DOI:
10.1039/C6RA07244E
(Paper)
RSC Adv., 2016,
6, 61725-61731
A gadolinium MOF acting as a multi-responsive and highly selective luminescent sensor for detecting o-, m-, and p-nitrophenol and Fe3+ ions in the aqueous phase†
Received
19th March 2016
, Accepted 12th June 2016
First published on 13th June 2016
Abstract
A microporous Gd-MOF, [Gd6(L)3(HL)2(H2O)10]·18H2O·x(solvent) (1), has been successfully synthesized by a solvothermal reaction between Gd(NO3)3·6H2O and the multidentate π-conjugated ligand, H4L, which has a Lewis basic pyridyl site (H4L = 5,5′-(pyridine-2,5-diyl)-isophthalic acid). The crystal structure shows that compound 1 consists of Gd3 units, which are further interlinked by multicarboxylate ligands to form a 2D network. A solid sample of 1 emits bright blue light, which can be assigned to H4L ligand-centered emission. Interestingly, the luminescence of finely ground particles of 1 dispersed in water shows high sensitivity and selectivity towards trace amounts of o-, m-, and p-nitrophenol (NP) and Fe3+ ions with good linearity, which indicates that 1 can be used as a multi-responsive luminescence sensor for the detection of o-, m-, and p-NP and Fe3+ ions in an aqueous system.
Introduction
Sensitive and selective detection of nitroaromatic compounds, which are both environmental pollutants and explosives that are a threat to public security, is crucial with respect to homeland security, environmental protection, military applications and mine-field analyses.1,2 Among the nitroaromatic compounds, nitrophenols have been included in the US Environmental Protection Agency (USEPA) list of priority pollutants, due to their pollution of water and the environment.3 o-, m-, and p-nitrophenols (NP) represent some of the most persistent and highly toxic nitrophenols; their rapid and trace detection is crucial to environmental protection and homeland security. However, relatively less effort has been focused on the detection of o-, m-, and p-NP than other hazardous substances until now,4 while related reports concerning the simultaneous detection of o-, m-, and p-NP are rare.5
Current commercial and technique-based instrumental methods, such as surface-enhanced Raman spectroscopy,6 cyclic voltammetry,7 and gas chromatography coupled with mass spectrometry,8 are expensive and are not always available. Recently, fluorescence-based detection methods have attracted great attention due to their low cost, high sensitivity, portability, short response times, and dual applicability in solid and solution media.1,2 A great number of fluorescence-based materials, such as conjugated polymers,1,9 nanoparticles,1b,e,f,10 and microporous metal–organic frameworks (MOFs),2a,d,e,11,12 have been applied to the detection of nitroaromatic compounds. The combination of accessible porosity within microporous MOFs and fluorescence provides these methods with the ability to transform host–guest interactions into detectable fluorescence changes, which also makes them potential candidates for sensing applications. Furthermore, the field selective detection in aqueous media of nitroaromatic compounds present in soil and ground water is highly desirable. To the best of our knowledge, related reports of microporous MOFs acting as sensors of nitroaromatic compounds in aqueous media are relatively rare,2e especially for sensing o-, m-, and p-NP.
The detection of Fe3+ ions in aqueous solution with high sensitivity and selectivity is also very important, since iron is an indispensable element in the biosphere and the environment.13 In biological processes, both Fe2+ and Fe3+ ions are essential and play different major roles.14 Therefore, it is crucial to be able to detect one from the other. In this context, some MOF-based fluorescent sensors for Fe3+ ions have been reported.13c,15,16 However, examples of the detection of Fe3+ ions in aqueous systems are relatively rare,16 especially those with good linearity, which limits their practical sensing application in environmental and biological systems.17
Herein we report the fluorescence properties and sensing behaviour of [Gd6(L)3(HL)2(H2O)10]·18H2O·x(solvent) (1) (H4L = 5,5′-(pyridine-2,5-diyl)-isophthalic acid, Scheme 1). The ligand, H4L, was chosen as a π-conjugated ligand to construct MOFs with respect to the following considerations: (i) its extreme rigidity, which is an advantage in the formation of porous structures and (ii) its π-conjugated structure and available Lewis-base sites, which may facilitate the sensing properties through particular host–guest interactions. We have obtained a functional Eu-MOF, which was isomorphous with 1, by using this ligand in our previous work.5 The Eu-MOF exhibits high sensitivity and selectivity towards o-, m-, and p-nitrophenol (NP), but the performance with respect to quantitative detection is poor due to the low linear dependency. However, compound 1 not only shows good sensitivity and selectivity towards o-, m-, and p-NP but also can sense Fe3+ ions in aqueous media along with good liner dependency, which is unprecedented. This also means that compound 1 could be used as a multi-responsive luminescence sensor for the quantitative detection of o-, m-, and p-NP and Fe3+ ions in a real aquatic system.
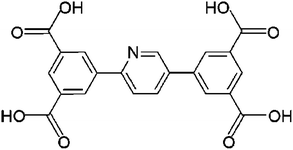 |
| | Scheme 1 The organic ligand, H4L. | |
Experimental section
General procedure
The reagents and the organic ligand, H4L (5,5′-(pyridine-2,5-diyl)-isophthalic acid), were obtained from commercial sources and used without further purification.
Synthesis of [Gd6(L)3(HL)2(H2O)10]·18H2O·x(solvent) (1)
Gd(NO3)3·6H2O (0.045 g, 0.1 mmol) and 5,5′-(pyridine-2,5-diyl)-isophthalic acid (0.041 g, 0.1 mmol) were dissolved in a mixed solvent of DEF/H2O (0.2 mL/6 mL) and transferred to a 23 mL Teflon-lined autoclave; thereafter, 5 drops of 0.1 mol L−1 NaOH aqueous solution were added to the mixture. After stirring in air for 20 min, the mixture was heated to 150 °C at a rate of 37.5 °C h−1 and kept for 4 days. After cooling to room temperature at a rate of 5 °C h−1, colourless block crystals of 1 were collected in 70% yield based on H4L. Elemental analyses—found: C, 38.33; H, 3.48; N, 3.00. IR bands (KBr, cm−1): 3423 s, 1622 s, 1571 s, 1457 s, 1388 s, 1300 w, 1045 m, 931 m, 755 s, 710 m.
Physical measurements
Fourier transform infrared (FTIR) spectra (KBr disk) were measured with a TENSOR 27 FT-IR spectrophotometer. The TG curve was recorded on an SDT Q600 thermal analyzer under an N2 atmosphere at a heating rate of 10 °C min−1 over the temperature range 25–1067 °C. Powder X-ray diffraction (PXRD) was carried out on a EMPYREAN PANALYTICAL apparatus. The luminescence spectra for the powdered solid samples were measured at room temperature on a Hitachi F-7000 fluorescence spectrophotometer. The UV-vis absorption spectra were measured with a U-3900 spectrophotometer.
Fluorescence measurements
The fluorescence properties of 1 were measured in the solid state and in various analyte suspensions at room temperature. Suspensions of 1 were prepared by dispersing 2 mg of fully ground sample 1 in 3 mL water solution containing different analytes, (2,4-dinitrotoluene (2,4-DNT), phenol, terephthaldehyde (TPAH), benzonitrile (BZNT), m-aminopyridine (m-APYD), p-toluenesulfonic acid (p-TSFA), o-hydroxy-benzoyl hydrazine (o-HBHDZ), o-nitrophenol (o-NP), m-nitrophenol (m-NP), and p-nitrophenol (0.1 mol L−1) (p-NP)) at room temperature under ultrasonication for 30 min and stirring for 3 h. For the fluorescence quenching titrations in dispersed media, the fluorescence of compound 1 was measured by dispersing 2 mg of fully ground sample 1 in 3 mL water at room temperature under ultrasonication for 30 min and stirring for 3 h, subsequently placing in a quartz cell of 1 cm width. All titrations were carried out by gradually adding water solutions of o-, m-, and p-NP in an incremental fashion. Each titration was repeated several times to obtain concordant values. For all measurements, dispersed solutions of 1 were excited at λex = 392 nm (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 510 cm−1) and their corresponding emission wavelength was monitored from 410 nm (24390 cm−1) to 560 nm (17857 cm−1). There was no change in the shape of the emission spectra as a result of gradual addition of o-, m-, or p-NP to a dispersed solution of 1 in water; only quenching of the initial fluorescence emission intensity was observed. The Fe3+ ion fluorescence detection experiment was conducted using a similar procedure, except that the analytes were Cd2+, Ca2+, Co2+, Fe2+, Fe3+, Mg2+, Mn2+, Ni2+, and Pb2+ ions instead.
510 cm−1) and their corresponding emission wavelength was monitored from 410 nm (24390 cm−1) to 560 nm (17857 cm−1). There was no change in the shape of the emission spectra as a result of gradual addition of o-, m-, or p-NP to a dispersed solution of 1 in water; only quenching of the initial fluorescence emission intensity was observed. The Fe3+ ion fluorescence detection experiment was conducted using a similar procedure, except that the analytes were Cd2+, Ca2+, Co2+, Fe2+, Fe3+, Mg2+, Mn2+, Ni2+, and Pb2+ ions instead.
Crystal data collection and refinement
Diffraction intensity data were collected at 293 K on a Bruker APEX II diffractometer equipped with a CCD area detector and graphite-monochromated Cu Kα radiation (λ = 1.54184 Å). Empirical absorption corrections were applied using the SADABS program.18 The structures were solved by the direct method and refined by the full-matrix least-squares method on F2, with all non-hydrogen atoms refined with anisotropic thermal parameters.19 All the hydrogen atoms attached to carbon atoms were placed in calculated positions and refined using the riding model. The hydrogens attached to water molecules were located from the difference Fourier maps and refined isotropically. From the difference Fourier map of 1, a number of diffuse scattered peaks with electron density were observed, which can be attributed to disordered solvent molecules. Attempts to model these peaks were unsuccessful because the residual electron density peaks obtained were diffused. So, PLATON/SQUEEZE was used to refine the structure further. Crystallographic data for 1 have been deposited at the Cambridge Crystallographic Data Center with the deposition number CCDC 1457726. Experimental details for the structural analyses of 1 are given in Table 1. Selected bond lengths and angles are given in Table S1.†
Table 1 Crystal data and structure refinements for compound 1
| Compound |
1 |
| Formula |
C105H103Gd6N5O68 |
| Mr |
3466.42 |
| Crystal system |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a, Å |
10.9788(5) |
| b, Å |
15.8121(10) |
| c, Å |
20.9085(13) |
| α, deg |
108.914(6) |
| β, deg |
91.050(5) |
| γ, deg |
106.141(5) |
| V, Å3 |
3274.8(3) |
| Z |
1 |
| Dc, g cm−3 |
1.758 |
| μ, mm−1 |
20.186 |
| Unique reflns/Rint |
11716/0.1011 |
| R1 [I > 2σ(I)] |
0.0772 |
| wR2(all data) |
0.1991 |
| GOF |
1.001 |
Results and discussion
Crystal structure
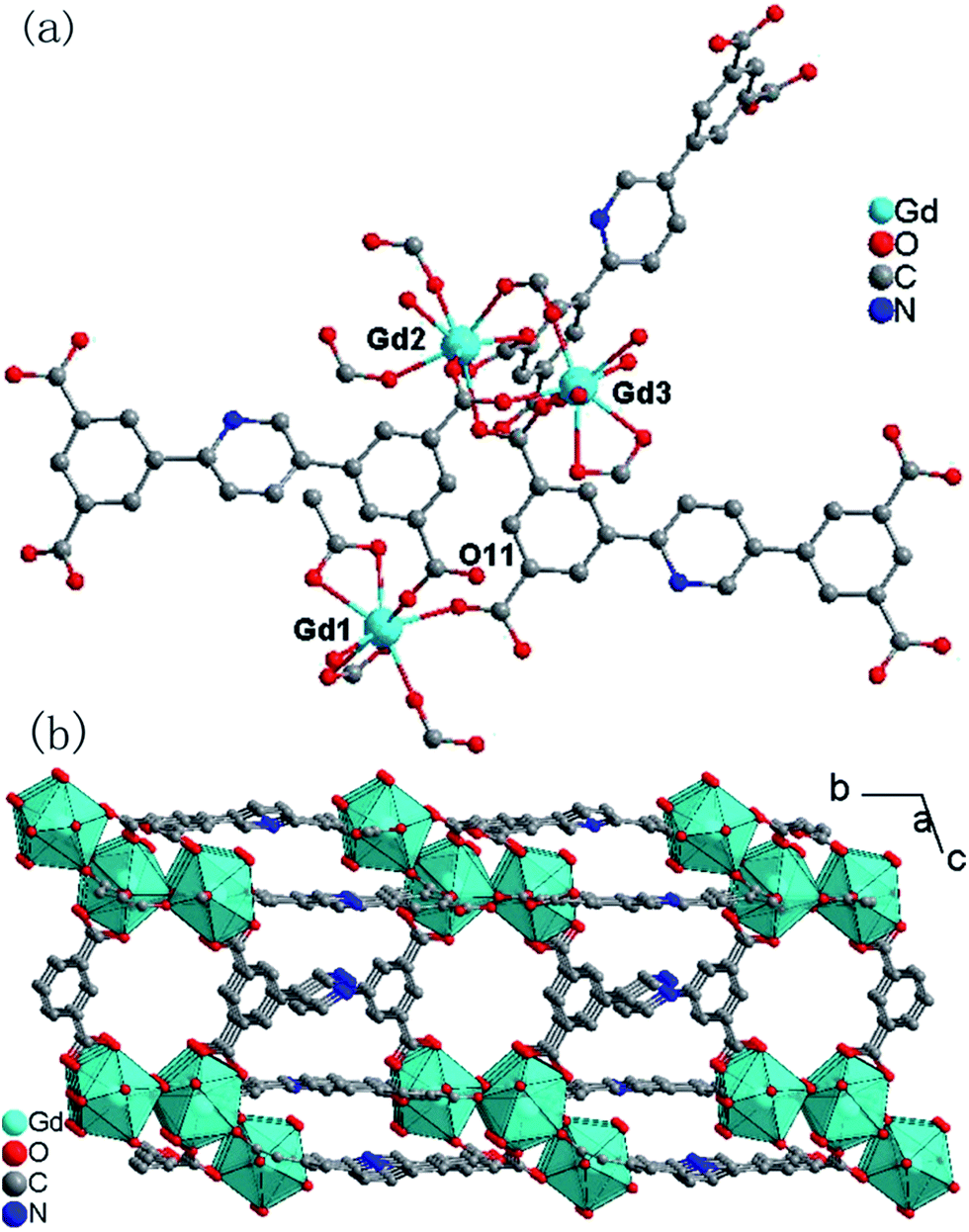
The single crystal X-ray analyses indicated that the structure of 1 (Fig. 1) was isomorphous with our previously reported Eu-MOF.5 The asymmetric unit contains three crystallographically independent eight-coordinated Gd3+ ions, one and a half completely deprotonated L4− anions, one incompletely deprotonated HL3− anion, in which one oxygen atom coordinates with a Gd13+ ion and the other oxygen atom is protonated (O11), five coordinated water molecules and nine lattice water molecules (Fig. 1a). The Gd1, Gd2 and Gd3 ions are all eight-coordinated by eight oxygen atoms. For Gd1, seven oxygen atoms from five different L4− ligands and one from a water molecule; for Gd2, seven from six different L4− ligands and one from a water molecule; for Gd3, five from four different L4− ligands and three from water molecules. The local coordination geometries of all three Gd3+ ions are trigonal dodecahedral (Fig. 1b and S1, ESI†). The Gd–O bond lengths range from 2.268(9) to 2.577(10) Å, which are comparable to those of previously reported Gd–O complexes.20 The two L4− ligands and one HL3− ligand act as μ7, μ4, and μ6 bridges, respectively (Fig. S2, ESI†), in which one carboxylate group adopts a μ1-η1-bridging mode, while the other adopts a μ2-η1:η1-bridging mode for the first L4− ligand (according to the numbering order), all four carboxylate groups adopt μ1-η2-bridging modes for the second L4− ligand, one carboxylate group adopts μ1-η1, two adopt μ2-η1:η1, and one adopts a μ1-η2-bridging mode for the HL3− ligand.
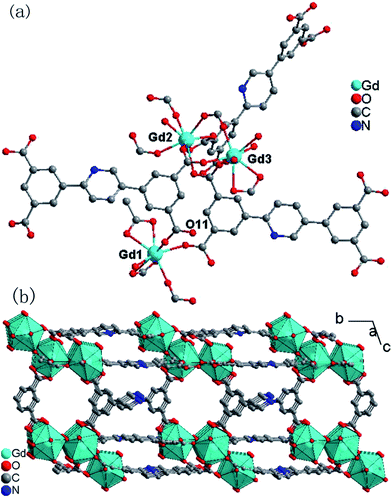 |
| | Fig. 1 (a) Local coordination environments of the Gd3+ centers in 1 (hydrogen atoms are omitted for clarity). (b) The 2D layer along the a direction (the hydrogen atoms are omitted) in 1. | |
The neighbouring Gd3+ ions are bridged into trinuclear Gd3 units by carboxylate groups with Gd⋯Gd distances ranging from 4.6539(1) to 9.2433(1) Å. Each Gd3 unit is interlinked by L4− and HL3− ligands acting as μ7, μ4 and μ6 bridges into 2D layers along the a direction (Fig. 1b). The free water molecules locate between the layers and the solvent-accessible volume of 1 is estimated by the PLATON program21 to be about 17.7% of the total crystal volume. The thermogravimetric analyses (TGA) indicated that the framework of 1 has good stability, as shown in Fig. S3, ESI,† the two-step weight losses before 300 °C correspond to the release of lattice water molecules, coordinated water molecules, and other solvent molecules, and subsequent weight losses correspond to the decomposition of compound 1. The thermal stability was also verified by PXRD of 1 heated at 100 °C, 200 °C, and 300 °C (Fig. S4, ESI†).
The solid-state luminescence spectra of 1
The solid-state luminescence spectra of 1 were investigated upon excitation at 392 nm (25510 cm−1). Compound 1 exhibits a single broad luminescence emission between 410 nm (24390 cm−1) and 560 nm (17857 cm−1) with a maximum peak at 459 nm (21786 cm−1) at room temperature. This luminescence emission behaviour is quite similar to that of the free H4L ligand (Fig. S5, ESI†). This could be attributed to the fact that the lowest excited state of Gd3+, 6P7/2, is too high to accept the energy from the ligand; therefore, its characteristic 4f–4f transition at 311 nm (32154 cm−1) is not visible,20b,22 and the fluorescence of 1 is ligand-centered. The solid-state luminescence emission mechanism of 1 is different from our previously reported Eu-MOF,5 for which the luminescence emission peaks could be attributed to the characteristic transitions of the Eu3+ ions.
Detection of nitroaromatic compounds in the aqueous phase
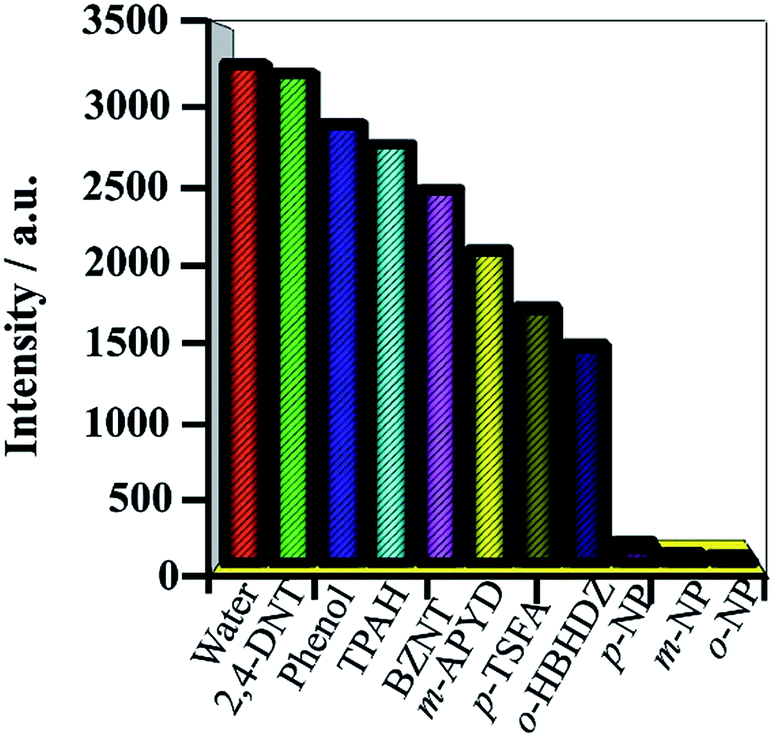
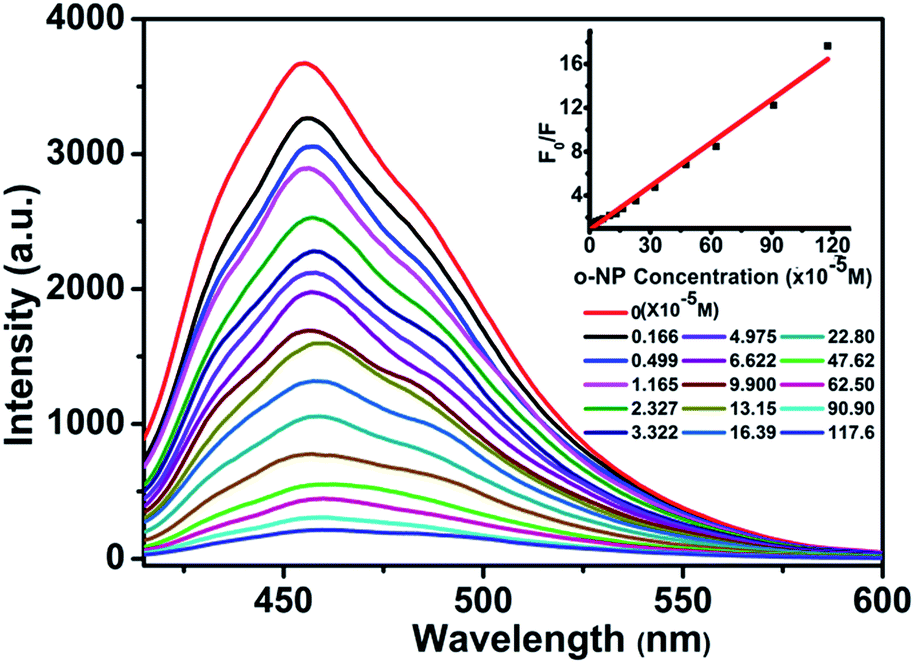
It is noteworthy that the detection of nitroaromatic compounds in the aqueous phase is relatively rare and highly desirable for practical applications.2e,23 Luminescence detection experiments were carried out with a water suspension of 1; 2 mg finely ground sample 1 was dispersed in 0.1 mol L−1 of different selected analyte solutions (2,4-DNT, phenol, TPAH, BZNT, m-APYD, p-TSFA, o-HBHDZ, o-NP, m-NP, and p-NP). These analytes were selected due to their solubilities in water. As shown in Fig. 2, o-, m-, and p-NP could almost completely quench the emission of 1. Such observations indicated that 1 could be used as a fluorescence sensor for o-, m-, and p-NP with high sensitivity and selectivity. Furthermore, the PXRD spectra (Fig. S6, ESI†) of the immersed samples of o-, m-, and p-NP were measured to confirm the stability of 1 in aqueous solutions. The observed patterns for the immersed samples gave the same diffraction peaks as the as-synthesized samples and the simulated patterns from a single crystal structure, which suggests that the basic framework remains during the sensing process. To further examine the sensitivity of sensing towards o-, m-, and p-NP, fluorescence-quenching titrations were performed. A series of suspensions of 1 in water with incremental additions of aqueous o-, m-, and p-NP solutions was prepared to monitor the emissive response (Fig. 3, S7 and S8†). In all the titration experiments, fast and high fluorescence quenching was observed upon incremental addition of o-, m-, and p-NP solution (0.01 M). Good linear relationships were achieved between the relative luminescence intensities and the o-, m-, and p-NP concentrations: for o-NP, R = 0.989, in the range 0–1176 μM; for m-NP, R = 0.986, in the range 0–2000 μM; for p-NP, R = 0.995, in the range 0–1428 μM. The quenching efficiency can be quantitatively analysed using the Stern–Volmer (SV) equation, (F0/F) = Ksv[Q] + 1, where F0 and F are the fluorescence intensities before and after addition of the analyte, respectively, Ksv is the quenching constant (M−1), and [Q] is the concentration of the analyte (M). The quenching constants are 1.14 × 104, 1.39 × 103, and 8.4 × 103 for o-, m-, and p-NP, respectively, which are comparable with a previously reported value.24 We note that the pKa values increase from o-NP (6.4) to p-NP (7.0) to m-NP (8.3) as the quenching constants decrease. This could be attributed to the hydrogen bonding interactions between NP and the organic ligands, where the interactions are supported by the ability of NP to act as an acid. The detection limits were 1.67, 1.67, and 1.67 ppm for o-, m-, and p-NP, respectively, which were nearly the same as the detection limits (1.7, 1.7 and 1.7 ppm) of our previously reported Eu-MOF sensing of o-, m-, and p-NP in a water system.5 Based on the above facts, for 1, the thermal stability, the high sensitivity and selectivity towards o-, m-, and p-NP, especially in aqueous systems, and good linear relationships indicate that compound 1 could be used as a fluorescent sensor for quantitative detection of o-, m-, and p-NP.
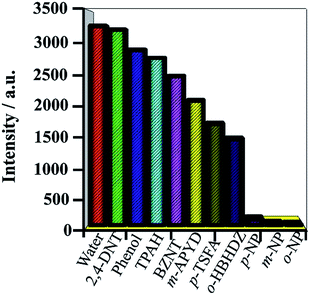 |
| | Fig. 2 Luminescence intensity of 1 dispersed in water upon the addition of different analytes (0.1 M). The emission intensity at 459 nm was selected. | |
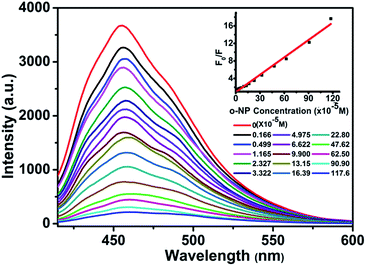 |
| | Fig. 3 Effect on the emission spectra of 1 dispersed in water of the incremental addition of an o-NP aqueous solution (λex = 392 nm). Inset: Stern–Volmer plot of F0/F versus the o-NP concentration. | |
Detection of metal ions in the aqueous phase
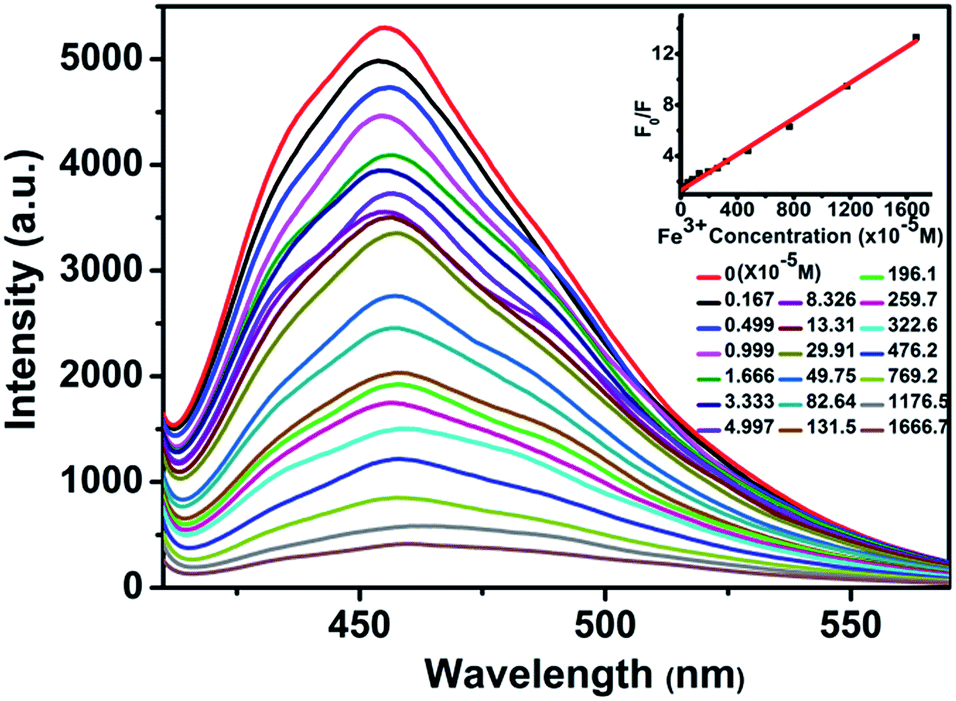
It is very easy to detect Fe3+ ions in aqueous solution with high sensitivity and selectivity, owing to the indispensability of elemental iron in the biosphere and environment.13 The luminescence detection experiments were performed with a water suspension of 1; 2 mg finely ground sample 1 was dispersed in 0.01 mol L−1 aqueous solutions of different selected metal ions (Cd2+, Ca2+, Co2+, Fe2+, Fe3+, Mg2+, Mn2+, Ni2+, and Pb2+; the corresponding anions were all nitrate ions). As shown in Fig. 4, Fe3+ ions could almost completely quench the emission of 1. Such observations indicated that 1 could be used as a fluorescence sensor for Fe3+ ions with high sensitivity and selectivity. Additionally, the PXRD spectra of the immersed samples of Fe3+ ions were measured to confirm the stability of 1 in aqueous solutions. To explore the sensitivity, further fluorescence quenching titrations were performed with the Fe3+ ions (Fig. 5). The fluorescence intensity decreased continuously upon incremental addition of an aqueous solution of Fe3+ ions (0.01 M, 0.1 M) to a standard suspension of 1 in water. As for sensing o-, m-, and p-NP, another good linear relationship was reached between the relative luminescence intensity and the concentration of Fe3+ ions with R = 0.995 in the range 0–16667 μM. The quenching efficiency was analysed by the SV equation and the quenching constant is 7.98 × 102 (M−1). The fluorescence quenching by Fe3+ ions could be detected at a very low concentration, 1.67 ppm, which is comparable to some previously reported fluorescence sensors for Fe3+ ions.16,17c Furthermore, the effect of anions in the case of the Fe3+ system has also been studied using 0.01 M FeCl3·6H2O, Fe2(SO4)3 and Fe(NO3)3·9H2O. As shown in Fig. S9, ESI,† the experiment showed that a sample of 1 immersed in an Fe(NO3)3·9H2O aqueous solution could almost completely quench the emission of 1, while the luminescence intensities of 1 immersed in FeCl3·6H2O and Fe2(SO4)3 aqueous solutions were much higher than in Fe(NO3)3·9H2O aqueous solution. It was postulated that nitrate ions more easily form hydrogen bonding interactions with the terminally coordinated water molecules and the uncoordinated carboxylate oxygen atoms (O11) of the ligands, which can serve as potential coordination sites to bind foreign Fe3+ ions. Therefore, the fluorescence quenching of 1 towards Fe3+ ions may be a synergistic effect of Fe3+ ions and nitrate ions in the aqueous system.
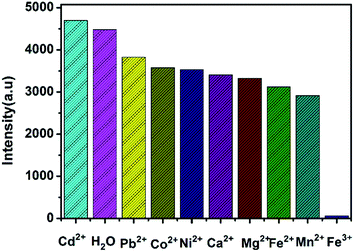 |
| | Fig. 4 Fluorescence intensity for 1 dispersed in aqueous solutions of selected inorganic ions. | |
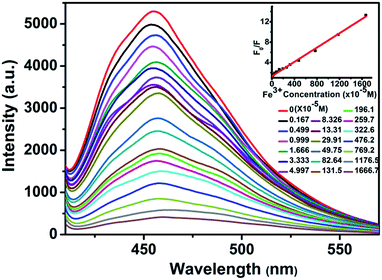 |
| | Fig. 5 Effect on the emission spectra of 1 dispersed in water of incremental addition of an aqueous solution of Fe3+ ions (λex = 392 nm). Inset: Stern–Volmer plot of F0/F versus the Fe3+ ion concentration. | |
The quenching mechanism
The quenching effect on the luminescence of 1 caused by o-, m-, and p-NP possibly originates from five aspects: (1) electron transfer from the conduction-band (CB) of 1 to the lowest unoccupied molecular orbitals (LUMOs) of the analytes (o-, m-, and p-NP);2e,11c,25 (2) the overlap between the excitation spectrum of 1 and the absorption spectrum of the analytes; (3) the collapse of the framework of 1; (4) the overlap between the absorption spectrum of the analytes and emission spectrum of fluorescence of 1; (5) the electronic static interactions between the free Lewis-base sites of pyridine rings in 1 and the hydroxyl groups of the analytes. Based on the above five aspects, further investigation was performed. The UV-vis absorption spectrum of the analytes in the aqueous phase were measured. As shown in Fig. S10, ESI,† o-, m-, and p-NP have no obvious absorption intensity at 392 nm (25510 cm−1) and 459 nm (21786 cm−1), which excludes the possibility of (2) and (4). Moreover, the PXRD patterns of 1, which is immersed in aqueous solutions of o-, m-, and p-NP, remain unchanged compared with the as-synthesized compound, which excludes the possibility of (3). Additionally, considering the microporous framework of the host material, the easily accessible micropores and the free Lewis-base sites of the pyridine rings of the H4L ligands in 1 provide facile diffusion routes and binding sites, which can enhance the host–guest interactions by many orders of magnitude,1d,2e,2f resulting in higher sensitivity and a fluorescence quenching effect. To detect the interactions between 1 and o-, m-, and p-NP, UV-vis spectra of 1 were measured upon addition of increasing amounts of o-, m-, and p-NP, and showed that the UV-vis absorption intensities were enhanced as the analyte concentrations increased (Fig. S11–13, ESI†). Based on our analyses, for 1, the above aspects, (1) and (5) should be the main possible fluorescence quenching mechanisms for 1 towards o-, m-, and p-NP in aqueous media. In order to further elucidate the quenching mechanism of the sensor, the luminescence lifetimes of suspensions of 1 in the absence and presence of o-, m-, and p-NP were measured and the characterization and analyses showed that the lifetimes changed to some extent (Fig. S14–16, ESI†), indicating that the dynamic quenching process was dominant for 1 towards o-, m-, and p-NP in the aqueous system. The dynamic quenching was due to collisional encounters between the π-conjugated moieties of the organic ligands and the analytes (o-, m-, and p-NP). The decay dynamics of the emission band at 459 nm for 1 dispersed in water in the absence and presence of o-, or m-, or p-NP can be fitted by a monoexponential curve, and their lifetimes are, 1.71 μs (for blank), 1.62 μs (for 0.33 μM o-NP), 1.62 μs (for 0.67 μM o-NP), 1.63 μs (for 0.33 μM m-NP), 1.30 μs (for 0.67 μM m-NP), 1.66 μs (for 0.33 μM p-NP), and 1.49 μs (for 0.67 μM p-NP).
The quenching effect of 1 towards Fe3+ ions possibly originates from the following two factors: (1) the interaction between the metal ions and the ligands. As mentioned above, in the structure of 1, there are uncoordinated carboxylate oxygen atoms (O11), which can serve as potential coordination sites to bind foreign metal ions; this was verified by monitoring the increase in the UV-vis absorption intensity with the Fe3+ concentration (Fig. S17, ESI†); (2) the overlap between the excitation spectrum of 1 and the absorption spectrum of the analytes. As shown in Fig. S18,† the UV-vis absorption spectra of different metal ions in the aqueous phase show that there is an observable absorption intensity of Fe3+ ions at 392 nm, while other metal ions do not absorb at this wavelength. There is competition between the absorption of Fe3+ ions and the excitation of 1, resulting in a decrease (even quenching) in luminescence.
Conclusions
In summary, a highly fluorescent microporous MOF [Gd6(L)3(HL)2(H2O)10]·18H2O·x(solvent) (1) has been designed and successfully synthesized with highly π-conjugated ligands containing Lewis basic pyridyl sites under solvothermal conditions. The fluorescence of 1, which is ligand-centered, shows high sensitivity and selectivity towards trace amounts of o-, m-, and p-NP and Fe3+ ions in aqueous media, and the luminescence intensities of the experimental system show good linear dependence on the concentrations of o-, m-, and p-NP and Fe3+ ions. The origin of the highly selective sensing of 1 towards o-, m-, and p-NP in aqueous systems can be attributed to electron transfer from the ligands of 1 to o-, m-, and p-NP and static electronic interactions between the free Lewis-base sites of pyridine rings in 1 and the hydroxyl groups of the analytes. And the mechanism of the highly selective sensing by 1 of Fe3+ ions in aqueous systems may be due to the interaction between the metal ions and the ligands and the overlap between the excitation spectrum of 1 and the absorption spectrum of the Fe3+ ions. Generally, the porosity of the MOF and the nature of the electronic structures of the MOF and the analytes are synergistic with respect to the observed fluorescence quenching behaviour. Generally, 1 can be used as a multi-responsive luminescence sensor for the detection of o-, m-, and p-NP and Fe3+ ions in a real aqueous system.
Acknowledgements
We are thankful for the financial support from NSFC (21301087, 21361016), the Inner Mongolia Autonomous Region Natural Science Fund Project (2013MS0206), and Programs of Higher-level Talents of Inner Mongolia University (SPH-IMU-30105-125135).
Notes and references
-
(a) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS PubMed;
(b) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed;
(c) Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261 RSC;
(d) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC;
(e) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543 RSC;
(f) L. Senesac and T. G. Thundat, Mater. Today, 2008, 11, 28 CrossRef CAS.
-
(a) X. H. Zhou, H. H. Li, H. P. Xiao, L. Li, Q. Zhao, T. Yang, J. L. Zuo and W. Huang, Dalton Trans., 2013, 42, 5718 RSC;
(b) S. A. Boyd, G. Sheng, B. J. Teppen and C. T. Johnston, Environ. Sci. Technol., 2001, 35, 4227 CrossRef CAS PubMed;
(c) P. S. Majumder and S. K. Gupta, Water Res., 2003, 37, 4331 CrossRef CAS PubMed;
(d) Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273–274, 76 CrossRef CAS;
(e) J. H. Qin, B. Ma, X. F. Liu, H. L. Lu, X. Y. Dong, S. Q. Zang and H. W. Hou, J. Mater. Chem. A, 2015, 3, 12690 RSC;
(f) D. Banerjee, Z. Hu and J. Li, Dalton Trans., 2014, 10668 RSC.
-
(a) Ö. Aktaş and F. Çeçen, J. Hazard. Mater., 2010, 177, 956 CrossRef PubMed;
(b) M. B. Cassidy, H. Lee, J. T. Trevors and R. B. Zablotowicz, J. Ind. Microbiol. Biotechnol., 1999, 23, 232 CrossRef CAS PubMed.
-
(a) K. K. Wang, L. Wang, W. Jiang and J. T. Hu, Talanta, 2011, 84, 400 CrossRef CAS PubMed;
(b) H. Y. Chen, L. W. Ruan, X. Jiang and L. G. Qiu, Analyst, 2015, 140, 637 RSC;
(c) D. Patra and A. K. Mishra, Sens. Actuators, B, 2001, 80, 278 CrossRef CAS;
(d) R. H. Yang, K. M. Wang, D. Xiao, K. Luo and X. H. Yang, Analyst, 2000, 125, 877 RSC.
- Y. Q. Wang, Q. H. Tan, H. T. Liu, W. Sun and Z. L. Liu, RSC Adv., 2015, 5, 86614 RSC.
- J. M. Sylvia, J. A. Janni, J. D. Klein and K. M. Spencer, Anal. Chem., 2000, 72, 5834 CrossRef CAS PubMed.
- M. Krausa and K. Schorb, J. Electroanal. Chem., 1999, 461, 10 CrossRef CAS.
- K. Hakansson, R. V. Coorey, R. A. Zubarev, V. L. Talrose and P. Hakansson, J. Mass Spectrom., 2000, 35, 337 CrossRef CAS.
-
(a) S. Shanmugaraju, S. A. Joshi and P. S. Mukherjee, J. Mater. Chem., 2011, 21, 9130 RSC;
(b) J. H. Li, C. E. Kendig and E. E. Nesterov, J. Am. Chem. Soc., 2007, 129, 15911 CrossRef CAS PubMed;
(c) Y. Y. Long, H. B. Chen, Y. Yang, H. M. Wang, Y. F. Yang, N. Li, K. Li, J. Pei and F. Liu, Macromolecules, 2009, 42, 6501 CrossRef CAS;
(d) N. N. Sang, C. X. Zhan and D. P. Cao, J. Mater. Chem. A, 2015, 3, 92 RSC.
-
(a) Y. Jiang, H. Zhao, N. N. Zhu, Y. Q. Lin, P. Yu and L. Q. Mao, Angew. Chem., Int. Ed., 2008, 47, 8601 CrossRef CAS PubMed;
(b) Y. M. Guo, F. P. Cao, X. L. Lei, L. H. Mang, S. J. Cheng and J. T. Song, Nanoscale, 2016, 8, 4852 RSC;
(c) A. Saini, N. Kaur and N. Singh, Sens. Actuators, B, 2016, 228, 221 CrossRef CAS;
(d) G. Z. Yue, S. Su, N. Li, M. B. Shuai, X. C. Lai, D. Astruc and P. X. Zhao, Coord. Chem. Rev., 2016, 311, 75 CrossRef CAS;
(e) X. P. Fan, Q. Y. He, S. G. Sun, H. J. Li, Y. X. Pei and Y. Q. Xu, Chem. Commun., 2016, 52, 1178 RSC.
-
(a) S. N. Zhao, X. Z. Song, M. Zhu, X. Meng, L. L. Wu, J. Feng, S. Y. Song and H. J. Zhang, Chem.–Eur. J., 2015, 21, 9748 CrossRef CAS PubMed;
(b) Q. B. Bo, H. T. Zhang, H. Y. Wang, J. L. Miao and Z. W. Zhang, Chem.–Eur. J., 2014, 20, 3712 CrossRef CAS PubMed;
(c) X. Z. Song, S. Y. Song, S. N. Zhao, Z. M. Hao, M. Zhu, X. Meng, L. L. Wu and H. J. Zhang, Adv. Funct. Mater., 2014, 24, 4034 CrossRef CAS;
(d) Y. Guo, X. Feng, T. Han, S. Wang, Z. Lin, Y. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485 CrossRef CAS PubMed;
(e) X. G. Liu, H. Wang, B. Chen, Y. Zou, Z. G. Gu, Z. Zhao and L. Shen, Chem. Commun., 2015, 51, 1677 RSC.
-
(a) P. Y. Wu, Y. H. Liu, Y. Liu, J. R. Wang, Y. Li, W. Liu and J. Wang, Inorg. Chem., 2015, 54, 11046 CrossRef CAS PubMed;
(b) L. L. Zhang, Z. X. Kang, X. L. Xin and D. F. Sun, CrystEngComm, 2016, 18, 193 RSC;
(c) L. Qin, L. X. Lin, Z. P. Fang, S. P. Yang, G. H. Qiu, J. X. Chen and W. H. Chen, Chem. Commun., 2016, 52, 132 RSC;
(d) L. H. Cao, F. Shi, W. M. Zhang, S. Q. Zang and T. C. W. Mark, Chem.–Eur. J., 2015, 21, 15705 CrossRef CAS PubMed;
(e) D. K. Singha, P. Majee, S. K. Mondal and P. Mahata, Eur. J. Inorg. Chem., 2015, 8, 1390 CrossRef;
(f) D. K. Singha, P. Majee, S. K. Mondal and P. Mahata, RSC Adv., 2015, 5, 102076 RSC;
(g) D. K. Singha, S. Bhattacharya, P. Majee, S. K. Mondal, M. Kumar and P. Mahata, J. Mater. Chem. A, 2014, 2, 20908 RSC.
-
(a) C. X. Yang, H. B. Ren and X. P. Yan, Anal. Chem., 2013, 85, 7441 CrossRef CAS PubMed;
(b) L. N. Xu, W. Mao, J. R. Huang, S. H. Li, K. Huang, M. Li, J. L. Xia and Q. Chen, Sens. Actuators, B, 2016, 230, 54 CrossRef CAS;
(c) W. Sun, J. Z. Wang, G. N. Zhang and Z. L. Liu, RSC Adv., 2014, 4, 55252 RSC.
- J. L. Chen, S. J. Zhuo, Y. Q. Wu, F. Fang, L. Li and C. Q. Zhu, Spectrochim. Acta, Part A, 2006, 63, 438 CrossRef PubMed.
-
(a) H. Xu, H. C. Hu, C. S. Cao and B. Zhao, Inorg. Chem., 2015, 54, 4585 CrossRef CAS PubMed;
(b) C. Q. Zhang, Y. Yan, Q. H. Pan, L. B. Sun, H. M. He, Y. L. Liu, Z. Q. Liang and J. Y. Li, Dalton Trans., 2015, 44, 13340 RSC;
(c) J. C. Jin, L. Y. Pang, G. P. Yang, L. Hou and Y. Y. Wang, Dalton Trans., 2015, 44, 17222 RSC;
(d) Z. H. Xiang, C. Q. Fang, S. H. Leng and D. P. Cao, J. Mater. Chem. A, 2014, 2, 7662 RSC.
-
(a) Y. L. Wu, G. P. Yang, X. Zhou, J. Li, Y. Ning and Y. U. Wang, Dalton Trans., 2015, 44, 10385 RSC;
(b) L. H. Cao, F. Shi, W. M. Zhang, S. Q. Zang and T. C. W. Mak, Chem.–Eur. J., 2015, 21, 15705 CrossRef CAS PubMed;
(c) Y. Zhou, H. H. Chen and B. Yan, J. Mater. Chem. A, 2014, 2, 13691 RSC;
(d) M. Zheng, H. Q. Tan, Z. G. Xie, L. Q. Zhang, X. B. Jing and Z. C. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 1078 CrossRef CAS PubMed.
-
(a) Z. Chen, Y. Sun, L. Zhang, D. Sun, F. Liu, Q. Meng, R. Wang and D. Sun, Chem. Commun., 2013, 49, 11557 RSC;
(b) Q. Tang, S. Liu, Y. Liu, J. Miao, S. Li, L. Zhang, Z. Shi and Z. Zheng, Inorg. Chem., 2013, 52, 2799 CrossRef CAS PubMed;
(c) X. H. Zhou, L. Li, H. H. Li, A. Li, T. Yang and W. Huang, Dalton Trans., 2013, 42, 12403 RSC.
- G. M. Sheldrick, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL Version 5.1, Bruker Analytical X-ray Instruments Inc., Madison, Wisconsin, USA, 1998 Search PubMed.
-
(a) L. Zhai, W. W. Zhang, X. M. Ren and J. L. Zuo, Dalton Trans., 2015, 44, 5746 RSC;
(b) L. L. Shen, L. Yang, Y. Fan, L. Wang and J. N. Xu, CrystEngComm, 2015, 17, 9363 RSC;
(c) S. Z. Zou, Q. P. Li and S. W. Du, RSC Adv., 2015, 5, 34936 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
-
(a) M. D. Allendorf, C. A. Bauer, R. K. Bhaktaa and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC;
(b) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC;
(c) Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed;
(d) H.-B. Zhang, X.-C. Shan, Z.-J. Ma, L.-J. Zhou, M.-J. Zhang, P. Lin, S.-M. Hu, E. Ma, R.-F. Li and S.-W. Du, J. Mater. Chem. C, 2014, 2, 1367 RSC.
-
(a) B. Joarder, A. V. Desai, P. Samanta, S. Mukherjee and S. K. Ghosh, Chem.–Eur. J., 2015, 21, 965 CrossRef CAS PubMed;
(b) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881 CrossRef CAS PubMed;
(c) S. S. Nagarkar, A. V. Desai and S. K. Ghosh, Chem. Commun., 2014, 50, 8915 RSC.
- J. X. Liu, H. Chen, Z. Lin and J. M. Lin, Anal. Chem., 2010, 82, 7380 CrossRef CAS PubMed.
-
(a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153 CrossRef CAS PubMed;
(b) K. S. Asha, K. Bhattacharyya and S. Mandal, J. Mater. Chem. C, 2014, 2, 10073 RSC;
(c) S. J. Toal, J. C. Sanchez, R. E. Dugan and W. C. Trogler, J. Forensic Sci., 2007, 52, 79 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1457726. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra07244e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.