion of CO2 into usable fuels and/or value-added chemicals14,15 have been widely investigated. From a chemical point of view, cycloaddition of CO2 with epoxides represents one of the most promising strategies to generate valuable chemicals.16–18 By using CO2 as the renewable and nontoxic one-carbon (C1) feedstock,19 cyclic carbonates are produced with 100% atom-economy efficiency.20 Cyclic carbonates can be directly employed in industry as solvents or intermediates, such as carbamates,21 polycarbonates22 and spiro compounds.23 Numerous advances in homogeneous (e.g., Co,24,25 Ni,26 Al27 and Cu-based28 monometallic or bimetallic complexes) and heterogeneous (e.g., metal–organic-frameworks (MOFs),29–31 mesoporous polymers,32 and zeolites33) catalysts have been made for the cycloaddition of CO2 with epoxides. However, most of the state-of-the-art catalysts can work fairly well to yield cyclic carbonates only at an elevated temperature, thus leading to an increase of the energy cost.
Transition-metal carbides (TMCs) show great potential in the field of catalysis owing to their outstanding electronic conductivity, good chemical stability, abundant surface active sites and high photo-to-thermal conversion efficiency.34–36 Cobalt carbide (Co2C), for example, has been demonstrated to be an effective catalyst in various transformations (e.g., hydrogen evolution and Fischer–Tropsch synthesis).37,38 However, the application of cobalt carbide in cycloaddition of CO2 with epoxides is still elusive. Considering these attributes, we are inspired to explore Co2C as a novel catalyst for cycloaddition of CO2 with epoxides by using solar energy to replace external thermal energy input for following reasons: (i) the high specific surface area and tuneable chemical composition provide abundant surface active sites to activate CO2 and/or epoxides; (ii) the effective CO2 adsorption ability on TMCs favours the very first step of CO2 conversion;39 (iii) the huge amount of heat released during the excellent photo-to-thermal conversion process would promote the endothermic reaction (high C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O bond energy of 750 kJ mol−1);40 and (iv) the feasible separation and reusability of the heterogeneous catalysts is beneficial for large-scale applications.41 To our delight, flower-like Co2C facilitates the catalysis of CO2-epoxide cycloaddition with yields of up to ∼95% under visible light (Scheme 1), showing the first example of using TMCs for photothermal-driven CO2 conversion.
O bond energy of 750 kJ mol−1);40 and (iv) the feasible separation and reusability of the heterogeneous catalysts is beneficial for large-scale applications.41 To our delight, flower-like Co2C facilitates the catalysis of CO2-epoxide cycloaddition with yields of up to ∼95% under visible light (Scheme 1), showing the first example of using TMCs for photothermal-driven CO2 conversion.
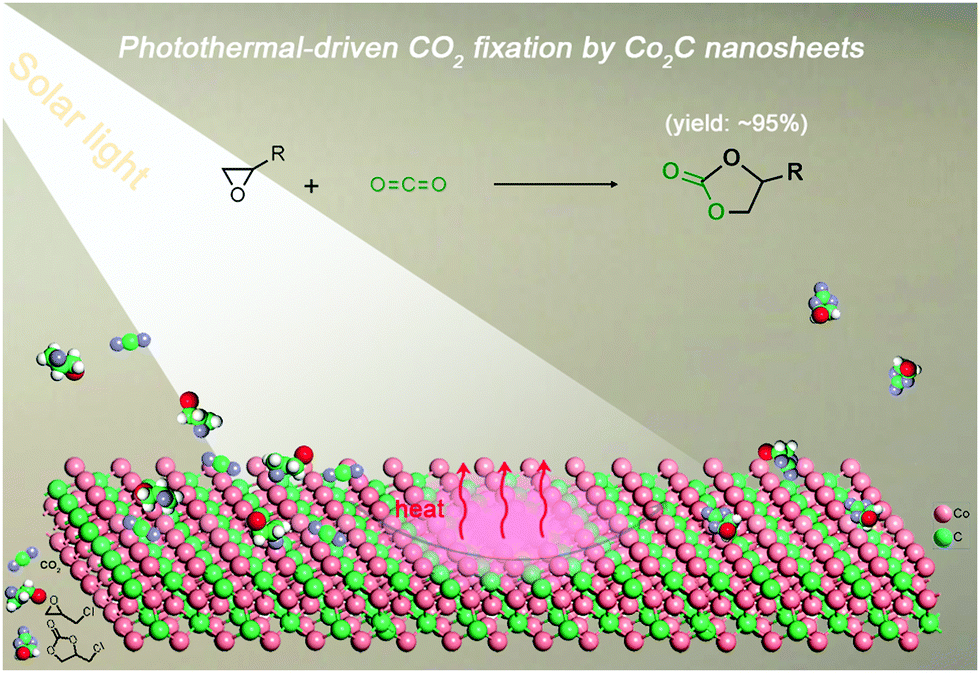 | ||
| Scheme 1 Illustration of the photothermal-driven cycloaddition of CO2 with epoxides catalyzed by Co2C nanosheets of the nanoflower. | ||
Flower-like Co2C was synthesized through a modified method of solution pyrolysis at high temperature (Fig. 1a),42–44 see experimental details in the ESI.† Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) clearly showed the flower-like morphology of the synthesized material (Fig. 1b and c). The average diameter of the nanoflowers was determined to be ∼500 nm by TEM characterization (Fig. 1c). The nearly apparent nanosheet on the outside of the nanoflowers indicated that the obtained nanoflower was a result of self-assembly of individual nanosheets. The thickness of individual nanosheet was determined to be ∼2.4 nm by atomic force microscopy (AFM) (Fig. S1, ESI†). In the high-resolution TEM image, a lattice distance of ∼2.41 Å was observed, indicating the exposure of the (101) facet of Co2C (Fig. 1d and e). Moreover, selective area electron diffraction (SAED) patterns in Fig. 1d confirmed the polycrystalline nature of the synthesized materials. In addition, elemental mapping analysis indicated the coexistence of Co, C and O elements (Fig. S2, ESI†), which matched well with the X-ray photoelectron spectroscopy (XPS) results (see below).
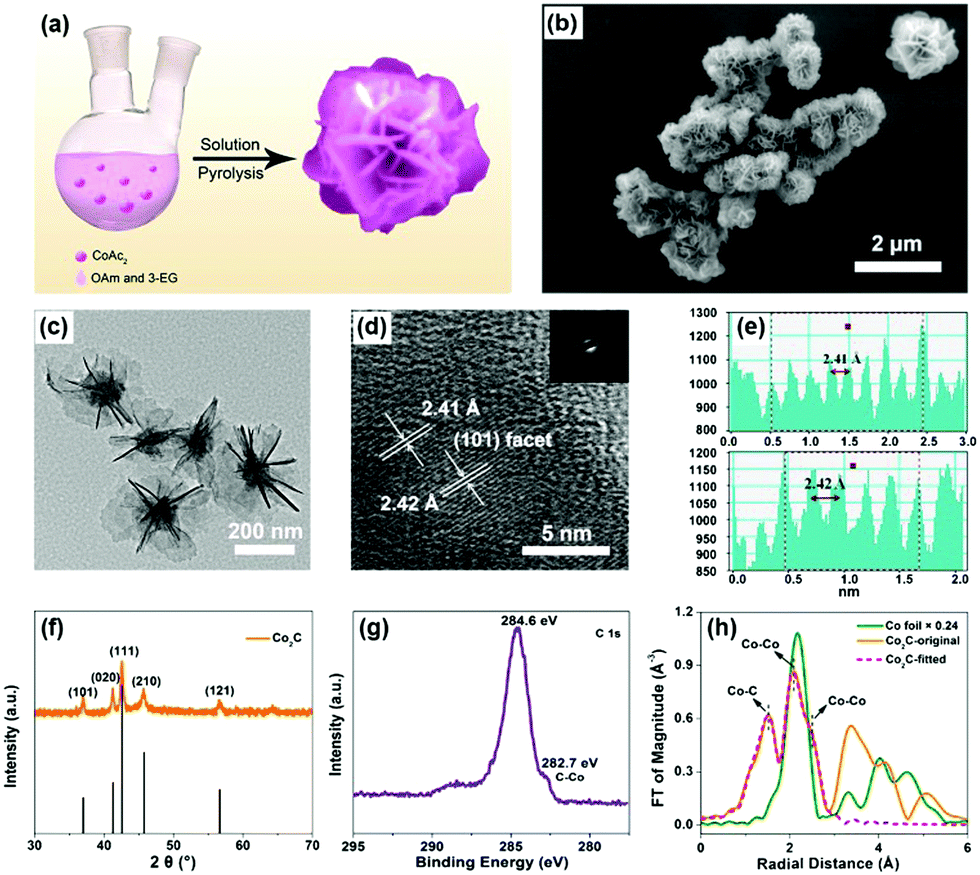 | ||
| Fig. 1 (a) Synthetic process of flower-like Co2C. (b) SEM image (the inset panel is the high-resolution SEM image) and (c) TEM image of Co2C nanoflowers. (d) High-resolution TEM image of Co2C (the inset panel is the corresponding SAED pattern). (e) The corresponding lattice distances of the exposed (101) plane of Co2C. (f) XRD pattern and (g) C 1s XPS spectrum of Co2C nanoflowers. (h) Magnitude of k2-weighted Fourier transform of the Co K-edge EXAFS spectra of Co foil and the obtained Co2C nanoflowers with corresponding curve-fitting results. | ||
The powder X-ray diffraction (XRD) (Fig. 1f) pattern of the as-prepared sample was in good agreement with the standard pattern of Co2C (Joint Committee on Powder Diffraction Standards (JCPDS) Powder Diffraction File (PDF) No. 65-1457),42 indicating the successful formation of Co2C. The peaks at 1630, 1420 and 1050 cm−1 corresponding to CC, C–H, and C–N bonds, respectively, in the Fourier transform infrared (FTIR) spectra (Fig. S3, ESI†) almost disappeared after calcination, suggesting the removal of surface organic ligands. Full XPS survey confirmed the coexistence of Co, C and O elements in the obtained sample (Fig. S4, ESI†). High-resolution XPS spectra of C 1s (Fig. 1g) and Co 2p (Fig. S5, ESI†) show the characteristic peaks of carbide and carbidic Co at 282.7 and 778.1 eV,36,45,46 respectively. Combined with the peak at 531.6 eV in the O 1s XPS spectrum (Fig. S6, ESI†), the peak at 781.0 eV in the Co 2p XPS spectrum was attributed to the Co(OH)2 species formed in the process of Co2C preparation, proving the fact that solution pyrolysis under high temperature inevitably leads to the formation of trace amounts of the hydroxide impurity.46,47Fig. 1h shows the X-ray absorption spectra (XAS) of the obtained sample. The Co K-edge extended X-ray absorption fine structure (EXAFS) of the synthesized sample exhibits two peaks in the R-space, which could be assigned to the first Co–C shell (1.89 Å) and the second Co–Co shell (2.54 Å) of Co2C, see the fitting details in Table S1, ESI.†
Then, photo-to-thermal conversion effects of Co2C nanoflowers were experimentally examined. UV-vis-NIR diffuse reflectance spectroscopy (DRS) measurement indicated that the synthesized Co2C nanoflowers showed strong absorption in the range of 300 to 1200 nm (Fig. S7, ESI†), directly confirming its excellent light-harvesting properties. When the water suspension of Co2C nanoflowers (0.6 mg mL−1) was exposed to a 635 nm laser at varied power densities (0.1, 0.5, 1.0 and 2.0 W cm−2), the solution displayed apparent temperature elevation (Fig. 2a). For instance, under 635 nm laser irradiation (0.5 W cm−2), the temperature of the Co2C nanoflowers water suspension reached 42.8 °C in 13 minutes, while the temperature of pure water only increased to 24 °C under the same conditions (Fig. S8, ESI†). Huge amount of heat released from Co2C nanoflowers was also monitored by an IR camera in the solid state. As shown in the inset panel of Fig. 2b, the local temperature of the powder Co2C sample quickly increased to 116.8 °C upon exposure to a 635 nm laser (0.5 W cm−2) for 4.0 min, further confirming the in situ conversion of solar light into heat.
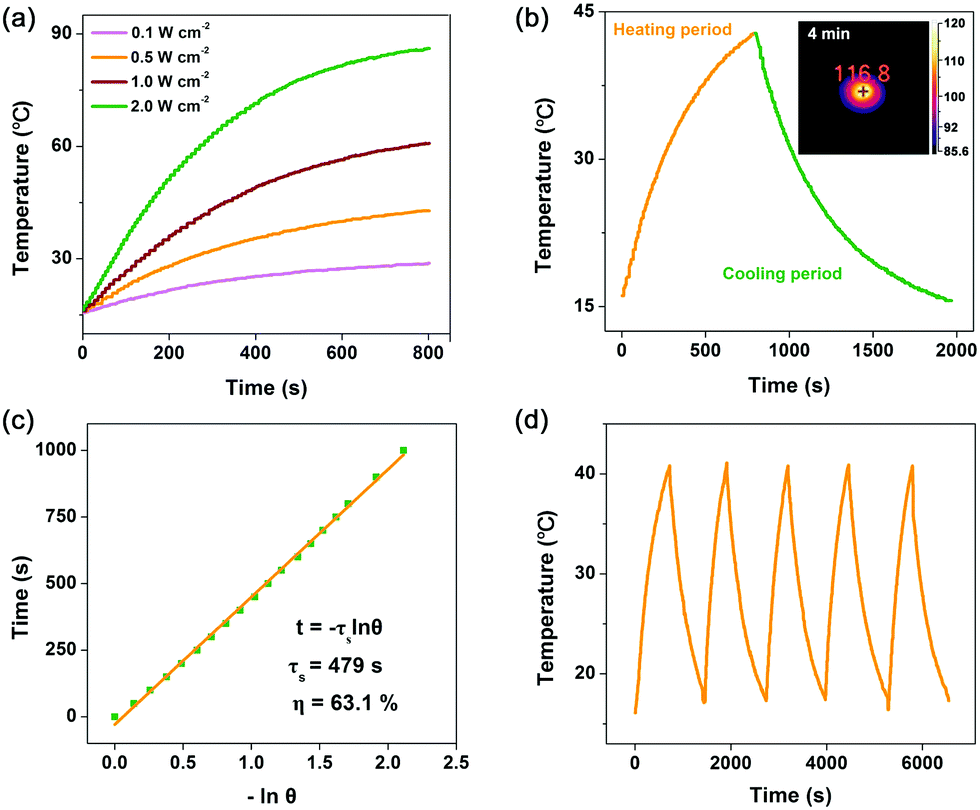 | ||
| Fig. 2 (a) Photothermal heating curves of Co2C nanoflowers dispersed in water under 635 nm laser irradiation at varied power densities (0.1, 0.5, 1.0 and 2.0 W cm−2). (b) Photothermal effect of Co2C aqueous dispersion under 635 nm laser irradiation (0.5 W cm−2) and the cooling process after laser off. The inset panel in (b) is the IR image of Co2C powder under 635 nm laser irradiation (0.5 W cm−2) for 4.0 min. (c) The corresponding time constant (τs) for the heat transfer from the system determined by applying the linear time data from cooling period. (d) Recycling-heating curves of the Co2C aqueous suspension with 635 nm laser irradiation at 0.5 W cm−2 for five laser on/off cycles. | ||
On the basis of the time constant for heat transfer and the maximal steady-state temperature, photo-to-thermal conversion efficiency (η), regarded as a major parameter in evaluating the performance in converting light to heat of a given material,48 of the flower-like Co2C was calculated to be as high as ∼63.1% at 635 nm (Fig. 2b and c, see details in the ESI†). The exceptional photo-to-thermal conversion performance of Co2C nanoflowers obtained here was comparable with those of the reported materials (Table S2, ESI†). To further evaluate its photothermal stability, temperature variations of Co2C nanoflowers suspension were recorded under light irradiation (laser on) followed by natural cooling to room temperature (laser off). As shown in Fig. 2d, negligible changes in temperature elevation were observed during 5 cycles, which highlighted the potential application of Co2C nanoflowers as durable photothermal materials.
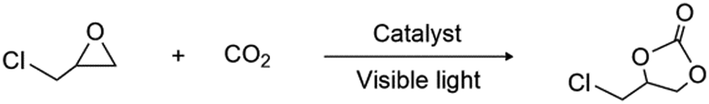
To investigate the catalytic performance of Co2C nanoflowers, 3-chloropropylene oxide was chosen as the model substrate under visible light (Table 1). A water/fan-cooling system was employed to maintain the outside temperature of the reactor at room temperature, see experimental details in the ESI.† Increase of Co2C nanoflowers from 0 to 25 mg obviously improved the yields up to ∼93.5% (entries 1–4; Table 1). Control experiments showed that all components were essential for the conversion. Trace or negligible amount of the product was detected without light, tetrabutylammonium bromide (TBAB) or CO2 (entries 5–7; Table 1). When tetrabutylammonium chloride (TBAC) was employed as the co-catalyst, a significantly declined yield of ∼27% was observed (entry 8; Table 1). This result showed that easier dissociation and stronger nucleophilicity of Br− compared to Cl− dramatically benefited the ring-opening reaction of epoxides.29 To verify the photothermal effects of Co2C nanoflowers, we monitored the temperature variation of the reaction solution by using a thermometer. Upon light irradiation, the temperature of the solution was significantly elevated to ∼60 °C in a period of 35 min (Fig. S9, ESI†), indicating the conversion of light into heat. Moreover, very similar yields were observed under either visible-light irradiation or external heating (60 °C), implying that the photothermal effects could promote the cycloaddition reaction between CO2 and epoxide (Fig. S10, ESI†).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Additive | Yieldb (%) |
| a Reaction conditions: 0.15 mmol 3-chloropropylene oxide, 0.25 mmol TBAB, 3 mL CH3CN as the solvent, blue LEDs (λ = 450 nm) as the light source, 15 h. b The carbonate product was quantified by 1H NMR with diphenylmethanol as the internal standard. The yield was calculated by the equation, [η (%) = n(carbonate)/n(3-chloropropylene oxide) × 100%]. c 0.1 mmol TBAB. d No light irradiation. e N2 instead of CO2. f 0.1 mmol TBAC. g Co2C nanoflowers treated with HCl (6 mol L−1). h 0.50 mmol TBAB, AM 1.5 (100 mW cm−2) as the light source, 24 h. | |||
| 1 | Co2C (0 mg) | TBAB | <5 |
| 2 | Co2C (15 mg)c | TBAB | 54 |
| 3 | Co2C (15 mg) | TBAB | 73.3 |
| 4 | Co2C (25 mg) | TBAB | 93.5 |
| 5 | Co2C (25 mg)d | TBAB | <5 |
| 6 | Co2C (25 mg) | — | 0 |
| 7 | Co2C (25 mg)e | TBAB | 0 |
| 8 | Co2C (15 mg)f | TBAC | 26.7 |
| 9 | Co2C (25 mg)g | TBAB | 81.9 |
| 10 | Co(OH)2 (25 mg) | TBAB | 64 |
| 11 | CoO (25 mg) | TBAB | 63.6 |
| 12 | Co3O4 (25 mg) | TBAB | 48.6 |
| 13 | Co2C (50 mg)h | TBAB | 66.4 |
Furthermore, the obtained Co2C sample was treated with hydrochloric acid (HCl; 6 mol L−1) to exclude the contribution of trace amounts of cobalt hydroxide (i.e., Co(OH)2) and/or cobalt oxide (i.e., CoO and Co3O4) on the surface of Co2C nanoflowers in CO2 fixation. After treating the sample with HCl, the slightly declined yield of the target product was still much higher than those of pure Co(OH)2, CoO, or Co3O4 under the same conditions (entries 9–12; Table 1). These results confirmed that Co2C, but not the hydroxide or oxide impurities, served as the real active sites for CO2 fixation. More importantly, the Co2C catalysed cycloaddition reaction of CO2 and epoxides with electron-donating/withdrawing groups could also give rise to products in good to excellent yields (∼95%) (Table S3, ESI†). Even under AM1.5 irradiation (entry 13; Table 1), this reaction proceeded with good yields. The activity was well preserved after three-time recycling (Fig. S11, ESI†), indicating the potential use of sunlight as the energy source for enhanced photothermal catalysis.
Based on the above experimental results, a plausible mechanism of the Co2C nanoflowers catalysed CO2-epoxide cycloaddition reaction is proposed (Fig. 3). Epoxide molecules adsorb on the surface exposed Co sites via Co–O interaction with an adsorption energy of −0.53 eV (Fig. S12a, see details of density functional theory (DFT) calculations in the ESI†), thereby leading to the elongation of C–O bond from 1.956 to 2.086 Å. Then, the nucleophilic Br− ion attacks the adsorbed epoxide at the less hindered carbon atom to generate the metal alkoxide intermediate via the ring-opening reaction. At the same time, CO2 adsorbs on the surface Co atom to give a bent molecular configuration (O1–C–O2 angle 146.99°) with a concerted interaction of C–Co and O–Co coordination (Fig. S12b, ESI†). With the aid of photothermal Co2C nanoflowers, the oxygen anion of the alkoxide intermediate combines with the adjacent highly activated CO2 molecules to yield the cyclic carbonate product, which is eventually released into the solvent to regenerate the catalyst. As a huge amount of heat is released due to the excellent photo-to-thermal conversion effects of Co2C, the endothermic CO2 cycloaddition reaction proceeds with high yields, which is comparable with the reported results (Table S4, ESI†).
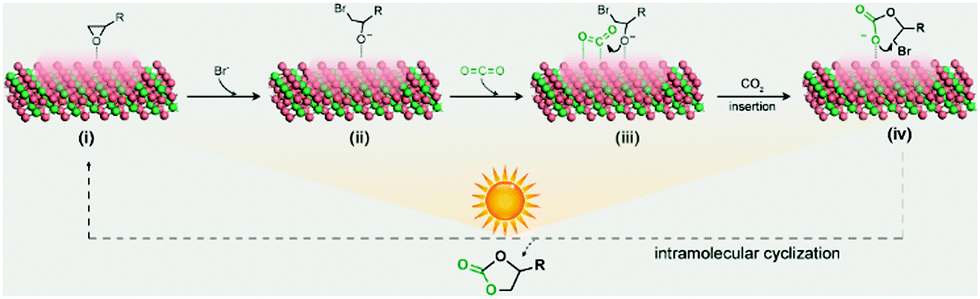 | ||
| Fig. 3 The proposed mechanism of Co2C catalysed CO2 cycloaddition with epoxides under light irradiation. | ||
In summary, an efficient CO2 cycloaddition reaction with epoxides is achieved on low-priced photothermal catalyst of Co2C nanoflowers. The yield of cyclic carbonates is up to ∼95% with visible-light irradiation, owing to the excellent photothermal effects of Co2C nanoflowers in converting light to heat. Besides, the high specific area as well as efficient CO2 adsorption on the exposed Co atoms of the catalysts can simultaneously activate the adsorbed CO2 and epoxides, thus promoting the CO2 fixation reaction. This work provides new insights into the utilization of TMCs in the field of advanced photothermal-driven catalysis. Moreover, the influence of the morphology, size and thickness of TMCs on the catalytic performance will be further investigated by us.
We are grateful for financial support from the National Key Research and Development Program of China (2017YFA0206903), the National Science Foundation of China (21861132004 and 21971251), the Strategic Priority Research Program of the Chinese Academy of Science (XDB17000000), the Key Research Program of Frontier Science of the Chinese Academy of Sciences (QYZDY-SSW-JSC029), the Youth Innovation Promotion Association of Chinese Academy of Sciences (2018031) and the K. C. Wong Education Foundation. We especially thank the Beijing Synchrotron Radiation Facility (BSRF, Beamline 1W2B) for X-ray absorption measurements. We thank Prof. Jiechao Ge and Dr. Qingyan Jia at the Technical Institute of Physics and Chemistry for photothermal-conversion measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS PubMed.
- S. C. Peter, ACS Energy Lett., 2018, 3, 1557–1561 CrossRef CAS.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- Q. Jiang, Z. Chen, J. Tong, M. Yang, Z. Jiang and C. Li, Chem. Commun., 2017, 53, 1188–1191 RSC.
- S. Wang, B. Y. Guan and X. W. D. Lou, J. Am. Chem. Soc., 2018, 140, 5037–5040 CrossRef CAS PubMed.
- Y. Wang, Z. Zhang, L. Zhang, Z. Luo, J. Shen, H. Lin, J. Long, J. C. S. Wu, X. Fu, X. Wang and C. Li, J. Am. Chem. Soc., 2018, 140, 14595–14598 CrossRef CAS PubMed.
- X. Meng, S. Ouyang, T. Kako, P. Li, Q. Yu, T. Wang and J. Ye, Chem. Commun., 2014, 50, 11517–11519 RSC.
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS.
- Y. Bai, J. Zhao, S. Feng, X. Liang and C. Wang, Chem. Commun., 2019, 55, 4651–4654 RSC.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- G. Kupgan, L. J. Abbott, K. E. Hart and C. M. Colina, Chem. Rev., 2018, 118, 5488–5538 CrossRef CAS PubMed.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 10804–10811 CrossRef CAS PubMed.
- C. Lu, J. Yang, S. Wei, S. Bi, Y. Xia, M. Chen, Y. Hou, M. Qiu, C. Yuan, Y. Su, F. Zhang, H. Liang and X. Zhuang, Adv. Funct. Mater., 2019, 29, 1806884 CrossRef.
- F. Liu, K. Huang, Q. Wu and S. Dai, Adv. Mater., 2017, 29, 1700445 CrossRef PubMed.
- M. Ding and H.-L. Jiang, ACS Catal., 2018, 8, 3194–3201 CrossRef CAS.
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- Y. Fan, M. Tiffner, J. Schörgenhumer, R. Robiette, M. Waser and S. R. Kass, J. Org. Chem., 2018, 83, 9991–10000 CrossRef CAS PubMed.
- R. Zevenhoven, S. Eloneva and S. Teir, Catal. Today, 2006, 115, 73–79 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- Y.-X. Zhang, L. Guo, Y.-H. Wang, L.-L. Zhu and Z.-I. Chen, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2009, 39, 445–448 CAS.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- X. Jiang, F. Gou, F. Chen and H. Jing, Green Chem., 2016, 18, 3567–3576 RSC.
- J. Honores, D. Quezada, G. Chacón, O. Martínez-Ferraté and M. J. C. L. Isaacs, Catal. Lett., 2019, 149, 1825–1832 CrossRef CAS.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- Q. Yang, C.-C. Yang, C.-H. Lin and H.-L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 3511–3515 CrossRef CAS PubMed.
- H. He, J. A. Perman, G. Zhu and S. Ma, Small, 2016, 12, 6309–6324 CrossRef CAS PubMed.
- P.-Z. Li, X.-J. Wang, J. Liu, J. S. Lim, R. Zou and Y. A. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS PubMed.
- G. Ji, Z. Yang, H. Zhang, Y. Zhao, B. Yu, Z. Ma and Z. Liu, Angew. Chem., Int. Ed., 2016, 55, 9685–9689 CrossRef CAS PubMed.
- D. Liu, G. Li, J. Liu, Y. Wei and H. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 22119–22129 CrossRef CAS PubMed.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- R. Zhao, M. Wang, D. Zhao, H. Li, C. Wang and L. Yin, ACS Energy Lett., 2018, 3, 132–140 CrossRef CAS.
- J. C. Mohandas, M. K. Gnanamani, G. Jacobs, W. Ma, Y. Ji, S. Khalid and B. H. Davis, ACS Catal., 2011, 1, 1581–1588 CrossRef CAS.
- Q. Guo, F. Liang, X.-Y. Gao, Q.-C. Gan, X.-B. Li, J. Li, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, ACS Catal., 2018, 8, 5890–5895 CrossRef CAS.
- I. Persson, J. Halim, H. Lind, T. W. Hansen, J. B. Wagner, L. A. Naslund, V. Darakchieva, J. Palisaitis, J. Rosen and P. O. A. Persson, Adv. Mater., 2019, 31, 1805472 CrossRef PubMed.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- J. Peng, X. Chen, W.-J. Ong, X. Zhao and N. Li, Chem, 2019, 5, 18–50 CAS.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84 CrossRef CAS PubMed.
- Z. J. Huba and E. E. Carpenter, CrystEngComm, 2014, 16, 8000–8007 RSC.
- M. Zamanpour, S. Bennett, P. Taheri, Y. Chen and V. G. Harris, J. Appl. Phys., 2014, 115, 17A747 CrossRef.
- H. Wang, S. P. Wong, W. Y. Cheung, N. Ke, W. F. Lau, M. F. Chiah and X. X. Zhang, Mater. Sci. Eng., C, 2001, 16, 147–151 CrossRef.
- S. Li, C. Yang, Z. Yin, H. Yang, Y. Chen, L. Lin, M. Li, W. Li, G. Hu and D. Ma, Nano Res., 2017, 10, 1322–1328 CrossRef CAS.
- D. Gazzoli, M. Occhiuzzi, A. Cimino, D. Cordischi, G. Minelli and F. Pinzari, J. Chem. Soc., Faraday Trans., 1996, 92, 4567–4574 RSC.
- L. Yuwen, J. Zhou, Y. Zhang, Q. Zhang, J. Shan, Z. Luo, L. Weng, Z. Teng and L. Wang, Nanoscale, 2016, 8, 2720–2726 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/d0cc01091j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |