
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The electronic structure of iridium oxide electrodes active in water splitting†
V.
Pfeifer
ab,
T. E.
Jones
*a,
J. J.
Velasco Vélez
ac,
C.
Massué
ac,
M. T.
Greiner
a,
R.
Arrigo
d,
D.
Teschner
a,
F.
Girgsdies
a,
M.
Scherzer
ac,
J.
Allan
a,
M.
Hashagen
a,
G.
Weinberg
a,
S.
Piccinin
e,
M.
Hävecker
ac,
A.
Knop-Gericke
a and
R.
Schlögl
ac
aFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: trjones@fhi-berlin.mpg.de
bHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Elektronenspeicherring BESSY II, Albert-Einstein-Str. 15, 12489 Berlin, Germany
cMax-Planck-Institut für Chemische Energiekonversion, Stiftstr. 34-36, 45470 Mülheim a. d. Ruhr, Germany
dDiamond Light Source Ltd, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX 11 0DE, UK
eInstituto Officina dei Materiali (CNR-IOM), c/o SISSA – Scoula Internazionale Superiore di Studi Avanzati, Via Bonomea 267, 34136 Trieste, Italy
First published on 21st December 2015
Abstract
Iridium oxide based electrodes are among the most promising candidates for electrocatalyzing the oxygen evolution reaction, making it imperative to understand their chemical/electronic structure. However, the complexity of iridium oxide's electronic structure makes it particularly difficult to experimentally determine the chemical state of the active surface species. To achieve an accurate understanding of the electronic structure of iridium oxide surfaces, we have combined synchrotron-based X-ray photoemission and absorption spectroscopies with ab initio calculations. Our investigation reveals a pre-edge feature in the O K-edge of highly catalytically active X-ray amorphous iridium oxides that we have identified as O 2p hole states forming in conjunction with IrIII. These electronic defects in the near-surface region of the anionic and cationic framework are likely critical for the enhanced activity of amorphous iridium oxides relative to their crystalline counterparts.
Water splitting presents an attractive solution to store excess energy from intermittent renewable sources in the form of hydrogen.1 Iridium oxide is the only known anode electrocatalyst that is both active and stable in the sluggish oxygen evolution reaction (OER) in acidic media,2 which has prompted significant research efforts focused on this conducting oxide. Recently, these efforts have revealed that amorphous forms of iridium oxide have higher activities than their rutile-type crystalline counterparts,3,4 which we refer to simply as rutile IrO2. An explanation for this increased activity has, however, proven elusive because of the challenges associated with understanding the combined influence of band structure, electron correlation, and spin–orbit coupling on the electronic structure of iridium oxide.5 For example, even the Ir 4f spectrum of rutile IrO2 has a line shape that cannot be fit using the standard theoretically derived function, making the origin of this unusual shape a matter of debate.6–8 Thus, while X-ray photoemission and absorption spectroscopies (XPS & XAS) have been used to search for OER relevant surface species in iridium and its oxides, both in situ9–13 and ex situ,4,14,15 unambiguous speciation has remained challenging. Herein, we combine XPS measurements and theory to uncover the origin of the peculiar Ir 4f line shape of rutile IrO2. We use this finding along with the Near-Edge X-ray Absorption Fine Structure (NEXAFS) of the O K-edge to identify the species present in a catalytically more active, amorphous, hydrated IrOx.
All experiments were performed on two commercially available iridium oxide powders. Prior to photoemission experiments, the powders were thoroughly characterized. X-ray diffraction (XRD) confirmed the rutile structure and phase purity of the IrO2 reference material whereas the IrOx powder was found to be X-ray amorphous with a minor metallic contribution (Fig. S1, ESI†). Energy-dispersive X-ray spectroscopy (EDX) yielded the expected stoichiometry of the rutile sample (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2/Ir:O). In contrast, the amorphous powder had a significantly higher oxygen concentration (Table S1, ESI†) giving rise to a total water loss of ∼6 wt% in thermogravimetric (TG) analysis (Fig. S2, ESI†). From temperature-programmed reduction (TPR) (Fig. S3, ESI†) of rutile IrO2, a formal Ir oxidation state of 4.1 ± 0.1 is deduced, demonstrating that iridium and oxygen in rutile IrO2 can be viewed as IrIV and OII−, despite the distorted octahedral environment around each Ir atom and the covalent character of the Ir–O interaction.16 In contrast, the formal oxidation state of oxygen-containing iridium species in the amorphous IrOx is only found to be 3.6 ± 0.1, suggesting that multiple iridium species are present in the amorphous IrOx, some of which are in an oxidation state of less than IV. This result is in line with titration experiments done on hydrated IrOx colloid catalysts, for which an average oxidation state of 3.2 was found.17 The presence of mixed Ir-valence states has been argued to be essential for the OER on amorphous IrOx.12,18 In support of this view, activity tests of the two powders confirmed the expected higher OER activity of the amorphous IrOx with respect to rutile IrO2 (Fig. S4, ESI†). The enhanced activity of amorphous iridium oxides is likely due to surface species that have an intrinsically higher OER activity than those of the rutile IrO2.3 To uncover the nature of these species, we turn to XPS and NEXAFS.
:2/Ir:O). In contrast, the amorphous powder had a significantly higher oxygen concentration (Table S1, ESI†) giving rise to a total water loss of ∼6 wt% in thermogravimetric (TG) analysis (Fig. S2, ESI†). From temperature-programmed reduction (TPR) (Fig. S3, ESI†) of rutile IrO2, a formal Ir oxidation state of 4.1 ± 0.1 is deduced, demonstrating that iridium and oxygen in rutile IrO2 can be viewed as IrIV and OII−, despite the distorted octahedral environment around each Ir atom and the covalent character of the Ir–O interaction.16 In contrast, the formal oxidation state of oxygen-containing iridium species in the amorphous IrOx is only found to be 3.6 ± 0.1, suggesting that multiple iridium species are present in the amorphous IrOx, some of which are in an oxidation state of less than IV. This result is in line with titration experiments done on hydrated IrOx colloid catalysts, for which an average oxidation state of 3.2 was found.17 The presence of mixed Ir-valence states has been argued to be essential for the OER on amorphous IrOx.12,18 In support of this view, activity tests of the two powders confirmed the expected higher OER activity of the amorphous IrOx with respect to rutile IrO2 (Fig. S4, ESI†). The enhanced activity of amorphous iridium oxides is likely due to surface species that have an intrinsically higher OER activity than those of the rutile IrO2.3 To uncover the nature of these species, we turn to XPS and NEXAFS.
Fig. 1 shows a superposition of the measured Ir 4f spectra of the rutile and the amorphous powders normalized by area. Photoemission of an Ir 4f7/2 (4f5/2) electron in IrO2 gives rise to the intensity at 61.8 eV (64.8 eV) with the anomalous, highly asymmetric line shape typical of rutile IrO2.19 Though the peak positions in the Ir 4f spectrum of amorphous IrOx are similar to that of rutile IrO2, the line shape is not, as highlighted by the difference spectrum. The amorphous IrOx can be seen to have less intensity where the main peaks of rutile IrO2 are located and more intensity around 62.4 eV and 65.4 eV. This additional intensity is not related to the metallic component seen in XRD, which would appear at 60.9 eV,20 suggesting that the metal detected with XRD is covered by a thick oxide layer and does not contribute to the OER activity. The lack of a metallic contribution in XPS makes it tempting to assign the features at 62.4 eV and 65.4 eV to the low oxidation state of iridium seen in TPR. However, the ambiguity in the line shape of IrO2 makes such an assignment questionable without further evidence.
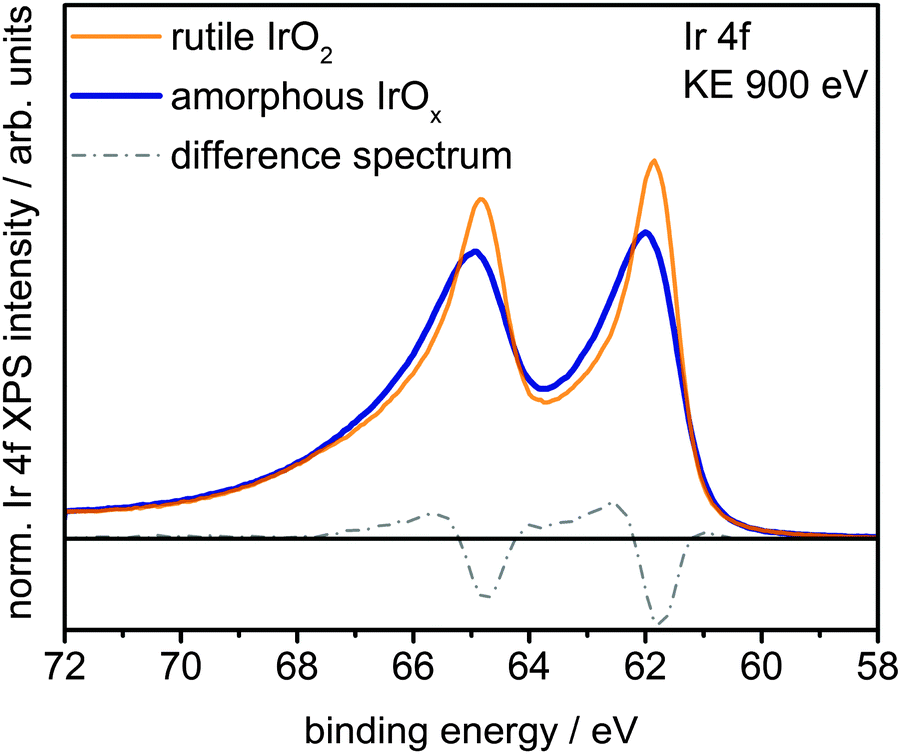 | ||
| Fig. 1 Ir 4f spectra and difference spectrum of rutile IrO2 and amorphous IrOx measured in UHV at a kinetic energy of the photoelectrons of 900 eV. | ||
It is well known from many-body theory that, as a conductor, IrO2 has asymmetric core level spectra.19 However, the peculiar asymmetry of the Ir 4f spectrum cannot be fit with only one component when employing the standard Doniach–Šunjic (DS) line shape.6,19 Although a variety of hypotheses have been put forward to explain this apparent failure,6–8,21,22 it should be noted that the DS function is formally exact only at the edge singularity. The widespread applicability of the DS line shape over a wider energy range is simply a consequence of the weakly structured electron–hole pair excitation spectra common in many metals.23 Clearly such weak structure is not required, nor is it always present. While the true line shape can be computed by perturbation theory,19 such an approach requires detailed knowledge of the electron–hole pair excitation spectra, hence the atomic structure, and offers little chemical insight. As such, we have chosen to employ a one-electron picture wherein the structured electron–hole pair excitation spectra give rise to “shake-up” satellites, simultaneous core ionization and monopole excitation of a valence electron. While approximate, this approach can be used to understand the experimental spectra and develop robust fit models for complex systems where we lack detailed knowledge of the structure.
To investigate the possibility of shake-up satellites in the Ir 4f spectrum of rutile IrO2, we turned to density functional theory (DFT) calculations of the states involved in the shake-up process. The d projected density of states (PDOS(d)) of an Ir atom with an Ir 4f core hole in rutile IrO2 has a strong narrow feature at 1 eV above the Fermi energy (Ef) (see inset in Fig. 2 and Fig. S9, ESI†) to which valence electrons can be excited. Because the ground state is metallic and, assuming constant excitation matrix elements, the probability of valence electron excitation scales with the inverse square of the excitation energy (1/ΔE2), the most intense shake-up satellites originate from occupied states near Ef. Weaker satellites will originate from states with energies below Ef, the most prominent of these will be due to shake-up from localized non-bonding Ir d states at 2 eV binding energy (Fig. S10, ESI†). Thus, the strongest shake-up satellite will appear at 1 eV higher binding energy than the main line in the Ir 4f spectrum and a weaker satellite may be seen at 3 eV above the main line.
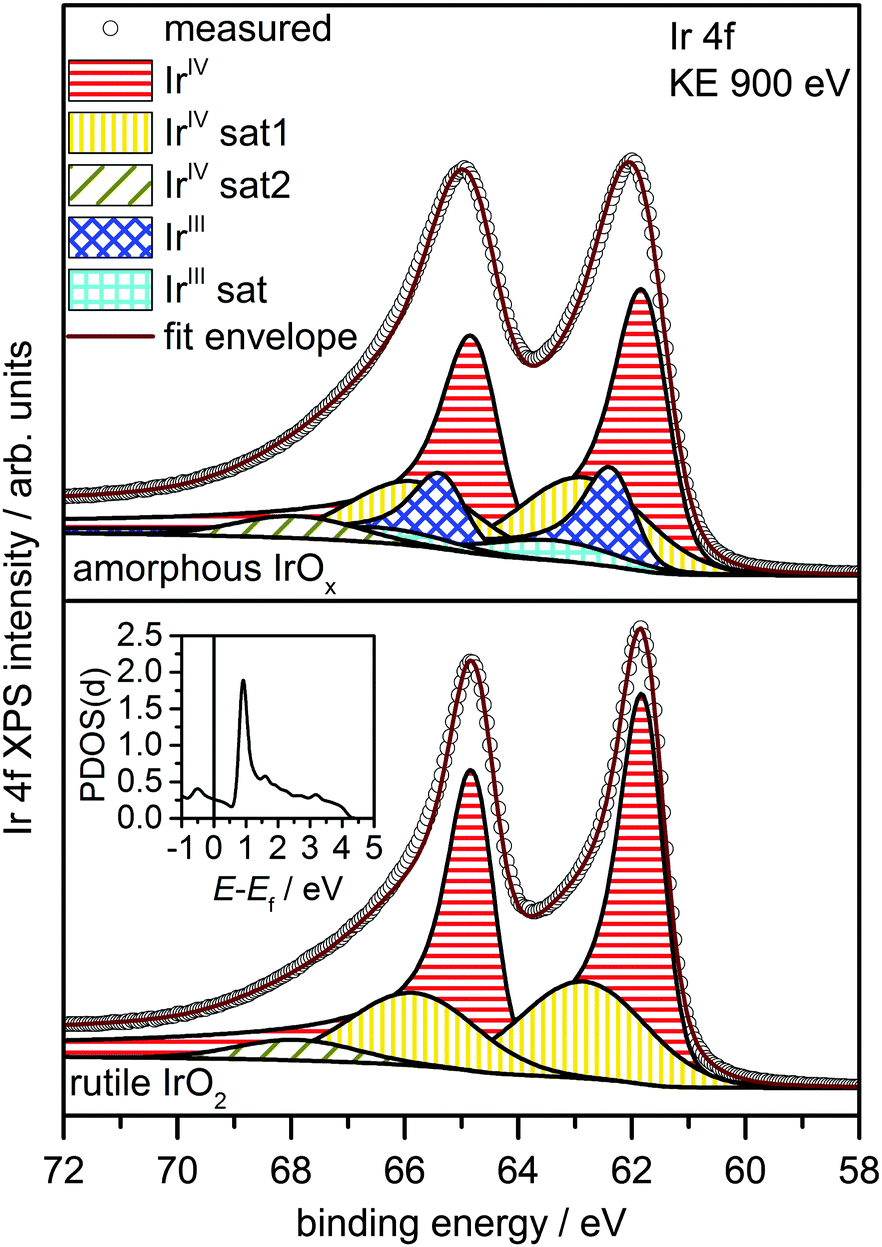 | ||
| Fig. 2 Theory-based fit models for rutile IrO2 (below) and amorphous IrOx (above). The inset shows the unoccupied PDOS(d) of rutile IrO2 in presence of an Ir 4f core hole. The sharp feature ∼1 eV above Ef causes the shake-up satellite to appear at ∼1 eV above the main line. | ||
Using this shake-up picture, the IrIV atoms in the IrO2 rutile structure can be fit as shown in Fig. 2. The fit consists of a DS function with the peak maximum at 61.8 eV and a Gaussian satellite at 1.0 eV higher binding energy. To slightly improve the fit, a small component at 3.1 eV higher binding energy than the Ir 4f5/2 line was included to account for shake-up from the localized non-bonding states. The respective satellite corresponding to the Ir 4f7/2 line was not included in the fit as it is buried by the main Ir 4f5/2 line. It is important to note that, while other authors have also suggested the presence of a 1 eV satellite in the Ir 4f spectrum due to iridium in an oxidation state greater than IV,22 our results show that the main lines and their shake-up satellites all originate from a single type of iridium in the oxidation state IV. All fit parameters can be found in Table S2 (ESI†).
The presence of principally IrIV and OII− in rutile IrO2 is further corroborated by NEXAFS measurements. The strong resonances at 530 eV and 533 eV are in excellent agreement with the computed O K-edge spectrum of rutile IrO2, where all oxygen is formally OII− (Fig. 3). In the O K-edge spectrum of the amorphous IrOx, however, a new resonance appears at ∼529 eV, which is also reflected in the O 1s spectrum (Fig. S5, ESI†). This type of pre-edge feature is commonly observed in other transition metal oxides with strong covalent interactions.24,25 While not an exact comparison, such a pre-edge is perhaps most well-known to emerge when hole doping superconducting cuprates, where it is associated with the formation of O 2p holes.24,25
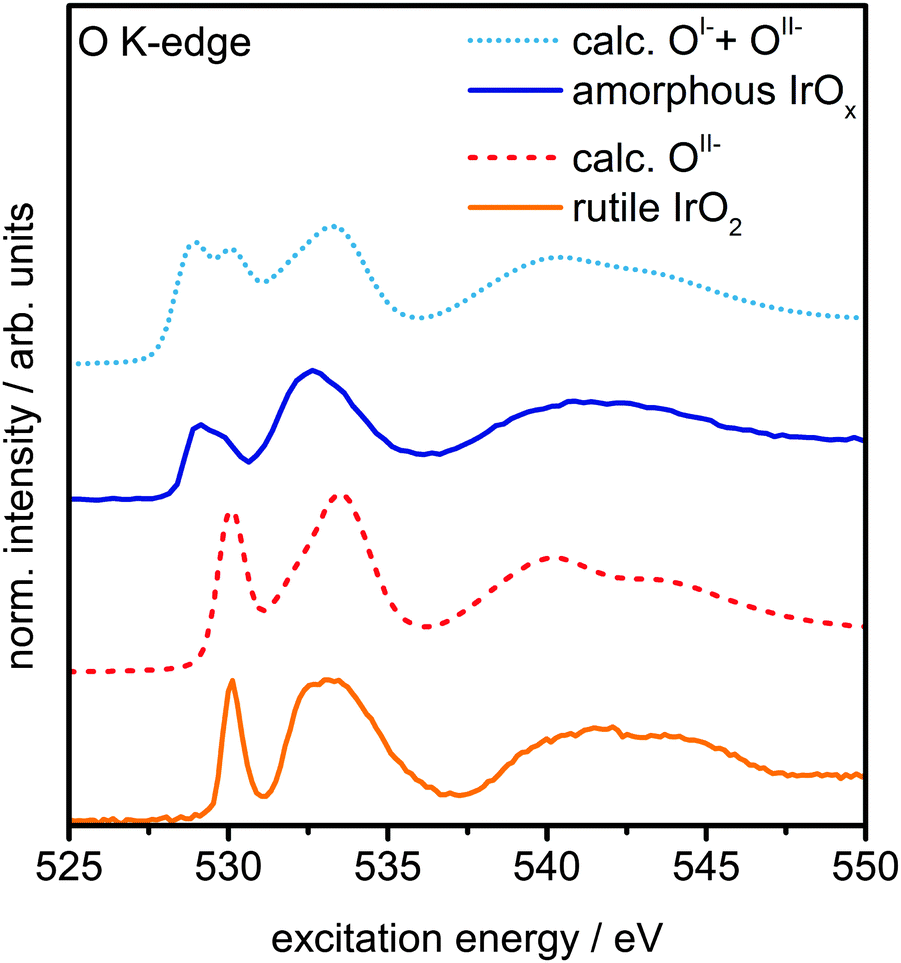 | ||
| Fig. 3 Measured O K-edge spectra of rutile IrO2 and amorphous IrOx and calculated O K-edges of OII− and a linear combination of 60% OII−and 40% OI− species. | ||
We can computationally test if such formally OI− species also lead to a resonance at ∼529 eV in iridium oxide by introducing iridium vacancies to create hole states in IrO2 (Fig. S6 and S8, ESI†). Doing so, we find that the holes localize on oxygen, which, unlike hole localization on iridium (see ESI†), leads to results consistent with experiment. The OI− species give rise to a new resonance at 529 eV in the calculated O K-edge, though they lack the resonance at 530 eV seen in the OII− spectrum (Fig. S7, ESI†).
Though this model structure cannot describe all aspects of the amorphous IrOx, by assuming that the electronic structure of the computed OI− in IrO2 is similar to that of the analogous coordination defect in the amorphous IrOx, we are able to recover the experimentally observed O K-edge spectrum. Under this assumption, we can take the model computed spectra as representatives of OI− and OII−, allowing us to conclude that amorphous IrOx contains both oxygen species within the probing depth of the measurement. This suggestion is supported by the fact that a linear combination of the two types of computed spectra, using 2:3 for the ratio of OI− to OII−, yields good agreement with experiment (Fig. 3). The physisorbed and chemisorbed water content of the IrOx sample, which was ignored in our computed spectra, leads to a smoothing of the measured O K-edge spectrum at excitation energies between 537 eV and 545 eV,26 along with additional intensity at higher binding energy in the O 1s spectrum (see Fig. S5, ESI†).
The electronic defects in the oxygen seen in NEXAFS also lead to the formation of electronic defects in the iridium framework. Six OI− form with each Ir vacancy, leaving two free electrons that reduce neighboring IrIV to IrIII to maintain local charge neutrality (Fig. 4). Our calculations reveal that this formally IrIII species has an Ir 4f7/2 binding energy of ∼62.2 eV, which is in line with binding energy values measured for IrIII in IrCl3.27 As before, assuming that the iridium vacancy modeled in our calculations is representative of coordination defects in the amorphous IrOx, we can use our computed result to conclude that IrIII can account for the increased intensity at higher binding energy in the Ir 4f spectrum (see Fig. 1). As with the IrIV species, analysis of the PDOS suggests that a shake-up satellite will appear at ∼1.5 eV above the Ir 4f main line of the IrIII species (see Fig. S11, ESI†).
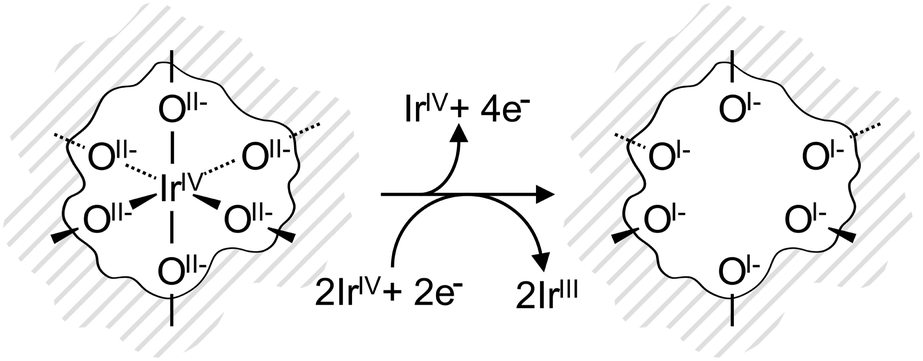 | ||
| Fig. 4 IrO6 octahedron in the near-surface region of the IrOx framework (grey lines). Initially, Ir and O are in the formal oxidation states IV and II−, respectively. Upon the creation of an Ir vacancy, surrounding oxygen atoms are oxidized to the formal oxidation state I−. The two additional electrons are distributed among neighboring IrIV reducing them to IrIII. | ||
Based on these theoretical findings, we obtain the fit model shown in Fig. 2 for the amorphous IrOx. The Ir 4f spectrum can be deconvoluted into a contribution of formally IrIV and IrIII species along with their respective satellites. An increased full width at half maximum of IrIV is used to recover the fact that the local environment around the IrIV atoms in the amorphous structure is expected to be less regular than in the crystalline rutile structure. All fit parameters can be found in Table S3 (ESI†).
In conclusion, we report on three major findings. First, we have presented theoretical calculations that offer a convincing explanation for the unusual Ir 4f line shape of phase-pure rutile IrO2. From these results, we developed a fit model, in which a Doniach–Šunjic line shape is combined with Gaussian satellites to account for electron shake-up from Ef into unoccupied states 1 eV above Ef. In the second part of the manuscript we have shown that caution is required for the speciation of iridium oxides. For though we observe an increase in intensity at higher binding energy in the Ir 4f spectrum of the catalytically more active amorphous iridium oxide powder, we see little indication for the presence of Ir in oxidation state V. Both TPR and our calculations suggest that while two Ir components are present in the amorphous iridium oxide, the lower binding energy feature is identified as formally IrIV, as in rutile IrO2, whereas the higher binding energy species is attributed to formally IrIII, which is associated with the formation of O 2p hole states. Finally, these O 2p hole states give rise to a formally OI− species. As a chemically electrophilic oxygen, OI− is expected to be susceptible to nucleophilic attack during OER, consistent with Fierro et al.'s28 findings that the OER is likely based on the consumption and regeneration of lattice oxygen from the near surface region of the catalyst. Our finding that an electrophilic oxygen can be identified by a resonance at ∼529 eV in the O K-edge of the amorphous IrOx, in addition to the OII− resonance at 530 eV, then allows the study of, not only the active metal centers, but the simultaneous presence of electronic defects in the anionic and cationic framework. These mixed valences of iridium and oxygen likely play a key role in OER catalysis, a finding that might not be restricted to iridium oxides but pertain to OER-active catalysts in general. To aid in our understanding of the intricate roles these species play in reactions, we are investigating their formation and dynamics as a function of applied potential using in situ XPS and NEXAFS.
Experimental and computational methods
Experimental methods
The samples investigated were a rutile IrO2 powder (Sigma Aldrich, 99.9% purity, calcined at 1073 K in 105 Pa O2 for 50 h to ensure phase purity) and an amorphous IrOx powder (AlfaAesar, 99.99% purity, as received). Prior to the photoemission measurements, the powders were characterized by XRD, EDX, TG, BET, and TPR. The OER performance was tested in a rotating disc electrode setup. Photoemission measurements were performed with the Near-Ambient-Pressure XPS setup at the ISISS beamline at BESSYII/HZB (Berlin, Germany). Further details can be found in the ESI.†Computational methods
All Density Functional Theory (DFT) calculations were performed with the Quantum ESPRESSO package29 using the Perdew, Burke, and Ernzerhof (PBE) exchange and correlation potential30 with spin polarization and scalar relativistic corrections. We employed ultrasoft pseudopotentials with a 50 Ry (500 Ry) kinetic energy (charge density) cutoff and a k-point mesh equivalent to (16 × 16 × 16) for the crystallographic unit cell along with Marzari–Vanderbilt cold smearing31 with a smearing parameter of 0.005 Ry. Core level binding energies (BE) were computed using the ΔSCF method to accurately recover initial and final state effects.32 Oxygen K-edge spectra were computed from Fermi's golden rule using the XSpectra package.33,34 Further details are given in the ESI.†Acknowledgements
The authors gratefully acknowledge BESSYII/HZB for beam time allocation under the proposal #14201159.References
- J. P. Barton and D. G. Infield, IEEE Trans. Energy Convers., 2004, 19, 441–448 CrossRef.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. Bernicke, E. Ortel, T. Reier, A. Bergmann, J. F. de Araujo, P. Strasser and R. Kraehnert, ChemSusChem, 2015, 8, 1908–1915 CrossRef CAS PubMed.
- T. Reier, D. Teschner, T. Lunkenbein, A. Bergmann, S. Selve, R. Kraehnert, R. Schlögl and P. Strasser, J. Electrochem. Soc., 2014, 161, F876–F882 CrossRef CAS.
- S. K. Panda, S. Bhowal, A. Delin, O. Eriksson and I. Dasgupta, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 155102 CrossRef.
- J. M. Kahk, C. G. Poll, F. E. Oropeza, J. M. Ablett, D. Geolin, J. P. Rueff, S. Agrestini, Y. Utsumi, K. D. Tsuei, Y. F. Liao, F. Borgatti, G. Panaccione, A. Regoutz, R. G. Egdell, B. J. Morgan, D. O. Scanlon and D. J. Payne, Phys. Rev. Lett., 2014, 112, 117601 CrossRef CAS PubMed.
- M. Hara, K. Asami, K. Hashimoto and T. Masumoto, Electrochim. Acta, 1983, 28, 1073–1081 CrossRef CAS.
- M. Peuckert, Surf. Sci., 1984, 144, 451–464 CrossRef CAS.
- H. G. S. Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169–7172 CrossRef PubMed.
- Y. B. Mo, I. C. Stefan, W. B. Cai, J. Dong, P. Carey and D. A. Scherson, J. Phys. Chem. B, 2002, 106, 3681–3686 CrossRef CAS.
- A. R. Hillman, M. A. Skopek and S. J. Gurman, Phys. Chem. Chem. Phys., 2011, 13, 5252–5263 RSC.
- A. Minguzzi, O. Lugaresi, E. Achilli, C. Locatelli, A. Vertova, P. Ghigna and S. Rondinini, Chem. Sci., 2014, 5, 3591–3597 RSC.
- M. Hüppauff and B. Lengeler, J. Electrochem. Soc., 1993, 140, 598–602 CrossRef.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- R. Kötz, H. J. Lewerenz, P. Brüesch and S. Stucki, J. Electroanal. Chem. Interfacial Electrochem., 1983, 150, 209–216 CrossRef.
- Y. Ping, G. Galli and W. A. Goddard, J. Phys. Chem. C, 2015, 119, 11570–11577 CAS.
- G. S. Nahor, P. Hapiot, P. Neta and A. Harriman, J. Phys. Chem., 1991, 95, 616–621 CrossRef CAS.
- A. Minguzzi, C. Locatelli, O. Lugaresi, E. Achilli, G. Cappelletti, M. Scavini, M. Coduri, P. Masala, B. Sacchi, A. Vertova, P. Ghigna and S. Rondinini, ACS Catal., 2015, 5, 5104–5115 CrossRef CAS.
- G. K. Wertheim and H. J. Guggenheim, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 4680–4683 CrossRef CAS.
- J. F. Moulder and J. Chastain, Handbook of X-ray photoelectron spectroscopy a reference book of standard spectra for identification and interpretation of XPS data, Perkin-Elmer, Eden Prairie, Minn., 1992 Search PubMed.
- J. Augustynski, M. Koudelka, J. Sanchez and B. E. Conway, J. Electroanal. Chem., 1984, 160, 233–248 CrossRef CAS.
- L. Atanasoska, R. Atanasoski and S. Trasatti, Vacuum, 1990, 40, 91–94 CrossRef CAS.
- G. D. Mahan, Many-particle physics, Springer Science & Business Media, 2013 Search PubMed.
- N. Nücker, M. Merz, P. Schweiss, E. Pellegrin, S. Schuppler, T. Wolf, V. Chakarian, J. Freeland, Y. U. Idzerda, M. Klaser, G. Müller-Vogt, G. Er, S. Kikkawa and G. Liu, J. Supercond., 1999, 12, 143–146 CrossRef.
- C. T. Chen, F. Sette, Y. Ma, M. S. Hybertsen, E. B. Stechel, W. M. C. Foulkes, M. Schluter, S. W. Cheong, A. S. Cooper, L. W. Rupp, B. Batlogg, Y. L. Soo, Z. H. Ming, A. Krol and Y. H. Kao, Phys. Rev. Lett., 1991, 66, 104–107 CrossRef CAS PubMed.
- H. Bluhm, D. F. Ogletree, C. S. Fadley, Z. Hussain and M. Salmeron, J. Phys.: Condens. Matter, 2002, 14, L227 CrossRef CAS.
- B. Folkesson, Acta Chem. Scand., 1973, 27, 287–302 CrossRef CAS.
- S. Fierro, T. Nagel, H. Baltruschat and C. Comninellis, Electrochem. Commun., 2007, 9, 1969–1974 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. d. Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- N. Marzari, D. Vanderbilt, A. De Vita and M. C. Payne, Phys. Rev. Lett., 1999, 82, 3296–3299 CrossRef CAS.
- E. Pehlke and M. Scheffler, Phys. Rev. Lett., 1993, 71, 2338–2341 CrossRef CAS PubMed.
- C. Gougoussis, M. Calandra, A. P. Seitsonen and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 075102 CrossRef.
- M. Taillefumier, D. Cabaret, A. M. Flank and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 195107 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Further details on the experiments, measurement results and the DFT calculations are presented. See DOI: 10.1039/c5cp06997a |
| This journal is © the Owner Societies 2016 |