
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a gas phase source for perfluoroalkyl acids to examine atmospheric sampling methods
John J.
MacInnis
a,
Trevor C.
VandenBoer
b and
Cora J.
Young
*a
aDepartment of Chemistry, Memorial University, St. John's, NL, Canada. E-mail: cora.young@mun.ca; Tel: +709-684-7280
bDepartment of Earth Science, Memorial University, St. John's, NL, Canada
First published on 11th April 2016
Abstract
An inability to produce environmentally relevant gaseous mixing ratios of perfluoroalkyl acids (PFAAs), ubiquitous global contaminants, limits the analytical reliability of atmospheric chemists to make accurate gas and particulate measurements that are demonstrably free of interferences due to sampling artefacts. A gas phase source for PFAAs based on the acid displacement mechanism using perfluoropropionate (PFPrA), perfluorobutanoate (PFBA), perfluorohexanoate (PFHxA), and perfluorooctanoate (PFOA) has been constructed. The displacement efficiency of gas phase perfluorocarboxylic acids (PFCAs) is inversely related to chain length. Decreasing displacement efficiencies for PFPrA, PFBA, PFHxA, and PFOA were 90% ± 20%, 40% ± 10%, 40% ± 10%, 9% ± 4%, respectively. Generating detectable amounts of gas phase perfluorosulfonic acids (PFSAs) was not possible. It is likely that lower vapour pressure and much higher acidity play a role in this lack of emission. PFCA emission rates were not elevated by increasing relative humidity (25%–75%), nor flow rate of carrier gas from 33–111 sccm. Overall, reproducible gaseous production of PFCAs was within the error of the production of hydrochloric acid (HCl) as a displacing acid (±20%) and was accomplished using a dry nitrogen flow of 33 ± 2 sccm. A reproducible mass emission rate of 0.97 ± 0.10 ng min−1 (n = 8) was observed for PFBA. This is equivalent to an atmospheric mixing ratio of 12 ppmv, which is easily diluted to environmentally relevant mixing ratios of PFBA. Conversely, generating gas phase perfluorononanoic acid (PFNA) by sublimating the solid acid under the same conditions produced a mass emission rate of 2800 ng min−1, which is equivalent to a mixing ratio of 18 ppthv and over a million times higher than suspected atmospheric levels. Thus, for analytical certification of atmospheric sampling methods, generating gas phase standards for PFCAs is best accomplished using acid displacement under dry conditions. This yields quickly stabilized, reproducible emissions and mixing ratios that are easily diluted to environmentally relevant levels. Gas phase PFBA from this source has also been shown to be quantitatively collected using an annular denuder coated with sodium carbonate (Na2CO3) according to Environmental Protection Agency (EPA) method Compendium I.O-4.2. Overall, producing gas phase PFAAs at constant atmospherically-relevant levels will enable the development of standard approaches in certifying gas and particle collection efficiencies for instruments interrogating the gas-particle partitioning and long-range transport of PFCAs in the atmosphere.
Introduction
Perfluoroalkyl acids (PFAAs) are persistent and bioaccumulative compounds found ubiquitously in abiotic and biotic matrices.1–8 Toxicological studies have demonstrated PFAA exposure in animals can cause endocrine disruption, physical developmental delays, and cancer.9–12 PFAAs are present in the atmosphere from a few13 to few hundred14–16 pg m−3, distributed among gas and particle measurements. It is expected PFAA exposures at environmentally relevant concentrations can negatively impact human health.10 PFAAs can be transported to remote locations, such as the Arctic, through a long-range transport mechanism in the atmosphere.3,8,17 Gas phase PFAAs can form in the atmosphere by oxidation of volatile precursors, such as fluorotelomer alcohols.18–20 PFAAs are volatile in the neutral form, but due to low pKa values,21 they are expected to be ionized under most environmental conditions. PFAAs have low volatility in the anionic state, so it is likely the condensed phase, including atmospheric particles, plays a role in long-range transport.It has been an analytical challenge to quantify mixing ratios of PFAAs in atmospheric samples due to an absence of gas standards across the relevant environmental range. Therefore, it is not currently possible to validate sampling methods for their measurement accuracy and precision. It has also been reported that current sampling methods suffer from artefact biases.14,22,23 A popular approach to collect aerosols involves pulling a large volume of air through a filter to collect particles, while collecting gases downstream using a sorbent. This approach has two major drawbacks: (i) gas phase PFAAs irreversibly sorb to some filters;22 and (ii) gas phase species present in the atmosphere, like hydrochloric acid (HCl), could react with perfluorocarboxylic acids (PFCAs) salts on particles releasing them to the gas phase, similar to other known chemistry.24 Improvements have been made over recent years to mitigate positive biases by deactivating the surfaces of filters, which has been found to minimize gas phase sorption.22 However, gas phase artefacts are not limited to sorption on filter material. The reaction of gas phase PFAAs with sampled particulates likely leads to sampling artefacts despite the presence of a deactivated filter. To prevent this, gases should be collected prior to particles, as is standard practice for measuring atmospheric acids such as HCl, nitric acid (HNO3), and sulphur dioxide (SO2).25,26 Annular denuders can provide such an option and have been demonstrated to effectively collect gas phase species during atmospheric sampling.25 This is accomplished by applying a coating to the concentric ring surfaces within the denuder, which is selective for the target compounds. One test of denuders for collection of PFAAs upstream of particle collection has been made. The sorbent used was XAD-4, which was shown to be unsuitable for collecting gas phase PFAAs, with recoveries from 50–72% for C8–C12 PFCAs.14 An examination of sampling artefacts was also performed by comparing particle-associated fractions of filters collected using a high-volume air sampler to those collected downstream from a denuder coated with XAD-4 resin.14 Since neither method has been fully validated for the collection of gas or particle-phase PFAAs, method intercomparison provides limited information. A full understanding of atmospheric mixing ratios of PFAAs, as well as their gas-particle partitioning can only be achieved if sampling artefacts are measured quantitatively and minimized.
Acid displacement has been used extensively to produce atmospherically relevant mixing ratios of standard gas phase nitrous acid (HONO).27–30 The most robust undertaking is described by Febo et al.,30 using controlled amounts of HCl for displacement of sodium nitrite (NaNO2) (1):
| HCl(g) + NaNO2(s) → NaCl(s) + HONO(g) | (1) |
The objective of this work was to develop a constant-output and controllable gas phase source for PFAAs at atmospherically relevant mixing ratios using a similar approach (2):
| HX(g) + MPFAA(s) → MX(s) + PFAAH(g) | (2) |
Materials and methods
Chemicals
Sodium salts of perfluoropropionic acid (PFPrA), perfluorobutanoic acid (PFBA), perfluorohexanoic acid (PFHxA), perfluorooctanoic acid (PFOA); potassium salts of perfluorobutane sulfonic acid (PFBS), and perfluorooctane sulfonic acid (PFOS); and solid perfluorononanoic acid (PFNA) were obtained from SynQuest Laboratories (Alachua, FL, USA). The purities of PFAA salts and acid were greater than or equal to 98%. A standard mixture of (C4 to C14) perfluorocarboxylic acids (PFC-MXA) and PFOS (L-PFOS) were obtained from Wellington Laboratories (Guelph, ON). Gravimetrically certified HCl and hydrobromic acid (HBr) permeation devices were purchased from Vici-Valco Instruments (Brockville, ON). High purity (Omnisolv) methanol and water were purchased from VWR (Oakville, ON) and used as the mobile phases for liquid chromatography analysis. A.C.S grade ammonium acetate (98.2%) was obtained from Fischer Scientific (Fair Lawn, NJ, USA) and was used in mobile phase buffers. Experiments used deionized water generated by a Barnstead Infinity™ Ultrapure Water System (Thermo Scientific; Waltham, MA, USA; 18.2 MΩ cm−1).Liquid chromatography-mass spectrometry analysis
Sample analysis was carried out using an Agilent 6230 liquid chromatography-electrospray ionization-time-of-flight mass spectrometer (LC-ESI-ToF). The analysis of PFAAs used ESI operated in negative mode at 325 °C, with a nebulizer gas flow of 11 L min−1, and a nebulizer pressure of 30 psi. The capillary voltage was held at 1500 V. A sample volume of 10 μL was injected onto a Kinetex C18 core–shell column (3.9 × 150 mm, 5 μm). The mobile phase was delivered at a flow of 200 μL min−1. Water and methanol with 100 μM ammonium acetate (pH 7) were used in combination as mobile phases for the elution of PFAAs. The solvent gradient for aqueous PFPrA extracts started with 90% water, held for 0.5 min, linearly decreased to 40% over 3.0 min, held for 1.5 min, then returned to initial conditions to re-equilibrate for 2 min. The remaining PFAAs were analysed using a solvent gradient starting with 90% water, linearly decreasing to 5% over 3 min, held for the next 6 min, then returned to initial conditions and equilibrated for 3 min.Quantitation of PFAAs was carried out through extracted ion chromatograms of molecular ion peaks. Additional characteristic peaks were used to confirm the identity of PFAAs (Table 1). Instrument limits of detection (LOD) and quantitation (LOQ) were defined as concentrations having signal-to-noise ratios of 3 and 10, respectively. Signal-to-noise ratios across the calibration range were determined by dividing peak heights by the standard deviation of the blank signal at the compound-specific retention time windows (Table 1). All calibration curves had coefficients of determination greater than 0.995.
| Analyte | pKa | Quantitative ion (m/z) | Confirmation ion (m/z) | LOD (ng mL−1) | LOQ (ng mL−1) |
|---|---|---|---|---|---|
| Perfluoropropionate (PFPrA) | 0.531 | 163 | 119 | 20 | 50 |
| Perfluorobutanoate (PFBA) | 0.432 | 213 | 169 | 7 | 20 |
| Perfluorohexanoate (PFHxA) | 0.932 | 313 | 269 | 3 | 10 |
| Perfluorooctanoate (PFOA) | 0.532 | 413 | 369 | 2 | 6 |
| Perfluorobutane sulfonate (PFBS) | −3.932 | 299 | 219 | 1 | 5 |
| Perfluorooctane sulfonate (PFOS) | −3.332 | 499 | 419 | 0.8 | 2 |
Ion chromatography analysis
PFPrA, chloride, and bromide samples were analysed by ion chromatography (IC) using an ICS-2100 (Thermo Scientific). An eluent gradient increasing from 1 to 60 mM potassium hydroxide was used to separate the anions at a flow rate of 1.5 mL min−1 using an AG19 guard column (4 mm × 50 mm) and AS19 analytical column (4 mm × 250 mm) over 32 minutes. The eluent was suppressed with an AERS 500 before the analytes were detected by conductivity.Acid permeation device certification
Acid emission rates of the HCl and HBr permeation devices were determined using IC since certified emission rates were generated by mass balance, not quantitative analysis of emitted material. The permeation devices were housed in an oven system constructed in-house (Fig. 1, with H removed). Permeation devices were held at a constant temperature inside tubing inserted into a thermostated aluminium block kept at 40.0 ± 0.1 °C. A dry nitrogen flow of 33 ± 2 standard cubic centimeters per minute (sccm) was produced using a critical orifice assembly (Lennox Laser; Glen Arm, MD, USA) with 12 psi head pressure provided from a compressed cylinder (Praxair Canada Inc.; Mississauga, ON; high purity). This critical orifice is a gasket with a 1 μm pinhole, and is housed in a ½′′ Swagelok VCR fitting held between two stainless steel glands. The nitrogen was then passed over the permeation device. Flow measurements (n = 4) over the 20 experiments were used to confirm system flow measured at the critical orifice was leak-free by measuring flow at the exit of the glass impinger using a DryCal Definer 220 (Mesa Labs, Lakewood, CO). These measurements are assumed to be representative of the flow conditions during all experiments. The acid emitted from each device was collected in an impinger containing 25 mL of deionized water. Multiple acid samples (n = 13) were collected with varying sampling durations. HCl and HBr mass and mole emission rates at 40.0 °C were determined by IC analysis of chloride and bromide using the method described above. The IC-validated permeation rates of two HCl permeation devices were 0.40 ± 0.04 ng min−1 and 1.0 ± 0.2 ng min−1, respectively (n = 5, each). The IC-validated permeation rate of the HBr permeation device was 1.0 ± 0.2 ng min−1 (n = 3). These were consistently lower than the gravimetrically certified rates. The HCl permeation rates were 16 and 7 times lower than those provided by the manufacturer, and the permeation rate of HBr was 14 times lower. The errors of IC-validated permeation rates correspond to one standard deviation of replicate acid collections over different durations (2–6 days). The uncertainty is a combination of variable permeation acid emissions and IC measurement accuracy, with the former accounting for the majority of the reported variability and the latter having an accuracy of 5% or better for the concentrations measured. Molar displacement efficiencies for PFAAs were calculated using the IC-validated permeation rates and not those of the manufacturer.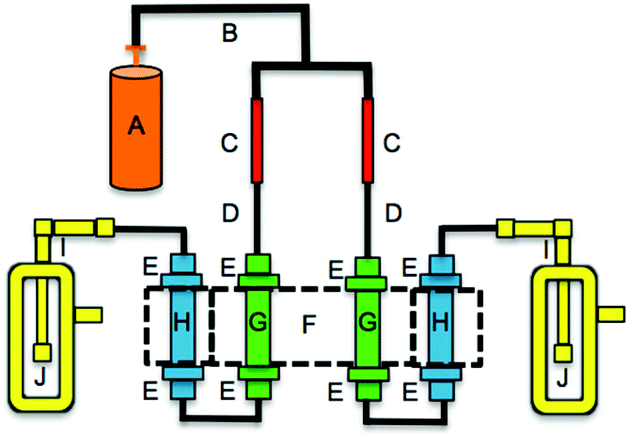 | ||
| Fig. 1 Schematic of the PFAA gas phase source for dry reactions. A: compressed nitrogen cylinder; B: ¼′′ polyethylene tubing; C: Swagelok® VCR assembly; D: ⅛′′ PFA tubing; E: ⅛–½′′ PTFE union; F: aluminium heating block; G: ½′′ PFA tubing housing acid permeation devices; H: ½′′ PFA tubing housing PFAA salt beds; I: ⅛–¼′′ PTFE elbow; J: glass impinger. PFAA salt beds can be placed inside and outside the aluminium heating block. The critical orifice (1 μm pinhole) is housed in C, held between two stainless steel glands. | ||
Permeation oven parameter optimization for PFAA emission
A schematic of the PFAA gas phase source is illustrated in Fig. 1. The HCl or HBr permeation device was thermostated at 40.0 ± 0.1 °C to maintain constant emission rates. The entrained acid was passed through a PFAA salt bed, which could also be thermostated in additional channels in the aluminium block. The PFAA salt bed was prepared by adding 0.5 g of PFAA salt into a pre-cut polytetrafluoroethylene (PTFE) tube (0.5′′ O.D. × 6′′), with the salt interspersed with small pieces of 0.25′′ and 0.125′′ O.D. PTFE tubing to ensure ease of gas flow. Two salt beds can be exposed to acid from two permeation sources simultaneously with a four channel heating block. PFAA salt beds were maintained at the same temperature as the permeation devices or room temperature. A perfluorononanoic acid (PFNA) bed was prepared in the same way as the salt beds described above. The salt or acid bed was placed downstream from the HCl or HBr permeation device, and displaced gas phase PFAA was bubbled through a glass impinger, collecting it into 25 mL of deionized water. Aqueous impinger samples were transferred into clean 30 mL polypropylene bottles (see QA/QC). Impinger samples were stored at room temperature until analysis. All tubing and fittings downstream of the acid permeation device were made of perfluoroalkoxy (PFA) and PTFE materials. Displacement efficiencies for each PFAA were determined by the relative ratio in the known HCl or HBr molar emission rates and the number of moles of PFAA collected in the impinger.Carrier gas flow effects on the PFAA displacement efficiencies were assessed by increasing flows to 41, 62, and 111 sccm. RH effects on displacement efficiencies were explored at 25, 50, and 75%. The RH and flow experiments were carried out interchangeably. To minimize carryover effects the system was flushed overnight to equilibrate with the starting conditions of the next experiment when switching from dry to wet conditions, or vice versa.
High flow rates and RH were explored by modifying the permeation oven setup as shown in Fig. 2, with three increasing flows being synonymous to the three increasing RH levels to decouple the RH effect from flow. Briefly, a PTFE tee was introduced upstream of the salt bed to add humidified air to the dry nitrogen flow using a mass flow controller (MFC) (0.01–0.5 L min−1, MKS GE50A, Andover, MA) to regulate the flow of nitrogen through an impinger of deionized water. The humidified flow mixed with the dry nitrogen and passed through the salt bed (dashed blue line, Fig. 2). A humidity probe from Vaisala (HMP 110, Helsinki, Finland) measured RH. For high flow experiments (i.e. flow >40 sccm) the flow from the MFC bypassed the humidifying glass impinger (red dashed line, Fig. 2).
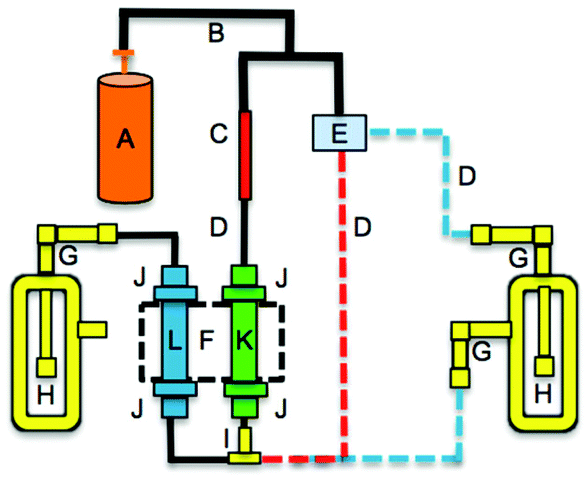 | ||
| Fig. 2 Schematic of one channel of the PFAA gas phase source with variable RH and flow reactions. A: compressed nitrogen cylinder; B: ¼′′ polyethylene tubing; C: Swagelok® VCR assembly; D: ⅛′′ PFA tubing; E; mass flow controller; F: aluminium heating block; G: ⅛–¼′′ PTFE elbow; H: glass impinger; I: ⅛′′ PTFE tee; J: ⅛–½′′ PTFE union; K: ½′′ PFA tubing housing an acid permeation device; L: ½′′ PFA tubing housing a PFAA salt bed. The critical orifice (1 μm pinhole) is housed in C, held between two stainless steel glands. | ||
Annular denuder PFAA collection efficiency
Sodium carbonate (Na2CO3) annular denuder coatings have been used to selectively scrub acidic gases such as HNO3, HONO, HCl, and SO2 effectively with quantitative removal efficiencies ranging from 97.7–99.5%.26 To evaluate the effectiveness of a Na2CO3 coating for gas phase PFAA collection, an annular denuder (URG-2000-30 × 150-3CSS) was coated with a mixed solution of glycerol and ethanol (Commercial Alcohols Inc., Brampton, ON). A volume of 10 mL of 0.1 M solution of Na2CO3 in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ethanol and water with 1% (w/v) glycerol was used. The solution was applied evenly to the denuder surface by rotation and inversion, followed by removing the excess solution and drying the coating under a stream of dry nitrogen. The annular denuder was placed between the permeation oven and the impinger (Fig. 2). A flow of 111 sccm from the permeation oven of PFBA was directed through the annular denuder for 67 hours and the outflow was collected into an impinger. The denuder was extracted with 10 mL of deionized water and the amount of PFBA in this extract and the impinger solution were quantified using the LC method described above. The protocol for coating the denuder was based on the atmospheric acid sampling method of the US-EPA (Compendium I.O-4.2.).
:50 ethanol and water with 1% (w/v) glycerol was used. The solution was applied evenly to the denuder surface by rotation and inversion, followed by removing the excess solution and drying the coating under a stream of dry nitrogen. The annular denuder was placed between the permeation oven and the impinger (Fig. 2). A flow of 111 sccm from the permeation oven of PFBA was directed through the annular denuder for 67 hours and the outflow was collected into an impinger. The denuder was extracted with 10 mL of deionized water and the amount of PFBA in this extract and the impinger solution were quantified using the LC method described above. The protocol for coating the denuder was based on the atmospheric acid sampling method of the US-EPA (Compendium I.O-4.2.).
QA/QC
Polypropylene bottles used for storing samples were washed three times with deionized water prior to sample transfer. After experimental samples were collected, the impinger was cleaned by rinsing all surfaces exposed to analyte(s) eight times with deionized water. Analytical, sampling, and method blanks were collected to identify and minimize any systematic error. The analytical blank was an aliquot of deionized water used for sampling and method blanks. The sampling blank consisted of adding 25 mL of deionized water to a glass impinger using a clean 25 mL polypropylene volumetric flask. The method blank consisted of passing HCl from the permeation oven over a bed of cut PFTE tubing that did not contain PFAA salt. The water-soluble components of the gas flow were collected downstream in the glass impinger. Target analytes were not observed above the limit of detection in any of the blanks.Quality control experiments were conducted to assess: (i) sorption losses of PFAAs to the impinger; (ii) losses via volatilization from deionized water; and (iii) efficacy of gas phase collection.
It is possible that PFAAs in the anionic state can adsorb to glass surfaces.33 To assess losses to the glass impinger during sample collection, known amounts of PFPrA, PFBA, PFHxA, PFOA, PFBS, and PFOS were spiked into 25 mL of deionized water contained in the glass impinger. The concentrations (200, 100, 50 ng mL−1) of spiked solutions corresponded to PFAA concentrations observed in typical experimental samples collected over 1–5 days. Two sets of conditions were tested using the spiked solutions added to the glass impingers: (i) PFAA solutions were immediately collected into a pre-cleaned polypropylene bottle; (ii) PFAA solutions were allowed to interact with the glass walls and all 25 mL was collected at times corresponding to experimental sampling durations (1–5 days). The impinger samples at each time were transferred into a pre-cleaned polypropylene bottle and analysed using LC-ESI-ToF to quantify losses.
To assess PFAA losses due to volatilization (direct or via aerosol formation) from the deionized water in the impinger, known amounts of PFBA were spiked into 25 mL of deionized water in a polypropylene volumetric flask. The spiked solutions were transferred into the glass impinger. Nitrogen was then bubbled through the impinger solution at 33 ± 2 sccm. The entire volume of the PFBA solution was removed from the impinger at times corresponding to the typical experimental sampling durations (1–5 days). To determine if losses were due to volatilization, an additional experiment with a second impinger in tandem was added to the first using PFBA quantified by LC-ESI-ToF.
A collection efficacy experiment using the tandem impinger design was also conducted. Gas phase PFBA was generated using acid displacement and collected using the tandem impingers. Evidence of inefficient PFBA gas phase collection would be indicated if PFBA was measured in the second impinger.
Results and discussion
QA/QC
An assessment of negative biases during sample collection was determined. PFAAs were measured using LC-ESI-MS methods described above. Results from QA/QC experiments are summarized in Table 2.The average recoveries (n = 3) for collected PFAA solutions were: 82%, 77%, 77%, 84%, 105%, 85% for PFPrA, PFBA, PFHxA, PFOA, PFBS, and PFOS, respectively. For PFAA solutions exposed to bubbling through the impinger (n = 3; 1–5 days) average recoveries were: 90%, 79%, 80%, 90%, 101%, 89% for PFPrA, PFBA, PFHxA, PFOA, PFBS, and PFOS, respectively. This indicates that losses of PFAAs to the glass surfaces of the impinger are at most 23%. The extent of PFAA loss from deionized water due to volatilization or particle export was equal to or less than sorption losses (90%, n = 3). Negligible losses of PFAAs by volatilization from deionized water were further supported by the collection efficacy experiments. An average recovery of 77% for PFBA (n = 3), in the first impinger was observed. No detectable concentrations of PFBA were found in the second impinger in series. This recovery is consistent with the 77% recovery due to impinger sorption losses. It can be concluded that losses of PFBA, and by proxy the other PFAAs, as a result of volatilization from deionized water are not significant.
Collecting gas phase PFBA was also used to ensure mass emission measurements were quantitative using the tandem impinger setup (n = 1). After a collection period of 65 hours, a concentration of 156 ng mL−1 was observed in the first impinger, which is equivalent to a mass emission rate of 0.84 ng min−1. This emission rate is in good agreement with the other PFBA emission measurements in this work (see Table 4). No detectable amounts of PFBA were found in the second impinger in series. The scrubbing efficacy of other PFAAs is expected to be more complete than PFBA as a result of decreasing air–water partitioning coefficient values with increasing molecular weight.32 It can be concluded that collecting gas phase PFBA is quantitative using the single impinger approach, and can be reliably extended to other PFAAs with similar physicochemical properties. Data was not corrected for recoveries presented in Table 2.
Acid displacement of PFCA salts
In order to evaluate the efficiency of the gas phase system, a mole balance of emission rates was used (3):| MT (PFAA) = E·MD (HX) | (3) |
All PFCA salts tested formed their corresponding acids through acid displacement (4):
| HX(g) + NaPFCA(s) → NaX(s) + PFCAH(g) | (4) |
To evaluate the relative efficacy of generating different gas phase PFCAs using acid displacement, a suite of PFCA congeners were examined (Fig. 3).
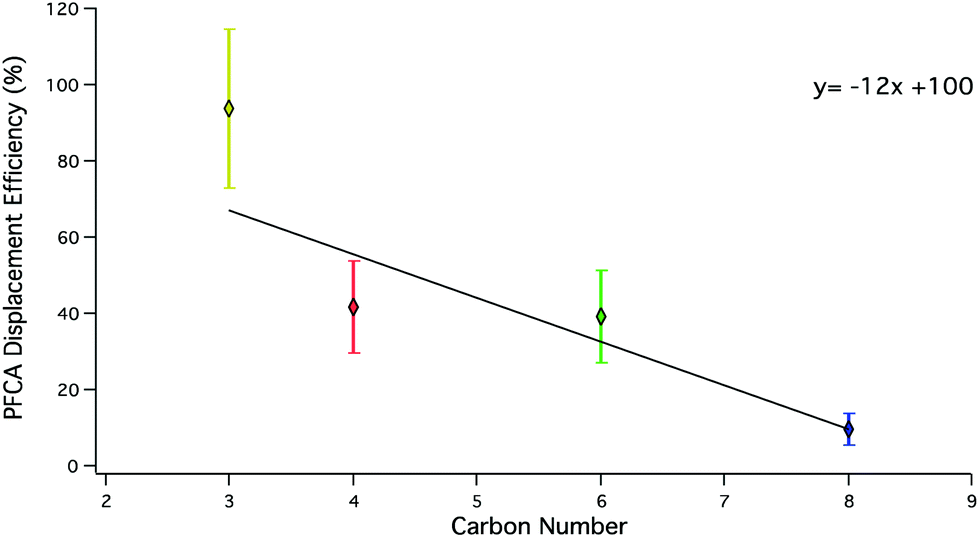 | ||
| Fig. 3 Mean displacement efficiencies of PFCA congeners under dry conditions. Error bars represent one standard deviation of congener measurements from each experiment. PFPrA (yellow; n = 5), PFBA (red; n = 8), PFHxA (green; n = 8), PFOA (blue; n = 3). The solid black line corresponds to a standard deviation error-weighted linear regression. | ||
The displacement efficiency of PFPrA was the highest in the series of PFAAs with an average of 90 ± 20%, followed by PFBA (40 ± 10%), PFHxA (40 ± 10%), and PFOA (9 ± 4%). PFBA (n = 2), PFHxA (n = 1), and PFOA (n = 3) samples were generated using a HBr permeation device. The remaining samples were generated using HCl permeation devices (Table 3). To validate LC-ESI-ToF quantification for PFPrA, samples were also analysed by IC. At the 95% confidence level (t-test, p < 0.05), the displacement efficiency determined by either analysis was not statistically different. A weighted linear regression using the standard deviation of displacement efficiencies from multiple displacement setups shows for every additional CF2 unit, the displacement efficiency decreases by 12% (R2 = 0.61) for this range of PFCA congeners.
| Congener | Displacing acid | Temperature (°C) | Displacing acid emission rate (ng min−1) | PFAA mass emission rate (ng min−1) | PFAA molar emission rate (mol min−1) | Displacement efficiency (%) | Average (%) | Standard deviation (%) |
|---|---|---|---|---|---|---|---|---|
| PFPrA | HCl | 40 | 0.40 | 1.9 | 8.8 × 10−12 | 100 | 90 | 20 |
| HCl | 40 | 0.40 | 2.2 | 1.0 × 10−11 | 120 | |||
| HCl | 40 | 0.40 | 1.6 | 7.3 × 10−12 | 86 | |||
| HCl | 40 | 0.40 | 1.6 | 7.4 × 10−12 | 87 | |||
| HCl | 40 | 0.40 | 1.2 | 5.8 × 10−12 | 69 | |||
| PFBA | HBr | 40 | 1.0 | 1.0 | 4.8 × 10−12 | 43 | 40 | 10 |
| HBr | 40 | 1.0 | 0.9 | 4.0 × 10−12 | 37 | |||
| HCl | 25 | 0.40 | 0.8 | 3.6 × 10−12 | 33 | |||
| HCl | 25 | 0.40 | 0.7 | 3.5 × 10−12 | 32 | |||
| HCl | 25 | 0.40 | 0.7 | 3.1 × 10−12 | 28 | |||
| HCl | 25 | 0.40 | 1.0 | 4.9 × 10−12 | 45 | |||
| HCl | 25 | 0.40 | 1.6 | 7.3 × 10−12 | 67 | |||
| HCl | 25 | 0.40 | 1.1 | 5.1 × 10−12 | 46 | |||
| PFHxA | HCl | 40 | 1.0 | 4.4 | 2.0 × 10−11 | 56 | 40 | 10 |
| HCl | 40 | 0.40 | 1.2 | 5.8 × 10−12 | 36 | |||
| HCl | 25 | 0.40 | 2.0 | 9.2 × 10−12 | 57 | |||
| HCl | 25 | 0.40 | 1.6 | 7.4 × 10−12 | 46 | |||
| HCl | 25 | 0.40 | 0.9 | 4.2 × 10−12 | 26 | |||
| HCl | 25 | 0.40 | 1.1 | 5.2 × 10−12 | 32 | |||
| HBr | 25 | 1.0 | 1.0 | 4.9 × 10−12 | 26 | |||
| PFOA | HBr | 25 | 1.0 | 0.7 | 3.4 × 10−12 | 14 | 9 | 4 |
| HBr | 25 | 1.0 | 0.3 | 1.3 × 10−12 | 5 | |||
| HBr | 25 | 1.0 | 0.5 | 2.2 × 10−12 | 9 | |||
| PFBS | HBr | 25 | 1.0 | <6.2 × 10−2 | <2.1 × 10−13 | <0.97 | 0.6 | 0.5 |
| HBr | 25 | 1.0 | <2.2 × 10−2 | <7.5 × 10−14 | <0.36 | |||
| HBr | 25 | 1.0 | <9.4 × 10−2 | <3.1 × 10−13 | <1.5 | |||
| HCl | 25 | 0.40 | <9.2 × 10−2 | <3.9 × 10−14 | <0.22 | |||
| HCl | 25 | 0.40 | <1.7 × 10−2 | <5.6 × 10−14 | <0.31 | |||
| HCl | 25 | 0.40 | <3.7 × 10−3 | <1.2 × 10−14 | <0.067 | |||
| PFOS | HBr | 25 | 1.0 | <7.0 × 10−3 | <1.4 × 10−14 | <0.11 | ||
Partitioning of a compound from the condensed to gaseous phase in this type of permeation and reaction system is likely influenced by the acidity and volatility of the displacing acid and targeted salt compounds. However, it is not always clear which property takes precedence in acid displacement. For example, it has been demonstrated that less-acidic HNO3 (pKa ∼ −5), can displace more-acidic HCl (pKa ∼ −7) from sodium chloride salts on atmospheric particles originating from sea spray.24,26 The aqueous acidities of the aforementioned acids do not appear to influence the outcome of acid displacement on atmospheric particles with low water content. Considering these acids have vapour pressures within a factor of two,34 the effectiveness of acid displacement is likely a complex function of their relative atmospheric mixing ratios and aerosol water content.35
The observed trends in acid displacement from this work are consistent with the acidity and volatility of PFCAs. The pKa of this suite of PFCAs has been reported21 as approximately 0.5, while the pKas of HCl and HBr are −7 and −9, respectively. Simply put, the difference in acidity would favour the transfer of a proton from the strong acids to the PFCAs. However, as described above, it is unlikely that acid strength alone explains the order of observed decreasing PFCA displacement efficiencies because the difference in pKa values between PFCA congeners is small.36 The relationship between PFCA displacement efficiencies and their predicted vapour pressures37 (Fig. 4) shows an inverse relationship (R2 = 0.68) to that observed for chain length. A weighted linear regression shows the displacement efficiency increases by 24% for every order of magnitude increase in vapour pressure (Pa) for this range of PFCA congeners. Estimations of displacement efficiencies of other PFCA congeners may be possible using these relationships.
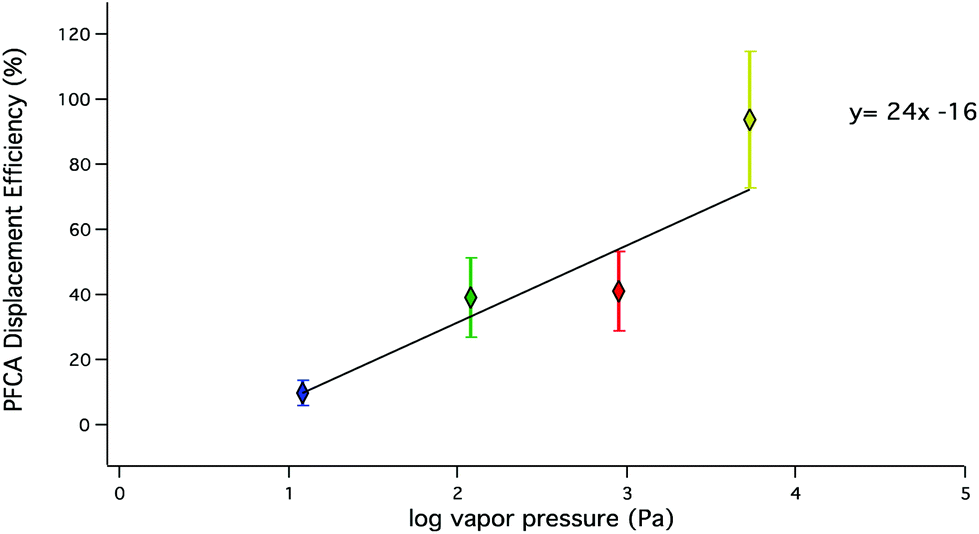 | ||
| Fig. 4 PFCA displacement efficiencies under dry conditions as a function of vapour pressure. PFPrA (yellow; n = 5) PFBA (red; n = 8), PFHxA (green; n = 8), PFOA (blue; n = 3). Predicted vapour pressures (C4–C8) are from Bhhatarai et al.37 and PFPrA from Sigma Aldrich.38 | ||
This suggests the efficiency of gas phase PFCA generation using acid displacement is driven by the volatility of the PFCA congeners in addition to the favourable transfer of a proton from either of the acids to the PFCA.
Acid displacement of PFSA salts
Acid displacement was attempted with PFSA salts. Potassium salts of PFBS and PFOS were tested under the same experimental conditions as the PFCAs. Gaseous HCl and HBr were used to modify acidity in the displacement of PFSAs. Attempts to quantify PFBS were performed using HCl (n = 3) and HBr permeation devices (n = 3) as acid sources. PFOS was tested using only the HBr permeation device (n = 1). Neither HCl nor HBr was able to displace detectable amounts of gaseous PFSAs from the potassium salts of PFBS or PFOS (Table 3). Difference in volatility is an unlikely explanation. We expect that the volatility of PFCAs and PFSAs would be similar because it is known for that the replacement of the carboxylic acid functional group on acetic acid with a sulfonic acid decreases vapour pressure only by approximately an order of magnitude.39,40 It has been demonstrated above that volatility contributes to PFCA displacement efficiency, so PFSA displacement efficiencies should encompass a similar range, albeit at lower values, if it were the driving property of successful acid displacement. Given the inability in this work to generate gas phase PFSAs, this suggests the greater acidity of the PFSAs may place a limitation on their ability to undergo acid displacement. The pKas of HCl and HBr are −7 and −9, respectively; while the pKas of these PFSAs have been calculated to lie in the range of −3 and −12.41,42 A similar lack of reaction between the weak acid HONO (pKa ∼ 3.16)43 and solid sodium sulphate (H2SO4, pKa2 ∼ 1.92) has been demonstrated, where the Gibb's energy the acid displacement reaction of this system at non-equilibrium conditions was calculated as 103 kJ mol−1, which demonstrates the non-spontaneity of the reaction.44 A Gibb's energy cannot be calculated for the PFSA system due to the lack of available physical data for the salts. It is likely that lower vapour pressure and stronger acidity both contribute to the lack of emission observed for PFSAs, with acidity more suspect in reducing displacement efficiency.It is also possible that PFSAs were generated, but not detectable. Thus, upper limits for emission rates of PFSAs at the instrument LOD were determined to be 34 pg min−1 for PFBS, and 7 pg min−1 for PFOS. These emission rate upper limits are about 10 and 100 times smaller than the measured PFOA emission rates for PFBS and PFOS, respectively.
Emission stability of PFBA
To assess the emission stability of the PFAA gas phase source, multiple PFBA samples were collected over 1–5 days using different amounts of HCl and HBr under dry conditions (Fig. 5). Measured source stability is critical to accurately assessing collection biases for atmospheric sampling of PFAAs.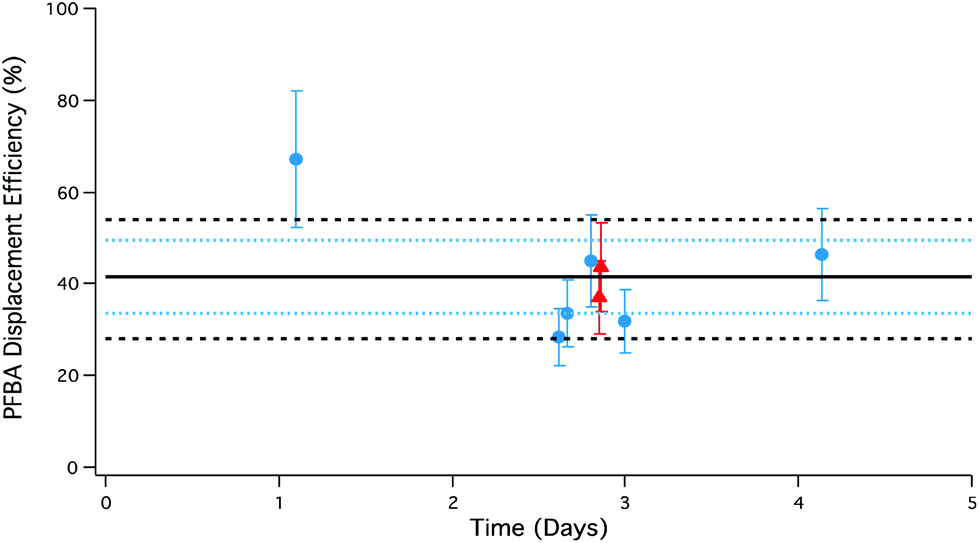 | ||
| Fig. 5 Temporal stability of PFBA displacement efficiencies using HCl (blue circles) and HBr (red triangles) permeation devices. Error bars represent the analytical accuracy of the LC-ESI-ToF for each measurement. The solid black line is the average displacement efficiency; blue dotted lines correspond to the measured 20% error in the HCl and HBr emission rates; black dashed lines correspond to the combined uncertainty in the mean of the PFBA measurements arising from: (i) active HCl and HBr emission rates; (ii) LC-ESI-ToF analysis; (iii) permeation oven temperature control; and (iv) critical orifice flow. | ||
Most PFBA measurements in Fig. 5 fall within the measured variability of the HCl and HBr acid emission rates from the permeation devices. This suggests displacement is limited by the transport of the displacing acid to the PFAA salt bed, and the probability of reaction between the gas and salt.
An additional consideration in controlling emission stability was modification of the PFAA salt bed temperature. The PFBA salt bed was operated at room temperature (n = 6; ∼25 °C), and thermostated at 40 °C (n = 2). The thermostated samples correspond to the HBr-generated PFBA represented as red triangles in Fig. 5. Based on the consistency of PFBA displacement efficiencies at different salt bed temperatures, the results suggest there is no temperature dependence between 25–40 °C, but there does appear to be a reduction in variability under the more stable temperature regime. The use of two separate thermostated aluminium blocks could therefore add further versatility to this system in controlling reagent and target analyte production rates.
Overall, the largest driver of variability in generating gas phase PFBA is the emission rate of the displacing acid from the permeation devices, ±20% for both HCl and HBr, thereby limiting the level of precision in the output of the PFBA source.
Comparison to direct volatilization of solid PFNA
This work has demonstrated that gas phase PFCAs can be consistently generated using acid displacement. It has been previously shown that gas-phase PFCAs can be obtained through sublimation of the solid acid.45 An output rate of solid PFNA (PFNA−H+) to the gas phase under identical conditions was determined. This was done in order to illustrate the importance of using salts for generating gas phase PFAAs that can be easily diluted to environmentally relevant amounts for bias characterization in atmospheric sampling. A PFNA concentration of 1.46 μg mL−1 was observed in the impinger used to collect the sublimed solid acid, which is equivalent to an atmospheric mixing ratio of 18 ppthv. This high concentration produced visible foam due to the surfactant nature of PFNA. This experiment was carried out at room temperature (25 °C ± 5 °C), which means to generate environmentally relevant mixing ratios of PFAAs13,46–48 at the parts-per-quadrillion (ppqv) level by reaction or sublimation of the solid acid, the flow would have to be diluted or the acid bed cooled to reduce the vapour pressure of PFNA. Given that a cooling system is inherently more complex than a heating system, it would be more difficult to generate consistent gas phase mixing ratios of PFAAs from the solid acid.To the knowledge of the authors, it is not possible to purchase salts of PFNA; therefore, a comparison of the emission rates from subliming the solid acid to the sodium salt cannot be made. However, the emission rate for a sodium salt of PFNA can be estimated according to the weighted linear regression from multiple displacement setups for PFCA congeners (Fig. 3). The expected emission rate of PFNA from a sodium salt was calculated to be 0.10 ng min−1 (E = 2%) and is approximately 104 lower than emission rates observed from the solid acid. This likely suggests that detectable quantities of PFNA and longer chain acids may be limited when using acid sources with mass emissions smaller than 0.40 ng min−1, and may not be possible at all for larger congeners if the assumed linear trend is valid.
Overall, using a salt is simple and provides emission rates that are easily diluted to atmospherically relevant levels, which is imperative in generating gas phase PFAA standards to test analytical instrumentation under representative sampling conditions.
Flow rate and RH effects
In a similarly-designed system, the displacement efficiency of a HONO gas phase source was reported to be contingent on chemical and physical conditions such as flow rate of the carrier gas and RH, which influences the equilibrium between the reacting salt bed and the carrier gas.30 Increasing flows in a gas phase HONO system minimized subsequent self-reactive decomposition of the target analyte by decreasing the mixing ratio, resulting in a higher purity output.To elucidate flow rate and RH dependencies of PFAA gaseous emissions, experiments were carried out using the PFBA sodium salt. PFBA was chosen as a proxy for all PFCAs because it is the most volatile PFCA that can be reliably quantified by LC-ESI-MS. Any observable flow or RH dependencies with PFBA is expected to be consistent across the larger PFCA congeners. All RH and flow experiments were carried out using the same HCl permeation device (0.40 ng min−1) and are summarized in Table 4.
| Experiment | Total production time (days) | Mass emission rate (ng min−1) | Flow-normalized emission (ng cm−3) |
|---|---|---|---|
| Flow (sccm) | |||
| 33 | 2.7 | 0.8 | 0.02 |
| 2.7 | 0.9 | 0.03 | |
| 4.1 | 1.1 | 0.05 | |
| 5.3 | 0.7 | 0.02 | |
| 6.0 | 1.0 | 0.03 | |
| 7.0 | 1.1 | 0.05 | |
| 8.0 | 1.6 | 0.02 | |
| 8.3 | 0.7 | 0.03 | |
| 41 | 3.0 | 1.0 | 0.02 |
| 5.9 | 1.3 | 0.03 | |
| 8.9 | 2.1 | 0.05 | |
| 62 | 3.9 | 0.7 | 0.01 |
| 4.9 | 1.3 | 0.02 | |
| 8.0 | 1.0 | 0.02 | |
| 111 | 1.9 | 1.1 | 0.01 |
| 7.9 | 1.2 | 0.01 | |
| RH (%) | |||
| 0 | 2.7 | 0.8 | 0.02 |
| 2.7 | 0.9 | 0.03 | |
| 4.1 | 1.1 | 0.03 | |
| 5.3 | 0.7 | 0.02 | |
| 6.0 | 1.0 | 0.03 | |
| 7.0 | 1.1 | 0.03 | |
| 8.0 | 1.6 | 0.05 | |
| 8.3 | 0.7 | 0.02 | |
| 25 | 3.9 | 0.1 | 0.003 |
| 5.9 | 0.3 | 0.006 | |
| 7.9 | 0.8 | 0.02 | |
| 50 | 3.9 | 0.1 | 0.002 |
| 7.9 | 0.2 | 0.003 | |
| 9.9 | 0.7 | 0.01 | |
| 75 | 3.0 | 1.0 | 0.009 |
| 11 | 1.3 | 0.01 | |
| 16 | 1.4 | 0.01 | |
For non-reactive compounds such as PFAAs, increasing flow rates should not increase the rate of mass transfer of the PFAAs generated at the solid surface to the gas phase, so long as the number of collisions between the HCl and salt are not reduced. No enhancement in PFBA emissions was observed with increasing flow relative to emissions under dry conditions, as seen in Table 4. Further evidence of this is shown in Fig. 6, where the mass emission rate (1.10 ± 0.08 ng min−1, n = 16) of PFBA was found to independent of flow and the total production time of the system.
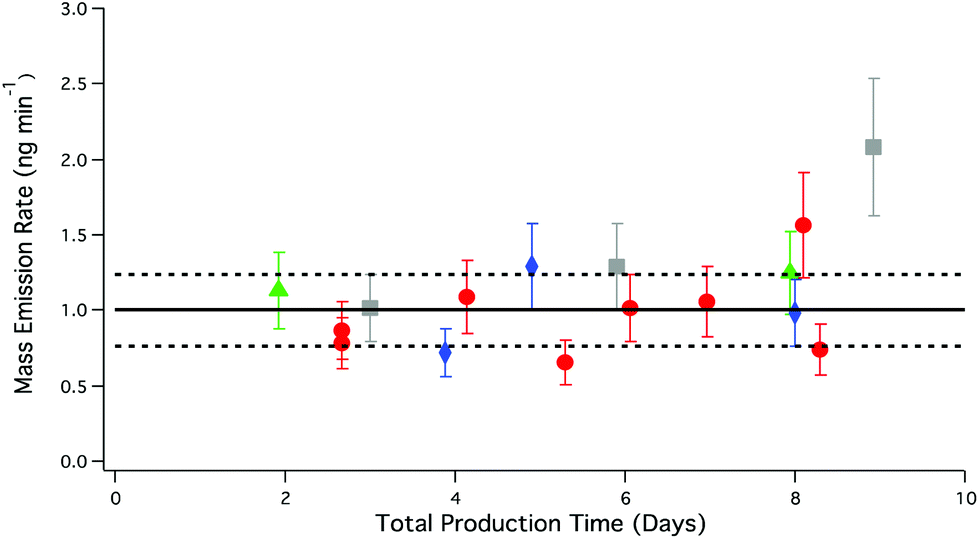 | ||
| Fig. 6 PFBA mass emission rate of different carrier gas flow rates through the salt bed as a function of total PFBA production time. Red circles, 33 sccm; grey squares 41 sccm; blue diamonds, 62 sccm; green triangles, 111 sccm. Error bars represent the analytical accuracy of the LC-ESI-ToF for each measurement. The solid black line represents the average mass emission rate of PFBA under dry conditions; dashed black lines correspond to the total uncertainty as described in Fig. 5. | ||
Flow-normalized emissions of RH experiments presented in Table 4 demonstrate decreases in PFBA emissions relative to experiments carried out in the absence of RH. The introduction of water into the salt bed at 25 and 50% RH resulted in a reduction of PFBA mass emission rate by 82% (0.18 ± 0.09 ng min−1, n = 4) for up to a week. The average mass emission rate at 0% RH was used as reference for determining the reduction in mass emission rate under humid conditions. Reduction in PFBA output was not observed for experiments conducted at 75% RH (Fig. 7).
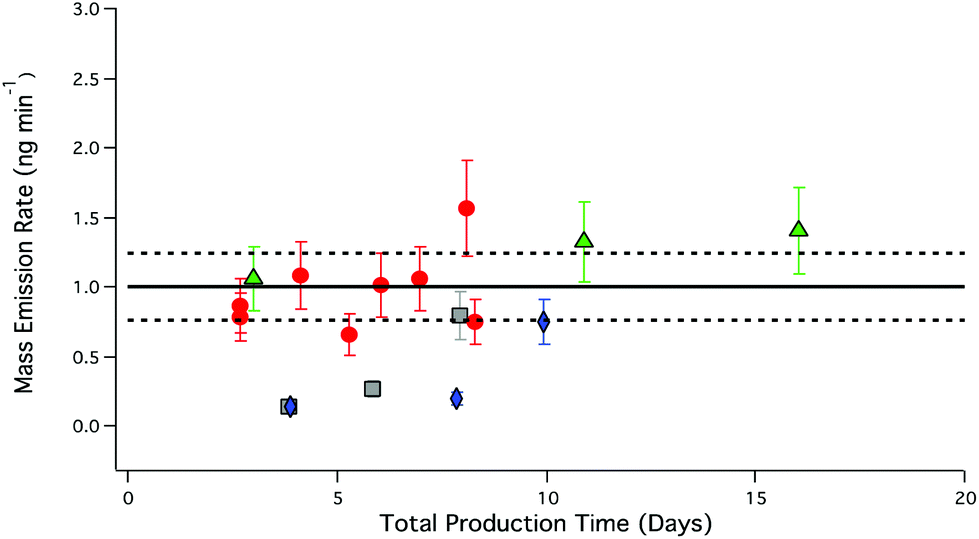 | ||
| Fig. 7 PFBA mass emission rate under different RH as a function of total PFBA production time. Red circles, 0% RH, grey squares, 25% RH; blue diamonds, 50% RH; green triangles, 75% RH. Error bars represent the analytical accuracy of the LC-ESI-ToF for each measurement. The solid black line represents the average mass emission rate of PFBA under dry conditions; dashed black lines correspond to the total uncertainty as described in Fig. 5. | ||
The introduction of water into the PFBA salt bed increases the complexity of the system and decreases PFBA mass emission rates relative to experiments at 0% RH, as seen in Fig. 7. Generating reproducible gas phase emissions of PFBA using RH requires longer equilibration periods compared to dry conditions. Over time, mass emission outputs eventually reached values (1.1 ± 0.3 ng min−1, n = 5) consistent with mass emission rates observed under dry conditions (1.10 ± 0.08 ng min−1, n = 16). Potential equilibration times of a week are not convenient unless a thermostated permeation system has sufficient channels to accommodate and continuously produce the full suite of targeted PFAA salts and precursor acid permeation devices. Whether displacement systems with water produce more stable emissions once equilibrated was therefore not determined. For most applications, it can be concluded that PFCAs are most reliably and simply produced by acid displacement under dry conditions.
Utility of annular denuder for quantitation
Annular denuders are used to selectively scrub gases from airflow for subsequent quantitative analysis, and to prevent reactive gases or those with strong sorption properties from causing biases in particulate matter filter measurements. This is accomplished by applying a coating on the surfaces of the three concentric rings to generate a reactive or sorptive surface. Collecting gas phase species prior to particle collection can significantly improve the accuracy of measurements by reducing sampling biases.14Given the strong acid nature of PFAAs, we assessed the likely ability of a Na2CO3 coating to quantitatively scrub gas phase PFAAs. The annular denuder was placed between the exit of a PFBA salt bed and the glass impinger (Fig. 1).
A flow rate of 111 sccm was used, providing a mixing ratio of a few ppmv for 67 hours. While this mixing ratio is much higher than expected ppq levels,13,48 dilution of this output is easily accomplished using MFCs and, based on this work, 1000 times closer than approaches that would utilize sublimation of solid acids under the same conditions. The measured denuder extract had a PFBA concentration equivalent to a mass emission rate of 1.2 ng min−1. The observed mass emission rate falls within the standard error of the 111 sccm mean measurements (±0.1 ng min−1), which suggests a quantitative recovery.
No detectable breakthrough of PFBA was observed in the glass impinger solution. Thus, the Na2CO3 coating is suitable for quantitative gas phase PFCA collection at parts per million mixing ratios and will therefore be effective at collecting lower PFAA amounts expected in real atmospheric samples. Archived samples that followed this method from standard monitoring networks therefore should be possible to quantitatively analyse for PFCAs and possibly PFSAs.
Conclusions
Acid displacement with HCl and HBr can generate gas phase PFCAs, however, they are unsuitable for generating gas phase PFSAs. The displacement efficiency of gas phase PFCAs is a function of chain length, with smaller PFCAs produced more efficiently. Because the acidities of PFCA congeners are similar, displacement efficiencies are likely driven by differences in volatility. Gas phase PFCA emission rates were not enhanced by carrier gas flow rates or RH. Reductions in PFCA emission rates for non-equilibrated water-containing systems were observed to persist over the first week of production relative to dry systems. Acid displacement produces PFCA mixing ratios easily diluted to atmospherically relevant values, while sublimation of solid acids generates mixing ratios on the order of 104 times higher. Overall, stable and reproducible gas phase PFCAs can be produced most suitably under dry conditions. Annular denuders can also be used to quantitatively sample gas phase PFCAs using a standard Na2CO3 coating. The generation of stable and reproducible gas phase PFAA emissions using acid displacement facilitates the production of small quantities of targeted compounds, which can be used in validation of atmospheric sampling methods. Furthermore, it may enable future assessments of sampling artefacts by allowing a gas phase internal standard addition during atmospheric sampling. Overall, this will dramatically improve our certainty when atmospheric measurements of PFAAs are validated using such gas calibration systems.Acknowledgements
The authors thank the National Science and Engineering Research Council and Environment and Climate Change Canada for funding this work. TCV acknowledges the support of a Banting Postdoctoral Fellowship. Linda Winsor and Robert Di Lorenzo are thanked for analytical support.References
- K. Y. Kwok, S. Taniyasu, L. W. Y. Yeung, M. B. Murphy, P. K. S. Lam, Y. Horii, K. Kannan, G. Petrick, R. K. Sinha and N. Yamashita, Environ. Sci. Technol., 2010, 44, 7043–7049 CrossRef CAS PubMed.
- B. F. Scott, C. Spencer, S. A. Mabury and D. C. G. Muir, Environ. Sci. Technol., 2006, 40, 7167–7174 CrossRef CAS PubMed.
- C. J. Young, V. I. Furdui, J. Franklin, R. M. Koerner, D. C. G. Muir and S. A. Mabury, Environ. Sci. Technol., 2007, 41, 3455–3461 CrossRef CAS PubMed.
- P. Meng, S. Deng, X. Lu, Z. Du, B. Wang, J. Huang, Y. Wang, G. Yu and B. Xing, Environ. Sci. Technol., 2014, 48, 13785–13792 CrossRef CAS PubMed.
- V. Langer, A. Dreyer and R. Ebinghaus, Environ. Sci. Technol., 2010, 44, 8075–8081 CrossRef CAS PubMed.
- M. Shoeib, T. Harner, B. H. Wilford, K. C. Jones and J. Zhu, Environ. Sci. Technol., 2005, 39, 6599–6606 CrossRef CAS PubMed.
- S. Taniyasu, K. Kannan, L. W. Y. Yeung, K. Y. Kwok, P. K. S. Lam and N. Yamashita, Anal. Chim. Acta, 2008, 619, 221–230 CrossRef CAS PubMed.
- C. M. Butt, U. Berger, R. Bossi and G. T. Tomy, Sci. Total Environ., 2010, 408, 2936–2965 CrossRef CAS PubMed.
- C. Lau, J. L. Butenhoff and J. M. Rogers, Toxicol. Appl. Pharmacol., 2004, 198, 231–241 CrossRef CAS PubMed.
- K. S. Betts, Environ. Health Perspect., 2007, 115, 250–256 CrossRef.
- S. S. White, S. E. Fenton and E. P. Hines, J. Steroid Biochem. Mol. Biol., 2011, 127, 16–26 CrossRef CAS PubMed.
- A. Jo, K. Ji and K. Choi, Chemosphere, 2014, 108, 360–366 CrossRef CAS PubMed.
- A. Dreyer and R. Ebinghaus, Atmos. Environ., 2009, 43, 1527–1535 CrossRef CAS.
- L. Ahrens, M. Shoeib, T. Harner, D. A. Lane, R. Guo and E. J. Reiner, Anal. Chem., 2011, 83, 9622–9628 CrossRef CAS PubMed.
- K. Harada, S. Nakanishi and N. Saito, Bull. Environ. Contam. Toxicol., 2005, 74, 64–69 CrossRef CAS PubMed.
- C. a. Barton, L. E. Butler, C. J. Zarzecki, J. Flaherty and M. Kaiser, J. Air Waste Manage. Assoc., 2006, 56, 48–55 CAS.
- T. J. Wallington, M. D. Hurley, J. Xia, D. J. Wuebbles, S. Sillman, A. Ito, J. E. Penner, D. A. Ellis, J. W. Martin, S. A. Mabury, C. J. Nielsen and M. P. Sulbaek Andersen, Environ. Sci. Technol., 2006, 40, 924–930 CrossRef CAS PubMed.
- D. A. Ellis, J. W. Martin, A. O. De Silva, S. A. Mabury, M. D. Hurley, M. P. Sulbaek Andersen and T. J. Wallington, Environ. Sci. Technol., 2004, 38, 3316–3321 CrossRef CAS PubMed.
- J. C. D'eon, M. D. Hurley, T. J. Wallington and S. A. Mabury, Environ. Sci. Technol., 2006, 40, 1862–1868 CrossRef.
- C. J. Young and S. A. Mabury, Rev. Environ. Contam. Toxicol., 2010, 208, 1–110 CAS.
- J. Cheng, E. Psillakis, M. R. Hoffmann and A. J. Colussi, J. Phys. Chem. A, 2009, 113, 8152–8156 CrossRef CAS PubMed.
- H. P. H. Arp and K.-U. Goss, Atmos. Environ., 2008, 42, 6869–6892 CrossRef CAS.
- H. P. H. Arp and K. U. Goss, Environ. Sci. Technol., 2009, 43, 8542–8547 CrossRef CAS PubMed.
- M. J. Rossi, Chem. Rev., 2003, 103, 4823–4882 CrossRef CAS PubMed.
- P. Kulkarni, P. A. Baron and K. Willeke, Aerosol Measurement: Principles, Techniques, and Applications, Wiley, 3rd edn, 2011 Search PubMed.
- A. Ianniello, H. J. Beine, M. S. Landis, R. K. Stevens, G. Esposito, A. Amoroso and I. Allegrini, Atmos. Environ., 2007, 41, 1604–1615 CrossRef CAS.
- Z. Vecera and P. Dasgupta, Environ. Sci. Technol., 1991, 255–260 CrossRef CAS.
- M. Taira and Y. Kanda, Anal. Chem., 1990, 62, 630–633 CrossRef CAS.
- R. S. Braman and M. A. Cantera de la, Anal. Chem., 1986, 58, 1533–1537 CrossRef CAS.
- A. Febo, C. Perrino, M. Gherardi and R. Sparapani, Environ. Sci., 1995, 29, 2390–2395 CrossRef CAS PubMed.
- A. L. Henne and C. J. Fox, J. Am. Chem. Soc., 1951, 73, 2323–2325 CrossRef CAS.
- Z. Wang, M. MacLeod, I. T. Cousins, M. Scheringer and K. Hungerbühler, Environ. Chem., 2011, 8, 389–398 CrossRef CAS.
- E. Poboży, E. Król, L. Wójcik, M. Wachowicz and M. Trojanowicz, Mikrochim. Acta, 2011, 172, 409–417 CrossRef PubMed.
- T. E. Daubert and R. P. Danner, Physical and Thermodynamic Properties of Pure Chemicals Data Compilation, Taylor and Francis, Washington, DC, 1989 Search PubMed.
- R. G. Hynes, M. A. Fernandez and R. A. Cox, J. Geophys. Res.: Atmos., 2002, 107, 4797 CrossRef.
- S. Rayne, K. Forest and K. J. Friesen, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 317–326 CrossRef CAS PubMed.
- B. Bhhatarai and P. Gramatica, Environ. Sci. Technol., 2011, 45, 8120–8128 CrossRef CAS PubMed.
- Safety data sheet for perfluoropropionic acid, http://www.sigmaaldrich.com/catalog/product/sial/77290?lang=en (accessed January 23, 2016).
- Safety data sheet for methanesulfonic acid, http://www.sigmaaldrich.com/catalog/product/sial/471356?lang=en®ion=SK (accessed January 23, 2016).
- Safety data sheet for acetic acid, http://www.sigmaaldrich.com/catalog/product/sial/320099?lang=en®ion=SK (accessed January 23, 2016).
- E. Raamat, K. Kaupmees, G. Ovsjannikov, A. Trummal, A. Kuett, J. Saame, I. Koppel, I. Kaljurand, L. Lipping, T. Rodima, V. Pihl, I. a. Koppel and I. Leito, J. Phys. Org. Chem., 2013, 26, 162–170 CrossRef CAS.
- D. Brooke, A. Footitt and T. Nwaogu, Environmental risk evaluation report: Perfluorooctanesulphonate (PFOS), 2004 Search PubMed.
- G. da Silva, E. M. Kennedy and B. Z. Dlugogorski, J. Phys. Chem. A, 2006, 110, 11371–11376 CrossRef CAS PubMed.
- T. C. VandenBoer, C. J. Young, R. K. Talukdar, M. Z. Markovic, S. S. Brown, J. M. Roberts and J. G. Murphy, Nat. Geosci., 2015, 8, 55–60 CrossRef CAS.
- M. A. Kaiser, B. S. Larsen, C.-P. C. Kao and R. C. Buck, J. Chem. Eng. Data, 2005, 50, 1841–1843 CrossRef CAS.
- J. L. Barber, U. Berger, C. Chaemfa, S. Huber, A. Jahnke, C. Temme and K. C. Jones, J. Environ. Monit., 2007, 9, 530–541 RSC.
- S. K. Kim and K. Kannan, Environ. Sci. Technol., 2007, 41, 8328–8334 CrossRef CAS PubMed.
- L. Ahrens, M. Shoeib, T. Harner, S. C. Lee, R. Guo and E. J. Reiner, Environ. Sci. Technol., 2011, 45, 8098–8105 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |