
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanoresponsive liquid crystals exhibiting reversible luminescent color changes at ambient temperature†
Masato
Mitani
a,
Shuhei
Ogata
b,
Shogo
Yamane
a,
Masafumi
Yoshio
a,
Miki
Hasegawa
b and
Takashi
Kato
*a
aDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kato@chiral.t.u-tokyo.ac.jp
bCollege of Science and Engineering, Aoyama Gakuin University, 5-10-1 Fuchinobe, Chuo-ku, Sagamihara, Kanagawa 252-5258, Japan
First published on 30th November 2015
Abstract
Mechanochromic luminescent liquid crystals containing oligothiophene derivatives have been obtained. These liquid-crystalline (LC) molecules having bi-, ter-, and quarterthiophene moieties exhibit shear-induced phase transitions accompanied by changes in their luminescent colors at ambient temperature. The shear-induced LC phases and their luminescent colors spontaneously revert back to the initial LC phases and the initial luminescent colors during the thermal aging processes at ambient temperature because the shear-induced LC phases are metastable phases. These three compounds show various luminescent colors and cover a wide visible region. The terthiophene and quaterthiophene derivatives recover their initial luminescent colors faster than the bithiophene derivative, indicating that not only the luminescent colors but also the stimuli-responsive properties are tunable through simple change of the π-conjugated length of the luminescent mesogen.
Introduction
Luminescence properties of organic molecules in condensed states are largely dependent on their assembled structures of π-conjugated moieties.1 If the assembled structures are transformed by applying external stimuli, the luminescence properties of the molecular assemblies may change. In this decade, the mechanochromic luminescent materials that change their molecular arrangements and luminescent colors in response to mechanical stimuli have been intensively studied because of their potential application in memories, sensor devices, and informational displays.1,2 Use of liquid crystals3 is one of the promising approaches to the development of mechanochromic luminescent materials because of their ordered structures and dynamic nature. Liquid-crystalline (LC) molecules change their orientations4 and show phase transitions in response to external stimuli such as heat, UV light,5 electric field,6 and mechanical force.1,7After the application of mechanical stimuli, most of the mechanochromic luminescent molecules need to be thermally annealed or exposed to solvent vapor for the recovery of their initial luminescence.1,2 These materials serve as long-term memory media, because they keep the mechanically induced luminescent colors stable until subsequent treatments are applied. On the other hand, if the materials spontaneously recover their initial luminescent colors after applying mechanical stimuli, short-term memory media is developed. Mechanochromic luminescent materials that show a reversible change in the luminescent colors at ambient temperature were reported.8,9 However, the tuning of their stimuli-responsive properties and luminescent colors is still difficult because most of these mechanochromic luminescent materials have been found serendipitously, and chemical derivatization of the parent compounds often causes the loss of their mechanoresponsive properties.
We have developed mechanochromic luminescent liquid crystals showing various emission colors.7 These molecules contain bulky dendritic moieties at both sides of the π-conjugated moieties such as pyrene,7a anthracene,7b and naphtharene derivatives.7c They need a heating process from the LC phases to the isotropic liquids and subsequent cooling in order to recover their initial luminescent colors because the shear-induced LC phases are thermodynamically more stable than their initial states. It has been demonstrated that LC phases and the stability of the dendritic molecules are tunable by changing the molecular shapes and the generation number of dendritic moieties.10,11a In this context, we focused on the substituents attached to the π-conjugated moieties to tune the stimuli-responsive properties of LC molecules. Herein, we report the luminescent liquid crystals containing modified substituents attached to the luminescent cores that show reversible luminescent color changes at ambient temperature.
Results and discussion
Molecular design
Compounds 1–3 containing oligothiophene derivatives as the luminescent cores were designed and synthesized (Fig. 1). Oligothiophene derivatives have been intensively studied for their electro- and photo-functions,11,12 and they have been widely used as building blocks to construct well-ordered molecular assemblies.11,13 Their absorption and emission properties can be controlled easily and systematically by changing the number of thiophene rings.14 Compounds 1–3 possess less bulky substituents attached to the luminescent cores than compounds in our previous reports.7 Racemic branched alkyl chains were introduced to enhance solubility, stabilize the LC phases, and decrease the clearing points.15 | ||
| Fig. 1 Molecular structures of compounds 1–3. | ||
Liquid-crystalline properties and mechanochromic behavior of compounds 1–3
Compound 1 based on the bithiophene moiety exhibits different phases at 25 °C depending on the cooling process from the isotropic liquid (Fig. 2). On cooling at a scanning rate of 5 K min−1, compound 1 shows an optically isotropic (OI) LC phase (Fig. 3a, G-form). Compound 1 in the OI phase (G-form) emits green photoluminescence under UV irradiation (Fig. 4a, left). On the other hand, a blue-green emissive crystalline (Cryst) phase is obtained by annealing at 85 °C (Fig. S1, ESI†). Crystallization of the OI phase has not been observed for more than one year under ambient conditions. These results suggest that the flexible branched alkyl chains stabilize the OI phase (G-form) and suppress crystallization.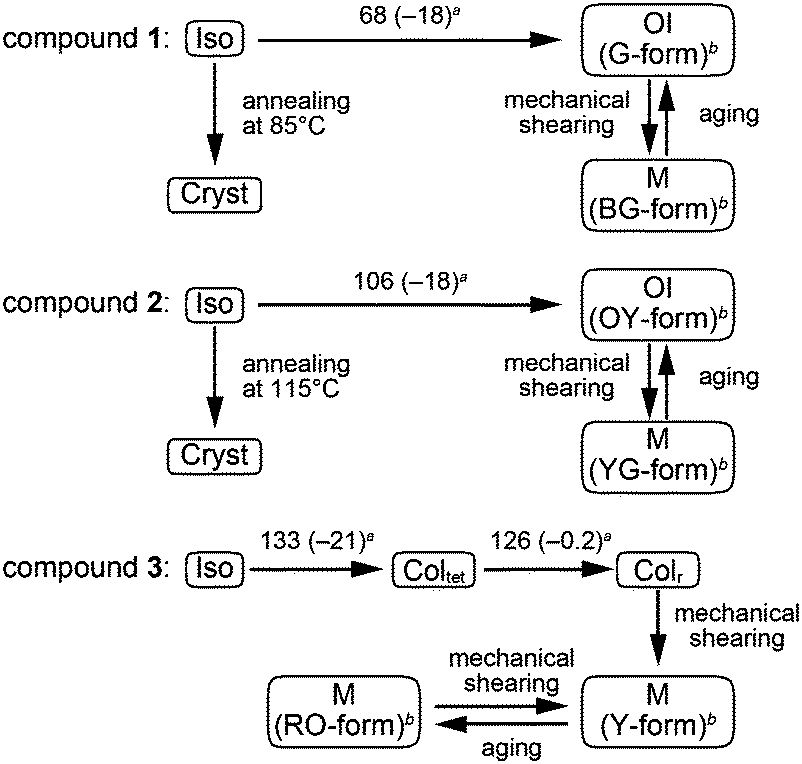 | ||
| Fig. 2 Thermal properties and mechanochromic behavior of compounds 1–3. a Transition temperatures (°C) taken at the onset points of the transition peaks and transition enthalpies (kJ mol−1, given in parentheses) determined by DSC on 1st cooling at a scanning rate of 5 K min−1. Iso: isotropic liquid; Cryst: crystalline; OI: optically isotropic LC; M: unidentified mesophase; Coltet: tetragonal columnar; Colr: rectangular columnar. b Capital letters in parentheses denote luminescent colors in each LC state: (G) green; (BG) blue-green; (OY) orange-yellow; (YG) yellow-green; (Y) yellow; (RO) red-orange. | ||
 | ||
| Fig. 3 Polarized optical microscopic images: (a) compound 1 in the optically isotropic LC phase (OI phase, G-form) at 25 °C; (b) compound 1 in the shear-induced mesophase (M phase, BG-form) at 25 °C; (c) compound 3 in the Coltet phase at 130 °C; and (d) compound 3 in the Colr phase at 25 °C. Scale bar: 200 μm. Arrows indicate the directions of polarizer and analyzer axes. | ||
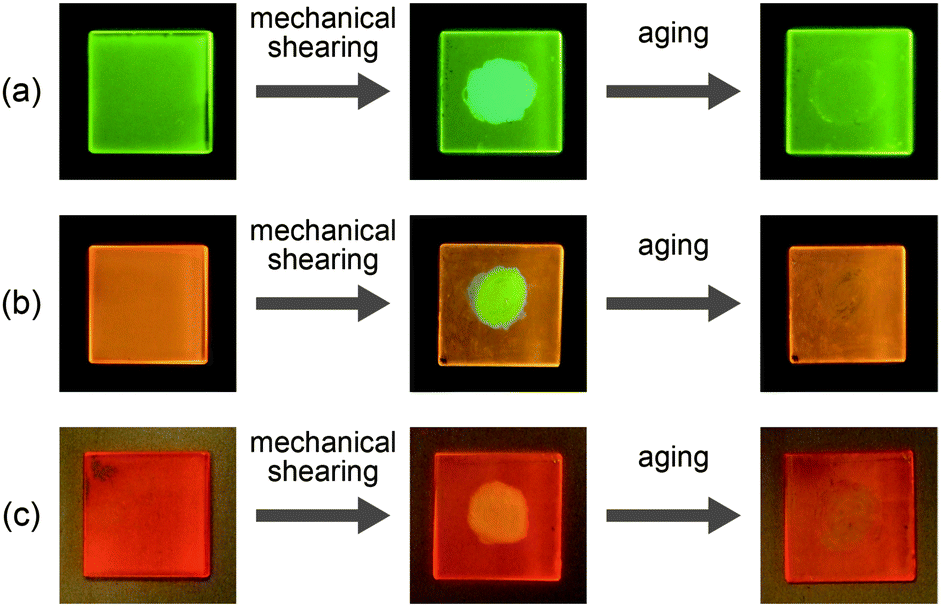 | ||
| Fig. 4 Luminescence images of compounds 1 (a), 2 (b), and 3 (c) under UV irradiation (λex = 365 nm) before shearing (left), after shearing (center), and after aging at ambient temperature (right). Mechanical shearing is applied to the central part of the sample coated on a quartz substrate. | ||
The XRD pattern of compound 1 in the OI phase (G-form) shows only two broad diffraction peaks at 44.3 and 24.2 Å in the small angle region (Fig. 5a, top). It was reported that molecules composed of the rigid part and the flexible part form micellar structures and show triclinic,16 tetragonal,17 cubic,7,11a and random micellar mesophases.18 Compound 1 may also form micelles because it consists of the bithiophene-based π-conjugated moiety as a rigid part and branched alkyl chains as a flexible part. The broad peaks in the small angle region of the XRD pattern of compound 1 in the OI phase (Fig. 5a, top, G-form) suggest the liquid-like short-range order of the micelles.18 The black image obtained by using a polarized optical microscope (Fig. 3a) may support the formation of micelles and their random arrangement (random micellar phase).18 The wide-angle region of the XRD pattern of compound 1 shows a diffused halo around 4.7 Å due to the disorder of the terminal branched alkyl chains.
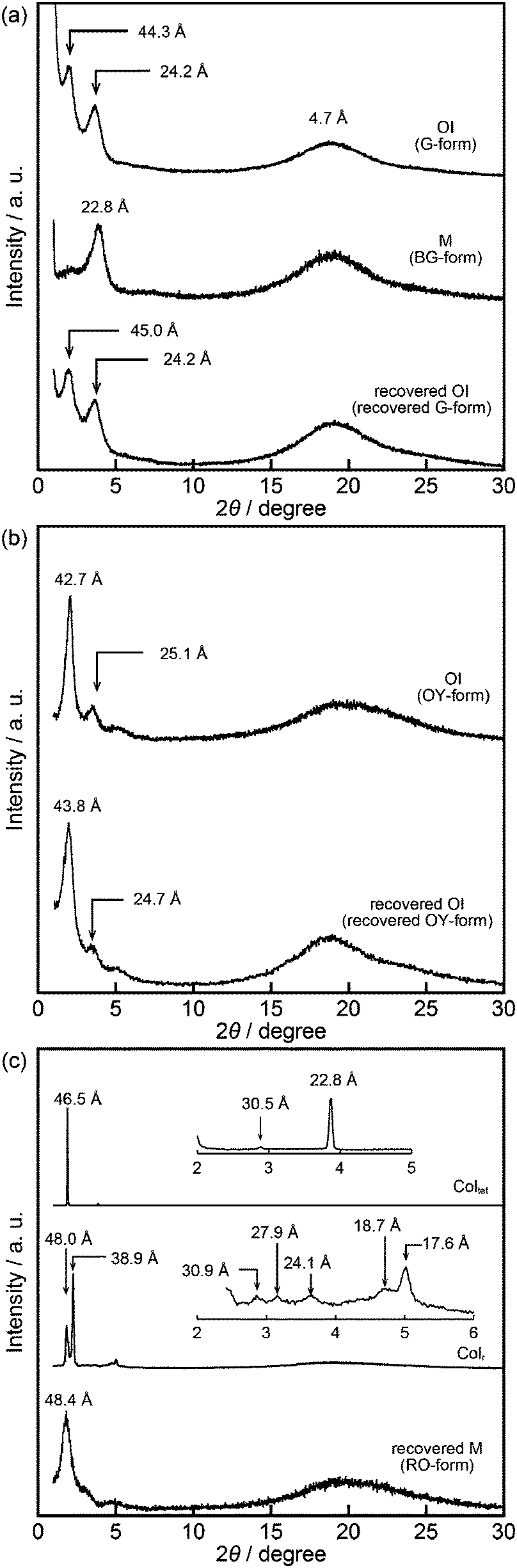 | ||
| Fig. 5 XRD patterns of compounds 1–3: (a) compound 1 in the OI phase (G-form at 25 °C, top), in the shear-induced M phase (BG-form at 25 °C, center), and in the recovered OI phase (recovered G-form at 25 °C, bottom); (b) compound 2 in the OI phase (OY-form at 25 °C, top) and in the recovered OI phase (recovered OY-form at 25 °C, bottom); (c) compound 3 in the Coltet phase at 130 °C (top), in the Colr phase at 25 °C (center), and in the recovered M phase (RO-form at 25 °C, bottom). | ||
Compound 1 shows reversible luminescent color changes triggered by mechanical shearing and subsequent thermal aging at ambient temperature. Upon applying mechanical shearing to compound 1 in the OI phase (G-form), a birefringent mesomorphic state (M phase, BG-form) is induced as observed in a polarized optical microscopic image (Fig. 3b), and the luminescent color changes from green to blue-green (Fig. 4a, center). Interestingly, the birefringence spontaneously becomes weaker at ambient temperature, and the luminescent color also reverts back to green. A photoluminescence image under UV irradiation at 365 nm for compound 1 aged for two days after mechanical shearing is shown in Fig. 4a, right. In the shear-induced M phase (BG-form), one broad peak is detected at 22.8 Å in the small-angle region of the XRD pattern (Fig. 5a, center), which is completely different from the OI phase (Fig. 5a, top, G-form). Furthermore, after aging the M phase (BG-form) at ambient temperature for two days, compound 1 exhibits an almost identical XRD pattern (Fig. 5a, bottom) as that of the initial OI phase (Fig. 5a, top, G-form). From these results, it is confirmed that mechanical shearing induces the OI–M phase transition, and subsequently the OI phase gradually recovers through the aging process at ambient temperature.
Compound 2 containing a terthiophene moiety shows LC properties and stimuli-responsive behavior which are similar to those of compound 1. Compound 2 shows an LC phase or a crystalline phase depending on the cooling process from the isotropic liquid (Fig. 2). Rapid cooling leads to the formation of an optically isotropic (OI) LC phase (Fig. S2, ESI,† OY-form) exhibiting orange-yellow luminescence (Fig. 4b, left). The Iso–OI phase transition is observed at 106 °C, which is higher than the Iso–OI phase transition temperature of compound 1 at 68 °C (Fig. 2 and Fig. S3, ESI†). The increase in the phase transition temperature is ascribed to the effects of the longer π-conjugated moiety of compound 2 compared to compound 1. In the XRD pattern of compound 2 in the OI phase (OY-form), two broad peaks at 42.7 and 25.1 Å are detected in the small-angle region (Fig. 5b, top), suggesting that compound 2 may form the random micellar LC phase. On the other hand, a crystalline phase emitting yellow-green luminescence is obtained by annealing compound 2 in the isotropic liquid phase at 115 °C (Fig. S2, ESI†).
Compound 2 also shows mechanochromic luminescence at ambient temperature. After applying mechanical shearing to compound 2 in the OI phase (OY-form), the luminescent color changes from orange-yellow to yellow-green due to the phase transition to a birefringent mesomorphic (M) phase (Fig. 4b, center, YG-form). Interestingly, the luminescent color immediately turns back to orange-yellow (Fig. 4b, right). It is noteworthy that compound 2 needs a shorter time to recover the luminescent color than compound 1. The recovery of the initial OI phase (OY-form) was confirmed by the XRD pattern measured after the aging process (Fig. 5b, bottom). However, the XRD pattern of the M phase (YG-form) could not be obtained because of the fast M (YG-form)–OI (OY-form) phase transition after the application of mechanical shearing.
Compound 3 shows two columnar phases on cooling from the isotropic liquid (Fig. 2) while compounds 1 and 2 exhibit optically isotropic LC phases. A tetragonal columnar LC (Coltet) phase is formed in the higher temperature region (133–126 °C, Fig. 3c). The XRD pattern of compound 3 at 130 °C shows three small-angle diffraction peaks at 46.5 (100), 30.5 (110), 22.8 Å (200) with the reciprocal d-spacing ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√2:2, confirming the tetragonal arrangement of the columns (Fig. 5c, top). Compound 3 in the Coltet phase emits red-orange luminescence. Further cooling to 126 °C induces a rectangular columnar LC (Colr) phase (Fig. 3d). The presence of two peaks in the small-angle region of the XRD pattern of compound 3 at 25 °C (Fig. 5c, center) is characteristic of a rectangular lattice. Although several peaks are detected in the small-angle region, the rectangular symmetry cannot be determined due to the lack of higher-order diffractions. Compound 3 in the Colr phase shows red-orange luminescence (Fig. 4c, left).
:√2:2, confirming the tetragonal arrangement of the columns (Fig. 5c, top). Compound 3 in the Coltet phase emits red-orange luminescence. Further cooling to 126 °C induces a rectangular columnar LC (Colr) phase (Fig. 3d). The presence of two peaks in the small-angle region of the XRD pattern of compound 3 at 25 °C (Fig. 5c, center) is characteristic of a rectangular lattice. Although several peaks are detected in the small-angle region, the rectangular symmetry cannot be determined due to the lack of higher-order diffractions. Compound 3 in the Colr phase shows red-orange luminescence (Fig. 4c, left).
Compound 3 in the Coltet phase does not exhibit the shear-induced phase transition, but compound 3 in the Colr phase shows the mechanochromic behavior. After applying mechanical shearing to compound 3 in the Colr phase at ambient temperature, the luminescent color changes from red-orange to yellow (Fig. 4c, center, M phase, Y-form), but the luminescent color immediately turns back to red-orange during the aging process at ambient temperature (Fig. 4c, right, M phase, RO-form). The XRD pattern of the M phase (RO-form) obtained after aging for three hours shows a broad peak at 48.4 Å (Fig. 5c, right), indicating that the M phase (RO-form) is an LC phase different from the Colr phase. It should be noted that the rectangular symmetry does not recover during the aging process, even though the luminescent color recovers. However, the reversible color changes from red-orange (M phase, RO-form) to yellow (M phase, Y-form) can be repeated.
Differential scanning calorimetry (DSC) measurements were performed to study the reversible phase transitions of compound 1. Fig. 6 shows the DSC traces of compound 1 on first heating. The OI phase (G-form), the M phase (BG-form), and the recovered OI phase (G-form) of compound 1 were used as the starting LC phases for Run 1–3, respectively. The thermal properties are presented in Table 1. In Run 1, an OI (G-form)–Iso phase transition is observed at 71 °C (Fig. 6, Run 1). On further heating, compound 1 crystallizes at 109 °C, and melts at 124 °C. The crystallization temperature is dependent on the heating rate. As the scanning rate increases, the crystallization temperature shifts to higher temperature (Fig. S4, ESI†). On cooling from the isotropic liquid state, compound 1 shows only one transition peak corresponding to the Iso–OI phase transition at 68 °C (Fig. 2 and Fig. S3, ESI†), which occurs at lower temperature than that of the Cryst–Iso phase transition at 124 °C (Table 1). These results indicate that compound 1 exhibits a supercooled liquid state on cooling from the isotropic liquid, and then forms the metastable OI phase (G-form) at the cooling rate of 5 K min−1. The phase transition behavior observed for compound 1 during the cooling and heating process is known as monotropic phase transitions.19 Flexible branched alkyl chains of compound 1 may play an important role in the inhibition of crystallization, leading to the formation of the OI phase (G-form).
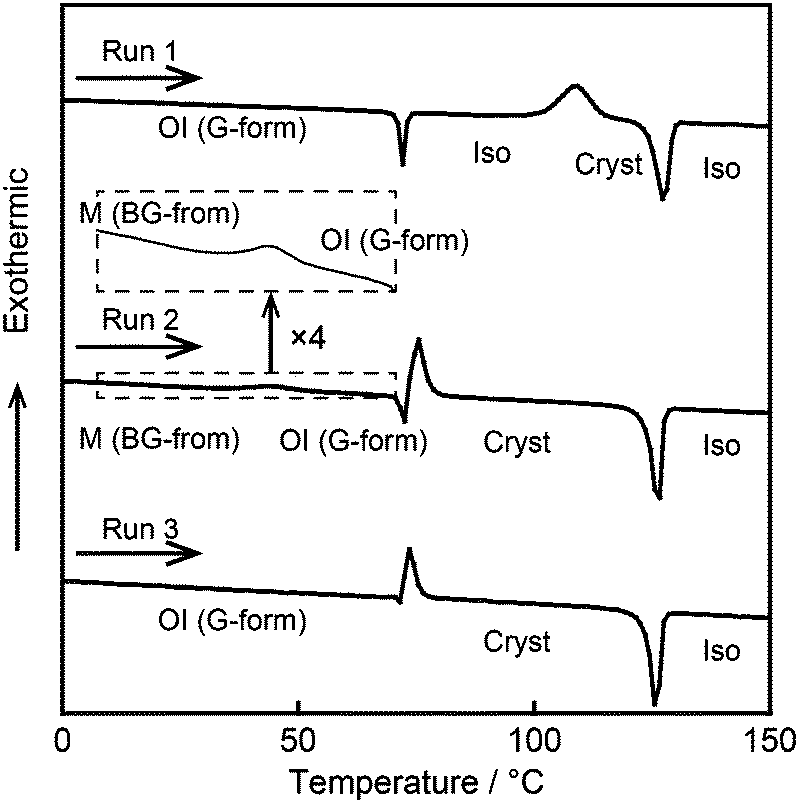 | ||
| Fig. 6 DSC traces of compound 1 on 1st heating: (Run 1) starting from the OI phase (G-form) obtained by cooling from the isotropic liquid phase; (Run 2) starting from the shear-induced M phase (BG-form) obtained by shearing compound 1 in the OI phase (G-form); (Run 3) starting from the recovered OI phase (G-form) obtained by aging compound 1 in the M phase (BG-form) at ambient temperature for two days. | ||
| Run | Phase transition behaviora |
|---|---|
| a Transition temperatures (°C) taken at the onset points of the transition peaks and transition enthalpies (kJ mol−1, given in parentheses) determined by DSC on 1st heating at a scanning rate of 5 K min−1. b Transition temperatures were taken at the maximum in the transition peaks. | |
| Run 1 | OI (G-form) 71 (19) Iso 109b (−60) Cryst 124 (67) Iso |
| Run 2 | M (BG-form) 44b (−10) OI (G-form) 71 (−31) Cryst 122 (69) Iso |
| Run 3 | OI (G-form) 71 (−29) Cryst 122 (66) Iso |
It should be noted that compound 1 in the shear-induced M phase (BG-form) shows an exothermic peak at 44 °C on heating with an enthalpy change of −10 kJ mol−1 corresponding to the phase transition to the OI phase (G-form) (Fig. 6, Run 2). This phase transition is accompanied by a change of the luminescent color from blue-green to green, and the transition is thermally irreversible. These results imply that the OI phase (G-form) is thermodynamically more stable than the M phase (BG-form). Subsequently, compound 1 crystallizes at 71 °C, and this crystallization temperature in Run 2 is lower than that observed in Run 1. It is known that the crystallization temperature of metastable phases varies with the sample preparation procedure.20 In Run 2, compound 1 may show quick crystallization due to the effects of the applied mechanical force. In Run 3, which uses the recovered OI phase (G-form) obtained by aging the shear-induced M phase (BG-form) at ambient temperature for two days as the starting LC phase, the exothermic peak at 44 °C is not detected (Fig. 6, Run 3), suggesting that the M (BG-form)–OI (G-form) phase transition has been completed during the aging process at ambient temperature.
DSC measurements were carried out for compounds 2 and 3 (Fig. S5 and S6, ESI†) as well as compound 1. However, compounds 2 and 3 show the M (YG-form)–OI (OY-form) and M (Y-form)–M (RO-form) phase transitions, respectively, which are much faster than the M (BG-form)–OI (G-form) phase transition of compound 1. Thus, these phase transitions could not be detected by DSC traces of compounds 2 and 3. The kinetics of phase transitions is governed by the activation energy for the phase transitions between shear-induced LC phases and recovered LC phases. Stronger π–π interactions may occur between adjacent luminescent cores in the initial LC states of compounds 1–3 compared to their shear-induced LC phases, because compounds 1–3 in the initial LC phases show red-shifted emission. Given that π–π interactions are essential driving force to recover the initial LC phases, the rate of phase transitions to the initial LC phases will increase with the increase of the π-conjugation length due to the stronger π–π interactions and decrease of the activation energy needed for the phase transitions. This is one possible explanation for the fast recovery of the luminescent colors for compounds 2 and 3 after mechanical shearing.
Absorption and emission properties of compounds 1–3
Fig. 7 shows the absorption and emission spectra of compounds 1–3 and the photophysical properties are summarized in Table 2. A diluted chloroform solution of compound 1 (c = 1 × 10−5 M) exhibits an absorption band centered at 396 nm (Fig. 7a, left) and structured emission at 454 nm and 482 nm (Fig. 7a, right). In the OI phase (G-form), the absorption maximum is slightly blue-shifted as compared to that of the diluted chloroform solution, and a shoulder around 440 nm is observed, indicating that ground-state electronic interactions between the π-conjugated moieties occur in the OI phase (G-form). As for the shear-induced M phase (BG-form), a broad absorption peak is detected at 396 nm. In contrast with similarities in the absorption spectra, the emission properties of compound 1 in the OI phase (G-form) and the M phase (BG-form) show completely different features. In the OI phase (G-form), a broad and structureless emission band is obtained at 540 nm (Fig. 7a, right). In the M phase (BG-form), compound 1 exhibits a structured and blue-shifted emission band at 492 nm. During the aging process at 25 °C, the emission spectrum of the M phase (BG-form) gradually changes, and the spectrum becomes almost identical to that of the initial OI phase (G-form) in two days of the aging process (Fig. S7, ESI†). In order to further examine the origin of the luminescence, the luminescence lifetimes of compounds 1–3 were measured (Table 2). Compound 1 in the chloroform solution shows a luminescence lifetime of 0.40 ns (Fig. S8, ESI†), which is independent of the monitored emission wavelength. This observation indicates that compound 1 in the chloroform solution mainly possesses a single emissive component. Lifetimes of 2.81 and 0.55 ns were obtained in the OI phase (G-form). The luminescence lifetime of 2.81 ns in the OI phase (G-form) is longer than that in the chloroform solution, suggesting that green luminescence of compound 1 in the OI phase (G-form) can be assigned to the excimer emission of bithiophene moieties.21 In the M phase (BG-form), compound 1 shows shorter lifetimes (0.80 and 0.14 ns, Fig. S8, ESI†) compared to those in the OI phase. These results imply that the shear-induced luminescent color change is due to the inhibition of the excimer formation in the M phase (BG-form).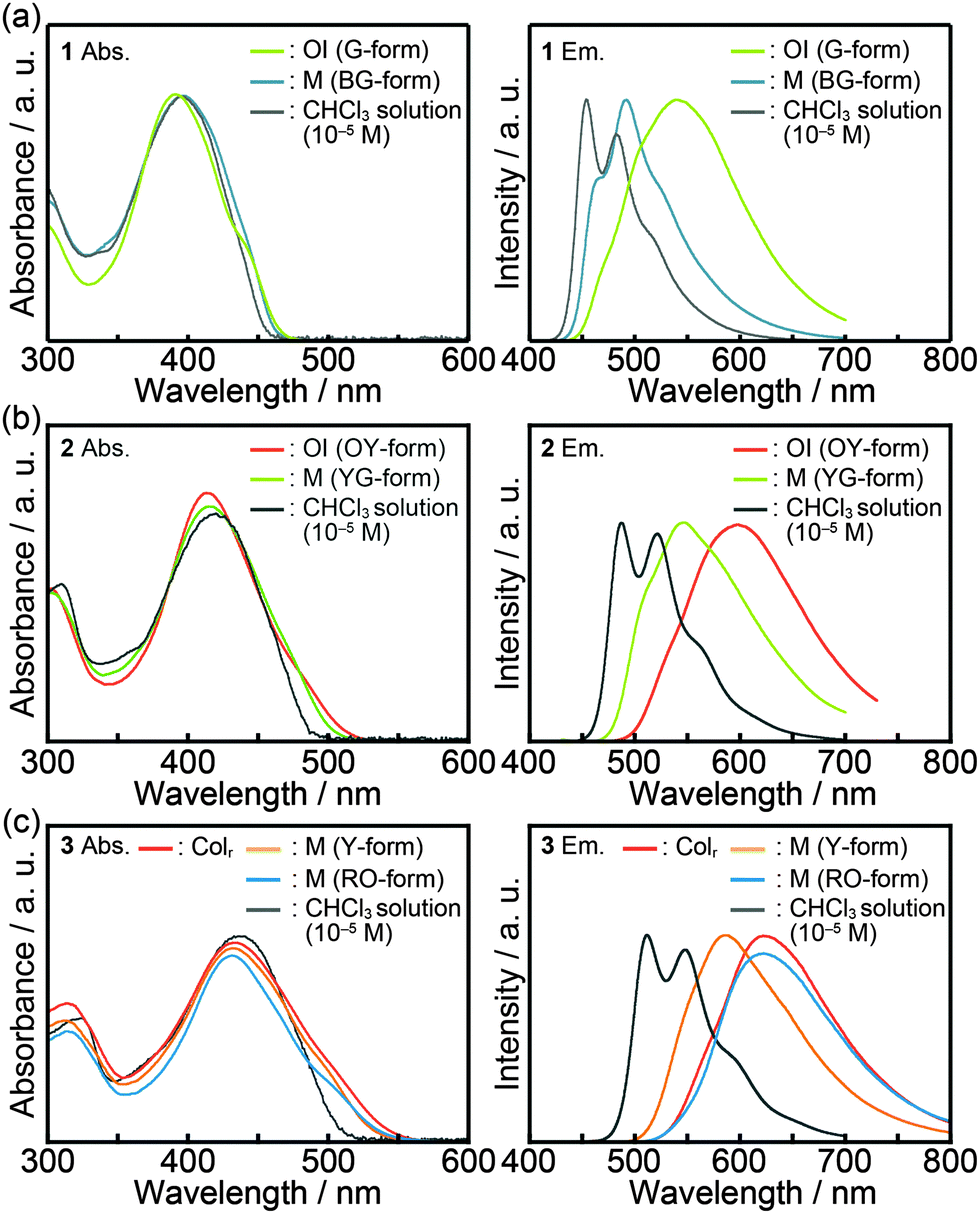 | ||
| Fig. 7 Absorption (left column) and emission spectra (right column): (a) compound 1; (b) compound 2; (c) compound 3. Spectra of compound 3 in the Colr phase were taken at 130 °C. All the other spectra were obtained at ambient temperature. λex = 380 nm (compounds 1 and 2) and λex = 430 nm (compound 3). | ||
| State | λ abs/nm | λ em /nm | τ 1/ns | τ 2/ns | |
|---|---|---|---|---|---|
| a λ ex = 380 nm for compounds 1 and 2, and λex = 430 nm for compound 3. b c = 1 × 10−5 M. c λ abs and λem are taken from the absorption and emission spectra of samples right after applying mechanical shearing. d Lifetime components shorter than 0.1 ns. e The values in parentheses are amplitudes of exponential decay components. | |||||
| 1 | Chloroform solutionb | 396 | 454, 482 | 0.40 | —d |
| OI phase (G-form) | 391 | 540 | 2.81 (0.38)e | 0.55 (0.62) | |
| M phase (BG-form) | 397 | 492 | 0.80 (0.12) | 0.14 (0.88) | |
| 2 | Chloroform solutionb | 420 | 487, 522 | 0.51 | —d |
| OI phase (OY-form) | 414 | 598 | 1.91 (0.45) | 0.24 (0.55) | |
| M phase (YG-form)c | 416 | 547 | — | — | |
| 3 | Chloroform solutionb | 438 | 512, 548 | 0.73 | —d |
| Colr phase | 434 | 624 | 1.45 (0.53) | 0.20 (0.47) | |
| M phase (Y-form)c | 432 | 587 | — | — | |
| M phase (RO-form) | 432 | 622 | 1.62 (0.56) | 0.55 (0.44) | |
As the number of thiophene rings increases, absorption and emission spectra of compounds 1–3 in diluted chloroform solution states are red-shifted (Table 2), indicating the enhancement of delocalization of π electrons and the reduction of the π–π* transition energy with increasing conjugation length. Luminescence lifetimes become longer as the number of thiophene rings increases (Table 2 and Fig. S9, ESI†). This relationship between the conjugation length and the luminescence lifetime is similar to that observed for the typical α-oligothiophenes.22
Compound 2 in the OI phase (OY-form) and compound 3 in the Colr phase possess longer lifetime components as compared to their solution states (Table 2 and Fig. S10, ESI†), suggesting that the excimer emission of the oligothiophene moieties is observed in each state. Broad and structureless emission bands of compound 2 in the OI phase (OY-form) and compound 3 in the Colr phase also suggest the presence of intermolecular interactions inducing the excimer formation in excited states. After applying mechanical shearing, the luminescence properties of compounds 2 and 3 change quickly, which makes it difficult to obtain the emission spectra of compound 2 in the pure M phase (YG-form) and compound 3 in the pure M phase (Y-form). The observed spectral shifts for compounds 2 and 3 induced by mechanical shearing are 51 and 37 nm, respectively (Table 2).
The luminescence of compounds 2 and 3 recovers in three hours after mechanical shearing, which was confirmed by emission spectra (Fig. S11 and S12, ESI†). The ambient temperature mechanochromic properties of compounds 1–3 based on mechanical shearing and subsequent aging can be repeated at least ten times (Fig. 8). In each cycle, the samples are aged at ambient temperature for more than two days for compound 1, and more than three hours for compounds 2 and 3.
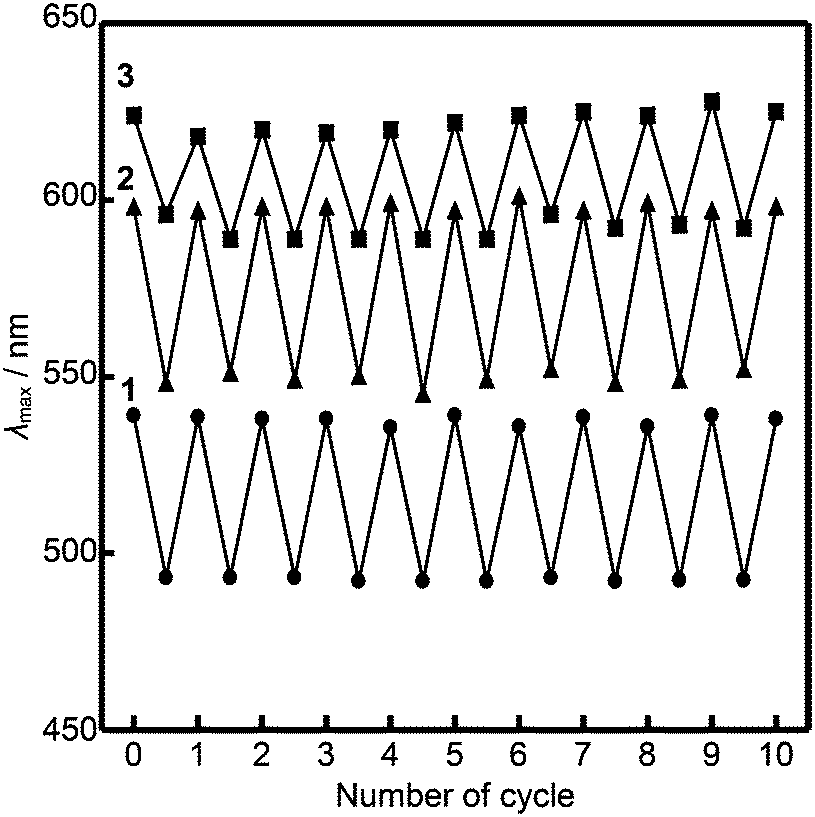 | ||
| Fig. 8 Change of the emission wavelength of compounds 1 (circle), 2 (triangle), and 3 (square) upon repeated cycles of mechanical shearing and aging at ambient temperature. λex = 380 nm (compounds 1 and 2) and λex = 430 nm (compound 3). | ||
Possible assembled structures
Infrared (IR) spectral measurements were conducted in order to obtain insight into the assembled structures of compounds 1–3. In chloroform solution (c = 1 × 10−3 M), compound 1 shows the absorption bands ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H stretching at 1677 and 3435 cm−1, respectively (Fig. 9a). On the other hand, in the OI phase (G-form) CO and N–H stretching bands are detected at 1646 and 3274 cm−1, respectively (Fig. 9b). These shifts to the lower wavenumber indicate that intermolecular hydrogen bonds are formed between the amide groups of adjacent molecules in the OI phase (G-form).7 Compound 1 in the M phase (BG-form) shows similar CO and N–H stretching bands (Fig. 9c) to those in the OI phase (G-form). These results suggest that the luminescent cores of compound 1 form ordered structures in both the OI phase (G-form) and the M phase (BG-form) through the intermolecular hydrogen bonds. Compounds 2 and 3 also form intermolecular hydrogen bonds between the adjacent molecules in the OI phase (OY-form) and the Colr phase, respectively (Fig. S13 and S14, ESI†).
O and N–H stretching at 1677 and 3435 cm−1, respectively (Fig. 9a). On the other hand, in the OI phase (G-form) CO and N–H stretching bands are detected at 1646 and 3274 cm−1, respectively (Fig. 9b). These shifts to the lower wavenumber indicate that intermolecular hydrogen bonds are formed between the amide groups of adjacent molecules in the OI phase (G-form).7 Compound 1 in the M phase (BG-form) shows similar CO and N–H stretching bands (Fig. 9c) to those in the OI phase (G-form). These results suggest that the luminescent cores of compound 1 form ordered structures in both the OI phase (G-form) and the M phase (BG-form) through the intermolecular hydrogen bonds. Compounds 2 and 3 also form intermolecular hydrogen bonds between the adjacent molecules in the OI phase (OY-form) and the Colr phase, respectively (Fig. S13 and S14, ESI†).
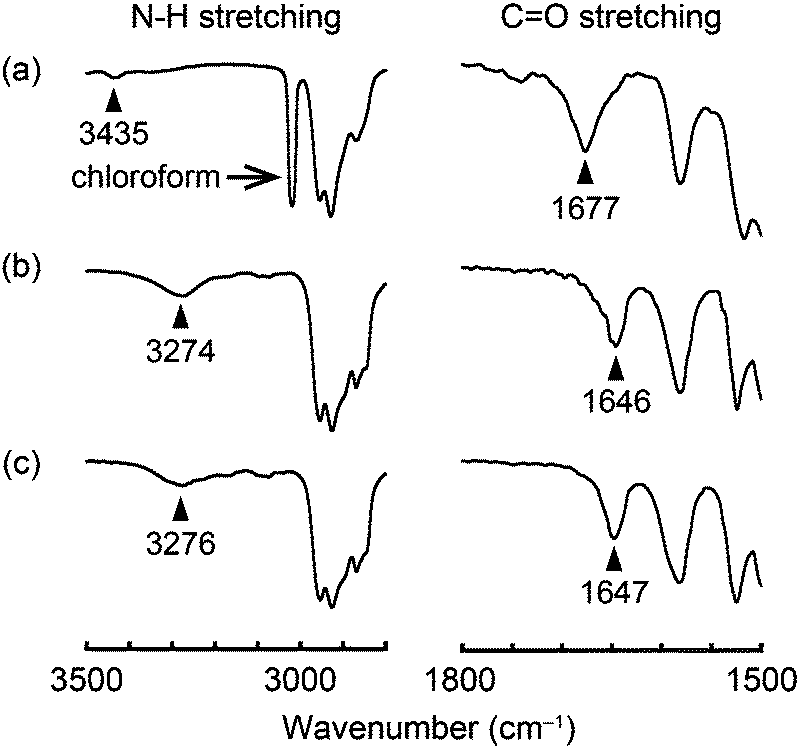 | ||
| Fig. 9 FT-IR spectra of compound 1 at 25 °C: (a) in chloroform solution (1 × 10−3 M); (b) in the OI phase (G-form); and (c) in the M phase (BG-form). | ||
Based on these results, possible assembled structures of compound 1 are proposed and shown in Fig. 10. In the OI phase (G-form), nanosegregation of π-conjugated moieties from the surrounding flexible alkyl chains occurs and compound 1 forms micelles (Fig. 10a). In each micelle, the luminescent cores stack in a face-to-face manner (Fig. 10b), leading to the formation of excimers.21 The arrangement of the stacking is disordered due to the incompatibility between the length of the π–π stacking and that of the H-bonded amide groups.7 In the M phase (BG-form), blue-shifted emission is observed, but the luminescent cores are not separated completely because the formation of intermolecular hydrogen bonds is retained even after mechanical shearing. Therefore the blue-green luminescence is caused by the insufficient overlap of the luminescent cores due to the tilted π-conjugated moieties to form the linear intermolecular hydrogen bonds (Fig. 10c). This tilted stacking mode inhibits the formation of the excimers, and as a result, compound 1 shows the blue-green emission.7
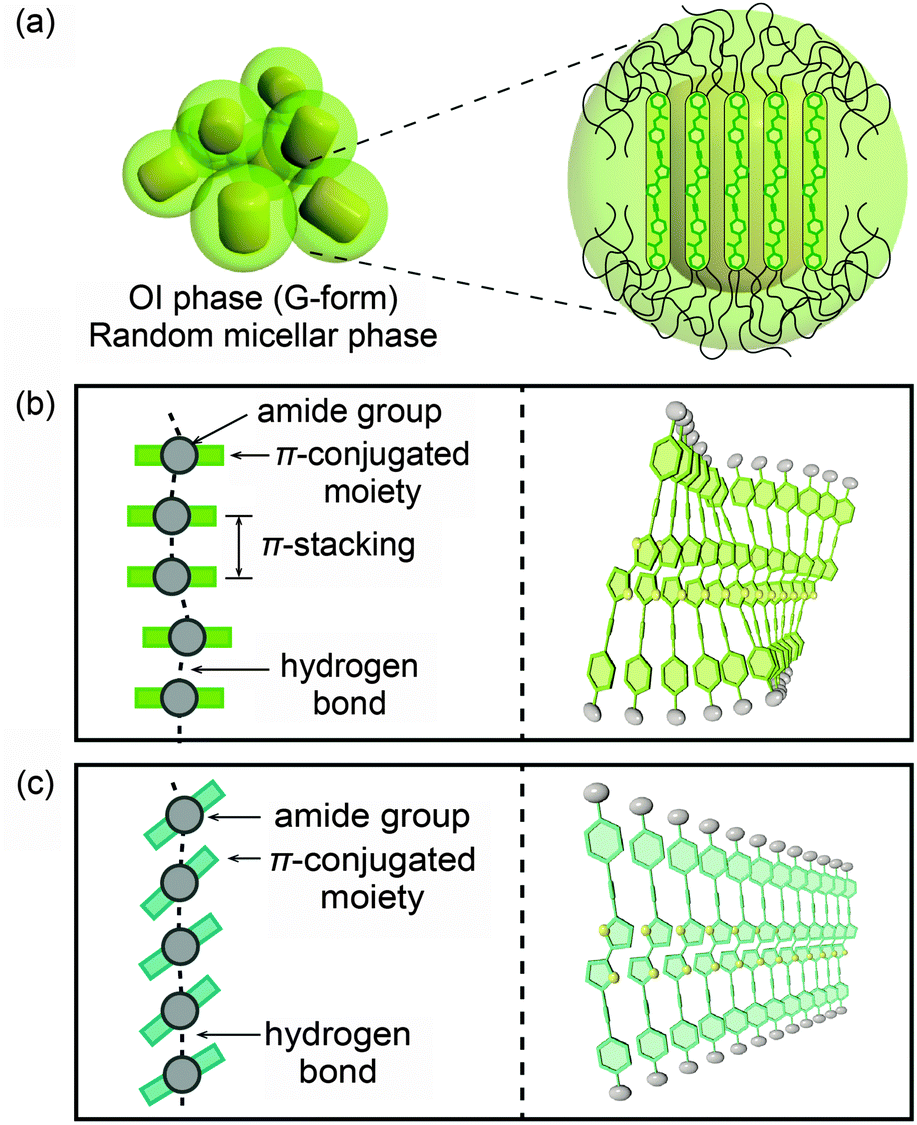 | ||
| Fig. 10 Possible schematic illustrations of assembled structures of compound 1: (a and b) in the OI phase (G-form); (c) in the M phase (BG-form). | ||
Conclusions
In summary, LC compounds 1–3 containing oligothiophene derivatives that show mechanochromic luminescence under ambient temperature conditions are reported. Luminescent colors of compounds 1–3 change in response to mechanical force, and their luminescent colors recover at ambient temperature without applying subsequent external stimuli. The shear-induced LC phases are unstable, and therefore they show spontaneous phase transitions to the initial LC phases even at ambient temperature. Compounds 1–3 exhibit various luminescent colors and cover a wide range of the visible region through simple tuning of the π-conjugated length of the luminescent mesogen. Our results also imply that fine-tuning of the stimuli-responsive properties can be achieved by molecular design, because the molecules with a longer π-conjugation length show fast recovery of their initial luminescent colors. This study provides material design strategy of mechanochromic luminescent molecules for application as switchable memory devices.Experimental section
Materials and syntheses
All reagents and solvents were purchased from Aldrich, Tokyo Kasei, and Kanto Chemicals. All of the reactions were carried out under an argon atmosphere in dry solvents. Silica gel column chromatography was carried out with silica gel 60 from Kanto Chemicals (silica gel 60, spherical, 40–50 μm). Recycling preparative GPC was carried out using a Japan Analytical Industry LC-9201 chromatograph.General procedures
1H and 13C NMR spectra were obtained using a JEOL JNM-LA400 spectrometer in CDCl3 solutions (400 and 100 MHz for 1H NMR and 13C NMR, respectively). Chemical shifts for 1H and 13C NMR were referenced to Me4Si (δ = 0.00) and CDCl3 (δ = 77.00) as internal standards, respectively, and are expressed in ppm (δ), multiplicity, coupling constant (Hz), and relative intensity. Mass spectra were obtained using a Bruker Daltonics Autoflex Speed using dithranol as the matrix. Elemental analyses were carried out using an Exeter Analytical Inc. CE-440 Elemental Analyzer. Polarized optical microscopic images were obtained with an Olympus BX51 equipped with a Mettler FP82 hot stage. Differential scanning calorimetry (DSC) measurements were performed on a NETZCH DSC204 Phoenix calorimeter at a scanning rate of 5 °C min−1. X-ray diffraction measurements were carried out on a Rigaku RINT2500 with a heating stage using Ni-filtered Cu Kα radiation (1.54 Å) and Rigaku Smartlab. The IR measurements were conducted on a JASCO FTIR-6100 and IRT-5000 using CaF2 plates. Absorption spectra were measured using a JASCO V-670 spectrophotometer equipped with an integrating sphere unit ISN-800T. Emission spectra were recorded on a JASCO FP-6500 spectrofluorometer equipped with a hot stage. The luminescence lifetime was measured using a Quantaurus-Tau (Hamamatsu C11367).Acknowledgements
This study was partially supported by a Grant-in-Aid for Scientific Research in the Innovative Area of “Fusion Materials” (Area no. 2206), from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (KAKENHI 22107003) for T. K. and (KAKENHI 23107528 and 25107730) for M. H. M. H. acknowledges partial support from the Supported Program for the Strategic Research Foundation at Private Universities, 2013–2017, with a matching fund subsidy from MEXT. M. M. is supported by Japan Society for the Promotion of Science (JSPS) through Program for Leading Graduate Schools (MERIT), and JSPS Research Fellowship for Young Scientists.References
-
(a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed
; (b) D. Yan and D. G. Evans, Mater. Horiz., 2014, 1, 46 RSC
-
(a) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC
-
(a)
Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH, Weinheim, 2nd edn, 2014 Search PubMed
-
(a) K. Ichimura, Chem. Rev., 2000, 100, 1847 CrossRef CAS PubMed
-
(a) Y. Norikane, Y. Hirai and M. Yoshida, Chem. Commun., 2011, 47, 1770 RSC
- A. Beneduci, S. Cospito, M. L. Deda, L. Veltri and G. Chidichimo, Nat. Commun., 2014, 5, 3105 Search PubMed
-
(a) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS PubMed
-
(a) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160 CrossRef CAS PubMed
-
(a) X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261 CrossRef CAS PubMed
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275 CrossRef CAS PubMed
-
(a) T. Yasuda, H. Ooi, J. Morita, Y. Akama, K. Minoura, M. Funahashi, T. Shimomura and T. Kato, Adv. Funct. Mater., 2009, 19, 411 CrossRef CAS
-
(a) I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281 CrossRef CAS
-
(a) E. Mena-Osteritz, A. Meyer, B. M. W. Langeveld-Voss, R. A. J. Janssen, E. W. Meijer and P. Bäueule, Angew. Chem., Int. Ed., 2000, 39, 2680 CrossRef CAS
-
(a) T. Noda, H. Ogawa, N. Noma and Y. Shirota, J. Mater. Chem., 1999, 9, 2177 RSC
-
(a) K. Ohta, Y. Morizumi, H. Ema, T. Fujimoto and I. Yamamoto, Mol. Cryst. Liq. Cryst., 1991, 208, 55 CrossRef CAS
- B. G. Kim, S. Kim, J. Seo, N.-K. Oh, W.-C. Zin and S. Y. Park, Chem. Commun., 2003, 2306 RSC
-
(a) M. Lee, B.-K. Cho, Y.-G. Jang and W.-C. Zin, J. Am. Chem. Soc., 2000, 122, 7449 CrossRef CAS
-
(a) M. Lee, D.-W. Lee and B.-K. Cho, J. Am. Chem. Soc., 1998, 120, 13258 CrossRef CAS
-
(a) V. Percec and A. Keller, Macromolecules, 1990, 23, 4347 CrossRef CAS
- O. Haida, H. Suga and S. Seki, J. Chem. Thermodyn., 1977, 9, 1133 CrossRef CAS
-
(a) A. Tsuge, T. Hara and T. Moriguchi, Tetrahedron Lett., 2009, 50, 4509 CrossRef CAS
-
(a) R. S. Becker, J. S. Melo, A. L. Maçanita and F. Elisei, Pure Appl. Chem., 1995, 67, 9 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, liquid-crystalline properties of compounds 1–3. See DOI: 10.1039/c5tc03578c |
| This journal is © The Royal Society of Chemistry 2016 |