
Bio-templated synthesis of hierarchical polypyrrole-coated VS4 for supercapacitors†
Yao
Ding
a,
Zhong Yi
Shi
a,
Kailin
Li
*ab,
Jinsong
Rao
a,
Xiaobin
Gong
a,
Shupei
Liu
a,
Bo
Yang
a and
Yu Xin
Zhang
 *a
*a
aCollege of Materials Science and Engineering, Chongqing University, Chongqing 400044, China. E-mail: 384369427@qq.com; zhangyuxin@cqu.edu.cn
bState Key Laboratory of Molecular Engineering of Polymers, Department of Chemistry, Fudan University, Shanghai 200433, China
First published on 12th November 2024
Abstract
Vanadium tetrasulfide (VS4) is increasingly acknowledged as a potential electrode material for supercapacitors, attributed to its unique one-dimensional structural characteristics and elevated sulfur content. However, its intrinsic low conductivity and the tendency of vanadium to dissolve in the electrolyte have severely hindered its cycling performance, resulting in limited specific capacity under practical application conditions. The realization of advanced energy storage materials predominantly hinges on the exploitation of multiple oxidation states, the design of rational nanostructures, and the achievement of high electrical conductivity. Consequently, we report the successful construction of VS4 and polypyrrole (PPy) cross-aligned nanostructures on the surface of bio-templated diatomite (De@VS4@PPy) using a two-step hydrothermal and oxidative polymerization technique, which has led to remarkable electrochemical performance (specific capacitance of 243.33 F g−1 at a current density of 1 A g−1) and outstanding energy storage capabilities (97.7% capacitance retention after 3000 cycles). The highly conductive and cross-aligned nanostructures facilitate efficient electrolyte ion diffusion and concurrently minimize charge transfer resistance. Notably, the De@VS4@PPy nanostructured electrode materials demonstrate significant specific capacitance, a broad potential window, and outstanding cycling stability. Furthermore, this strategy can be readily extended to practical applications, exemplified by the asymmetric supercapacitors assembled employing De@VS4@PPy nano-electrode materials, which can achieve potential windows and maximum energy densities up to 1.8 V and 21.75 W h kg−1 (at 899.94 W kg−1), respectively. This work serves as a valuable reference for future studies focused on the screening and optimization of superior electrode materials.
1. Introduction
The burning of traditional fossil energy sources has resulted in serious resource shortages and environmental pollution problems.1–4 Creating devices for storing and efficiently converting sustainable energy has turned into a critical necessity.5–7 Supercapacitors (SCs) have become favored for future energy storage because of their substantial power density, swift charge–discharge speed, good cycling stability (exceeding 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles), and excellent safety.8–10 Nonetheless, the widespread use of these methods is impeded due to their low energy density.11,12 Based on the composition of supercapacitors and their underlying energy storage mechanisms, it is evident that the strategic selection of electrodes with superior electrochemical properties, coupled with the rational design of their structure, constitutes an effective approach to achieving high energy density.3,13–15
000 cycles), and excellent safety.8–10 Nonetheless, the widespread use of these methods is impeded due to their low energy density.11,12 Based on the composition of supercapacitors and their underlying energy storage mechanisms, it is evident that the strategic selection of electrodes with superior electrochemical properties, coupled with the rational design of their structure, constitutes an effective approach to achieving high energy density.3,13–15
Among all electrode materials, transition metal sulfides such as TiS2, MoS2, NiS2 and VS4 have become a popular research topic because of their unique crystal structure, superior specific capacity and remarkable redox reversibility.10,16–20 Among them, VS4 has excellent electrochemical properties due to its one-dimensional crystal structure and high sulfur content. The one-dimensional crystal structure of VS4 resulted in weak interactions between neighboring bonds, which would effectively facilitate the charge transfer kinetics.21–23 However, it does not allow the conductivity of VS4 to reach a satisfactory level.24,25 Enhancing the conductivity of an electrode can be effectively achieved by coating a transition metal sulfide with a highly conductive material, such as polypyrrole (PPy).26,27 PPy, known for its notable electrical conductivity, is widely used as an electrode substance because of its straightforward preparation, elevated specific capacitance, and robust chemical stability.28–30 Hasanzadeh et al. constructed cauliflower-like PPy nanocomposites in nickel foam. The structure design and outstanding electrical conductivity of PPy have significantly enhanced the electronic conductivity. The electrode that has been prepared attains a distinct capacitance level of 605.2 C g−1 at a current density of 1 A g−1. Furthermore, the electrode material's remarkable stability in cycling is evidenced by its 92.12% capacitance retention following 5000 cycles.31 In addition, designing structures for transition metal sulfides is a way to improve energy storage. By designing specialized structures and downsizing the material, volume changes can be minimized, the amount of active sites at interfaces can be increased, and ion diffusion paths can be shortened.32 These factors contribute to enhanced electrode conductivity and improved cycling stability. To date, researchers have explored the preparation of various VS4 structures.16,33,34 Li et al. used a solvothermal technology to prepare biomimetic anemone-like VS4 nanostructures. When the current density reaches 0.4 A g−1, VS4, resembling an anemone-like structure, demonstrates an elevated specific capacitance, marked at 617 F g−1. In addition, assembled symmetrical capacitors show excellent cycling stability, maintaining a capacity of more than 93% after 12000 charge–discharge cycling tests at 6 Ag−1.35 In addition, Karuppasamy et al. prepared hierarchical nano-urchin-like, nanoflower, and polyhedron-structured VS4 by using ethylenediaminetetraacetic acid as a mediator. The unique morphology and substantial specific surface area of these materials provide effective pathways for ion diffusion within the electrolyte, consequently improving the electrochemical performance.17 Diatomite (De) is commonly used as an excellent scaffold for creating templates of three-dimensional structures. It has been demonstrated that incorporating diatomite markedly diminishes the electrode's volumetric enlargement throughout its charge–discharge cycles. Furthermore, the three-dimensional porous structure of this material improves the area where the electrode meets the electrolyte.14,36 Although the electrochemical performance of the electrode materials can be improved by both capping polypyrrole (PPy) and incorporating diatomite design structures. The assembly of ternary complexes of transition sulfides and PPy coating layers on the diatomite surface has not been reported so far.
Herein, we improved the electrochemical characteristics of electrode materials through the deliberate design of cross-layered nanostructures on robust stencils. In this paper, we successfully employed hydrothermal and oxidative polymerization techniques to synthesize VS4 and PPy cross-layered nanostructures on diatomite bio-templated (De@VS4@PPy). The diatomite acts as a scaffold facilitating the growth of VS4, mitigating the agglomeration of VS4 nanosheets, while VS4 offers abundant active sites and varied oxidation states, and PPy significantly enhances the electrical conductivity. The unique cross-aligned nanostructure, coupled with exceptional electrical conductivity, creates abundant pathways for electrolyte ion diffusion. This impressive synergy enables the synthesized De@VS4@PPy to deliver exceptional electrochemical performance in alkaline electrolyte. Consequently, De@VS4@PPy operates reliably within a potential window of 0–0.8 V. This study shows that it has good specific capacitance with 97.7% capacitance retention after 3000 charge/discharge cycles. Moreover, the proposed approach is applicable to the fabrication of an asymmetric supercapacitor, where De@VS4@PPy serves as the positive electrode and activated carbon (AC) as the negative electrode. This asymmetric supercapacitor achieves a peak energy density of 21.75 W h kg−1 at a power density of 899.94 kW kg−1. These results are remarkable compared to the performance of other transition metal sulfides.
2. Results and discussion
PPy and VS4 cross-aligned in diatomite (De@VS4@PPy) were obtained by hydrothermal and oxidative polymerisation as shown in Scheme 1. Briefly, the pretreated diatomite was initially homogenized with ammonium metavanadate and thioacetamide in a methanol solution. The VS4 was uniformly deposited onto the diatomite surface through a conventional solvothermal reaction. As depicted in Fig. 1b and S1b,† the flaky VS4 is vertically aligned on the diatomite surface, with its layered structure forming large pores that enhance the electrode's contact with the electrolyte. After a simple purification of the initial sample, the solid powder was subjected to oxidative polymerization in a solution containing pyrrole, resulting in the deposition a PPy film at the De@VS4 surface. The parallel PPy layer is interspersed within the pores created by the lamellar VS4 (Fig. 1c and S1c†). The integration of PPy enhances electron transfer between the electrode and the current collector, while simultaneously improving the conductivity of the electrode. To further examine the impact of the PPy-coating on the morphology and structure characteristics of the samples, De@PPy and De@VS4 comparison samples were synthesized and analyzed using SEM. As illustrated in Fig. 1a and S1a,† the PPy layer deposited on the diatomite surface reduces the number of nanopores and smooths the surface. Also shown from Fig. 1b and S1b,† the VS4 nanosheets were vertically and uniformly distributed on the diatomite surface. This phenomenon was confirmed by TEM images (Fig. 1d), where the low contrast of the polymer-formed coating layer rendered it easily discernible under the electron microscope. To confirm the constituents present in the sample, EDS was used to test the elemental composition of the final sample. The findings presented in Fig. 1e indicate that the elements C, O, V, N, S and Si are evenly distributed across the surface of the sample. The EDS mapping and atomic content analysis of De@VS4@PPy reveal that the V to S ratio is approximately 1:4, further confirming the successful synthesis of VS4 (Fig. 1e and Table S1).†
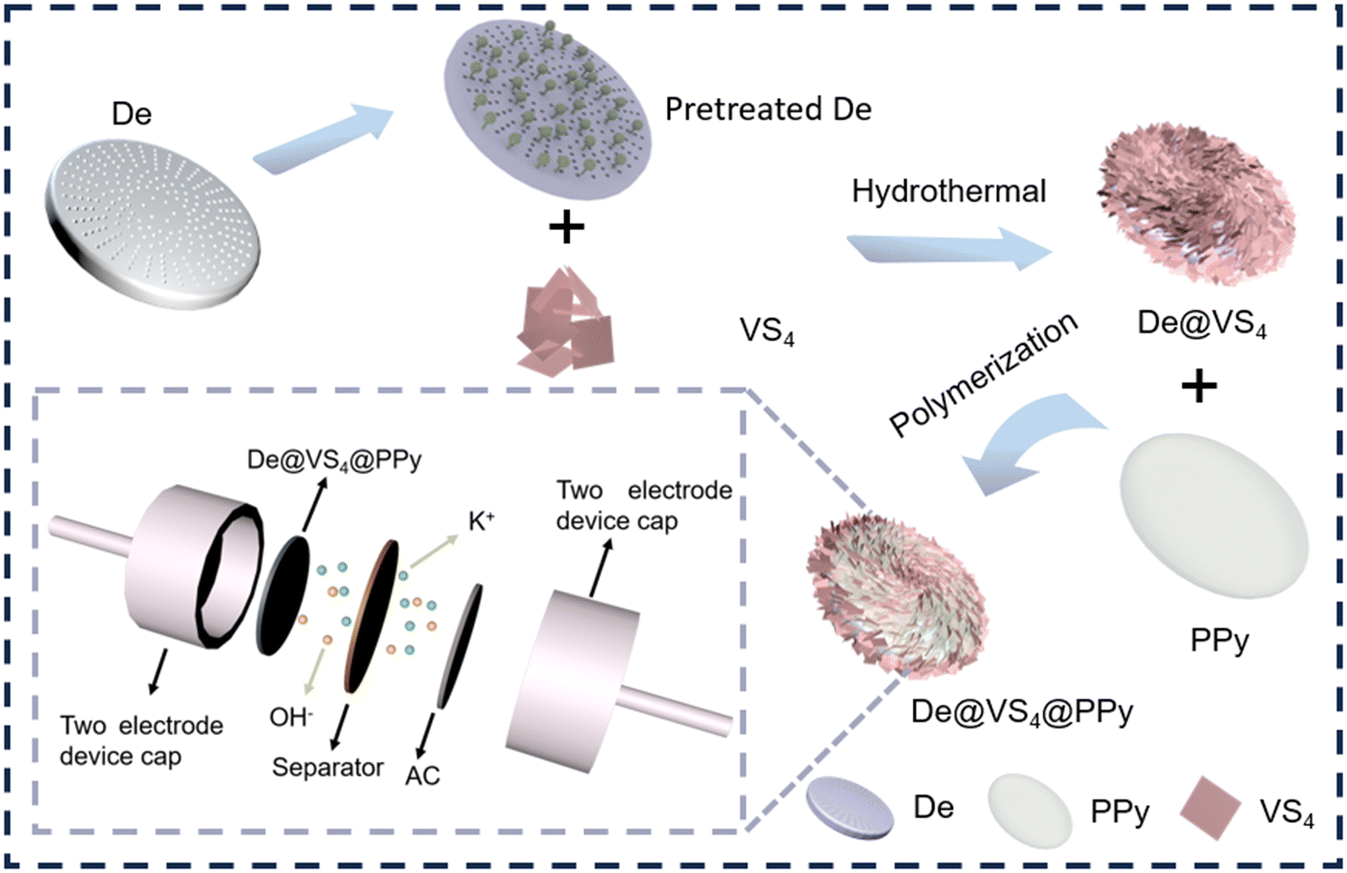 | ||
| Scheme 1 The schematic representation of the transition process from De to De@VS4@PPy. | ||
 | ||
| Fig. 1 Morphological and structural characterization of De@PPy, De@VS4 and De@VS4@PPy. Different magnification SEM images of (a and a1) De@PPy, (b and b1) De@VS4 and (c and c1) De@VS4@PPy. Different magnification TEM images of (d and d1) De@VS4@PPy. (e) EDS elemental mapping of C, O, V, N, S, and Si of De@VS4@PPy. | ||
To further validate the feasibility of the experimental program, spectroscopic characterization of the prepared samples was conducted (Fig. 2). Fig. 2a shows the XRD pattern of the De@PPy, De@VS4 and De@VS4@PPy samples to characterize the crystal structures. The diffraction patterns of the samples correspond to the standard PDF cards of VS4 (PDF#87-0603) and SiO2 (PDF#82-0512). The absence of additional impurity peaks in the diffractogram indicates that the synthesised version of the target product is indeed the expected one. The diffraction peaks at 21.7°, 31.2°, and 35.9° correspond to the (101), (102), and (112) planes of SiO2, whereas the peaks at 23.4° and 28.1° are assigned to the (112) and (−202) planes of VS4. Furthermore, there is no notable variation in the diffraction peaks between De@VS4 and De@VS4@PPy, indicating that the PPy coating does not affect the diatomite bio-templated structure. However, additional evidence is required to confirm the successful deposition of PPy. The more sensitive Raman spectroscopy was selected to further confirm the successful encapsulation of PPy. The peaks at 277, 360 and 558 cm−1 are characteristic of VS4, corresponding to the V–S, V2S4-cage and S–S bonds, respectively.17,35,37 The introduction of PPy results in a reduction of the distinct peaks associated with VS4, while simultaneously giving rise to new peaks at 1322 (C–N+), 1397 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), and 1584 cm−1 (CC).38 In contrast, the characteristic peaks of diatomite are observed in the range 850–1250 cm−1 and belong to the Si–O stretching in various polymerized silicate tetrahedra, respectively.39,40
N), and 1584 cm−1 (CC).38 In contrast, the characteristic peaks of diatomite are observed in the range 850–1250 cm−1 and belong to the Si–O stretching in various polymerized silicate tetrahedra, respectively.39,40
 | ||
| Fig. 2 Spectroscopic characterization of De@PPy, De@VS4 and De@VS4@PPy. XRD patterns (a) and Raman spectra (b) of De@PPy, De@VS4 and De@VS4@PPy. High-resolution (c) N 1s, (d) V 2p and (e) S 2p XPS spectra of De@VS4@PPy. | ||
Further investigation of the elemental composition and chemical valence states of De@VS4@PPy was conducted through XPS analysis (Fig. 2c and d and S3†). The full XPS survey spectrum depicted in Fig. S3a† uncovers elements C, N, Si, O, V, and S. Fig. 2d depicts a high-resolution vanadium (V) spectrum, displaying clear peaks at 516.7 eV and 524.4 eV, aligning with the V 2p3/2 and V 2p1/2 states of V4+.41,42 Additional signals at 514.1 eV and 521.8 eV are indicative of the formation of V–C bonds.43,44 The lack of extra peaks confirms that vanadium exists solely in the V4+ oxidation state. In the N 1s spectrum of De@VS4@PPy, the peaks at 399.8, 400.2, and 401.7 eV represent the emission centres corresponding to N+, –NH–, and N– of PPy, respectively (Fig. 2c).38 Additionally, the S 1s spectrum peaks at 163.7 and 164.3 eV typify the S22− dimers, reinforcing the successful synthesis of VS4.45 The O 1s spectrum peak at 285.4 eV (Fig. S3b†) correlates with the C–S bond, indicating a robust interaction between VS4 and PPy. Furthermore, the observed peaks at 284.8 and 287.8 eV are indicative of C–C and O–CC bonds.46 The presence of V and S, along with C and N, confirms the successful formation of VS4 and PPy. To verify that the PPy coating does not alter the diatomite bio-template, the XPS spectra of Si and O were examined. The Si 2p high-resolution spectrum at 102.5 eV corresponds to Si–O bonds. In the O 1s spectrum of De@VS4@PPy, the peaks at 530.1, 531.7, and 532.9 eV represent the emission centres corresponding to V–O, V–O–C, and C–O–C, where the V–O bonding is due to partial oxidation of V produced during hydrothermal processes.4,23,47 Overall, PPy and VS4 establish a stable architecture on the diatomite bio-template via the C–S bond, significantly enhancing electron transport and ensuring excellent cycling stability when utilized as supercapacitor electrodes.
As depicted in Fig. S4a,† the CV curves were recorded at different scan rates. The figure reveals that the CV curve shapes for De@VS4@PPy remain consistent with increasing scan rates, indicating its exceptional rate performance. Additionally, the pronounced redox peak within the 0–0.8 V potential window further supports this, leading to the selection of this potential window for measuring the CV of all subsequent samples in the three-electrode system. The aforementioned redox peaks are primarily attributed to the redox reactions between V4+/V5+ and S2−/S− and the electrolyte, as described in the following equation:
| VS4 + OH− + H2O ↔ VS4OH + e− | (1) |
Fig. S4b† illustrates the CC curves of De@VS4@PPy at different current densities, offering substantial evidence of its strong electrochemical performance. The CC curve of De@VS4@PPy exhibits a distinct nonlinear characteristic, which signifies its redox-active properties and is consistent with the findings derived from the CV measurements. At low current densities, the charging time exceeds the discharging time, likely due to the limited ion insertion on the electrode surface. To further substantiate the effectiveness of assembling metal sulfides and PPy on diatomite as a strategy for enhancing electrochemical performance, CV and CC curves were acquired for both De@VS4 and De@PPy. Fig. 3a depicts the CV curves for the De@VS4@PPy, De@VS4, and De@PPy electrodes at a scanning speed of 100 mV s−1. Clearly, the area beneath the CV curve for the De@VS4@PPy electrode significantly exceeds that of its comparative counterparts, indicating that De@VS4@PPy exhibits superior redox kinetics and specific capacitance relative to De@VS4 and De@PPy. The enhancement in electrochemical efficiency is due to the hastened movement of ions, aided by the augmented count of active sites, a consequence of the stratified composition of diatomite, VS4, and PPy. Moreover, a pronounced polarization is observed in both De@VS4 and De@PPy, suggesting that the simultaneous incorporation of VS4 and PPy on the diatomite surface effectively extends the potential window. This may be due to interfacial effects between VS4 and PPy.
 | ||
| Fig. 3 The (a) CV curves at 100 mV s−1 scan rate and (b) CC curves at 1 A g−1 current density of De@PPy, De@VS4 and De@VS4@PPy. (c) Rate capability of De@PPy, De@VS4 and De@VS4@PPy at different current densities. (d) Nyquist plot of De@PPy, De@VS4 and De@VS4@PPy. (e) CV curves with low scanning sweeps of De@VS4@PPy; (f) the percentage of capacitance contribution at different scan rates. (g) The logIp – logv linear fitting curves. (h) Cycling stability of De@VS4@PPy at 3 A g−1. | ||
At a current density of 1 A g−1, the CC curves demonstrate that the De@VS4@PPy electrode maintains a prolonged discharge duration in contrast to both De@VS4 and De@PPy electrodes. As demonstrated in Fig. 3b, this electrode attains a significant specific capacitance value of 243.33 F g−1. Despite a greater current density of 10 A g−1, the De@VS4@PPy electrode maintains its superiority, achieving a specific capacitance of 113.3 F g−1 and maintaining a capacitance retention rate of 46.56%. These values notably surpass those of the De@VS4 electrode (43.33 F g−1, 42.27%) and the De@PPy electrode (26.67 F g−1, 35.25%). These results indicate that De@VS4@PPy with a layered structure has good rate performance and is advantageous in electrochemical processes.
To further clarify the underlying factors for the capacitance enhancement of De@VS4@PPy relative to De@VS4 and De@PPy, we conducted impedance testing of De@VS4@PPy, De@VS4, and De@PPy using electrochemical impedance spectroscopy (EIS) under a stable voltage. The lack of a clear semicircle in the Nyquist plots' high-frequency area implies a negligible charge transfer resistance (Rct). Notably, in the low-frequency spectrum, De@VS4@PPy exhibits a sharper slope compared to De@VS4 and De@PPy, with the curve for De@VS4@PPy approaching a nearly vertical orientation. This behavior can be attributed to the cross-aligned nanostructures formed by VS4 and PPy, which enhance the electrode-to-electrolyte contact, thereby leading to high electrical conductivity. This finding is consistent with the inferences made in earlier research that relied on CV and GCD data. Fig. 3e presents the CV curves recorded at lower scan rates. As the scanning speed increases, the De@VS4@PPy electrode's oxidation and reduction peaks move more towards positive and negative potentials, a change linked to reduced electrochemical kinetics at elevated scanning speeds. For a precise evaluation of the electrode's charge storage characteristics, the correlation between the peak current (Ip) and the scanning rate (v) was ascertained through the subsequent eqn (2).
| logIp = blogν + loga | (2) |
In this equation, a and b represent adjustable constants. Fig. 3f shows linear fits for anodic and cathodic peaks of electrodes at low scan rates. These peak currents were derived from the relationship between current density and scan rate, and linear trends were obtained by fitting these data points. The parameter b determines the electrode's electrochemical behavior: when b = 0.5, the behavior is diffusion-controlled, while b = 1 indicates capacitive behavior. The b value between 0.5 and 1 suggests a hybrid behavior, combining both capacitive and diffusion-controlled mechanisms. Findings indicate that the b values for both the anodic and cathodic peaks of the De@VS4@PPy electrode lie in this spectrum, signifying a blend of capacitive and diffusion-controlled characteristics.22,48,49 Furthermore, the R2 values for the anodic and cathodic fits are 0.99844 and 0.9993.
Furthermore, the eqn (3) below calculates the capacitive impact of the electrode material at different scanning speeds:
| I/v1/2 = k1v1/2 + k2 | (3) |
To further assess the practical application potential of De@VS4@PPy, its electrochemical performance was systematically evaluated using a two-electrode device. The detailed construction schematic is presented in Scheme 1, where De@VS4@PPy serves as the positive electrode, activated carbon (AC) as the negative electrode, with 1 M KOH as the electrolyte, and a filter membrane as the diaphragm (De@VS4@PPy//AC). The electrochemical properties of De@VS4@PPy and AC in a three-electrode configuration were initially examined, as depicted in Fig. S8.† For the positive electrode De@VS4@PPy, the CV curve shows a functional range of 0–0.8 V, in contrast to the negative electrode AC, which varies from −1 to 0 V. From this analysis, it can be inferred that the maximum operating window of the two-electrode device extends from 0 to 1.8 V. Nevertheless, to prevent potential damage to the prepared De@VS4@PPy//AC device, the CV test was conducted across various voltage windows (0.8, 1.0, 1.2, 1.4, 1.6 and 1.8 V). As illustrated in Fig. 4a, the CV curves across all voltage windows exhibit a distorted rectangular shape, with no discernible polarization peaks. This observation suggests that De@VS4@PPy//AC device demonstrates stable energy storage performance within a potential range of 0–1.8 V, thereby justifying the selection of this potential window for subsequent tests. As illustrated in Fig. 4b, CV tests for the De@VS4@PPy//AC device were conducted at scanning rates ranging from 10 to 100 mV s−1. All resulting curves exhibit a distorted rectangular shape, likely due to surface capacitance and diffusion-controlled mixing. Notably, there is no obvious redox peak in the CV curve, which suggests the excellent rate performance of the De@VS4@PPy//AC device, which is consistent with the previous three-electrode test results. Fig. 4c presents the CC curves of De@VS4@PPy//AC device at various current densities. All curves display incomplete symmetrical triangles, confirming their capacitive properties. Moreover, the consistent shape of the CC curves at high current densities further demonstrates that the De@VS4@PPy//AC device exhibits good reversibility and high coulombic efficiency. Subsequently, the specific capacitance of the De@VS4@PPy//AC device was determined using eqn (1) in the ESI.†
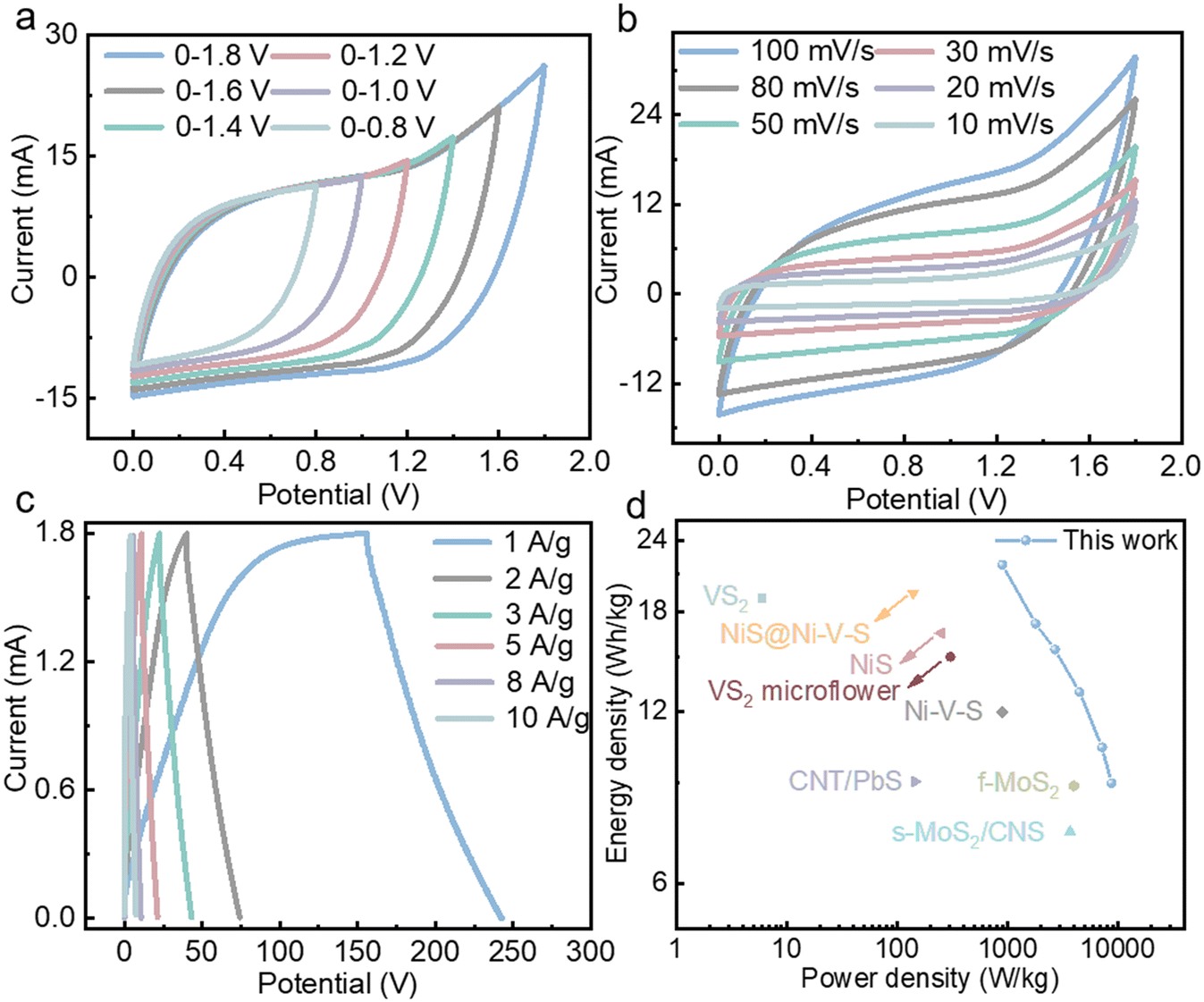 | ||
| Fig. 4 (a) CV curves of De@VS4@PPy//AC with different potentials. (b) CV curves of De@VS4@PPy//AC with different scan rates. (c) CC curves of AC//De@VS4@PPy//AC with different current densities. (d) Ragone plots of the AC//De@VS4@PPy asymmetric supercapacitor. | ||
As shown in Fig. S8,† the specific capacitance at 1 A g−1 was measured to be 48.33 F g−1. Although the capacitance decreases with increasing current density, it still retains an impressive 20 F g−1 at a high current density of 10 A g−1, which is significantly higher than the 33 F g−1 reported for MWCNT@VS2 by other researchers.50 High capacitance combined with broad potential windows is generally beneficial for improving the energy and power densities of supercapacitors. For the De@VS4@PPy//AC device, these energy and power densities were determined using eqn (2) and (3) in the ESI.† The asymmetric supercapacitor based on De@VS4@PPy//AC achieved an energy density of 21.75 W h kg−1 at a power density of 899.94 W kg−1, while still maintaining an energy density of 9 W h kg−1 as the power density increased to 8.76 kW kg−1. These values exceed those reported for vanadium sulfides in previous studies, such as VS2 (19 W h kg−1 at 6 W kg−1),51 Ni–V–S (12 W h kg−1 at 900 W kg−1)52 and VS2 microflowers (15 W h kg−1 at 304 W kg−1).53 Similarly, the De@VS4@PPy//AC device shows superior energy and power densities when compared to other transition metal sulfides, such as CNT/PbS (9.06 W h kg−1 at 145.5 W kg−1),54 NiS@Ni–V–S (19.4 W h kg−1 at 140 W kg−1),55 f-MoS2 (6.9 W h kg−1 at 4000 W kg−1),33 and s-MoS2/CNS (7.4 W h kg−1 at 3700 W kg−1).56 These findings suggest that De@VS4@PPy demonstrates excellent electrochemical performance and significant potential for practical applications.
3. Conclusion
In summary, this paper introduces a novel approach for constructing cross-aligned layered structures of VS4 and PPy on bio-templated diatomite for supercapacitor electrode applications. The distinct cross-aligned hierarchical morphology of the synthesized nanomaterials, combined with the high conductivity of PPy, facilitates the creation of extensive active channels for both electrolyte diffusion and electron transfer, thereby enhancing the electrochemical performance of De@VS4@PPy by reducing the diffusion time. Compared to materials solely loaded with VS4 (De@VS4) or coated with PPy (De@PPy), the fabricated layered De@VS4@PPy exhibits superior capacitance (243.33 F g−1 at 1 A g−1), an extended operating window, commendable rate performance, and outstanding cycling stability (97.7% capacitance retention after 3000 cycles). Utilizing the three-electrode study results, an asymmetric supercapacitor was assembled with De@VS4@PPy as the positive electrode and AC as the negative electrode. This device demonstrates a broad potential range of 0–1.8 V and achieves a notable energy density of 21.75 W h kg−1 at a power density of 899.94 W kg−1. These electrochemical results underscore that the cross-aligned nanostructures significantly contribute to the enhanced performance and prolonged cycling stability of the supercapacitor. Consequently, this study advocates for further exploration into mitigating agglomeration and enhancing the electrical conductivity of metal sulfide nanomaterials through strategic nanostructure design, thereby advancing the development of high energy density and power density supercapacitors.Data availability
Data will be made available on request.Author contributions
K. Li and Y. Zhang: conceptualization, methodology, supervision, and writing – review and editing. Y. Ding: synthesis, characterization, and investigation. Z. Shi and X. Gong: visualization. J. Rao, S. Liu, and B. Yang: formal analysis and writing – original draft preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the Graduate Scientific Research and Innovation Foundation of the financial support of the National Natural Science Foundation of China (Grant no. 22405052), and the China Postdoctoral Science Foundation (Grant no. 2024M750491). The authors are equally grateful for the financial support from the Fundamental Research Funds for the Central Universities (Grant no. 2024CDJQYJCYJ-001). The authors also thank the Electron Microscopy Center of Chongqing University and Ceshigo for their material characterization.References
- S. X. Zhou, Y. J. Zhao, K. X. Zhang, Y. R. Xun, X. Y. Tao, W. T. Yan, W. Zhai and J. Ding, Nat. Commun., 2024, 15, 6481 CrossRef CAS
.
- J. X. Huang, Y. Y. Hu, J. Z. Li, H. Wang, T. S. Wang, H. Wu, Y. J. Li, M. Wang and J. H. Zhang, ACS Energy Lett., 2023, 8, 2316–2324 CrossRef CAS
- Q. Sun, Z. Y. Guo, T. Shu, Y. F. Li, K. L. Li, Y. X. Zhang, L. Li, J. Y. Ning and K. X. Yao, ACS Appl. Mater. Interfaces, 2024, 16, 12781–12792 CrossRef CAS PubMed
- D. Li, J. S. Zhou, X. H. Chen and H. H. Song, ACS Appl. Mater. Interfaces, 2016, 8, 30899–30907 CrossRef CAS PubMed
- S. Ghosh, G. R. Rao and T. Thomas, Energy Storage Mater., 2021, 40, 426–438 CrossRef
- Z. Y. Xiong, P. J. Guo, Y. S. Yang, S. Y. Yuan, N. Z. Shang, C. Wang, Y. F. Zhang, H. Wang and Y. J. Gao, Adv. Energy Mater., 2022, 12, 2103226 CrossRef CAS
- G. Liu, Y. Xu, T. Yang and L. Jiang, Nano Mater. Sci., 2023, 5, 101–116 CrossRef CAS
- L. Wu, Y. Kang, X. Y. Shi, E. D. Yang, J. X. Ma, X. F. Zhang, S. X. Wang and Z. S. Wu, ACS Nano, 2023, 17, 22580–22590 CrossRef CAS PubMed
- H. Y. Liu, T. Xu, C. Y. Cai, K. Liu, W. Liu, M. Zhang, H. S. Du, C. L. Si and K. Zhang, Adv. Funct. Mater., 2022, 32, 2113082 CAS
- T. Wang, Y. Wang, J. Lei, K.-J. Chen and H. Wang, Exploration, 2021, 1, 20210178 Search PubMed
- K. L. Li, H. Teng, Q. Sun, Y. F. Li, X. X. Wu, X. J. Dai, Y. Wang, S. M. Wang, Y. F. Zhang, K. X. Yao, Z. H. Bao, J. S. Rao and Y. X. Zhang, J. Energy Storage, 2022, 53, 105094 Search PubMed
- L. Kang, S. D. Liu, Q. Zhang, J. X. Zou, J. Ai, D. H. Qiao, W. D. Zhong, Y. X. Liu, S. C. Jun, Y. Yamauchi and J. Zhang, ACS Nano, 2024, 18, 2149–2161 CrossRef CAS PubMed
- K. Karuppasamy, D. Vikraman, S. Hussain, P. Santhoshkumar, R. Bose, P. Sivakumar, A. Alfantazi, J. W. Jung and H. S. Kim, Small, 2022, 18, 2107284 CrossRef CAS PubMed
- K. L. Li, Z. Y. Guo, Q. Sun, X. J. Dai, Y. F. Li, K. X. Yao, X. Y. Liu, Z. H. Bao, J. S. Rao and Y. X. Zhang, Chem. Eng. J., 2023, 454, 140223 CrossRef CAS
- H. Song, Y. Wang, Q. Fei, D. H. Nguyen, C. Zhang and T. Liu, Exploration, 2022, 2, 20220006 CrossRef CAS PubMed
- D. Vikraman, S. Hussain, K. Karuppasamy, A. Feroze, A. Kathalingam, A. Sanmugam, S. H. Chun, J. Jung and H. S. Kim, Appl. Catal., B, 2020, 264, 118531 CrossRef
- K. Karuppasamy, D. Vikraman, S. Hussain, B. Thirumalraj, P. Santhoshkumar, H. Parangusan, H. C. Park, J. W. Jung and H. S. Kim, J. Energy Chem., 2023, 79, 569–580 CrossRef CAS
- Y. Liu, J. Sun, S. Y. Lin, Z. K. Xu and L. Li, J. Alloys Compd., 2020, 820, 153113 CrossRef CAS
- X. Chu, Y. Wang, L. Cai, H. Huang, Z. Xu, Y. Xie, C. Yan, Q. Wang, H. Zhang, H. Li and W. Yang, SusMat, 2022, 2, 379–390 CrossRef CAS
- C. Jing, J. Y. Ma, Q. Sun, Y. H. Li, X. Tang, F. L. Ling, Y. J. Wang, W. D. Zhang, X. J. Zhou and Y. X. Zhang, CrystEngComm, 2023, 25, 1941–1950 RSC
- Y. Niu, P. Luo, X. Chen, J. Song, X. R. He, H. L. Sun, Z. P. Li, C. Wang and J. Jiang, Chem. Eng. J., 2024, 493, 152372 CrossRef CAS
- X. F. Wang, Y. F. Zhang, J. Q. Zheng, H. M. Jiang, X. Y. Dong, X. Liu and C. G. Meng, J. Colloid Interface Sci., 2020, 574, 312–323 CrossRef CAS
- L. Q. Yu, S. X. Zhao, Q. L. Wu, J. W. Zhao and G. D. Wei, Adv. Funct. Mater., 2020, 30, 2000427 CrossRef CAS
- Y. Kim, T. Park, J. Na, J. W. Yi, J. Kim, M. Kim, Y. S. Bando, Y. Yamauchi and J. J. Lin, Nanoscale, 2020, 12, 8608–8625 RSC
- M. P. Harikrishnan and A. C. Bose, Energy Fuels, 2023, 37, 10799–10826 CrossRef
- Z. Zhang, N. Song, J. Wang, Y. Liu, Z. Dai and G. Nie, SusMat, 2022, 2, 646–657 CrossRef CAS
- J. Y. Ma, L. Y. Huang, W. Dong, C. Jing, X. Tang, N. Li, Y. J. Wang, S. Jiang, F. L. Ling, L. Feng and X. J. Zhou, J. Energy Storage, 2024, 100, 113770 CrossRef
- H. Liu, Q. Chen, H. C. Chen, S. Z. Zhang, K. F. Wang, Y. J. Chen, H. Z. Liu, C. Y. Zhang, L. Shi and H. Li, Small, 2024, 20, 2307308 CrossRef CAS
- K. Shi, G. M. Zhang, Z. F. Han, L. X. Ma, J. Q. Hou, D. S. Song, Z. G. Fu, W. Zhou, C. X. Guo, H. F. Shi, X. Y. Zhu and H. B. Lan, Adv. Eng. Mater., 2024, 26, 2301308 CrossRef CAS
- T. T. Zhou, S. W. Ling, S. X. Sun, R. X. Yuan, Z. Q. Wu, M. Y. Fu, H. N. He, X. L. Li and C. H. Zhang, J. Energy Chem., 2024, 95, 117–125 CrossRef CAS
- M. Hasanzadeh, R. Ansari and M. Farahpour, J. Alloys
Compd., 2023, 951, 169965 CrossRef CAS
- J. Y. Ma, Q. Sun, C. Jing, F. L. Ling, X. Tang, Y. H. Li, Y. J. Wang, S. Jiang, K. X. Yao and X. J. Zhou, CrystEngComm, 2023, 25, 3066–3078 RSC
- M. Ramu, J. R. Chellan, N. Goli, P. Joaquim, V. Cristobal and B. C. Kim, Adv. Funct. Mater., 2020, 30, 1906586 CrossRef CAS
- S. Ratha, S. R. Marri, J. N. Behera and C. S. Rout, Eur. J. Inorg. Chem., 2016, 2, 259–265 CrossRef
- H. Y. Li, J. K. Feng, L. Xiang, J. Huang and B. Xie, J. Power Sources, 2020, 457, 228031 CrossRef CAS
- C. Z. Zhang, K. L. Li, T. Sun, X. Y. Liu, X. J. Dai, Q. Zhou, D. S. Wang, X. F. Zhang, J. H. Ding, X. H. Huang, J. S. Rao, Y. Hou, P. A. Yang, K. C. Liu and Y. X. Zhang, ACS Appl. Nano Mater., 2024, 7, 3001–3011 CrossRef CAS
- Y. H. Man, A. Li, H. W. Tang, J. L. Sun, Y. T. Fei, Y. C. Du and X. S. Zhou, Sci. China: Chem., 2024, 67, 3153–3161 CrossRef CAS
- K. L. Li, S. H. Feng, C. Jing, Y. X. Chen, X. Y. Liu, Y. X. Zhang and L. Zhou, Chem. Commun., 2019, 55, 13773–13776 RSC
-
D. R. Neuville, D. de Ligny and G. S. Henderson, Spectroscopic Methods in Mineralology and Materials Sciences, 2014, vol. 78, pp. 509–541 Search PubMed
- G. A. Farfan, D. A. McKeown and J. E. Post, Geobiology, 2023, 21, 520–533 CrossRef
- X. C. Xie, H. L. Shuai, X. Wu, K. J. Huang, L. N. Wang, R. M. Wang and Y. Chen, J. Alloys Compd., 2020, 847, 156288 CrossRef CAS
- T. Q. Guo, Y. Z. Song, Z. T. Sun, Y. H. Wu, Y. Xia, Y. Y. Li, J. H. Sun, K. Jiang, S. X. Dou and J. Y. Sun, J. Energy Chem., 2020, 42, 34–42 CrossRef
- M. K. Naseem, M. Azmat, C. L. Du, M. Ismail, H. Baig, R. Jiang, A. Ali, M. S. Zou, Y. Q. Zhu and C. B. Cao, ACS Appl. Mater. Interfaces, 2024, 16, 41996–42006 CrossRef CAS PubMed
- S. H. Tian, J. J. Huang, H. C. Yang, G. Liu, Q. Zeng, D. Wang, X. Sun, K. Tao, G. H. Liu and S. L. Peng, Small, 2022, 18, 2205163 CrossRef CAS
- S. Z. Wang, H. Y. Chen, J. X. Liao, Q. Sun, F. P. Zhao, J. Luo, X. T. Lin, X. B. Niu, M. Q. Wu, R. Y. Li and X. L. Sun, ACS Energy Lett., 2019, 4, 755–762 CrossRef CAS
- Q. Pang, J. T. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef CAS PubMed
- Y. L. Zhou, P. F. Liu, F. Y. Jiang, J. Tian, H. Z. Cui and J. Yang, J. Colloid Interface Sci., 2017, 498, 442–448 CrossRef CAS
- H. T. Zang, F. Yang, S. L. Cao, M. Z. Yang, W. L. Liu, F. P. Cai, M. M. Ren and Y. Wang, J. Alloys Compd., 2022, 899, 163377 CrossRef CAS
- X. F. Wang, Y. F. Zhang, J. Q. Zheng, X. Liu and C. G. Meng, J. Colloid Interface Sci., 2019, 554, 191–201 CrossRef CAS
- E. Meyer, A. Bede, D. Mutukwa, R. Taziwa and N. Zingwe, J. Energy Storage, 2020, 27, 6021–6028 Search PubMed
- S. Ratha, S. R. Marri, N. A. Lanzillo, S. Moshkalev, S. K. Nayak, J. N. Behera and C. S. Rout, J. Mater. Chem. A, 2015, 3, 18874–18881 RSC
- Y. Y. Liu, L. Xu, X. T. Guo, T. T. Lv and H. Pang, J. Mater. Chem. A, 2020, 8, 20781–20802 RSC
- S. A. Patil, I. Rabani, S. Hussain, Y. S. Seo, J. Jung, N. K. Shrestha, H. Im and H. Kim, Nanomaterials, 2022, 12, 339 CrossRef CAS PubMed
- C. Gopi, S. Ravi, S. S. Rao, A. E. Reddy and H. J. Kim, Sci. Rep., 2017, 7, 46519 CrossRef PubMed
- R. Manikandan, C. J. Raj, K. H. Yu and B. C. Kim, Appl. Surf. Sci., 2019, 497, 143778 CrossRef CAS
- T. N. Y. Khawula, K. Raju, P. J. Franklyn, I. Sigalas and K. I. Ozoemena, J. Electrochem. Soc., 2016, 163, A1927–A1935 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ce01010h |
| This journal is © The Royal Society of Chemistry 2025 |