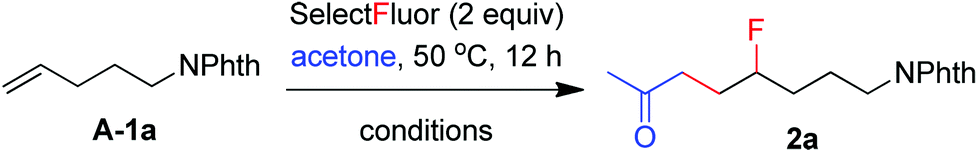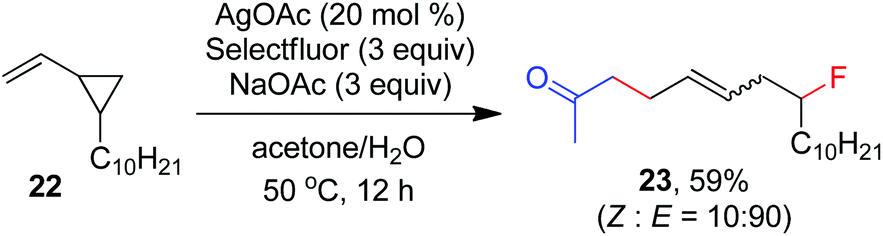DOI:
10.1039/C4QO00256C
(Research Article)
Org. Chem. Front., 2014,
1, 1299-1305
Formal fluorine atom transfer radical addition: silver-catalyzed carbofluorination of unactivated alkenes with ketones in aqueous solution†
Received
26th September 2014
, Accepted 12th November 2014
First published on 13th November 2014
Abstract
In this article, we report the first examples of carbofluorination of unactivated alkenes. Under catalysis by AgNO3, the reactions of unactivated alkenes with Selectfluor reagent and active methylene compounds such as acetoacetates or 1,3-dicarbonyls in CH2Cl2–H2O–HOAc solution afforded the corresponding three-component condensation products under mild conditions. Furthermore, with the promotion of NaOAc, the AgOAc-catalyzed carbofluorination of various unactivated alkenes with Selectfluor and acetone proceeded smoothly in aqueous solution at 50 °C. The carbofluorination was efficient and highly regioselective, and enjoyed a broad substrate scope and wide functional group compatibility. These formal fluorine atom transfer radical addition reactions provide a convenient entry to structurally divergent, polyfunctional organofluorine compounds as versatile synthetic intermediates.
Introduction
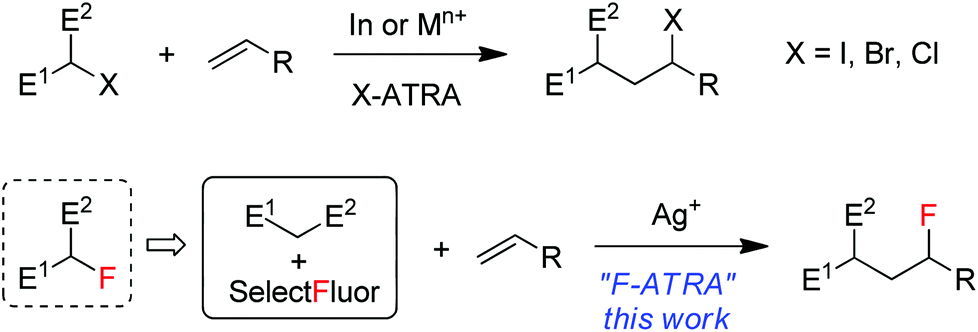
Atom transfer radical addition (ATRA) reactions, discovered by Kharasch1 and significantly promoted by Curran2 and others, have been demonstrated to be a versatile tool in organic synthesis.3 The most common ATRA processes are iodine-atom-transfer in which carbon–iodine bonds are added across alkenes or alkynes in the presence of a radical initiator. A representative example is the addition of iodinated active methylene compounds onto electron-rich alkenes, as shown in Scheme 1, a process driven partially by a radical polar effect.4 Bromine and chlorine ATRA reactions have also been developed and can be catalyzed by a number of transition metal complexes such as copper and ruthenium via transition-metal-assisted Cl (or Br)-atom-transfer mechanism.5 On the other hand, fluorine ATRA reactions are virtually unknown probably because of the much higher C–F bond dissociation energies. However, if one considers that monofluorinated active methylene compounds are typically prepared from the reactions of active methylene compounds with electrophilic fluorinating reagents such as Selectfluor (1-chloromethyl-4-fluoro-1,4-diazoniabicyclic[2.2.2]octane bis(tetrafluoroborate)),6 it would be an interesting variant of F-ATRA to use directly a combination of the two starting materials as a substitute for their products (monofluorinated active methylene compounds) (Scheme 1). In view of the widespread and growing use7 of organofluorine compounds in agrochemicals, pharmaceuticals and materials and the importance of 18F-labeled organic compounds in positron emission tomography (PET),8 this formal F-ATRA process should be advantageous in that it allows the rapid assembly of cheap and readily available substrates and fluorinating reagents into polyfunctional fluorinated molecules.9 Herein we report silver-catalyzed formal F-ARTA reactions in aqueous solution.
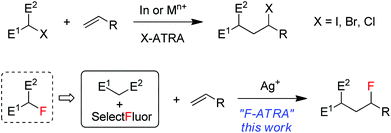 |
| | Scheme 1 ATRA versus formal ATRA. | |
Results and discussion
We recently reported10 that the combination of a catalytic amount of AgNO3 with Selectfluor resulted in the decarboxylative fluorination of aliphatic carboxylic acids,10a the intramolecular aminofluorination of alkenes10b and the intermolecular phosphonofluorination of unactivated alkenes.10c During the course of these studies, we noted that small amounts of hexane-2,5-dione could be detected by GC-MS in some cases when acetone was used as the co-solvent. This phenomenon implied that acetonyl radicals might be generated from the oxidation of acetone11 by reaction with AgNO3–Selectfluor. If this was the case, electrophilic α-keto radicals might be produced and trapped by electron-rich alkenes to give the adduct radicals as nucleophilic alkyl radicals. The subsequent fluorination of the adduct radicals would lead to the carbofluorination products. It should be pointed out that only a few examples of intermolecular carbofluorination have been reported and they dealt with activated alkenes such as styrenes or enamines.12,13
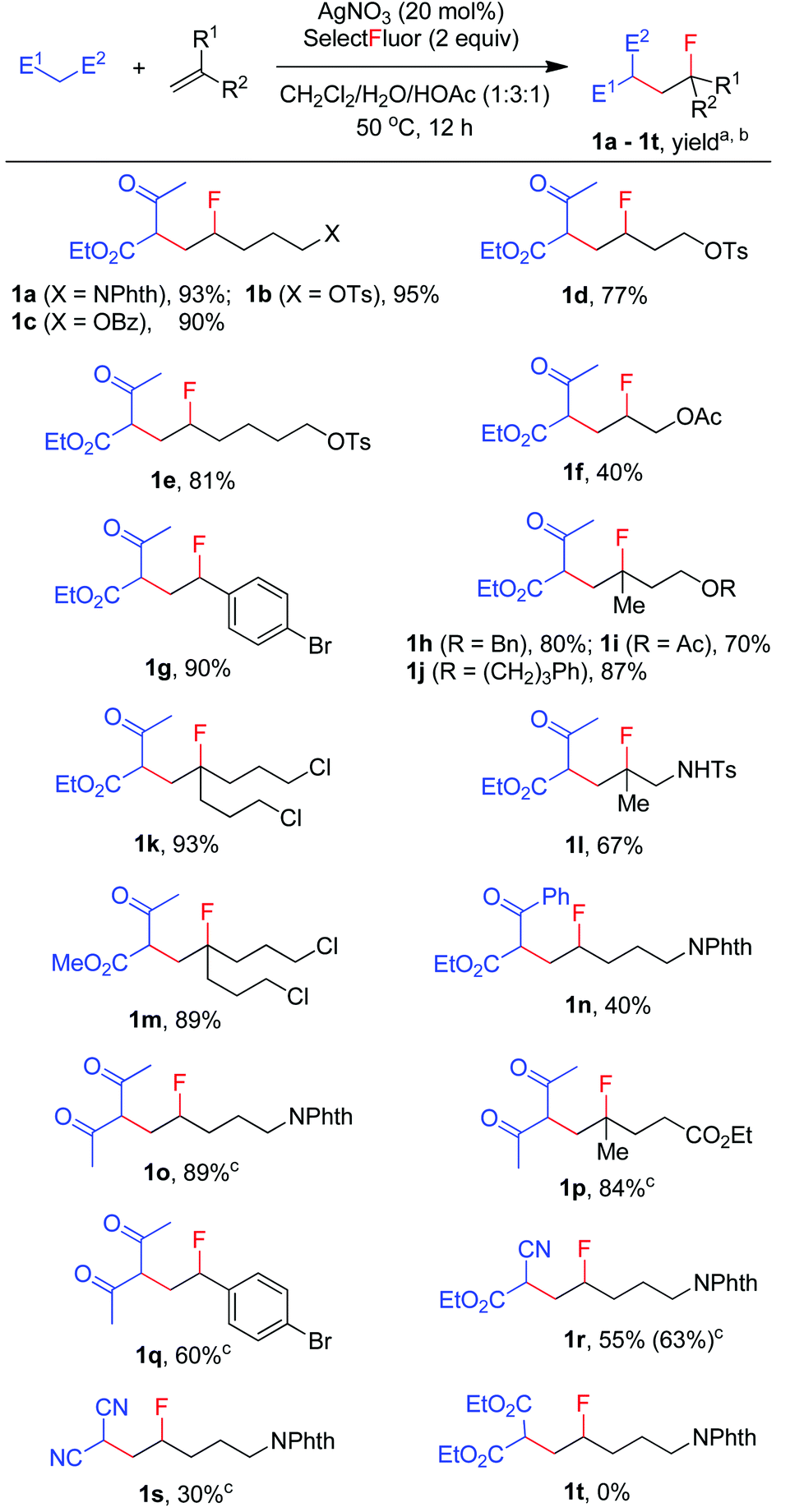
Thus, N-(pent-4-en-1-yl)phthalimide (A-1a) and ethyl acetoacetate, which is more prone to oxidation than acetone,14 were used as the model substrates in the search for optimal reaction conditions (see Table S1 in the ESI† for details). We were delighted to find that, with catalysis by AgNO3, the reaction of the two model substrates with Selectfluor proceeded smoothly in CH2Cl2–H2O–HOAc (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1) at reflux (∼50 °C) leading to the expected F-ATRA product 1a in 93% isolated yield. Control experiments indicated that AgNO3 was necessary to initiate the reaction, while other silver(I) salts such as AgOAc showed similar effect. No fluorination took place when Selectfluor was switched to N-fluorobis-(benzenesulfonyl)imide (NSFI).15 To the best of our knowledge, this is also the first example of intermolecular carbofluorination of unactivated alkenes.
:3:1) at reflux (∼50 °C) leading to the expected F-ATRA product 1a in 93% isolated yield. Control experiments indicated that AgNO3 was necessary to initiate the reaction, while other silver(I) salts such as AgOAc showed similar effect. No fluorination took place when Selectfluor was switched to N-fluorobis-(benzenesulfonyl)imide (NSFI).15 To the best of our knowledge, this is also the first example of intermolecular carbofluorination of unactivated alkenes.
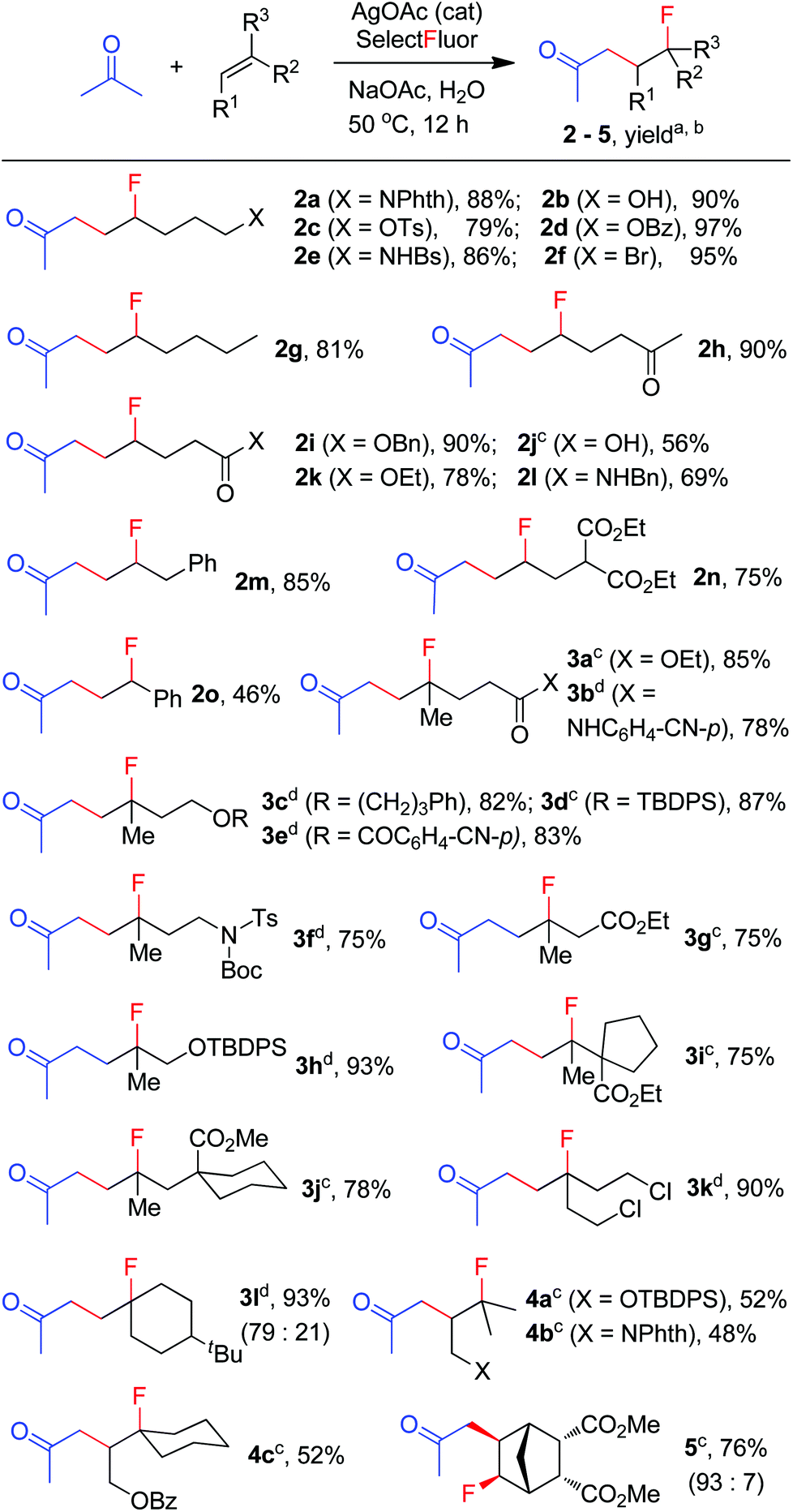
We then moved on to examine the scope and limitation of this new method. As illustrated in Scheme 2, a number of electron-rich mono-substituted alkenes underwent F-ATRA to afford the expected products 1a–1f in satisfactory yields. Styrenes were also excellent substrates for the condensation, as exemplified by the synthesis of 1g in 90% yield. Di-substituted alkenes also exhibited a high reactivity (1h–1l). Functional groups such as ester, tosylate, sulfonamide and aryl or alkyl halide, were well tolerated. Active methylene compounds other than acetoacetates could also be employed for the carbofluorination. While the reactions of acetylacetone proceeded sluggishly under the above conditions, they were speeded up by the addition of K2S2O8 (1 equiv.), furnishing the corresponding products (1o–1q) in high yields. The role of K2S2O8 remains unclear at this moment.16 Cyanoacetate was also a good substrate for the condensation, as proved by the synthesis of 1r in good yield. On the other hand, malononitrile showed a low efficiency and diethyl malonate failed to give any carbofluorination product.17 The results seem to indicate that the reactivity of active methylene compounds decreases in the order of acetoacetate > acetylacetone > cyanoacetate > malononitrile > malonate.
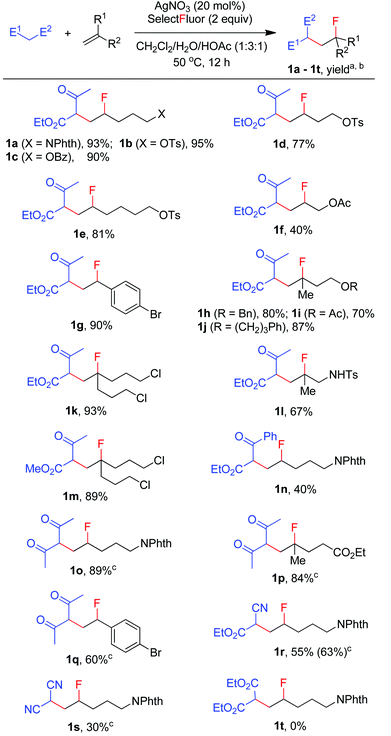 |
| | Scheme 2 Carbofluorination of alkenes with active methylene compounds. aReaction conditions: alkene (1 mmol), ketone (3 mmol), Selectfluor (2 mmol), AgNO3 (0.2 mmol), CH2Cl2 (2.5 mL), H2O (7.5 mL), HOAc (2.5 mL), 50 °C, 12 h. bIsolated yield based on the alkene. cK2S2O8 (1 equiv.) was used as the additive. | |
The above excellent performance of acetoacetates and 1,3-diketones urged us to examine the possibility of using ordinary ketones such as acetone in the carbofluorination. The condensation of acetone with alkene A-1a and Selectfluor under the above conditions gave only a trace amount of the expected carbofluorination product 2a. We then went on to optimize the reaction conditions (Table 1). When acetone was directly used as the co-solvent, all the alkene A-1a was consumed and 2a was isolated in 17% yield (entry 4, Table 1). Switching AgNO3 to AgOAc did not increase the product yield (entry 5, Table 1). However, a large portion (∼60%) of alkene A-1a was now recovered. This unusual difference prompted us to check the effect of NaOAc. Indeed, with 3 equivalents of NaOAc as the additive, the product yield was increased to 90% (entry 8, Table 1). The reaction also proceeded smoothly when the catalyst loading was lowered to 10 mol% (entry 9, Table 1). Notably, with water as the only solvent and 3 equivalents of acetone as the substrate, the reaction also proceeded to give the product 2a in 30% yield (entry 11, Table 1). While the role of NaOAc is not clear at this moment, it might act as a weak base to absorb the acid generated during the reaction. KOAc showed an effect similar to NaOAc (entry 13, Table 1). However, the reaction was significantly retarded when NaOAc was replaced by NaHCO3 (entry 14, Table 1).
Table 1 Optimization of conditions for the synthesis of 2a
|

|
| Entrya |
Conditions |
Yieldb (%) |
|
Conditions: A-1a (0.2 mmol), Selectfluor (0.4 mmol), acetone, H2O (2 mL), Ag(I) source, organic solvent and additive if applicable, 50 °C, 12 h.
Isolated yield based on A-1a.
Water (1.5 mL) was used.
|
| 1c |
AgNO3 (20 mol %), acetone (3 equiv.), CH2Cl2–H2O–HOAc (1:3:1) |
Trace |
| 2c |
AgNO3 (20 mol %), acetone (3 equiv.), CH2Cl2–H2O (1:3) |
Trace |
| 3c |
AgNO3 (20 mol %), acetone (3 equiv.), CH3CN–H2O (1:3) |
Trace |
| 4 |
AgNO3 (20 mol %), acetone–H2O (1:1) |
17 |
| 5 |
AgOAc (20 mol %), acetone–H2O (1:1) |
18 |
| 6 |
AgOAc (20 mol %), NaOAc (1 equiv.), acetone–H2O (1:1) |
59 |
| 7 |
AgOAc (20 mol %), NaOAc (2 equiv.), acetone–H2O (1:1) |
75 |
| 8 |
AgOAc (20 mol %), NaOAc (3 equiv.), acetone–H2O (1:1) |
90 |
|
9
|
AgOAc (10 mol %), NaOAc (3 equiv.), acetone–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1) |
88
|
| 10 |
AgOAc (10 mol %), NaOAc (3 equiv.), acetone (10 equiv.), H2O |
63 |
| 11 |
AgOAc (10 mol %), NaOAc (3 equiv.), acetone (3 equiv.), H2O |
30 |
| 12 |
NaOAc (3 equiv.), acetone–H2O (1:1) |
0 |
| 13 |
AgOAc (10 mol %), KOAc (3 equiv.), acetone–H2O (1:1) |
84 |
| 14 |
AgOAc (10 mol %), NaHCO3 (3 equiv.), acetone–H2O (1:1) |
0 |
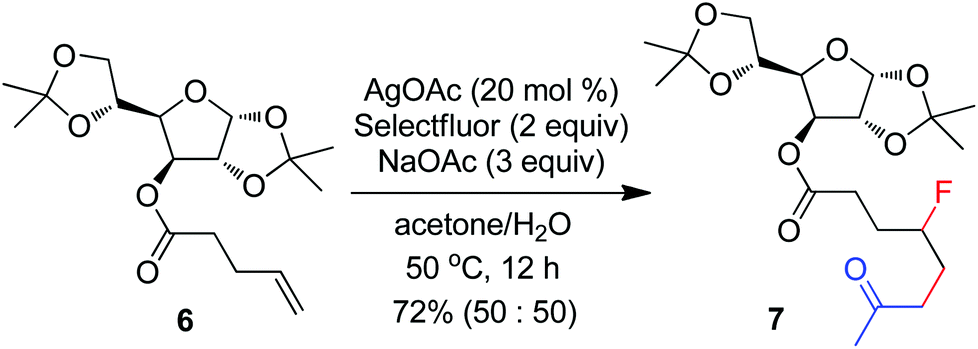
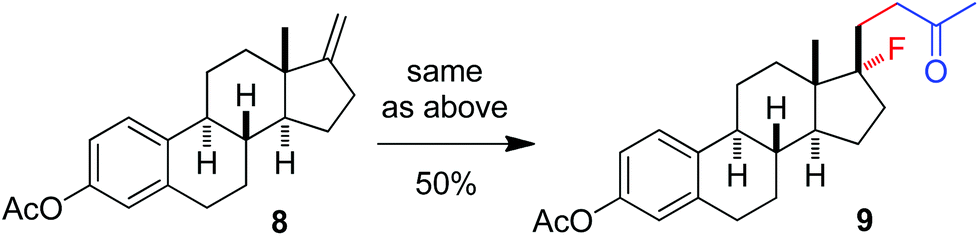
The above optimization revealed that the F-ATRA with acetone proceeded nicely under much milder conditions. The procedure was particularly advantageous in that no other organic solvent was required and the condensation was conducted under almost neutral conditions. As a result, the carbofluorination enjoyed a broad substrate scope and wide functional group compatibility, as demonstrated in Scheme 3. Mono-, di- and even tri-substituted alkenes were all good acceptors, furnishing the corresponding products 2, 3 and 4, respectively. Various functional groups were well tolerated, including labile free carboxylic acid, unprotected hydroxyl group and primary alkyl bromide (in 2b, 2j and 2f), a unique characteristic of ATRA reactions. In addition, the reactions were scalable and the products could be easily obtained in gram scale without any decrease in efficiency. Note that the reactions were not only efficient but also highly regioselective. The condensation could also be stereoselective, as indicated by the synthesis of bicyclic compound 5. This method could be utilized in the direct modification of complex molecules. For example, the condensation of carbohydrate 6 with acetone and Selectfluor gave the product 7 in 72% yield (eqn (1)). In another case, steroid 8 underwent stereoselective carbofluorination to give 9 as the only stereoisomer isolated (eqn (2)).
| | 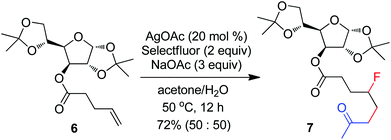 | (1) |
| | 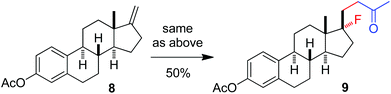 | (2) |
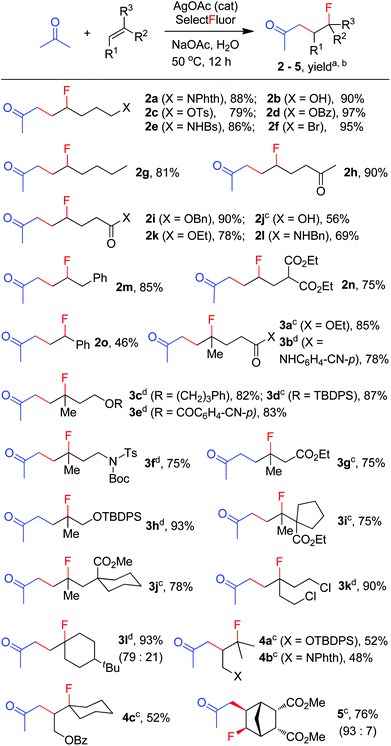 |
| | Scheme 3 Carbofluorination of alkenes with acetone. aReaction conditions: alkene (2 mmol), Selectfluor (4 mmol, 2 equiv.), AgOAc (0.2 mmol, 10 mol%), NaOAc (6 mmol), acetone (20 mL), H2O (20 mL), 50 °C, 12 h. bIsolated yield based on the alkene. cAgOAc (20 mol%) and H2O (10 mL) were used. dSelectfluor (3 equiv.) and AgOAc (15 mol%) were used. | |
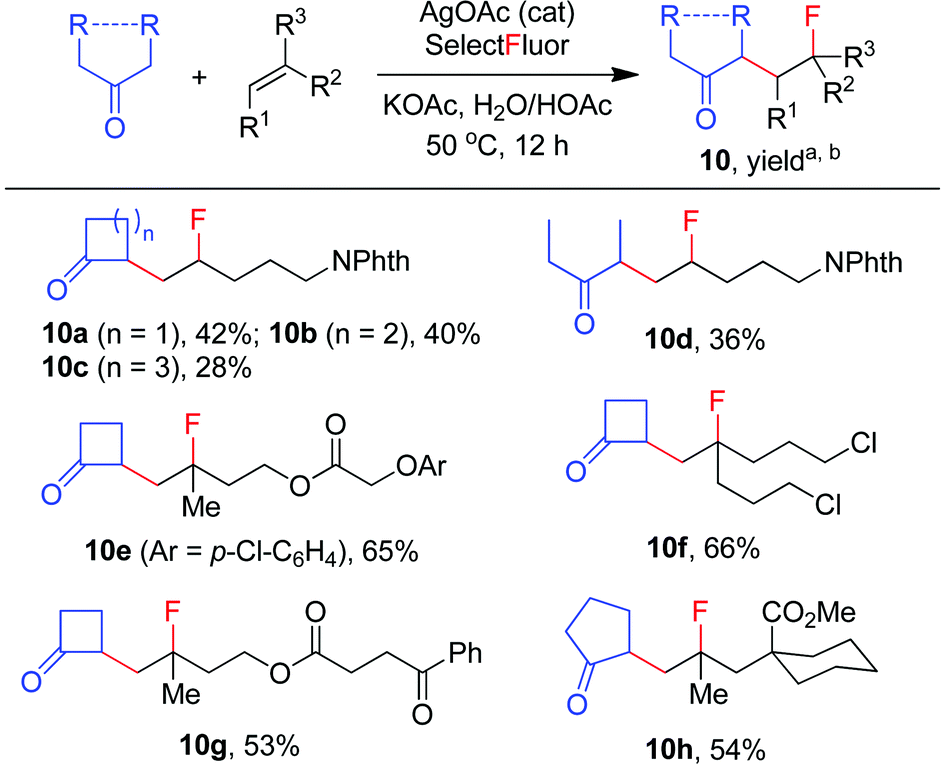
The carbofluorination could also be extended to the use of other ordinary ketones such as cycloalkanones and 3-pentanone, as depicted in Scheme 4. Under the re-optimized conditions, moderate to good efficiency was typically observed. Nevertheless, direct α-fluorination of ketones could now be detected in small extents, presumably due to the decreased electrophilicity of α-keto radicals (vide infra).
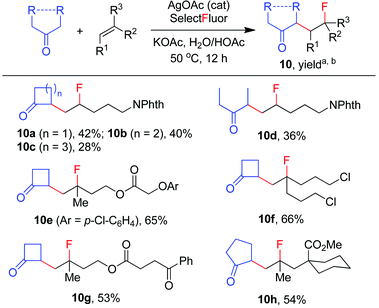 |
| | Scheme 4 Carbofluorination of alkenes with simple ketones. aReaction conditions: alkene (0.2 mmol), ketone (2 mmol), Selectfluor (0.4 mmol), AgOAc (0.04 mmol), KOAc (0.6 mmol), H2O (1 mL), HOAc (0.4 mL), 50 °C, 12 h. bIsolated yield based on the alkene. If applicable, the products were obtained as ∼1:1 mixture of stereoisomers. | |
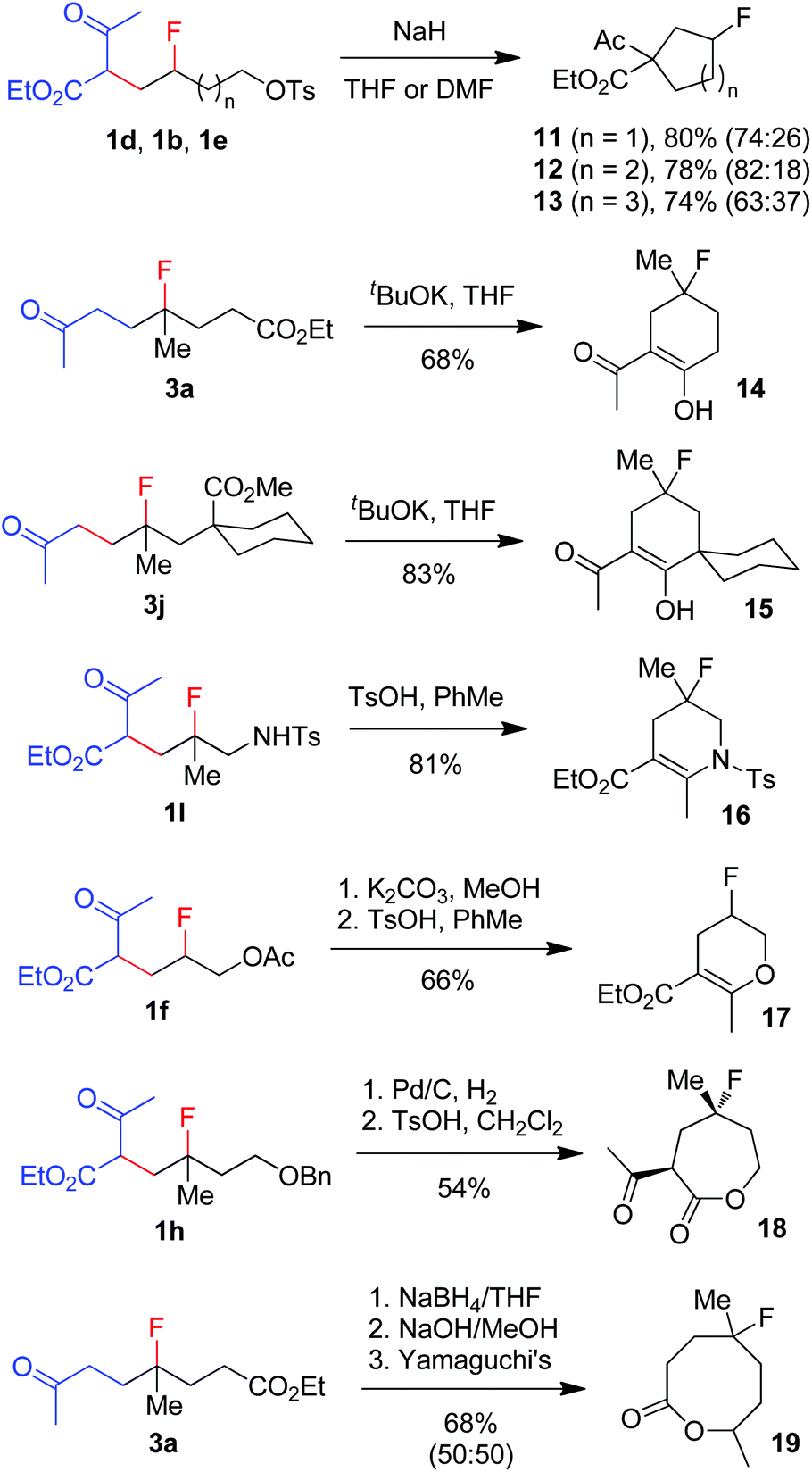
The broad substrate scope and excellent functional group compatibility of carbofluorination demonstrated above allow convenient access to divergent and polyfunctional organofluorine compounds, which should serve as versatile synthetic intermediates. For example, monofluorinated carbo- and heterocycles18 can be easily prepared from the carbofluorination products (Scheme 5). Thus, fluorinated carbocycles 11–13 were readily prepared in one step from compounds 1d, 1b and 1e, respectively, via intramolecular nucleophilic substitution. 4-Fluorocyclohexanones 14 and 15 were produced from 3a and 3jvia base-promoted Dieckmann condensation. Cyclic enamine 16 and enol ether 17 were readily obtained from 1l and 1fvia dehydration. Deprotection of benzyl ether 1h followed by ester exchange afforded 7-membered lactone 18 as a single stereoisomer. Reduction of ketone 3a followed by hydrolysis and subsequent Yamaguchi lactonization16 produced 8-membered lactone 19. It is conceivable that more new fluorinated cyclic compounds can be reached via similar strategies. Note that these cyclic compounds are also valuable building blocks in the synthesis of more complex fluorinated molecules.
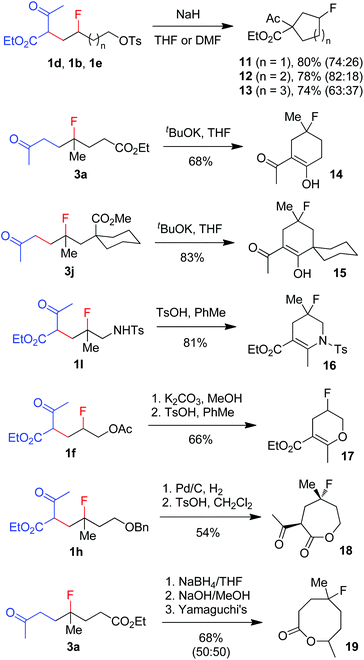 |
| | Scheme 5 Conversion of carbofluorination products to fluorinated carbo- and heterocycles. | |
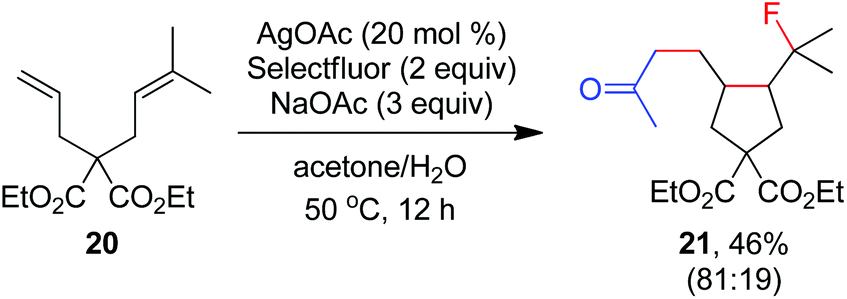
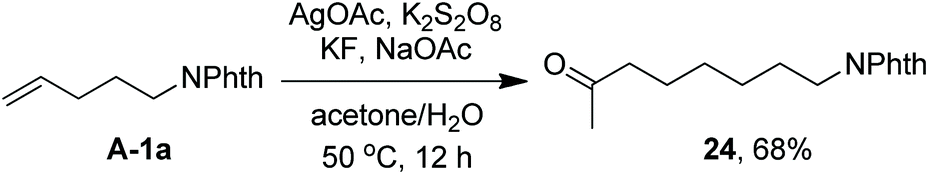
As indicated above, a radical fluorination10,12c,19,20 mechanism might be involved in the carbofluorination of unactivated alkenes. More direct evidence was the reaction of 1,6-diene 20 with acetone in which the cyclization product 21 was isolated in 46% yield along with the recovery of diene 20 in 23% yield (eqn (3)). Furthermore, vinylcyclopropane 22 was designed as a radical probe.21 The reaction of 22 with acetone under the above optimized conditions (Scheme 2) led to the ring-opening product 23 in 59% yield along with the recovery of 22 in 20% yield (eqn (4)). These experiments provide solid evidence for the intermediacy of α-carbonyl radicals in carbofluorination. To gain more insight into the mechanism, the following experiments were carried out. When Selectfluor was replaced by a combination of K2S2O8 (2 equiv.) and KF (2 equiv.), the reaction of acetone with alkene A-1a gave the addition–hydrogenation product 24 in 68% yield while no carbofluorination product 2a could be detected (eqn (5)). This implied that Ag(II)F alone was unlikely to initiate the carbofluorination. When AgOAc was switched to divalent silver–1,10-phenanthroline complex Ag(Phen)2S2O8, the reaction of acetone with A-1a and Selectfluor under various conditions also led to the formation of 24 rather than 2a, indicating that the adduct radicals could not directly abstract fluorine atoms from Selectfluor.
| | 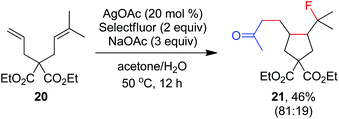 | (3) |
| | 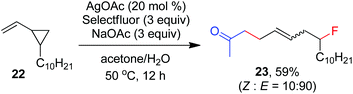 | (4) |
| | 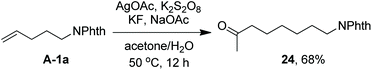 | (5) |
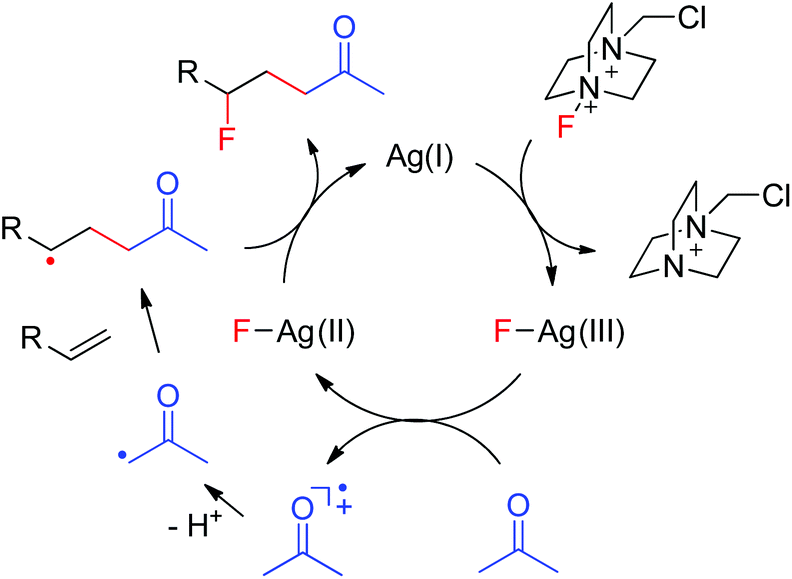
A plausible mechanism was thus proposed based on the above results as well as our previous findings.10 As shown in Fig. 1, the interaction of Ag(I) with Selectfluor generates the Ag(III)–F intermediate, presumably via oxidative addition. The single electron transfer between acetone and Ag(III)–F gives Ag(II)–F and acetonyl radical cation. Deprotonation of the acetonyl radical cation affords the electrophilic α-carbonyl radical, which adds selectively to the electron-rich C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to provide the adduct radical as a nucleophilic alkyl radical. The subsequent fluorine atom transfer from Ag(II)–F to the adduct radical leads to the carbofluorination product and regenerates the catalyst Ag(I), which enters into the next catalytic circle. This mechanism well explains the umpolung of ketones. Moreover, when α-keto radicals bear an alkyl substituent, they become less electrophilic and the efficiency of carbofluorination is lowered, consistent with our experimental observations. Note that the proposed mechanism is closely similar to that of transition-metal-assisted Cl-ATRA,3b,5 thus justifying its classification as a F-ATRA process.
C bond to provide the adduct radical as a nucleophilic alkyl radical. The subsequent fluorine atom transfer from Ag(II)–F to the adduct radical leads to the carbofluorination product and regenerates the catalyst Ag(I), which enters into the next catalytic circle. This mechanism well explains the umpolung of ketones. Moreover, when α-keto radicals bear an alkyl substituent, they become less electrophilic and the efficiency of carbofluorination is lowered, consistent with our experimental observations. Note that the proposed mechanism is closely similar to that of transition-metal-assisted Cl-ATRA,3b,5 thus justifying its classification as a F-ATRA process.
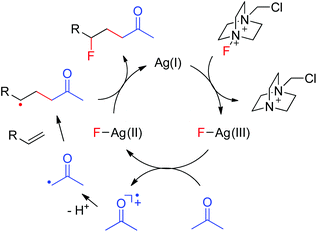 |
| | Fig. 1 Proposed mechanism of carbofluorination. | |
Conclusion
In conclusion, we have successfully developed silver-catalyzed formal F-ARTA reactions employing ketones and Selectfluor as a substitute for α-fluoroketones, allowing the three-component condensation of unactivated alkenes, ketones and Selectfluor in aqueous solution under mild conditions. The results significantly expand the scope of radical fluorination. Furthermore, they offer a new way of thinking for the practice of ATRA reactions, which should find important application both in radical chemistry and in organofluorine chemistry.
Experimental section
Typical procedure for silver-catalyzed carbofluorination of unactivated alkenes with active methylene compounds
N-(Pent-4-en-1-yl)phthalimide (A-1a, 215 mg, 1.0 mmol), AgNO3 (34 mg, 0.20 mmol) and Selectfluor (706 mg, 2.0 mmol) were placed in a Schlenk tube under nitrogen atmosphere. Dichloromethane (2.5 mL), water (7.5 mL), acetic acid (2.5 mL) and ethyl acetoacetate (0.38 mL, 3.0 mmol) were added successively at room temperature. The reaction mixture was gently refluxed (at ∼50 °C) with stirring for 12 h. The resulting mixture was cooled down to room temperature and extracted with CH2Cl2 (3 × 20 mL). The organic phases were combined and dried over anhydrous Na2SO4. After the removal of solvent under reduced pressure, the crude product was purified by column chromatography on silica gel with hexane–ethyl acetate (5:1, v:v) as the eluent to give the pure product 1a as a colorless oil. Yield: 338 mg (93%). Ketone 1a is in equilibrium with its enol form in CDCl3 (in ∼86:14 ratio at 20 °C), as indicated by 1H NMR. 1H NMR (400 MHz, CDCl3) δ 7.82–7.84 (m, 2H), 7.71–7.73 (m, 2H), 4.40–4.61 (m, 1H), 4.17–4.27 (m, 2H), 3.66–3.75 (m, 3H), 2.00–2.52 (m, 5H), 1.59–1.89 (m, 4H), 1.24–1.30 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 202.3/202.2, 169.2/169.0, 168.2, 133.9, 131.9, 123.1, 91.6/91.5 (2d, J = 167.5 Hz), 61.5, 55.5/55.2 (2d, J = 2.3 Hz), 37.3, 33.3/33.1 (2d, J = 19.3 Hz), 32.5 (d, J = 20.5 Hz)/32.4 (d, J = 20.0 Hz), 29.5/29.0, 24.2 (d, J = 1.5 Hz)/24.1 (2d, J = 1.2 Hz), 13.9; 19F NMR (282 MHz, CDCl3): δ −182.9/−183.5 (2m, 1F); IR (film): ν (cm−1) 1772, 1713; ESI-MS: (m/z) 386.1 (M+ + Na); HRMS calcd for C19H22FNNaO5 (M + Na): 386.1374, found: 386.1361.
Typical procedure for silver-catalyzed carbofluorination of unactivated alkenes
N-(Pent-4-en-1-yl)phthalimide (A-1a, 431 mg, 2.0 mmol), AgOAc (34 mg, 0.20 mmol), Selectfluor (1.41 g, 4.0 mmol) and sodium acetate (489 mg, 6.0 mmol) were placed in a Schlenk tube under nitrogen atmosphere. Water (20 mL) and acetone (20 mL) were then added successively at room temperature. The reaction mixture was then stirred at 50 °C for 12 h. The resulting mixture was cooled down to room temperature and extracted with CH2Cl2 (3 × 50 mL). The organic phases were combined and dried over anhydrous Na2SO4. After the removal of solvent under reduced pressure, the crude product was purified by column chromatography on silica gel with hexane–ethyl acetate (10:1, v:v) as the eluent to give the pure product 2a as a white solid. Mp: 56–58 °C. Yield: 512 mg (88%). 1H NMR (400 MHz, CDCl3): δ 7.73–7.77 (m, 2H), 7.62–7.66 (m, 2H), 4.35–4.52 (m, 1H), 3.61–3.65 (m, 2H), 2.46–2.57 (m, 2H), 2.07 (s, 3H), 1.45–1.87 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 207.7, 168.3, 133.9, 132.0, 123.1, 92.8 (d, J = 167.0 Hz), 38.7 (d, J = 3.8 Hz), 37.4, 32.4 (d, J = 20.5 Hz), 29.9, 28.8 (d, J = 21.2 Hz), 24.3 (d, J = 3.8 Hz); 19F NMR (282 MHz, CDCl3): δ −183.7 (m, 1F); IR (KBr): ν (cm−1) 1772, 1712, 1615; ESI-MS: (m/z) 314.1 (M+ + Na); HRMS calcd for C16H18FNNaO3 (M + Na): 314.1163, Found 314.1176.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (grants 21228202, 21272259, 21290180, 21472220 and 21361140377) and by the National Basic Research Program of China (973 Program) (grants 2015CB931900 and 2011CB710805).
References
- M. S. Kharasch, E. V. Jenson and W. H. Urry, Science, 1945, 102, 128 CAS.
- D. P. Curran, M.-H. Chen and D. Kim, J. Am. Chem. Soc., 1986, 108, 2489–2490 CrossRef CAS PubMed.
- For reviews, see:
(a)
J. Byers, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001, vol. 1, pp. 72–89 Search PubMed;
(b)
C. Li, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Chichester, UK, 2012, pp. 943–964 Search PubMed.
-
(a) D. P. Curran, Synthesis, 1988, 417–439 CrossRef CAS PubMed;
(b) D. P. Curran, Synthesis, 1988, 489–513 CrossRef CAS.
-
(a) T. Pintauer, Eur. J. Inorg. Chem., 2010, 2449–2460 CrossRef CAS;
(b) K. Severin, Curr. Org. Chem., 2006, 10, 217–224 CrossRef CAS;
(c) A. J. Clark, Chem. Soc. Rev., 2002, 31, 1–11 RSC.
-
(a) R. E. Banks, S. N. Mohialdin-Khaffaf, G. S. Lal, I. Sharif and R. G. Syvret, J. Chem. Soc., Chem. Commun., 1992, 595–596 RSC;
(b) R. P. Singh and J. M. Shreeve, Acc. Chem. Res., 2004, 37, 31–44 CrossRef CAS PubMed;
(c) P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
-
(a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed;
(b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC;
(c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC;
(d) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
-
(a) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed;
(b) S. M. Ametamey, M. Horner and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- For the latest selected reviews, see:
(a) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160–171 CrossRef CAS PubMed;
(b) T. Furuya, J. E. M. N. Klein and T. Ritter, Synthesis, 2010, 1804–1821 CAS;
(c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed;
(d) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
-
(a) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed;
(b) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640–4643 CrossRef CAS PubMed;
(c) C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082–14085 CrossRef CAS PubMed.
- For the latest example of oxidative generation of acetonyl radicals, see: B. Schweitzer-Chaput, J. Demaerel, H. Engler and M. Klussmann, Angew. Chem., Int. Ed., 2014, 53, 8737–8740 CrossRef CAS PubMed.
-
(a) A. D. Dilman, P. A. Belyakov, M. I. Struchkova, D. E. Arkhipov, A. A. Korlyukov and V. A. Tartakovsky, J.Org.
Chem., 2010, 75, 5367–5370 CAS;
(b) E. P. A. Talbot, T. A. Fernandes, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 4101–4104 CrossRef CAS PubMed;
(c) H. Wang, L.-N. Guo and X.-H. Duan, Chem. Commun., 2014, 50, 7382–7384 RSC.
- For intramolecular carbofluorination, see: J. R. Wolstenhulme, J. Rosenqvist, O. Lozano, J. Ilupeju, N. Wurz, K. M. Engle, G. W. Pidgeon, P. R. Moore, G. Sandford and V. Gouverneur, Angew. Chem., Int. Ed., 2013, 52, 9796–9800 CrossRef CAS PubMed.
- For reviews on oxidative generation of C-radicals, see:
(a) J. Iqbal, B. Bhatia and N. K. Nayyar, Chem. Rev., 1994, 94, 519–564 CrossRef CAS;
(b) B. B. Snider, Chem. Rev., 1996, 96, 339–363 CrossRef CAS PubMed;
(c)
T. Linker, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001, vol. 1, pp. 219–228 Search PubMed.
- E. Differding and H. Ofner, Synlett, 1991, 187–189 CrossRef CAS.
- P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955–5958 CrossRef CAS PubMed.
- A small amount of PhthN(CH2)3CHF(CH2)2CO2H was isolated as the carbofluorination of the alkene A-1a with the solvent acetic acid. This seems to indicate that diethyl malonate is less reactive than acetic acid. See SI for details.
-
Fluorinated Heterocyclic Compounds: Synthesis, Chemistry and Applications, ed. V. A. Petrov, Wiley, New Jersey, 2009 Search PubMed.
- For highlights, see:
(a) M. P. Sibi and Y. Landais, Angew. Chem., Int. Ed., 2013, 52, 3570–3572 CrossRef CAS PubMed;
(b) A. Lin, B. Huehls and J. Yang, Org. Chem. Front., 2014, 1, 434–438 RSC;
(c) J.-A. Ma and S. Li, Org. Chem. Front., 2014, 1, 712–715 RSC.
- For examples of radical fluorination reactions, see:
(a) M. Rueda-Becerril, C. C. Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed;
(b) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed;
(c) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588–13591 CrossRef CAS PubMed;
(d) S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580–10583 CrossRef CAS PubMed;
(e) J. C. T. Leung, C. Chatalova-Sazepin, J. G. West, M. Rueda-Becerril, J.-F. Paquin and G. M. Sammis, Angew. Chem., Int. Ed., 2012, 51, 10804–10807 CrossRef CAS PubMed;
(f) S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722–1724 CrossRef CAS PubMed;
(g) Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed;
(h) S. Mizuta,
et al.
, Org. Lett., 2013, 15, 2648–2651 CrossRef CAS PubMed;
(i) H. Shigehisa, E. Nishi, M. Fujisawa and K. Hiroya, Org. Lett., 2013, 15, 5158–5161 CrossRef CAS PubMed;
(j) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024–6027 CrossRef CAS PubMed;
(k) J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed;
(l) M. Rueda-Becerril, O. Mahe, M. Drouin, M. B. Majewski, J. G. West, M. O. Wolf, G. M. Sammis and J.-F. Paquin, J. Am. Chem. Soc., 2014, 136, 2637–2641 CrossRef CAS PubMed;
(m) S. Bloom, J. L. Knippel and T. Lectka, Chem. Sci., 2014, 5, 1175–1178 RSC;
(n) Z. Li, C. Zhang, L. Zhu, C. Liu and C. Li, Org. Chem. Front., 2014, 1, 100–105 RSC;
(o) J.-B. Xia, Y. Ma and C. Chen, Org. Chem. Front., 2014, 1, 468–472 RSC;
(p) S. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2014, 53, 4690–4693 CrossRef CAS PubMed;
(q) X. Huang, W. Liu, H. Ren, R. Neelamegam, J. M. Hooker and J. T. Groves, J. Am. Chem. Soc., 2014, 136, 6842–6845 CrossRef CAS PubMed;
(r) C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2014, 136, 9780–9791 CrossRef CAS PubMed;
(s) C. W. Kee, K. F. Chin, M. W. Wong and C.-H. Tan, Chem. Commun., 2014, 50, 8211–8214 RSC.
-
M. Newcomb, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001, vol. 1, pp. 317–336 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00256c |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.