
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emergence of tunable resistive pulse sensing as a biosensor
Emma L. C. J.
Blundell†
,
Laura J.
Mayne†
,
Emily R.
Billinge†
and
Mark
Platt
 *
*
Department of Chemistry, Centre for Analytical Science, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: m.platt@lboro.ac.uk
First published on 23rd January 2015
Abstract
The article is written as a guide and tutorial that focuses on the use of Tunable Resistive Pulse Sensing, TRPS, as a platform for the detection of biological analytes. Within the field of biosensors there is a continuous emergence of new technologies or adaptations to platforms that push the limits of detection or expand dynamic ranges. TRPS is both unique and powerful in its ability to detect a wide range of biological analytes; including metabolites, proteins, cellular vesicles, viruses and whole cells. Each analyte can be analysed on the same platform without modification by changing the pore size, and is simple enough to follow to allow users from a range of backgrounds to start developing their own assays. The instrument can provide information regarding analyte concentration, size, and charge. Here we hope to give an overview of where this technology is being used and provide some guidance to new users, in the hope it will inspire and enable future experiments.
 Emma L. C. J. Blundell | Emma graduated from Loughborough University with an upper second class hons MChem in Chemistry with Analytical Science in 2013. She is now working on a PhD at Loughborough in collaboration with Izon Scientific investigating resistive pulse sensing for the detection of viruses. Emma is also researching fields based around nanoparticle surface modifications and zeta potential analysis. |
 Laura J. Mayne | Laura Mayne is a PhD student at the centre for analytical science at Loughborough University working on development of new sensors for radionuclides and particle modification as part of the DISTINCTIVE consortium with Dr Mark Platt, Dr Steve Christie and Dr Nick Evans. She received her MChem in Medicinal and Pharmaceutical Chemistry from Loughborough University in 2014. |
 Emily R. Billinge | Emily-Rose Billinge received her BSc in Neuroscience with Pharmacology from the University of Nottingham, UK in 2012. She is currently a PhD student in analytical chemistry with Dr Mark Platt at Loughborough University, UK in the Centre for Analytical Science. Her research is focussed around the development of new biosensors incorporating aptamers and nano/microbeads. |
 Mark Platt | Mark Platt graduated with a degree in Chemistry and Analytical Chemistry from the University of Salford in 2001. He moved into the area of liquid/liquid electrochemistry with Prof. R. A. W. Dryfe and obtained his PhD in 2004 from the University of Manchester. He spent time as a postdoc in Penn State (USA), Cambridge then Manchester (UK) and finally Dublin (Ire) before taking up an academic position at Loughborough University. His research interests sit at the interface between materials, electrochemistry and analytical chemistry developing sensors for health and wellbeing. |
1. Introduction
In this tutorial review we highlight some of the current research and advances within the field of Resistive Pulse Sensing, RPS, focusing on an emerging variant of RPS using tunable pores, known as Tunable Resistive Pulse Sensing, TRPS. The review aims to provide an overview of its use of within the field of biosensors and provide hints and tips to encourage new users as they develop their own TRPS methods.Modern day resistive pulse sensors trace their origins back to the Coulter counter, created in the 1940s to count and size biological cells and microorganisms.1,2 The principle is remarkably simple; two reservoirs are filled with conductive solutions, each containing an electrode, which are then separated by an aperture “the pore”. The sample is added to one of the reservoirs and an ionic current is passed between the electrodes and through the pore. If an analyte passes through the pore it occludes the ionic current causing a transient current decrease known as a “blockade event”. The magnitude of the blockade event provides the information needed to determine the size of the analyte, and the number of blockades per unit time provides information on the analyte concentration, Fig. 1. The size of the pore ultimately determines the sensitivity of the technique and thus the analyte that can be analysed, as a significant occlusion event is only observed when the analyte is comparable in size to the pore.3
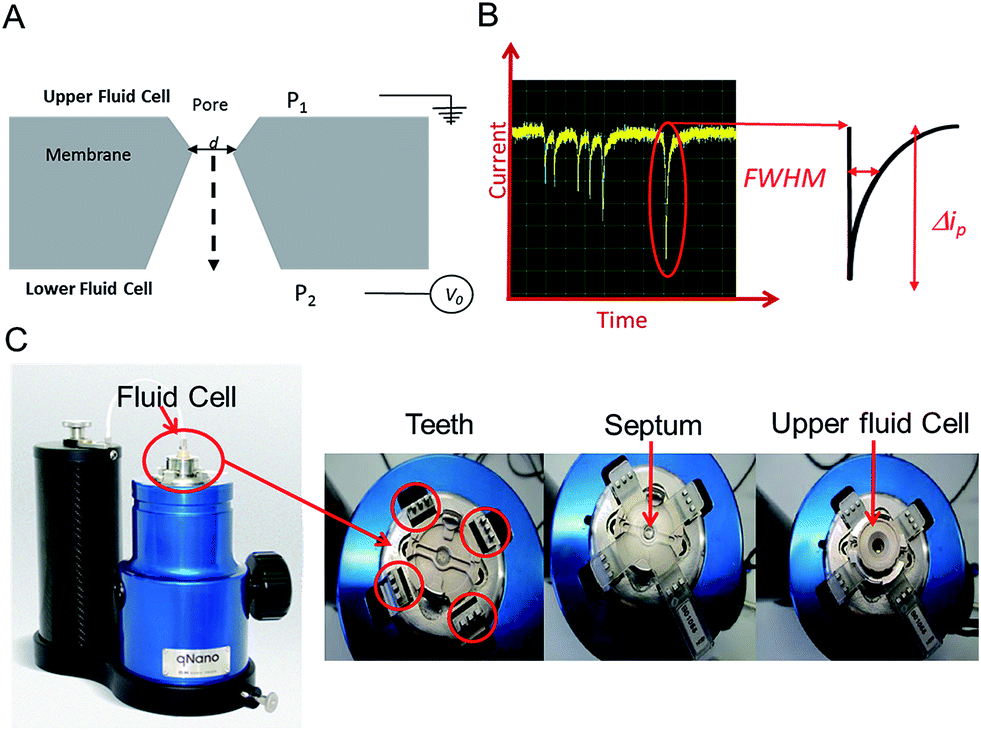 | ||
| Fig. 1 (A) Sectional schematic of a pore. The sample is typically placed into the upper fluid cell. (B) Example of baseline current and “blockade” events (current dips) that are each caused by an analyte traversing the pore. Each event is analysed for full width half maximum (FWHM) duration and Δip. (C) The Izon qNano instrument, showing the fluid cell, teeth and crucifix plastic membrane with aperture. | ||
In the 1990s the Coulter counter was revived in the form of a biological nanopore sensor with the use of the α-haemolysin protein for the detection of ssDNA.4,5 The diameter of the haemolysin-pores is around 1.4 nm at its narrowest point and as the ssDNA passes through the pore, each of the 4 different bases produces a unique signal allowing the sequence of the DNA to be determined.3 With improvements in manufacturing, characterisation and nanofabrication techniques it became possible to reproducibly produce pores from the microscale down to the nanoscale in a range of materials. Solid-state nanopores often support more chemical versatility than biological equivalents, with carbon nanotubes,6 PDMS,7 glass,8 silicon,9 polycarbonate,10 and graphene11 having been used as substrates. Some of these materials have also been incorporated into fluidic devices.2 Here we don't wish to review the synthetic procedures for preparing biological and solid state pores and we would direct the reader to reviews found elsewhere.2–4,9,12
The ability to tune the pore size to the analyte of interest has allowed the RPS technique to detect analytes that range from single molecules, DNA, proteins, cellular vesicles to whole cells including viruses and bacteria, and again detailed reviews on the types of analytes and applications can be found elsewhere.13,14 One property that all solid state and biological pores share is the fixed nature of the pore size. Once prepared and assembled the pore size cannot be changed, thus pores which can be reversibly manipulated in real time offer great advantages in this field.
An elegant and novel adaptation to RPS incorporates a tunable elastomeric pore, which allows for further versatility as the pore can be stretched in real time to suit the sample.15 Tunable pores are fabricated by mechanically puncturing a thermoplastic polyurethane membrane. The membrane is mounted onto “teeth” in the instrument and then stretched in a controlled bi-axial, reversible manner to change the pore geometry, see Fig. 1.
The technique is currently referred to as Tunable Resistive Pulse Sensing, TRPS, but has been previously known as Scanning Ion Occlusion Spectroscopy, SIOS,16 and variants such as size-tunable pore sensors, or tunable elastomeric pore sensors are found in the literature. We will use the term TRPS in this review as an umbrella term to cover all of these variants unless a specific quote from a publication is used. TRPS has been developed to accurately determine the concentration, size and surface charge of dispersed inorganic particles and whilst we concentrate on biological analytes within this review, we direct the reader to key papers for the characterisation and identification of inorganic particles.17–20
TRPS is much more versatile than solid state pore equivalents, but there are limitations to how much each pore can be stretched, thus users typically match a membrane with a pore size to the sample of interest. The company Izon Science Ltd supplies tunable pores, TPs, in a range of sizes, each with a wide optimal size range. This enables the user to match a pore to a specific sample. The pore used for a TRPS measurement determines the size of the particle it can measure, the smallest pore currently commercially available is the NP100 that has a size limitation of 70–200 nm. This lower limit is determined by the smallest analyte size that can produce detectable blockades in relation to the background current noise. As these TPs are relatively inexpensive (tens of euros per pore) it has enabled laboratories from different disciplines to develop methods of analysis using RPS without the usual concerns of synthesis/breaking or blocking the pores. Passing complex biological samples through a small aperture often leads to blockages, and if the blockage is not removed the pore is unable to perform any further analysis; this is often the end of life for many solid-state pores. Along with its versatility to change the pore size to match the analyte the tunable pore, TP, also allows users to pause data capture if a blockage occurs, stretch and open the pore dislodging the trapped analyte, and then reduce the pore size back to its original size before continuing with the experiment. Alternatively the TPs are robust enough that trapped analytes can also be dislodged by tapping the instrument or by applying a pressure to the upper fluid cell, P1 > P2 (Fig. 1), effectively forcing the buffer and blockage through.
Given the appearance of simplicity, reduced cost and versatility, new users could be understood for thinking that the data analysis is simple. In fact the translocation of a particle through the pore is complex and some assumptions are made to simplify the analysis. Current methodologies of analysis and interpretation owe their thanks to Willmott, Vogel, Kozak and Trau who have led the way in modelling and understanding the TRPS technology.15,20–23 Models and methodologies exist within the literature for studying particle shape, charge, orientation and direction of transport.17,20,23–25 Each translocation event reveals a large amount of information on the analyte, such as its size, zeta potential and shape. Whilst the data is capable of being extracted to interrogate it with third party software, the supplier of the instrument provides an interface (Izon Control Suite) for new users and where possible here in the tutorial we try to use the basic features of the software available to everyone.
For a new technology to be seen as enabling and to be adopted by the scientific community several key features are highly desirable; cost effectiveness, ease of use and accurate, reproducible data. In reality, detailed and accurate data can sometimes come with a cost, not always in price, but in effort required to extract the data. Setting up a TRPS measurement is simple, but getting reproducible data on consecutive runs requires the analyst to carry out the measurement carefully, attentively, and with a detailed level of understanding of the system. However, time and effort exerted during data collection is rewarded with the high quality of information. As can be found in cytometry technologies, particle-by-particle analysis of the sample leads to a much more accurate and sensitive assay.26 Since the first bioassay publications using TRPS in 2007 the number of applications and publications has doubled each year, herein we provide an introduction on how to set up the system, troubleshooting ideas and a review of the applications of the TRPS system.
2. Theory
The instrument most widely associated with TRPS is produced by the company Izon Science referred to as the qNano or qViro, and as with standard RPS equipment two fluid reservoirs are to be filled with a conducting electrolyte solution. In this setup the pore is mounted horizontally and reservoirs oriented above and below the pore membrane, with the sample typically placed into the top reservoir, see Fig. 1. The qNano uses an elastic size-tunable pore which is fabricated in a thermoplastic polyurethane membrane. The membranes are penetrated with a needle to create a single pore which is conically shaped.27 The size and geometry of the fabricated aperture can be modified by modifying the puncturing needle thus allowing the detection of particles ranging from 70 nm to 10 μm over the full range of manufactured pores.16 The cruciform TP is mounted by eyelets to teeth on the instrument, above the lower fluid cell, the system can be seen in Fig. 1C. The arms can be altered to increase the stretch on the pore, where it has been shown that applying a stretch of 10 mm to the membrane increases the pore opening by 54%.22.1 Analyte size
The conical pore gives rise to an asymmetric current pulse, Fig. 1, with resistance highest at the narrowest pore constriction resulting in a sharp drop in current which tails back toward the original baseline value as the resistance diminishes toward the base of the pore.20 For a conical pore, the change in the resistance, ΔR, across the length of the pore. L, is given by eqn (1),28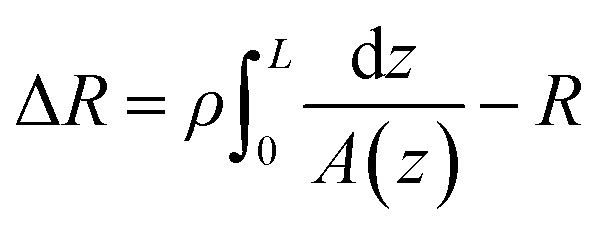 | (1) |
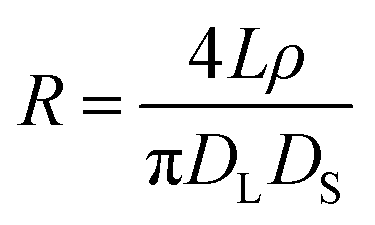 | (2) |
2.2 Analyte concentration
The frequency of the pulses, J, can be related to the concentration of the analyte, Cs, as well as the velocity of the traversing particle, vp. The velocity term is the sum of the fluidic, vF, electrophoretic, vE, and electroosmotic, vO, velocities, i.e. vp = vF + vE + vO. Here we typically ignore the contribution from diffusion due to the magnitude of other forces and end effects are not taken into account in the analysis. vp can be written as;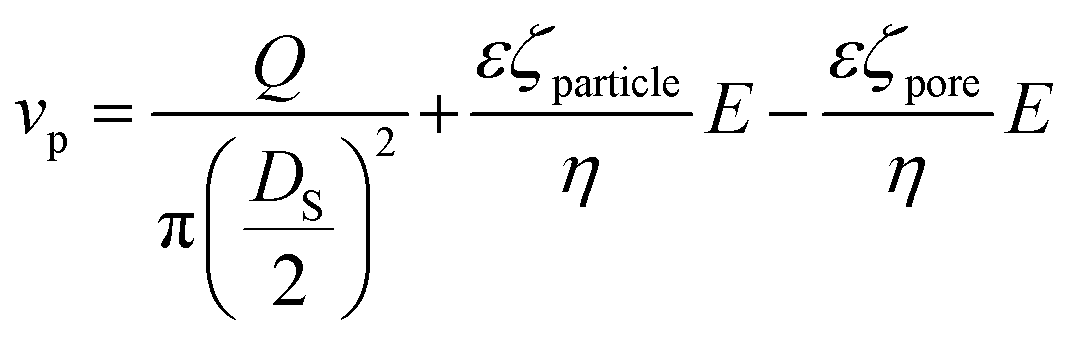 | (3) |
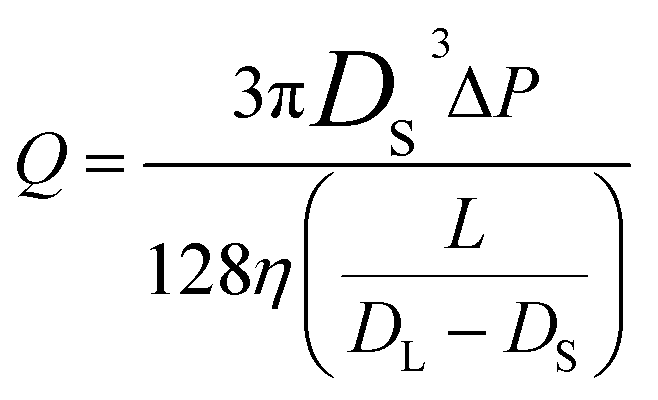 | (3a) |
2.3 Varying the pressure across the pore
The reader will note that the pulse frequency is related to the pressure difference across the pore. With the Izon system there is always an inherent pressure head guaranteed within the setup due to gravity, indicated with P1 and P2 in Fig. 1A. Using the variable pressure system supplied with the instrument users can vary the ratio between P1 and P2 across the pore. Willmott et al., showed that pressure-driven transport can be made dominant,23 which has advantages for studying zeta potential values. It also helps data analysis with samples that contain a low concentration of analyte. Such samples would typically have a low particle count rate, and thus would result in long run time. By applying a pressure to the cell, P1 ≫ P2, analytes can be driven through the pore more frequently reducing the total run time. Alternatively if a sample is too concentrated, producing blockade events that are not clearly resolved from one another, or the particles are moving at too great a velocity, reducing the information and signal within the peak width, it is possible to slow the blockade events by the application of vacuum, P2 ≫ P1. As a guide the particle rate should ideally not exceed 1000 particles per min, with an optimum range between 500–700 particles per min.Pressure ratios are controlled by the variable pressure module, VPM. The VPM has a mobile ‘arm’ with a scale of 0–20 cm. The amount of pressure applied is relative to the length of the arm e.g. if the arm is inserted 5 cm into the system, a pressure of ‘5 cm’ is applied. For a negative pressure, P2 > P1, the same arm is pulled out of the instrument to the required length in cm. Each cm of pressure is equivalent to approximately 1000 Pa.21 The unit of cm will be used for the remainder of the review when denoting a pressure applied to the TRPS system.
2.4 Zeta potential theory
Evident from eqn (3) is the relationship between particle velocity and its zeta potential. Zeta potential is defined as the electrostatic potential at the border between the diffuse layer and compact layer30 (also known as the Stern layer)31 of a colloidal system. Zeta potential is related to the surface charge of the particle and is often used as an indicator of colloidal stability. It also offers a unique and additional parameter to identify biological analytes when their sizes are comparable, as Martin et al., demonstrated by measuring the velocity of several proteins using a solid state pore system.32,33 The Smoluchowski approach34 defining zeta potential is displayed in eqn (4), where ζ is zeta potential, η is dynamic viscosity of the fluid, μ is particle mobility and ε is the dielectric constant.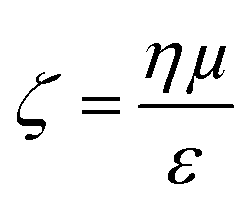 | (4) |
The Smoluchowski approach supports that a particle's zeta potential can be determined from its velocity taking into account convective and electro osmotic forces, as well as the electrophoretic mobility of the particle. The electrophoretic mobility is a measure of the translocation time of the analyte through the pore under an applied electric field, the convective forces are attributed to the flow of the solution through the pore due to gravity and any applied pressures; whereas the electro osmotic forces relate to the flow of liquid through the pore which arises from the charge on the pores surface and the movement of liquid in the electric field. In their seminal paper Vogel and Willmott et al., developed a method of balancing the electro osmotic and electrophoretic effects by balancing the pressure across the pore allowing zeta potential values to be extracted from the resistive pulse, their methodology is shown in Fig. 2.35
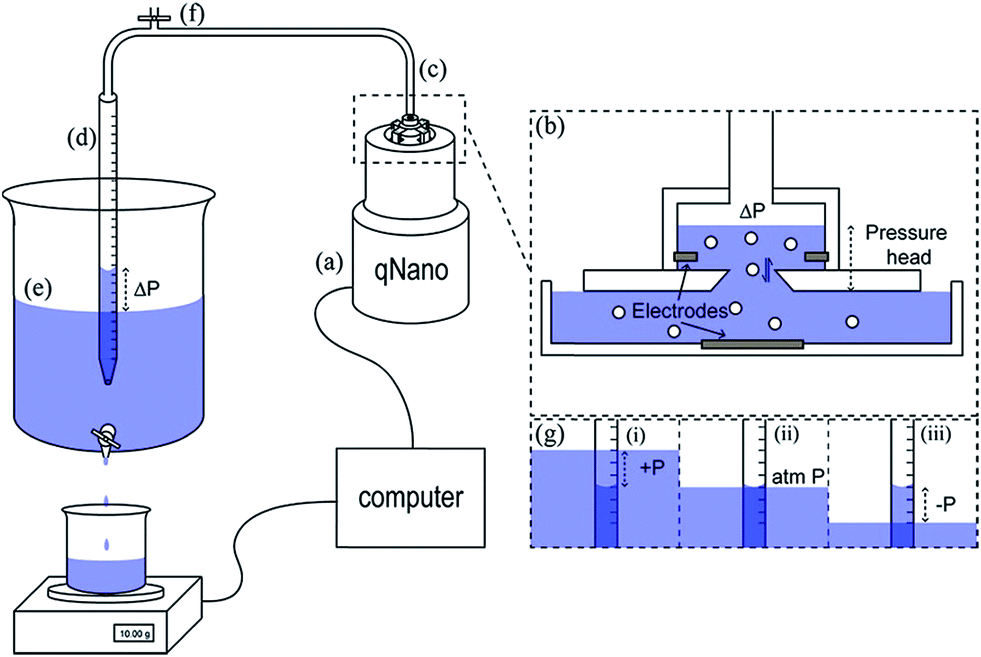 | ||
| Fig. 2 Schematic of the variable pressure set up used with the qNano pore sensor (a). Pressure in the top fluid cell (b) is precisely controlled via a flexible tubing connection (c) by varying the height difference between the water level in a partially submerged buret (d) and the water level of a large water reservoir (e). The buret was equilibrated with atmospheric pressure by opening a valve (f). Progression of an experiment where pressure is varied from positive to negative (g). Reprinted with permission from Anal. Chem., 2012, 84(7), 3125–3131. Copyright (2012) American Chemical Society. | ||
2.5 Setting up TRPS
TRPS is a relatively simple set up procedure, and users can find the protocol within the user manuals, here we include some useful points that we have adopted as part of our best practice.The pore membrane is connected to the system via the four arms, Fig. 1, and the desired pore stretch can be obtained (>43 mm, 43 being the smallest distance between the teeth applying no stretch to the membrane), the electrolyte is then placed in the lower fluid cell (75 μL). Care needs to be taken to ensure no bubbles are introduced into the electrolyte, if bubbles do occur the electrolyte needs to be replaced. If no bubbles are present, the upper fluid cell can be placed on top and twisted into place. Once the upper fluid cell is connected and before you place liquid into the upper cell switch on the instrument. The current should be ∼0.5 nA and stable. If the current is drifting or unstable, there may be a fluid leak in the system and the instrument needs to be turned off, taken apart, cleaned, and dried thoroughly. The electrolyte solution can then be placed into the upper fluid cell (40 μL). The Faraday cage is placed over the fluid cell to reduce background noise and the system is switched on using the computer software. Upon the application of a voltage a stable baseline should be observed. Sufficient voltage needs to be applied to ensure a baseline current > 50 nA. If the baseline is fluctuating rapidly or not settling at a current, it is not stable and a number of troubleshooting methods can be applied (Table 1).
| Problem | Solution |
|---|---|
| Introduced particles but can't see peaks | • Check pore classification is appropriate for size of interest |
| • Reduce stretch (minimum 43 mm) | |
• Sample may be too concentrated, clean pore and dilute sample 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100; if resulting particle rate is too slow apply pressure :100; if resulting particle rate is too slow apply pressure |
|
| Frequent blockages | • Clean pore thoroughly |
| • Check for bubbles in both fluid cells | |
| • Ensure sample is well dispersed – try vortexing, sonicating or the addition of a surfactant – or all three! | |
| • Increase pore stretch or change to a larger pore if necessary | |
| • Try additional pressure | |
| Unclean pore | • Increase stretch and voltage where possible |
| • Replace the sample/electrolyte with fresh electrolyte buffer several times | |
| • Apply pressure/vacuum to the pore to dislodge any particles ‘stuck’ in the pore | |
| Rate trace not linear | • Add pressure for calibration files |
| • Check for bubbles | |
| • Sonicate and vortex sample | |
| • Dilute sample to reduce risk of blockages | |
| Bubbles frequent | • Possibility of too much surfactant, reduce concentration |
| • Warm working conditions (>30 °C) can result in bubbles due to gradual evaporation of buffer | |
| Current rises rapidly/saturates | • Fluid leak, remove fluid cell from the TRPS instrument and dry thoroughly |
| • Decrease applied voltage | |
| Current drops rapidly/Current drops to zero | • Pore may be partially blocked, troubleshoot to remove the blockage, increasing the stretch and applying a pressure/vacuum can aid this process |
| • Air bubbles in the upper or lower fluid cell. Remove the liquid from the appropriate fluid cell and replace ensuring no bubbles are present | |
3. Applications of TRPS
3.1 Cells/bacteria
Optical density, OD, measurements are commonly used in monitoring bacterial growth due to its ease of implementation.36 OD measurements allow the bacterial growth to be followed in real time,37 however the accuracy of these measurements can be hindered by low sensitivity.37 OD measurements use an approximation of the cell number and cell size by changes in the light scattering from the sample.36 Plate counts and microscopes are employed when an account of the bacteria present is needed; these are usually time-consuming and laborious as a stain on the bacteria is usually needed and requires manual counting.38 Other modern bacterial growth monitoring methods such as flow cytometry26,39 and microscopy40 are also common in the literature. Allen et al., developed a method using TRPS to monitor the growth of two commonly used bacteria; Gram-positive Bacillus subtilis str.168 (BSU168) and Gram-negative Escherichia coli str. DH5α (DH5α).36 TRPS was used to monitor the bacterial cell concentration and cell volume, both of which can be analysed simultaneously,36 see Fig. 3. The cell volume dynamics and the level of bacterial cell chain formation can be observed by tracking the spread of the cell volume histogram. Coefficient of variation, CV, was used to measure the level of dispersion and precision of the concentration measured by TRPS where a CV lower than 5% is considered acceptable; CVs observed for the samples were lower than 2%.36 The low CV was a good indicator that TRPS is reliable for measuring the concentration of the bacteria present. When using TRPS, a measurement can be made within a few minutes which is in contrast to traditional colony-plating methods which involve long preparation steps of agar plates, incubation and counting.36 The concentration measured by TRPS was different than the concentration measured by OD and colony plating, which is highlighted in Fig. 3. The difference is due to TRPS counting all the cells present in the liquid and cannot differentiate between dead and live cells as colony plating does; in addition cell counting methods are often approximated due to the chosen field of view.36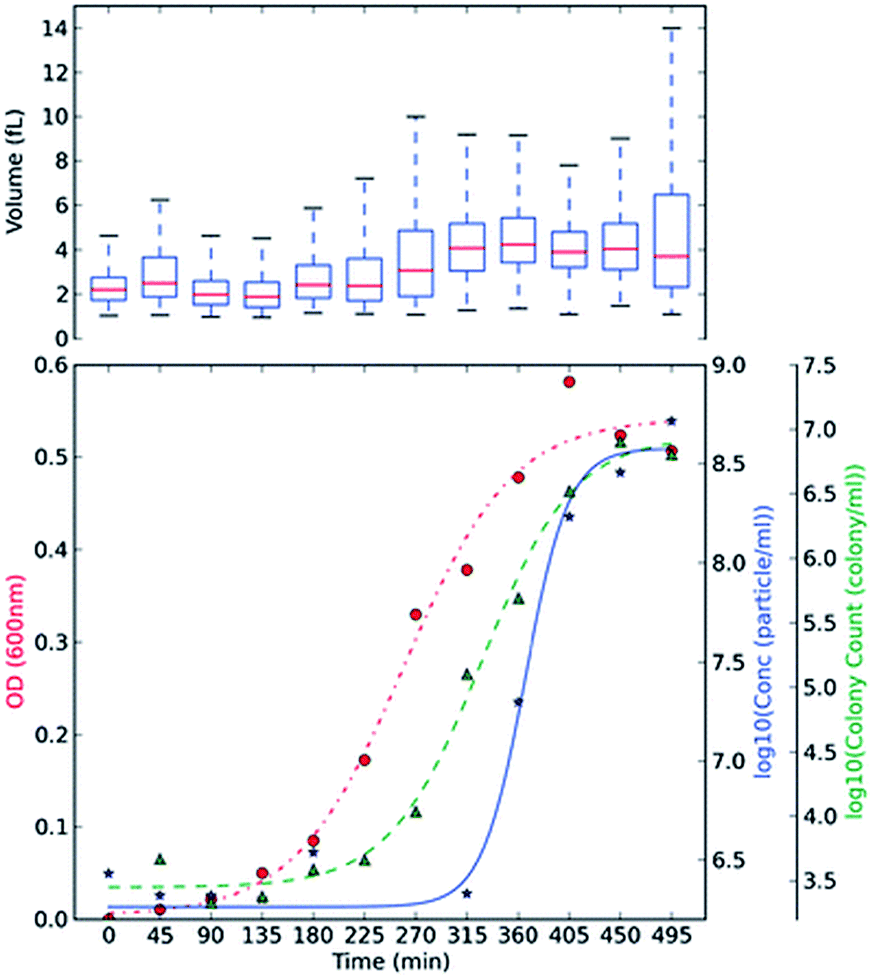 | ||
| Fig. 3 BSU168 and DH5α growth monitoring using plate counting, OD and TRPS methods. Culture BH BSU168 in MG-High. The OD changes (red, ●), particle counts by TRPS in log base 10 (blue, ★), and viable colony counts by plating method in log base 10 (green, ▲) were measured in 45 min intervals. The averaged triplicate measurements of concentration were shown, and boxplot of volume (fL) measured by TRPS was included at the top of each subsection, using at least ≥500 particles. Springer and Applied Microbiology and Biotechnology, 2013, 98, 855, Monitoring bacterial growth using tunable resistive pulse sensing with a pore-based technique, Allen. C. S et al., Fig. 2. ©Springer-Verlag Berlin Heidelberg 2013. With kind permission from Springer Science and Business Media. | ||
3.2 Viruses
Sub 100 nm sized particles are generally a challenge for many nanoparticle characterisation techniques. Viruses are infectious agents made up of nucleic acids and are particularly valuable in scientific and medical research. The virus' genetic material is contained within a protein shell (capsid) and this as a whole is known as a virion or virus particle, VP. Virus like particles, VLPs, are also available with similar capsid properties to VPs. VLPs are self-assembled protein structures with similar, and sometimes identical, structure to their resident virus41 and can be applied to fields including gene therapy and vaccine development. VLPs have gained further interest in the development of nanomaterials because of their small size (10–200 nm), construction flexibility and structural uniformity.41Analytical tools for the detection and characterisation of viruses and VLPs are constantly being developed and improved. These included ELISA,42 real-time PCR,43,44 loop-mediated isothermal amplification (LAMP),43,45 multiplex tools incorporating bead arrays, and next generation sequencing (NGS).42 One of the main challenges for virus detection comes from the small sizes of virions and VLPs. Small particle analysis by TRPS systems has been developed recently by Vogel et al., successfully detecting and sizing virions 70–95 nm in diameter, producing highly reproducible data in agreement with both optical methods and TEM (within 6%).22 Optical methods are common but are generally averaging techniques46 that don't allow for the versatility of particle-by-particle analysis available from TRPS. The ability to detect individual 70 nm particles is an exciting prospect for nanotechnology and current TRPS developments are allowing for even smaller particles to be analysed on a particle-by-particle basis.
3.3 Cellular vesicles
Cells release numerous types of membrane particles under physiological and pathological conditions. Originally thought to be an artefact of the body, research is now demonstrating that these microvesicles, MVs, may be part of the cell signalling process, facilitating cellular messaging and the exchange of RNAs which may even precede the release of protein inflammation markers. MVs have recently seen a surge in interest as they may act as clinically relevant biomarkers and their properties such as size, concentration and composition could provide important physiological information and be of potential use in early diagnostics.47–50 Although their prevalence or characteristics may be well tied to a range of disorders, a gold standard technique for their characterisation has yet to emerge. MVs range in size from around 20 nm to 1 μm and at current there is a lack of availability of techniques which can accurately characterise MVs in terms of their size and concentration51,52 with concentration analyses largely varying from instrument to instrument due to the working range of the method in question and differences in the minimum detected size.53For many years the gold standard of particle characterisation in terms of size accuracy has been electron microscopy. However, these methods have severely limited abilities to quantify their concentration. As the concentration of circulating MVs has been reported to vary in several different physiological states such as hypercholesterolaemia,54 atherosclerosis55 and even exposure to pollutants,56 the lack of accurate concentration analysis is a significant limitation, requiring a second method to attain concentration data.57 Flow cytometry is a high-throughput method for identifying and quantifying analytes based upon the scattering of light or the fluorescence signal of the particles. Whilst flow cytometry has previously been used to quantify MVs, a lack of resolution below 200 nm lends bias to any size or concentration data as many subtypes of MV are believed to lie below this threshold.54
The application of TRPS within the field of MV characterisation is highly advantageous due to the ability to easily fit a range of pores which can detect particles as small as 70 nm providing a particle-by-particle analysis to elucidate concentration. By using several pores sequentially it is possible to analyse a wide range of sizes of MVs and due to particle-by-particle analysis, generate an accurate size distribution despite the polydispersity of the sample.53,54,58 Using TRPS makes it possible to attain accurate size and concentration data for a wide range of vesicles, and in conjunction with complimentary techniques a rich level of information can be obtained. Szabó et al., investigated the cell-signalling potential of MVs and conducted an extensive study using a wide range of techniques to gain a full picture of the effects of MVs on the gene expression of recipient cells, see Fig. 4. TRPS was used to determine the concentration and size distribution of the MVs using two membranes. It was found that recorded size distributions by TRPS were comparable to values obtained by SEM and TEM with the mean diameter lying at around 350 nm.59
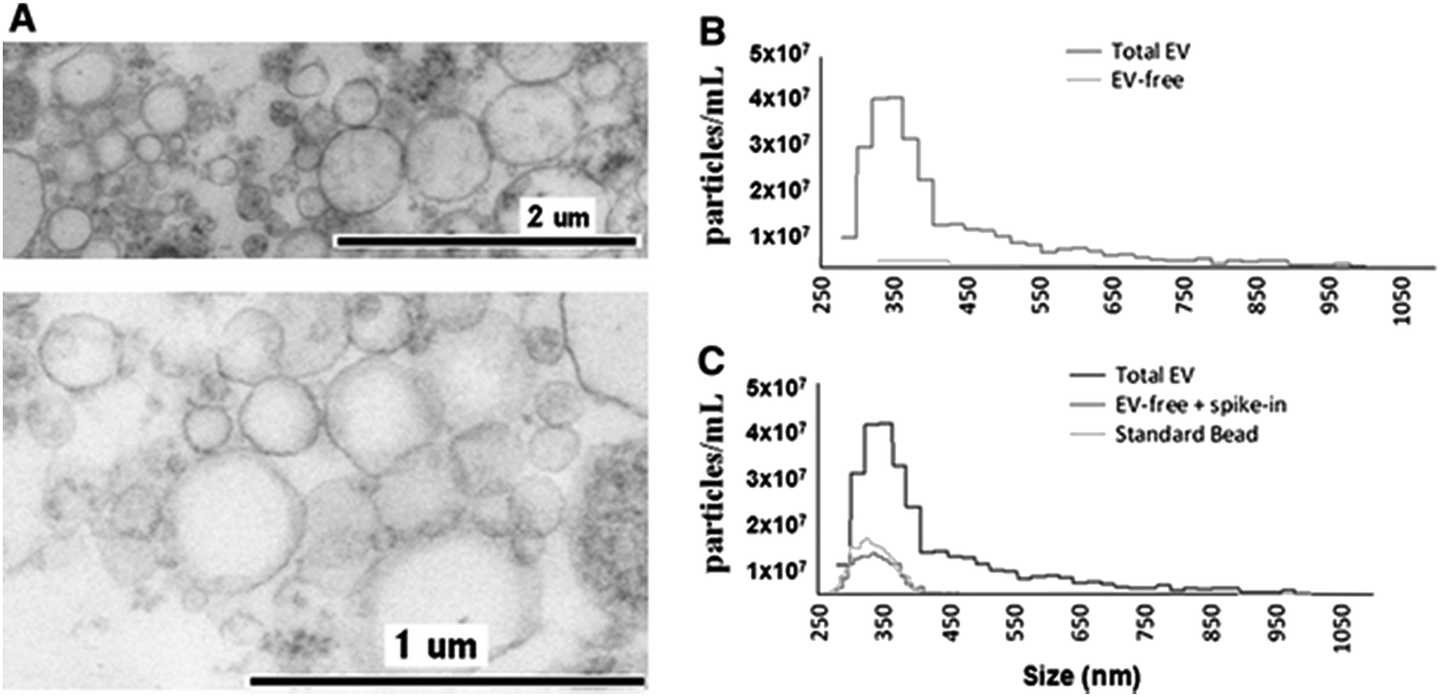 | ||
| Fig. 4 Determination of EV quality, composition and size distribution. (A) Transmission electron micrograph of CCRF-derived EVs. (B) Izon qNano analysis of the size distribution and concentration of extracellular vesicles. Cell-depleted CCRF supernatants, containing EVs, were analysed using an NP400 membrane from Izon Ltd, at a stretch value of 47 mm, 120 nA current, and 10 cm water pressure; at least 500 particles were detected. (C) Confirmation of EV removal by a known size bead spike. We spiked in the EV-depleted CCRF supernatant with known size reference beads (SKP400D at 4.5 × 107 particles per mL concentration) to confirm that the detected absence of positive signals in the EV-free supernatant was not due to a blockade of the nanopore. Springer and Cellular and Molecular Life Sciences, 2014, 71, 4055, Monitoring bacterial growth using tunable resistive pulse sensing with a pore-based technique, Allen. C. S et al., Fig. 1. ©Springer Basel 2014. With kind permission from Springer Science and Business Media. | ||
In further support of TRPS as a new tool in MV characterisation, Connolly et al., measured low-density-lipoprotein (LDL) cholesterol linked to familial hypercholesterolaemia using TRPS for the MV size and concentration, flow cytometry to determine MV origin, and gas chromatography to monitor the fatty acid composition of the MVs; this multifaceted approach is likely the best way to gain a full view of the vesicles being investigated. This study was able to monitor the clinically relevant progression of apheresis treatment for hypercholesterolaemia by monitoring MV concentration by TRPS.54
3.4 Particles based bioassays – DNA extraction/detection
Nanoparticle based bioassays are being increasingly developed for point-of-care assays.60 The immobilisation of biological components onto their surfaces allows them to be used as drug delivery agents, bioimaging substrates or to be incorporated into a range of sensing technologies. The development of superparamagnetic beads, SPBs, and magnetophoresis devices have made it possible to rapidly and efficiently separate cells, proteins and DNA from complex mixtures, such as, plasma, urine, and culture media, in a manner that does not require complicated equipment.61,62 By functionalising the SPBs surface with DNA they can be used to extract target DNA from solution. TRPS is particularly well suited to the detection of DNA on the surface of particles. Although there exists a wealth of available technologies to sequence and characterise DNA samples with high accuracy, at current many of these techniques require PCR, gel electrophoresis and fluorescence63–65 which are contributors to lengthy processing and expense involved in DNA analysis.Several groups have engaged in work with RPS and TRPS technology to quantify DNA hybridisation63,66 and to monitor particle surface modifications. The benefits of using TRPS technology over conventional techniques is its ability to generate a label-free signal, fast run time (in most cases sub 5 minutes). Booth et al., devised a method which incorporated the use of an applied vacuum to be able to elucidate zeta potential of beads under several different conditions.66 By applying a positive potential bias across the pore, polyanionic DNA lends negative charge to the beads which in turn increases electrophoretic mobility. As a vacuum is applied and is gradually increased, the frequency of particles moving through the pore slows, stops and then eventually reverses as beads are pulled back up from the underside of the pore. This inflection point is relative to the surface charge of the beads as when negative charge is increased the amount of vacuum applied must also be increased to overcome the electrophoretic mobility of each particle.21,67 Using this variable pressure method it was possible to measure the increased charge loading resultant from DNA hybridisation.
An additional method utilising the hybridisation of DNA concerns the specific creation of aggregates using complementary sequences to join groups of beads together.60 Agglutination assays are easily adaptable to TRPS, aggregate size is able to be easily determined by monitoring the increase in volume as each aggregate passes through the pore68,69via alterations to the magnitude of the peak and the frequency of particles through the pore and has been employed for the study of Au particle agglutination.70 Previous work employs fluorescence and light scattering that both require an additional label for analysis,71 whereas in TRPS the aggregate is in effect the label and there is no need for additional markers; in this sense it is “label-free”. An additional consideration in the use of aggregation assays is the ability to bring each of the components into close proximity, one such way to do this is the employ SPBs as outlined in a proof-of-concept assay in which 1 and 3 μm SPBs were coated in avidin and then incubated in the presence of a range of concentrations of biotin.69 It was found that the application of a permanent magnet and rolling of sample vials caused increased aggregation due to the beads being brought into closer proximity to each other, this action was termed magnet assisted aggregation (MAS).
A similar technique has been used via TRPS to monitor single nucleotide polymorphisms (SNPs) using the highly selective aggregation of AuNPs. This was completed by using probes with a controlled number per AuNP and with a specific sequence and length such that aggregation occurs only in the presence of a complete complement target,63 see Fig. 5. The use of TRPS was advantageous over the use of a solid state pore for several reasons worth discussing – firstly, the study of potentially large aggregates poses the threat of blockage; however, by being able to stretch the pore in real time it is possible to temporarily pause recording, open the pore, allow the blockage to pass, restore the desired stretch and resume recording. In addition, in this study it was possible to tune the applied stretch so that single particles are not visible above the level of baseline noise and only aggregates are visible. In theory this proof-of-concept method could be utilised to target any desired SNP by tailoring the capture probes of choice.
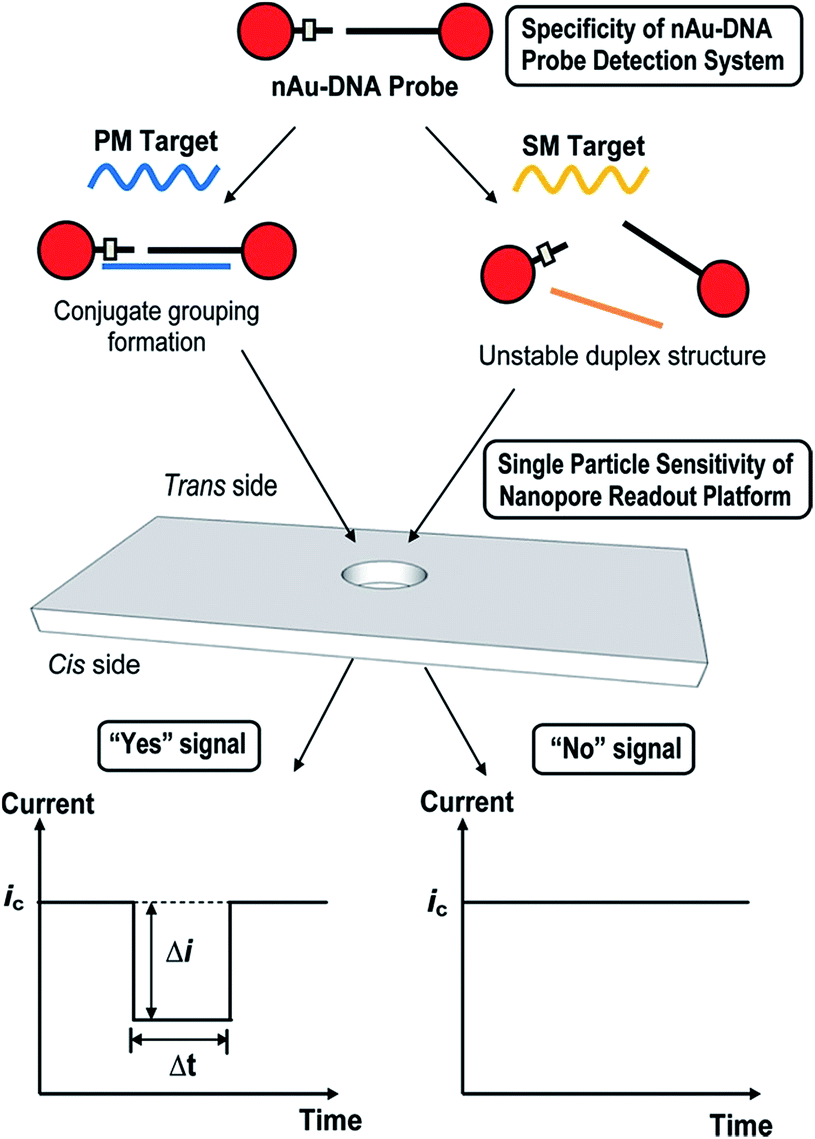 | ||
| Fig. 5 Schematic illustration of nanopore-based single-nucleotide detection using a nAu–DNA probe. The ssDNA sequences on nAu-100b and nAu-18b probes were designed to be complementary to the mutant (mut) sequence and single mismatched to the wild-type (wt) sequence. In the presence of a perfectly matched (PM) target, a well-defined nanoparticle assembly, termed conjugate grouping, forms. Each distinct conjugate grouping is picked up as an individual signal (“Yes” signal) when it translocates the pore of the membrane from the trans to the cis side. Every successful translocation activity is termed a blockade event, which is characterized by its blockade magnitude (Δi) and baseline translocation duration (Δt). When a single-mismatched (SM) target is added, the intermediate duplex structure is energetically unstable and fails to form an assembly structure. The smaller-sized nAu–DNA probe does not result in an appreciable dip in baseline current (ic) and is taken to produce a “No” signal. Reprinted with permission from ACS Nano, 2012, 6, 8815–8823. Copyright (2012) American Chemical Society. | ||
3.5 Confirming the DNA is on the particles
Due to the negative sugar–phosphate backbone, functionalising DNA onto nanoparticles can alter their behaviour within TRPS due to a change in their surface charge. As the surface charge of the bead directly impacts upon the pulse frequency, eqn (3), and the pulse width, FWHM, it is relatively simple to confirm the presence of DNA on the beads. To do this user must first ensure that the increase in pulse frequency is due to the presence of the DNA and not caused by a change in particle concentration. First their concentration is to be verified using calibration beads with the application of large pressure i.e. P1 ≫ P2Fig. 1, typically done using a pressure greater than 5 cm on the pressure module. This pressure ensures that the dominant force acting upon the beads is the fluidic component of eqn (3). Once the concentration has been found, a sample of beads before and after the modification of DNA is prepared to the same concentration. Both samples are then run under no additional pressure (0 cm) at a range of voltages to produce a plot of pulse frequency vs. voltage, see Fig. 6. The presence of DNA on the particle is confirmed when there is a larger change in rate as a function of voltage.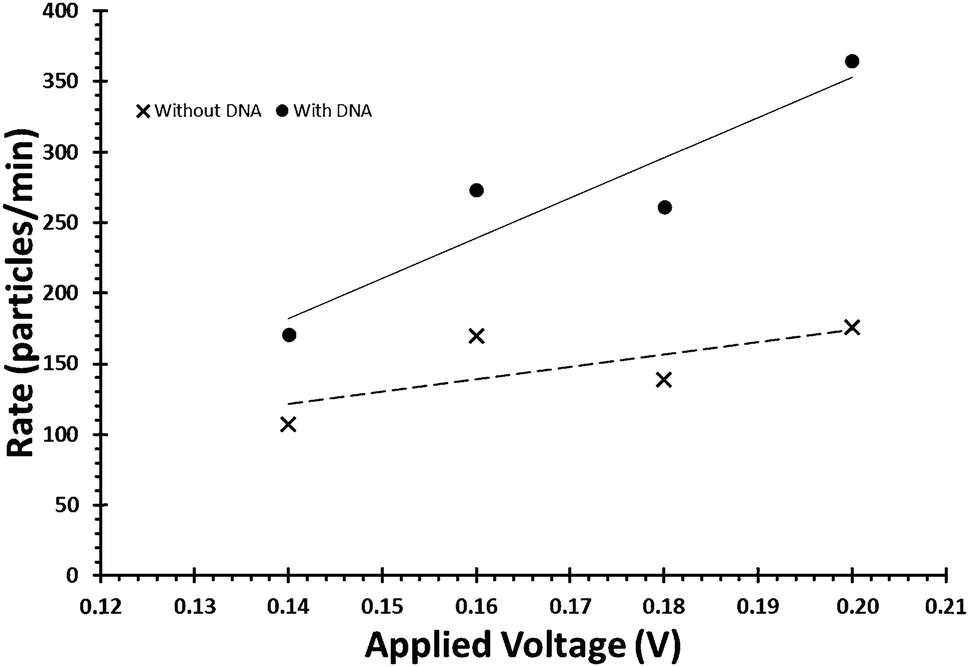 | ||
| Fig. 6 Example of the relationship between applied voltage and particle rate, for beads of identical concentrations with and without the DNA on their surface. | ||
3.6 Particle–aptamer–protein studies
Many traditional bead-based protein assays have revolved around the use of antibodies. Where previously antibodies have been the capture probe of choice, aptamer technologies are gaining interest.72–75 Aptamers are conventionally generated through the process known as SELEX76–79 (systematic evolution of ligands by exponential enrichment), where strong binding sequences are evolved/enriched from extensive libraries, or by CLADE (closed loop aptameric directed evolution) which produces the aptamers ‘on-chip’.80–82 Due to their comparable selectivity, stability and cost; over the last two decades, aptamers have started to challenge antibodies in their use on many technology platforms. As discussed above, the modification of particle surfaces with aptamers should be a process well suited to TRPS technologies.Billinge et al., demonstrated TRPS as a label-free detection platform for the detection of the thrombin protein using SPBs coated with thrombin aptamer.83 When the thrombin protein was introduced to the aptamer-coated beads, a decrease in pulse frequency was observed. The isoelectric point of thrombin lies at pH 7.1,84 suggesting that in pH 7.4 PBST buffer, as used in the study, the overall surface charge of the thrombin molecule should be largely neutral. The shielding of the negative DNA aptamer with the protein reduced the electrophoretic mobility of the bead moving through the pore, reflected in a reduced frequency, J, and increased FWHM.
Alsager et al., developed an assay to 17β-estradiol (E2) using aptamer-coated carboxyl beads monitored with Dynamic Light Scattering (DLS) and TRPS to observe changes in zeta potential. In addition to the changes in electrophoretic mobility observed by Billinge et al., a change in the size as the aptamer bound to its target was also observed. TRPS was able to resolve a 20 nm increase in the mode diameter of beads once the aptamer had been bound to the beads, followed by a reduction in size as the target analyte E2 was introduced.85
Additional aptamer-bead based assays are able to be designed such that if an analyte contains multiple binding epitopes, aggregate formation can be monitored. Platt et al., used a specific class of nanomaterial termed nanorods to detect femtomolar levels of homodimeric PDGF-BB by monitoring specific aggregation of nanorods.86 In the same study the authors also demonstrated the ability to generate aggregates of specific orientations and differentiate between these in the presence of different analytes; see Fig. 7.
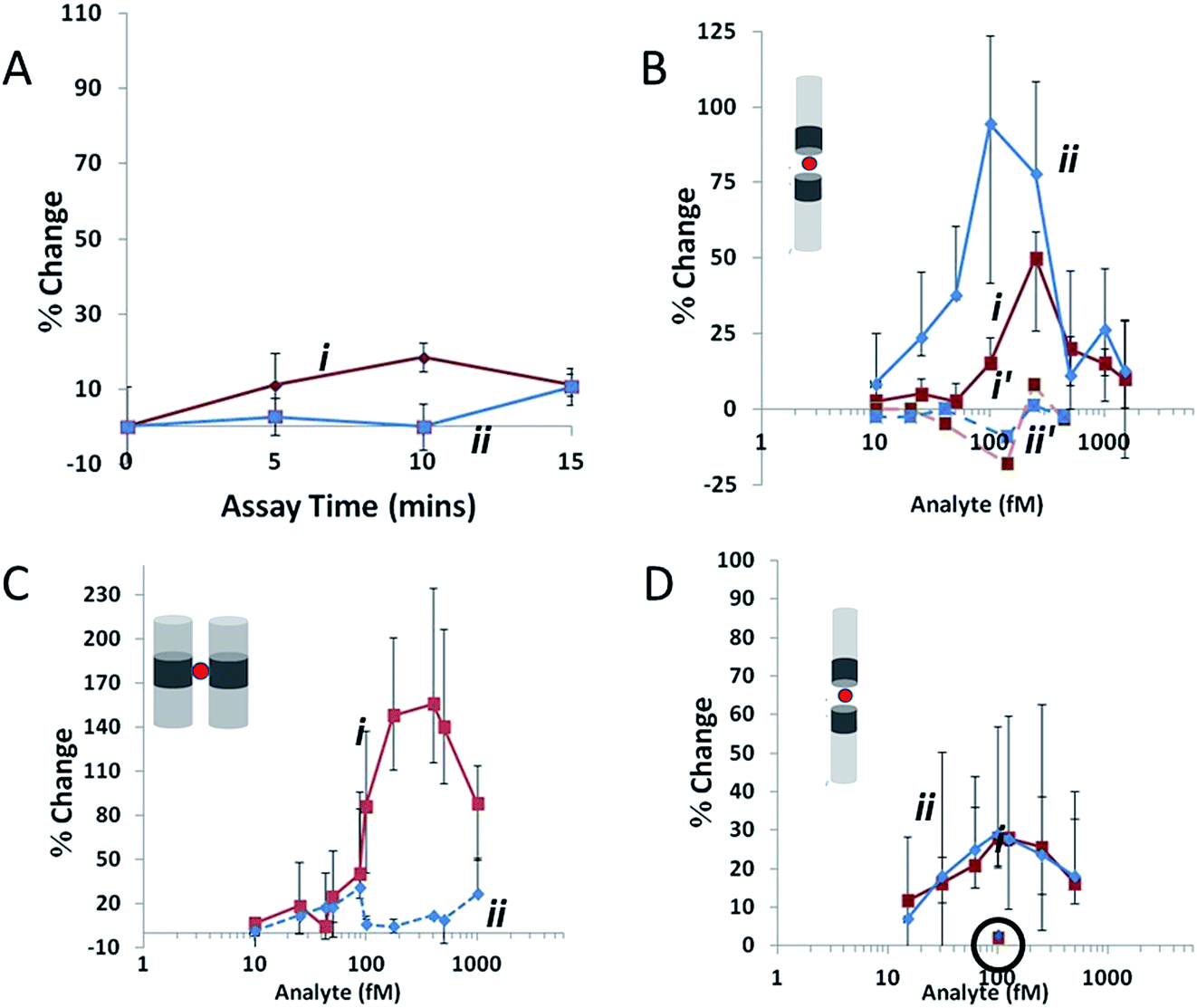 | ||
| Fig. 7 Agglutination assay data collected at a stretch of 45.5 mm and potential of 0.14 V, where red (i) indicates ip and blue (ii) indicates FWHM. (A) Variation in Δip and FWHM for 400 fM AuNi rods (1.23 μm long with CV 20%, Ni content 15% by length) as assay time is increased in the absence of an analyte. (B) The same rods as (A) at assay time 10 min. Ni segments are functionalized with avidin and the concentration of the biotinylated-BSA analyte is varied. Dashed lines represent a 10 min assay with a non-biotinylated target. (C) A biotin–avidin assay at 10 min as in (B), using 500 fM AuNiAu rods (0.82 μm long with CV 14%, Ni content 18% by length) in the side-on configuration. (D) 150 fM AuNi rods (1.1 μm long with CV 20%, Ni content 14% by length) at assay time 10 min. Ni segments are functionalized with PDGF aptamer, and the analyte is PDGF. The circled data points at 100 fM indicate the change in FWHM and Δip for the same rods using a control protein. Error bars show the d25 and d75, values for each data point, lines joining data points are drawn to guide the eye. Reprinted with permission from Small, 2012, 8, 2436–2444 ©2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
TRPS has also been used to obtain kinetic information for aptamer–target interactions, and recorded results similar to traditional SPR techniques.83 A recent development of this TRPS-based assay takes advantage of the excellent size discrimination inherent in a particle-by-particle system. Billinge and Platt have demonstrated the ability to successfully analyse the binding of two aptamers to their respective proteins simultaneously; by using the bead size as a label, two different populations of beads were selectively coated with aptamer and incubated with a combination of different protein concentrations.87 These key proof-of-concept works demonstrate the ability of one label-free approach to measure virtually any aptamer target by tailoring the aptamer of choice.
4. Conclusions
TRPS is a technology platform that is becoming more widely accepted amongst analytical laboratories as a common platform for analysis, one of the reasons being the many variants of samples and analytes that can be analysed. Current TRPS users have contributed greatly towards the validation of the technique for future studies with the technology. In our opinion we are at a tipping point where the data from TRPS no longer needs to be verified alongside DLS, TEM or other technologies. We predict that as this technique continues to develop within the individual fields of MV, protein and DNA analysis, TRPS will become more capable of multiplexing across different ohmic fields, and it is this exciting possibility that makes TRPS an exciting platform to work with.Acknowledgements
The authors thank the Centre for Analytical Science at Loughborough University. The work was supported by the European Commission for Research (PCIG11-GA-2012-321836 Nano4Bio) and Loughborough University Chemistry Department (Start-up fund). The research was supported by the National Institute for Health Research (NIHR) Diet, Lifestyle & Physical Activity Biomedical Research Unit based at University Hospitals of Leicester and Loughborough University. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. L. Mayne is supported by EPSRC EP/L014041/1 – DISTINCTIVE (http://distinctiveconsortium.org). Emma Blundell is sponsored by Izon Science Ltd. The author also thanks Dr Steve Christie for his support.References
- R. R. Henriquez, T. Ito, L. Sun and R. M. Crooks, Analyst, 2004, 129, 478–482 RSC.
- D. Kozak, W. Anderson, R. Vogel and M. Trau, Nano Today, 2011, 6, 531–545 CrossRef CAS PubMed.
- T. Albrecht, T. Gibb and P. Nuttall, Engineered Nanopores for Bioanalytical Applications, Elsevier, 2013 Search PubMed.
- M. Rhee and M. A. Burns, Trends Biotechnol., 2007, 25, 174–181 CrossRef CAS PubMed.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs, X. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS PubMed.
- T. Ito, L. Sun, R. R. Henriquez and R. M. Crooks, Acc. Chem. Res., 2004, 37, 937–945 CrossRef CAS PubMed.
- O. A. Saleh and L. L. Sohn, Nano Lett., 2002, 3, 37–38 CrossRef.
- R. An, J. D. Uram, E. C. Yusko, K. Ke, M. Mayer and A. J. Hunt, Opt. Lett., 2008, 33, 1153–1155 CrossRef PubMed.
- C. Dekker, Nat. Nanotechnol., 2007, 2, 209–215 CrossRef CAS PubMed.
- S. Lee, Y. Zhang, H. S. White, C. C. Harrell and C. R. Martin, Anal. Chem., 2004, 76, 6108–6115 CrossRef CAS PubMed.
- S. Nam, I. Choi, C. Fu, K. Kim, S. Hong, Y. Choi, A. Zettl and L. P. Lee, Nano Lett., 2014, 14, 5584–5589 CrossRef CAS PubMed.
- E. A. Heins, Z. S. Siwy, L. A. Baker and C. R. Martin, Nano Lett., 2005, 5, 1824–1829 CrossRef CAS PubMed.
- L. Sexton, L. Horne and C. Martin, in Molecular- and Nano-Tubes SE-6, ed. O. Hayden and K. Nielsch, Springer, USA, 2011, pp. 165–207 Search PubMed.
- H. Bayley and C. R. Martin, Chem. Rev., 2000, 100, 2575–2594 CrossRef CAS PubMed.
- G. S. Roberts, D. Kozak, W. Anderson, M. F. Broom, R. Vogel and M. Trau, Small, 2010, 6, 2653–2658 CrossRef CAS PubMed.
- G. S. Roberts, S. Yu, Q. Zeng, L. C. L. Chan, W. Anderson, A. H. Colby, M. W. Grinstaff, S. Reid and R. Vogel, Biosens. Bioelectron., 2012, 31, 17–25 CrossRef CAS PubMed.
- G. R. Willmott, R. Vogel, S. S. C. Yu, L. G. Groenewegen, G. S. Roberts, D. Kozak, W. Anderson and M. Trau, J. Phys.: Condens. Matter, 2010, 22, 454116 CrossRef CAS PubMed.
- G. R. Willmott and P. W. Moore, Nanotechnology, 2008, 19, 475504 CrossRef CAS PubMed.
- G. R. Willmott, M. Platt and G. U. Lee, Biomicrofluidics, 2012, 6, 14103–14115 CrossRef PubMed.
- G. R. Willmott and B. E. T. Parry, J. Appl. Phys., 2011, 109, 094307 CrossRef.
- R. Vogel, W. Anderson, J. Eldridge, B. Glossop and G. Willmott, Anal. Chem., 2012, 84, 3125–3131 CrossRef CAS PubMed.
- R. Vogel, G. Willmott, D. Kozak, G. S. Roberts, W. Anderson, L. Groenewegen, B. Glossop, A. Barnett, A. Turner and M. Trau, Anal. Chem., 2011, 83, 3499–3506 CrossRef CAS PubMed.
- G. R. Willmott, S. S. C. Yu and R. Vogel, 2010 Int. Conf. Nanosci. Nanotechnol., 2010, pp. 128–131 Search PubMed.
- D. Kozak, W. Anderson, R. Vogel, S. Chen, F. Antaw and M. Trau, ACS Nano, 2012, 6, 6990–6997 CrossRef CAS PubMed.
- G. S. Roberts, S. Yu, Q. Zeng, L. C. L. Chan, W. Anderson, A. H. Colby, M. W. Grinstaff, S. Reid and R. Vogel, Biosens. Bioelectron., 2011, 31, 17–25 CrossRef PubMed.
- H. M. Davey and D. B. Kell, Microbiol. Rev., 1996, 60, 641–696 CAS.
- S. J. Sowerby, M. F. Broom and G. B. Petersen, Sens. Actuators, B, 2007, 123, 325–330 CrossRef CAS.
- D. Kozak, W. Anderson and M. Trau, Chem. Lett., 2012, 41, 1134–1136 CrossRef CAS.
- G. R. Willmott, R. Vogel, S. S. C. Yu, L. G. Groenewegen, G. S. Roberts, D. Kozak, W. Anderson and M. Trau, J. Phys.: Condens. Matter, 2010, 22, 454116 CrossRef CAS PubMed.
- A. Sze, D. Erickson, L. Ren and D. Li, J. Colloid Interface Sci., 2003, 261, 402–410 CrossRef CAS PubMed.
- M. Kaszuba, J. Corbett, F. M. Watson and A. Jones, Philos. Trans. R. Soc., A, 2010, 368, 4439–4451 CrossRef CAS PubMed.
- L. T. Sexton, L. P. Horne and C. R. Martin, Mol. BioSyst., 2007, 3, 667–685 RSC.
- L. T. Sexton, H. Mukaibo, P. Katira, H. Hess, S. A. Sherrill, L. P. Horne and C. R. Martin, J. Am. Chem. Soc., 2010, 132, 6755–6763 CrossRef CAS PubMed.
- A. L. Loeb, J. T. G. Overbeek and P. H. Wiersema, J. Electrochem. Soc., 1961, 108, 269C–269C CrossRef.
- R. Vogel, W. Anderson, J. Eldridge, B. Glossop and G. R. Willmott, Anal. Chem., 2011, 84, 3125–3131 CrossRef PubMed.
- A. C. S. Yu, J. F. C. Loo, S. Yu, S. K. Kong and T.-F. Chan, Appl. Microbiol. Biotechnol., 2014, 98, 855–862 CrossRef CAS PubMed.
- C. Begot, I. Desnier and J. Daudin, J. Microbiol., 1996, 25, 225–232 Search PubMed.
- J. Maki and C. Remsen, Appl. Environ. Microbiol., 1981, 41, 1132–1138 CAS.
- M. K. Winson and H. M. Davey, Methods, 2000, 21, 231–240 CrossRef CAS PubMed.
- R. L. Kepner and J. R. Pratt, 1994, 603–615.
- A. Zeltins, Mol. Biotechnol., 2013, 53, 92–107 CrossRef CAS PubMed.
- A. Voller and A. Bartlett, J. Gen. Virol., 1976, 65–67 Search PubMed.
- F. Ronghui, H. Minling, W. Jianche and L. Yuanhua, Plant Dis. Pests, 2013, 4, 33–35 Search PubMed.
- K. Prabha, V. K. Baranwal and R. K. Jain, Indian J. Virol., 2013, 24, 157–165 CrossRef CAS PubMed.
- T. Notomi and H. Okayama, Nucleic Acids, 2000, 28, 63 CrossRef.
- W. I. Goldburg, Am. J. Phys., 1999, 67, 1152 CrossRef.
- J. Skog, T. Wurdinger, S. Van Rijn, D. Meijer, L. Gainche, M. Sena-esteves, W. T. Curry Jr, R. S. Carter, A. M. Krichevsky and X. O. Breakefield, Nat. Cell Biol., 2012, 10, 1470–1476 CrossRef PubMed.
- Y. Meng, S. Kang and D. A. Fishman, Cancer Immunol. Immunother., 2005, 54, 807–814 CrossRef CAS PubMed.
- L. G. Lima, R. Chammas, R. Q. Monteiro, M. E. C. Moreira and M. A. Barcinski, Cancer Lett., 2009, 283, 168–175 CrossRef CAS PubMed.
- C. Grange, M. Tapparo, F. Collino, L. Vitillo, C. Damasco, M. C. Deregibus, C. Tetta, B. Bussolati and G. Camussi, Cancer Res., 2011, 71, 5346–5356 CrossRef CAS PubMed.
- F. Momen-Heravi, L. Balaj, S. Alian, J. Tigges, V. Toxavidis, M. Ericsson, R. J. Distel, A. R. Ivanov, J. Skog and W. P. Kuo, Front. Physiol., 2012, 3, 354 Search PubMed.
- F. Momen-Heravi, L. Balaj, S. Alian, A. J. Trachtenberg, F. H. Hochberg, J. Skog and W. P. Kuo, Front. Physiol., 2012, 3, 162 CAS.
- E. van der Pol, F. A. W. Coumans, A. E. Grootemaat, C. Gardiner, I. L. Sargent, P. Harrison, A. Sturk, T. G. van Leeuwen and R. Nieuwland, J. Thromb. Haemostasis, 2014, 12, 1182–1192 CrossRef CAS PubMed.
- K. D. Connolly, G. R. Willis, D. B. N. Datta, E. A. Ellins, D. A. Price, I. A. Guschina, D. A. Rees and P. E. James, J. Lipid Res., 2014, 55, 2064–2072 CrossRef CAS PubMed.
- J. O. Burton, H. a. Hamali, R. Singh, N. Abbasian, R. Parsons, A. K. Patel, A. H. Goodall and N. J. Brunskill, PLoS One, 2013, 8, e72663 CAS.
- J. Emmerechts, L. Jacobs, S. Van Kerckhoven, S. Loyen, C. Mathieu, F. Fierens, B. Nemery, T. S. Nawrot and M. F. Hoylaerts, J. Thromb. Haemostasis, 2012, 10, 96–106 CrossRef CAS PubMed.
- Z. Varga, Y. Yuana, A. E. Grootemaat, E. van der Pol, C. Gollwitzer, M. Krumrey and R. Nieuwland, J. Extracell. Vesicles, 2014, 3, 23298 CrossRef PubMed.
- L. Musante, D. Tataruch, D. Gu, A. Benito-Martin, G. Calzaferri, S. Aherne and H. Holthofer, Sci. Rep., 2014, 4, 7532 CrossRef CAS PubMed.
- G. Szabó, B. Tarr, K. Pálóczi, K. Éder, E. Lajkó, Á. Kittel, S. Tóth, B. György, M. Pásztói, A. Németh, X. Osteikoetxea, É. Pállinger, A. Falus, K. Szabó-Taylor and E. Buzás, Cell. Mol. Life Sci., 2014, 1–13 Search PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- J. Llandro, J. Palfreyman, A. Ionescu and C. Barnes, Med. Biol. Eng. Comput., 2010, 48, 977–998 CAS.
- Y. S. Ang and L.-Y. L. Yung, ACS Nano, 2012, 6, 8815–8823 CrossRef CAS PubMed.
- C. M. Niemeyer and D. Blohm, Angew. Chem., 1999, 38, 2865–2869 CrossRef CAS.
- C. Niemeyer, Angew. Chem., Int. Ed. Engl., 2001, 40, 4128–4158 CrossRef CAS.
- M. A. Booth, R. Vogel, J. M. Curran, S. Harbison and J. Travas-Sejdic, Biosens. Bioelectron., 2013, 45, 136–140 CrossRef CAS PubMed.
- J. A. Somerville, G. R. Willmott, J. Eldridge, M. Griffiths and K. M. McGrath, J. Colloid Interface Sci., 2013, 394, 243–251 CrossRef CAS PubMed.
- V. Gubala, C. Crean, R. Nooney, S. Hearty, B. McDonnell, K. Heydon, R. O'Kennedy, B. D. MacCraith and D. E. Williams, Analyst, 2011, 136, 2533–2541 RSC.
- E. R. Billinge, J. Muzard and M. Platt, Nanomaterials and Nanosciences, 2013, 1, 1 CrossRef.
- C.-C. Huang, Y.-F. Huang, Z. Cao, W. Tan and H.-T. Chang, Anal. Chem., 2005, 77, 5735–5741 CrossRef CAS PubMed.
- J. Baudry, C. Rouzeau, C. Goubault, C. Robic, L. Cohen-Tannoudji, A. Koenig, E. Bertrand and J. Bibette, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16076–16078 CrossRef CAS PubMed.
- W. Rowe, M. Platt and P. J. R. Day, Integr. Biol., 2009, 1, 53–58 RSC.
- E. Levy-Nissenbaum, A. F. Radovic-Moreno, A. Z. Wang, R. Langer and O. C. Farokhzad, Trends Biotechnol., 2008, 26, 442–449 CrossRef CAS PubMed.
- G. Mayer, Angew. Chem., Int. Ed. Engl., 2009, 48, 2672–2689 CrossRef CAS PubMed.
- X. Fang and W. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS PubMed.
- M. Platt, W. Rowe, J. Knowles, P. J. Day and D. B. Kell, Integr. Biol., 2009, 1, 116–122 RSC.
- L.-L. Li, P. Ge, P. R. Selvin and Y. Lu, Anal. Chem., 2012, 84, 7852–7856 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- C. G. Knight, M. Platt, W. Rowe, D. C. Wedge, F. Khan, P. J. R. Day, A. McShea, J. Knowles and D. B. Kell, Nucleic Acids Res., 2009, 37, e6 CrossRef PubMed.
- M. Platt, W. Rowe, D. C. Wedge, D. B. Kell, J. Knowles and P. J. R. Day, Anal. Biochem., 2009, 390, 203–205 CrossRef CAS PubMed.
- W. Rowe, D. C. Wedge, M. Platt, D. B. Kell and J. Knowles, Bioinformatics, 2010, 26, 2145–2152 CrossRef CAS PubMed.
- E. R. Billinge, M. Broom and M. Platt, Anal. Chem., 2014, 86, 1030–1037 CrossRef CAS PubMed.
- P. G. Righettl and G. Tudor, J. Chromatogr., 1981, 220, 115–194 CrossRef.
- O. a. Alsager, S. Kumar, G. R. Willmott, K. P. McNatty and J. M. Hodgkiss, Biosens. Bioelectron., 2014, 57, 262–268 CrossRef CAS PubMed.
- M. Platt, G. R. Willmott and G. U. Lee, Small, 2012, 8, 2436–2444 CrossRef CAS PubMed.
- E. R. Billinge and M. Platt, artic. submitt., 2015.
Footnote |
| † Authors contributed equally to the paper. |
| This journal is © The Royal Society of Chemistry 2015 |