
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrelating active sites and oxidative species in single-atom catalyzed Fenton-like reactions
Jie
Miao
a,
Yunyao
Jiang
a,
Xixi
Wang
b,
Xue
Li
c,
Yuan
Zhu
d,
Zongping
Shao
 be and
Mingce
Long
*c
be and
Mingce
Long
*c
aSchool of Environmental Science and Engineering, Nanjing Tech University, Nanjing 211816, China
bState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing, 210009, China
cSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: long_mc@sjtu.edu.cn
dSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT7 1NN, UK
eDepartment of Chemical Engineering, Curtin University, Perth, 6845, Australia
First published on 3rd July 2024
Abstract
Single-atom catalysts (SACs) have gained widespread popularity in heterogeneous catalysis-based advanced oxidation processes (AOPs), owing to their optimal metal atom utilization efficiency and excellent recyclability by triggering reactive oxidative species (ROS) for target pollutant oxidation in water. Systematic summaries regarding the correlation between the active sites, catalytic activity, and reactive species of SACs have rarely been reported. This review provides an overview of the catalytic performance of carbon- and metal oxide-supported SACs in Fenton-like reactions, as well as the different oxidation pathways induced by the metal and non-metal active sites, including radical-based pathways (e.g., ·OH and SO4˙−) and nonradical-based pathways (e.g.1O2, high-valent metal-oxo species, and direct electron transfer). Thereafter, we discuss the effects of metal types, coordination environments, and spin states on the overall catalytic performance and the generated ROS in Fenton-like reactions. Additionally, we provide a perspective on the future challenges and prospects for SACs in water purification.
1. Introduction
Water is an indispensable resource for human survival. In recent years, with the rapid development of society and industrial economy, water pollution issues have become more prominent, and the demand for environmental protection has been increasing. Especially, emerging contaminants (ECs) are widely present in water.1 ECs are pollutants with potential physiological toxicity, including endocrine disrupting chemicals (EDCs), pharmaceutical and personal care products (PPCPs), pesticides and others. They have more stable chemical structures and are more difficult to be removed than conventional pollutants, thus posing serious health risks to humans.2 Advanced oxidation processes (AOPs), including electrochemical oxidation,3,4 ozone oxidation,5 wet catalytic oxidation,6 photocatalytic oxidation,7 Fenton and Fenton-like reactions,8–11 have emerged as a powerful way to eliminate ECs, by generating reactive oxidative species (ROS) with high oxidation potential.12,13 Among the diverse AOPs, Fenton-like reactions have gained continuous attention as a promising strategy to remove recalcitrant pollutants, due to their inherent simplicity, efficiency, cost-effectiveness and environmental friendliness.14,15The chemical oxidants employed in Fenton-like reactions involve peroxide (H2O2) and persulfates (such as peroxydisulfate (PDS, S2O82−) and peroxymonosulfate (PMS, HSO5−)), which can be activated to generate ROS using homogeneous and heterogeneous catalysts. Although metal ions (e.g., Co2+, Fe2+, Cu2+) and their soluble complexes are effectively applied in the homogeneous system,16–18 the difficult recovery of such soluble catalysts leads to secondary pollution, restricting their applications (Fig. 1). In contrast, the heterogeneous Fenton-like catalysts address these issues by their improved stability and easy separation.19–21 In particular, some metal-based heterogeneous catalysts, such as nano-metal oxides, metal nanoparticles (NPs), and metal single-atom catalysts (SACs), have attracted increasing attention in Fenton-like reactions due to their superior activities.22–24 However, the confined surface locations of metal active centers in heterogeneous NP catalysts result in inferior catalytic efficiency compared with their homogeneous counterparts, suffering from low metal atom utilization efficiency because of agglomeration of metal atoms and embedding in the bulk of NP catalysts.25,26 Additionally, most reported NP catalysts have uneven particle size distribution and versatile surface structure properties, posing a great challenge to explore the intrinsic catalytic mechanism and establish the structure–activity relationship in Fenton-like reactions.24,27,28
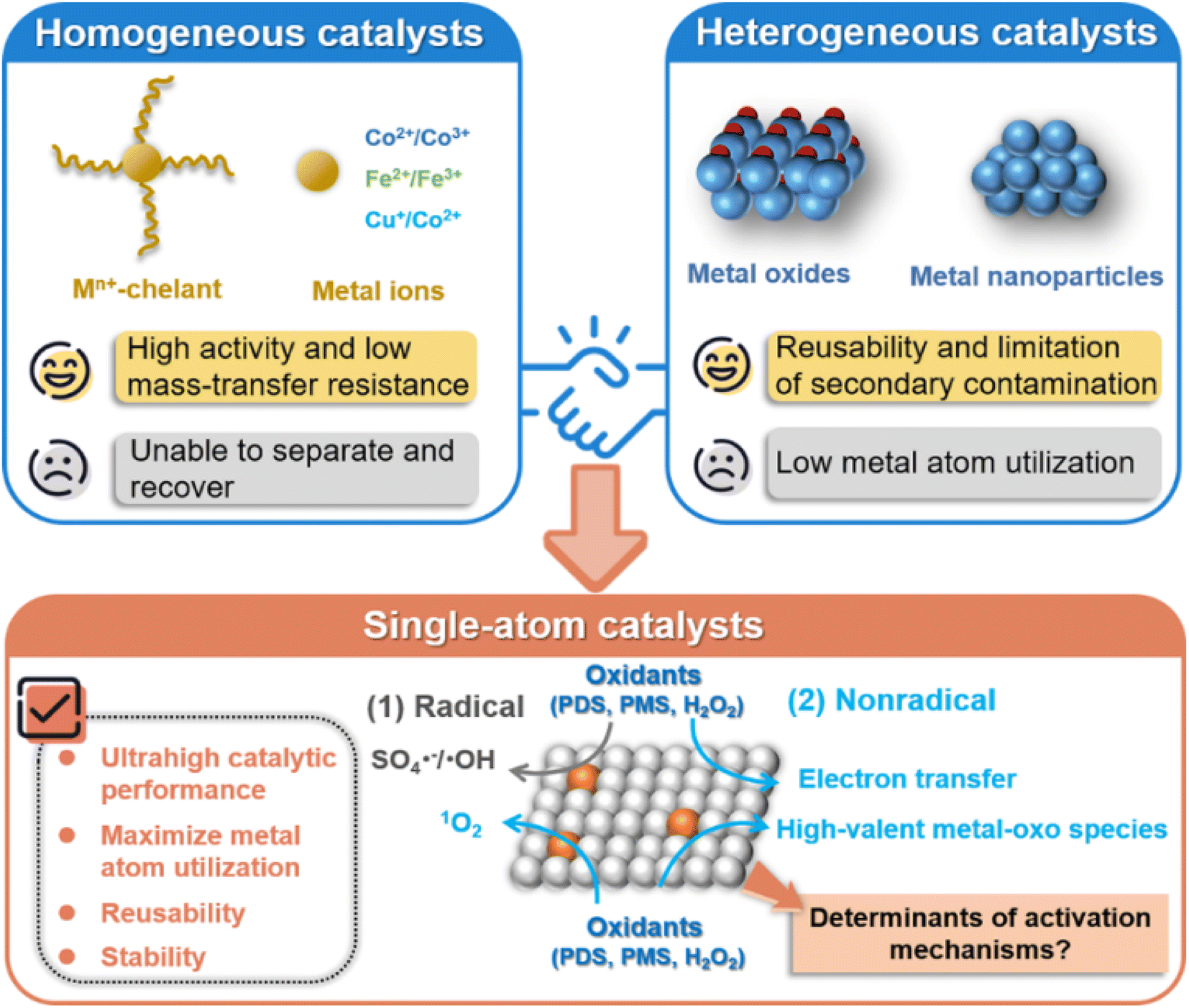 | ||
| Fig. 1 Comparison of the properties of homogeneous and heterogeneous metal catalysts, and SACs in Fenton-like reactions. | ||
In recent studies, SACs have been developed to downsize metal NPs to the atom-size level on the supporting materials, featuring nearly 100% metal atom utilization efficiency and inheriting the merits of both homogeneous and heterogeneous catalysts, which can overcome the limitations in the kinetics and catalytic activity of heterogeneous catalysts in Fenton-like reactions.29–32 In contrast to metal NPs, SACs also demonstrate enhanced structural robustness and catalytic stability as a result of the strong covalent interactions between their single metal atom sites and the supports.33 A comparison of the advantages and limitations of homogeneous and heterogeneous metal catalysts, and SACs is shown in Fig. 1. Moreover, the single metal sites in SACs with tunable electronic properties are significantly influenced by metal types, supports and coordination environments.34–37 This can lead to different interactions between isolated atomic sites and reactants, such as peroxides and organic pollutants, resulting in distinct Fenton-like activities and mechanisms.38,39
The timeline of the development of heterogeneous SACs as Fenton-like catalysts is presented in Fig. 2. The heterogeneous SACs used in Fenton-like reactions were first reported in 2018.40 Since then, various SACs with transition or noble metal atoms (e.g. Co, Fe, Ni, Cu, Cr, Mn, Au and Ag) confined in supports (such as carbon substrates and metal oxides) have been developed. These SACs presented exceptional catalytic activities for Fenton-like reactions and EC degradation.35,41–44 In addition, SACs with M–Nx configuration are widely investigated in Fenton-like catalytic reactions. Furthermore, substituting N with other heteroatoms (such as O, C, P, S, etc.) and altering the coordination number of N can modulate the electronic properties around single atoms, thus precisely tuning the catalytic performance and mechanism.45–50 For example, Co SACs with N/O dual coordination (Co–OCN) was 4.9 times more active than Co–CN for PMS activation and showed higher selectivity for Co(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O production.34 Co–N3C sites with low coordination surpassed Co–N4 sites in converting PDS for organic pollutant oxidation.48 Thus, heterogeneous SACs with precise coordination structures are crucial for understanding Fenton-like catalytic mechanisms, and for designing efficient heterogeneous catalysts.34,47,51
O production.34 Co–N3C sites with low coordination surpassed Co–N4 sites in converting PDS for organic pollutant oxidation.48 Thus, heterogeneous SACs with precise coordination structures are crucial for understanding Fenton-like catalytic mechanisms, and for designing efficient heterogeneous catalysts.34,47,51
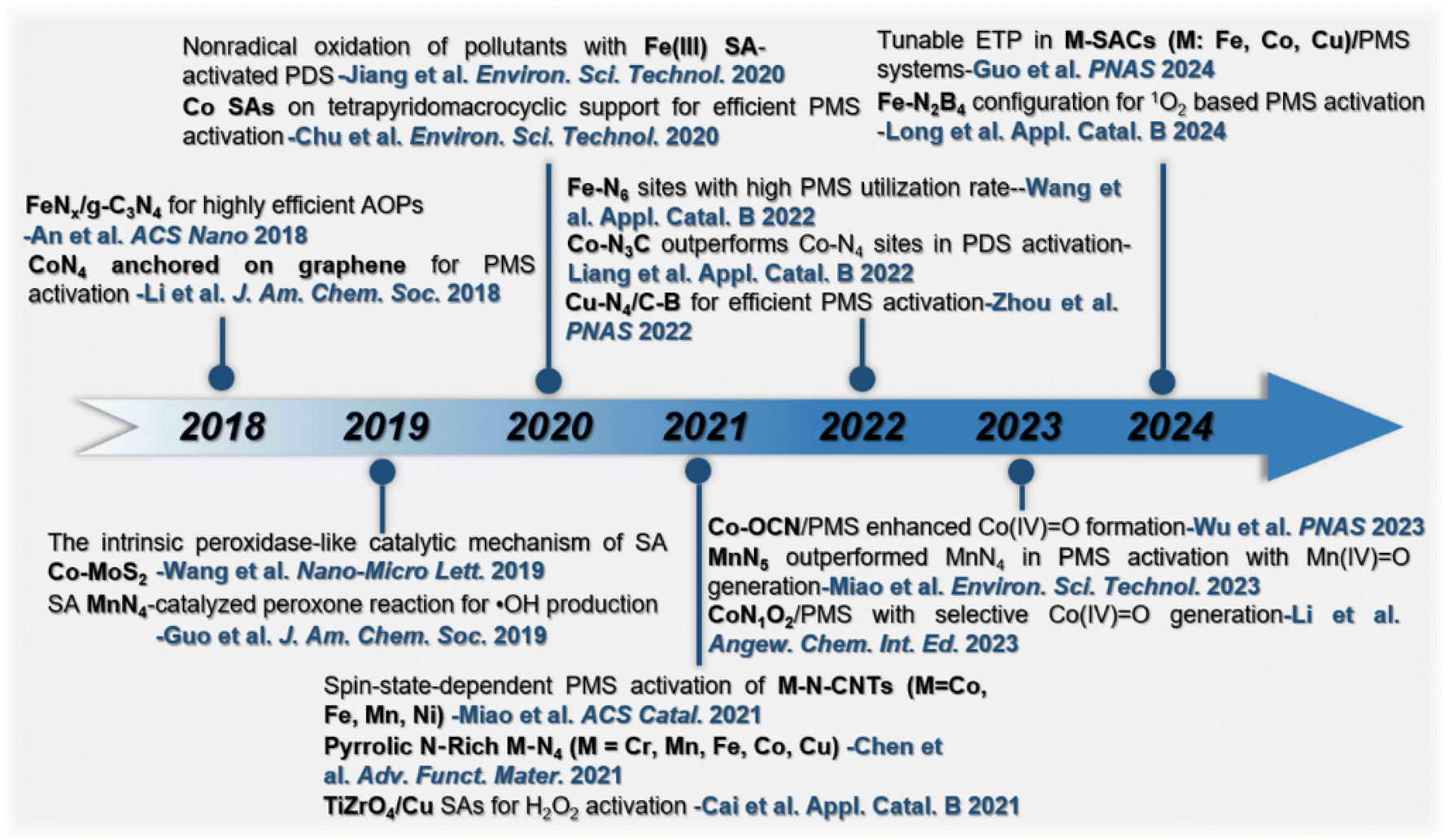 | ||
| Fig. 2 The timeline of SACs developed as heterogeneous Fenton-like catalysts. | ||
Although there are some recent reviews on the design, synthesis, characterization and performance of SACs applied in AOPs,39,52–54 a comprehensive review regarding the Fenton-like catalytic mechanism over heterogeneous SACs, especially the correlation between single-atom active sites and the generated ROS remains highly desirable. Herein, we examined the relevant literature on SACs with different metal types, supports and coordination environments for Fenton-like reactions in the recent five years to review the dominant ROS and the involved catalytic mechanisms. We systematically discuss the effects of single-atom metal sites, coordination environment and their electronic structures on the generation of oxidative species. These discussions will particularly focus on regulating the chemical properties of metal centers to produce desirable oxidative species for the directional degradation of ECs in wastewater treatment. Finally, we provide a brief summary and propose the challenges and opportunities to further advance the development of SACs-AOPs in water purification.
2. Different types of SACs in Fenton-like reactions
Several SACs have been applied in Fenton-like processes by using hydrogen peroxide (H2O2) or persulfate (PDS and PMS) as oxidizing agents. As illustrated in Table 1, different types of metal centers and supports play a crucial role in determining the catalytic performance of SACs and the generated ROS in Fenton-like reactions. More importantly, the selection of different supports critically influences the coordination and spatial configuration of single-atom metal sites serving as catalytic active sites, thereby tuning the intrinsic reactivity of SACs to enhance their catalytic activity.37,45,55 In this section, we will retrospectively examine recent research progress, categorizing SACs applied in Fenton-like processes based on the types of support, discussing their synthesis methods and their correlated catalytic performances. Until now, SAC based Fenton-like reactions are just in their infancy, with the relevant catalysts predominantly confined to carbon-based SACs and a smaller portion consisting of metal oxide-based SACs.47,56,57| Types of SACs | Catalysts | Oxidants | Metal loading | Pollutants | Major ROS | Active site | Mechanism | Ref. |
|---|---|---|---|---|---|---|---|---|
| a e—electron transfer process. | ||||||||
| Carbon-based SACs | SA-Cr/PN-g-C3N4 | H2O2 | — | Bisphenol A | ·OH | CrII–N4 | Radical | 62 |
| Cu-SA/NGO | H2O2 | 5.80 wt% | Phenol | ·OH | Cu–N4 | Radical | 42 | |
| Fe-PANI | H2O2 | 7.67 wt% | Acid Red G | ·OH | Fe–N | Radical | 63 | |
| rGO@NC/SA-Fe | H2O2 | 0.83 wt% | Rhodamine B | ·OH | Fe–N4 | Radical | 64 | |
| Fe-SAC | H2O2 | 1.68 wt% | Acyclovir | ·OH | Fe–N5 | Radical | 65 | |
| Bio-SA-Fe/g-C3N4 | H2O2 | 1.20 wt% | Sulfamethoxazole | ·OH, O2˙− | Fe-pyridinic N4 | Radical | 66 | |
| FeN5/NG | H2O2 | 1.97 wt% | Phenol | ·OH, O2˙− | Fe-pyridinic N5 | Radical | 67 | |
| Cu-N/C-SAC(S) | PDS | 1.90 wt% | Tetracycline | e−, O2˙−, 1O2 | Cu–C2N2 | Mixed | 68 | |
| DFeNC | PDS | 1.60 wt% | Sulfamethoxazole | SO4˙−, ·OH, 1O2 | Fe–N4 | Mixed | 69 | |
| SAZn@BC | PDS | 1.00 wt% | Trimethoprim | e− | Zn–N4 | Nonradical | 70 | |
| Co SA/CN-900 | PDS | 1.25 wt% | Tetracycline hydrochloride | e−, 1O2 | Co-pyrrolic N4 | Nonradical | 71 | |
| Fe-N-C | PDS | 5.00 wt% | 2,4-Dichlorophenol | FeIVO |
FeIII-pyridinic N4 | Nonradical | 72 | |
| CoSA-N3-C | PDS | 1.20 wt% | Bisphenol A | SO4˙−, ·OH | Co–N3 | Radical | 48 | |
| C3N4-Fe-rGO | PDS | 5.00 wt% | Tetracycline | SO4˙−, ·OH | Fe–N2O1 | Radical | 73 | |
| Co@MNC | PMS | 4.95 wt% | Bisphenol A | CoIVO, O2˙−, 1O2 |
Co–N2 | Mixed | 74 | |
| Fe-CN0.05 | PMS | 2.61 wt% | Bisphenol A | O2˙−, 1O2 | Fe–N3O1 | Mixed | 75 | |
| FeSA | PMS | 0.20 wt% | p-Nitrophenol | O2˙−, 1O2 | Fe–N4 | Mixed | 76 | |
| SA-Mn-NSC | PMS | 7.60 wt% | Enrofloxacin | O2˙−, 1O2 | Mn–N4 | Mixed | 77 | |
| Mn SAC | PMS | 1.70 wt% | Nitenpyram | O2˙−, 1O2 | Mn–N4 | Mixed | 78 | |
| SA-Zn-NC | PMS | 0.82 wt% | Acid Orange 7 | O2˙−, 1O2 | Zn–N4 | Mixed | 79 | |
| Co-CGBC | PMS | 11.30 mg g−1 | 2,4,4′-Trichlorobiphenyl | SO4˙−, ·OH, 1O2 | Co–N3S1 | Mixed | 80 | |
| NCoHCP | PMS | 3.32 wt% | Bisphenol A | SO4˙−, ·OH, 1O2 | Co-pyridinic N | Mixed | 81 | |
| Cu-SACs | PMS | 4.08 wt% | Oxytetracycline | SO4˙−, ·OH, 1O2 | Cu–Nx | Mixed | 82 | |
| FeSA-CN | PMS | 6.32 wt% | Sulfasalazine | SO4˙−, ·OH, 1O2 | Fe-pyridinic N4 | Mixed | 83 | |
| Co-N-C | PMS | 0.45 at% | 2,4-Dichlorophenol | SO4˙−, ·OH, e− | Co–N4 | Mixed | 84 | |
| Ni-SAs@CN | PMS | 0.26 wt% | Sulfamethoxazole | SO4˙−, ·OH, Ni-PMSa | Ni–N4 | Mixed | 85 | |
| FeUAC@FeSA-NC | PMS | 1.522 wt% | Sulfamethoxazole | SO4˙−, ·OH, O2˙−, 1O2, Fe-PMSa | FeIII–N4 | Mixed | 86 | |
| Co/C2N3-650 | PMS | 7.13 wt% | Sulfamethoxazole | 1O2 | Co–N3 | Nonradical | 87 | |
| ZIF-CoN3P-C | PMS | 1.65 wt% | Sulfadiazine | 1O2 | Co–N3P1 | Nonradical | 88 | |
| p-CoSi1N3@D | PMS | 0.05 wt% | Bisphenol A | 1O2 | Co–N3Si1 | Nonradical | 89 | |
| 0.5-Co-N@BC | PMS | 0.57 at% | Sulfamethoxazole | 1O2 | Co–N4 | Nonradical | 90 | |
| CoPc/G-NH2 | PMS | 0.75 wt% | Phenol | 1O2 | Co–N4 | Nonradical | 91 | |
| Co-SA/CMN | PMS | 0.70 wt% | 17β-Estradiol | 1O2 | Co–N4 | Nonradical | 92 | |
| CoSAC-NG | PMS | 0.36 wt% | Bisphenol A | 1O2 | Co–N4 | Nonradical | 93 | |
| SA-Co CNP | PMS | 0.10 wt% | Acetaminophen | 1O2 | Co–N4 | Nonradical | 94 | |
| SACoN/BCN | PMS | 5.10 wt% | Sulfamethazine | 1O2 | Co–N4B2 | Nonradical | 95 | |
| SACs Cu@C | PMS | 0.78 at% | Ibuprofen | 1O2 | Cu–C4Cl2 | Nonradical | 96 | |
| Curved Fe1-N4 | PMS | 1.00 wt% | Tetracycline | 1O2 | Curved Fe1–N4 | Nonradical | 97 | |
| Fe SA/NPCs | PMS | 2.17 wt% | Rhodamine B | 1O2 | Fe–N4 | Nonradical | 98 | |
| FeSA-NC | PMS | 0.43 wt% | Bisphenol A | 1O2 | Fe–N4 | Nonradical | 99 | |
| Fe1/CN | PMS | 11.20 wt% | 4-Chlorophenol | 1O2 | Fe–N4 | Nonradical | 100 | |
| FeSA-NGK | PMS | 2.57 wt% | Bisphenol A | 1O2 | Fe–N5 | Nonradical | 101 | |
| FeSA/AC-N-CPS | PMS | 0.94 wt% | Bisphenol A | 1O2 | Fe–Nx | Nonradical | 102 | |
| 1% Fe-N/C | PMS | 0.32 at% | Bisphenol F | 1O2 | Fe–Nx | Nonradical | 103 | |
| RuSA-N-C | PMS | 0.35 wt% | Orange II | 1O2 | Ru-pyridinic N4 | Nonradical | 104 | |
| SA Co-N-C(30) | PMS | 0.24 at% | Chloroquine phosphate | 1O2, e− | Co–N3 | Nonradical | 105 | |
| FeCNx-600 | PMS | 2.67 wt% | Bisphenol A | 1O2, surface-bound SO4˙− | Fe–N4 | Nonradical | 106 | |
| CoN3 | PMS | 2.90 wt% | Rhodamine B | CoIV O |
Co–N3 | Nonradical | 107 | |
| SAC-Co-2 | PMS | 2.56 wt% | 4-Chlorophenol | CoIV O |
Co–N4 | Nonradical | 108 | |
| CoSA-N3-C | PMS | 0.28 wt% | Norfloxacin | CoIV O, e− |
Co–N3 | Nonradical | 109 | |
| Co-N-CNTs | PMS | 0.06 wt% | Sulfamethoxazole | Co-PMSa | Co–N | Nonradical | 41 | |
| SACo-NGs | PMS | 1.96 wt% | Rhodamine B | ae− | Co2+–N–Cπ | Nonradical | 110 | |
| SA Co-N/C | PMS | 2.45 wt% | Naproxen | e− | Co–N3 | Nonradical | 111 | |
| ZIF-8@67-C | PMS | 27.27 wt% | Bisphenol A | e− | Co–N4 | Nonradical | 112 | |
| Co-N5/CNTs | PMS | 1.39 wt% | Sulfamerazine | e− | Co–N5 | Nonradical | 113 | |
| 3SACu@NBC | PMS | 3.41 wt% | Bisphenol A | e− | Cu–N4 | Nonradical | 114 | |
| FeSA-N/O-C | PMS | 1.28 wt% | Bisphenol A | e− | Fe–N4O1 | Nonradical | 115 | |
| CNFe2-0.6 | PMS | 16.64 wt% | Sulfamethoxazole | e− | Fe–N5 | Nonradical | 116 | |
| 5-SAFe-CN | PMS | 7.33 wt‰ | o-Phenylphenol | e− | Fe–N2O2 | Nonradical | 117 | |
| MnN5 | PMS | 2.80 wt% | 4-Chlorophenol | MnIV O |
MnII–N5 | Nonradical | 45 | |
| Mnsa-NCNTs | PMS | 0.47 wt% | 4-Chlorophenol | e− | Mn–N4 | Nonradical | 118 | |
| SAMn-G | PMS | 2.77 wt% | Benzaldehyde | e− | Mn–N4 | Nonradical | 119 | |
| Zn-N@C-10 | PMS | 0.36 wt% | Sulfamethoxazole | e− | Zn–N4 | Nonradical | 120 | |
| Fe-PNC | PMS | 1.42 wt% | Bisphenol A | e− | Fe–N4P2 | Nonradical | 121 | |
| FeSA-N-C | PMS | 0.93 wt% | Bisphenol A | FeIV O |
FeIII–N4 | Nonradical | 122 | |
| SA-Fe/CN | PMS | 0.60 wt% | Bisphenol A | FeIV O |
FeIII-pyrrolic N4 | Nonradical | 123 | |
| Fe-N3/C | PMS | 0.75 wt% | p-Nitrophenol | FeIV O, 1O2 |
Fe–N3 | Nonradical | 124 | |
| Fe-N-C | PMS | 1.27 wt% | Bisphenol A | FeIV O, 1O2 |
Fe–N4 | Nonradical | 125 | |
| Fe2-N-C | PMS | 0.51 wt% | Rhodamine B | FeIV O, 1O2 |
N3–Fe–Fe–N3 | Nonradical | 126 | |
| Cu-N4/C-B | PMS | 1.05 wt% | Bisphenol A | High-valent copper, e− | Cu–N4/C–B | Nonradical | 35 | |
| Mn-CN | PMS | 5.20 wt% | Oxalic acid | ·OH | Mn–N4 | Radical | 127 | |
| Cu/NV-CN | PMS | 5.00 wt% | Phenol | ·OH | Cu–N4 | Radical | 128 | |
| CoSA-PMOF | PMS | 6.10 wt% | Moxifloxacin | SO4˙− | Co–N4 | Radical | 129 | |
| SACo@g-C3N4 | PMS | 3.17 wt% | Ethylbenzene | SO4˙−, ·OH | Co–N2 | Radical | 130 | |
| Co-doped g-C3N4 | PMS | 1.00 wt% | Rhodamine B | SO4˙−, ·OH | Co–N4 | Radical | 131 | |
| SACo@NG | PMS | 4.10 wt% | Benzyl alcohol | SO4˙−, ·OH | Co–N4 | Radical | 132 | |
| Mn-ISAs@CN | PMS | 0.27 at% | Bisphenol A | SO4˙−, ·OH | Mn–N4 | Radical | 133 | |
| Metal oxide-based | Cu/Al2O3 | H2O2 | 1.00 wt% | 1,3-Dihyroxyacetone | ·OH | CuII | Radical | 134 |
| Au/VO2 | PDS | 3.70 wt% | Rhodamine B | SO4˙− | Au–V | Radical | 135 | |
| Pd/CeO2 | PDS | 0.25 wt% | Bisphenol A | SO4˙−, ·OH, e− | Pd0 | Mixed | 136 | |
| 2.5Cu/CeO2 | PMS | — | Tetracycline | 1O2 | Cu–N1O4 | Nonradical | 137 | |
| Co-CuO(Ov) | PMS | 0.187 wt% | Tetracycline | 1O2, e− | Ov | Nonradical | 138 | |
| CoN1O2 | PMS | 1.69 at% | Sulfamethoxazole | CoIVO |
Co–N1O2 | Nonradical | 47 | |
| Co/BiVO4 | PMS | 5.00 wt% | Tetracycline hydrochloride | SO4˙−, ·OH | CoII | Radical | 139 | |
2.1 Carbon-based SACs
Due to their abundant anchoring sites and excellent surface adsorption properties to stabilize single metal atoms, carbon-based materials are commonly employed as supports for SACs.30,54,57 Specifically, the carbon supports possess large surface areas, offering ample anchoring sites for single metal atoms. Moreover, their binding affinity to metal atoms can be adjusted using heteroatom dopants, and by means of defects, functional group grafting, and the coordination of SACs,56,58,59 which plays an essential role in their catalytic activities in the Fenton-like process. Table 1 summarizes the carbon-based SACs applied in Fenton-like reactions. Choosing the right carbon support can significantly boost the intrinsic activity and loading of single metal atoms, effectively enhancing the Fenton-like activity of SACs. The commonly used carbon supports include carbon nanotubes (CNTs), graphitic carbon nitride (g-C3N4), graphene oxide (GO), carbon derived from metal–organic framework (MOF) precursors and biomass, etc. Among them, MOFs and g-C3N4 are the two most frequently selected supports for Fenton-like reactions, owing to their rich nitrogen contents and adjustable structures (Fig. 3a).56,60,61 We therefore discuss carbon-based SACs applied to Fenton-like reactions based on these two supports. | ||
| Fig. 3 (a) The statistics on the supports for synthesizing SACs for Fenton-like reactions; (b) the statistics of publications on reaction pathways involved in Fenton-like reactions over SACs. | ||
As one precursor to three-dimensional carbon materials, MOFs are crystalline porous materials formed by the periodic arrangement of metal centers and organic ligands (Fig. 4a). Due to their ordered frameworks and abundant unsaturated metal active sites, MOFs and their composite materials can be easily transformed into carbon-based SACs for Fenton-like reactions through pyrolysis or post-treatment.60 During the pyrolysis, the nitrogen in the MOF can covalently interact with metal ions to form M–N configuration, significantly preventing metal aggregation as well as improving the stability of SACs. For example, Ye et al. developed a surfactant shell strategy to synthesize the Fenton-like catalyst, an Fe-based SAC with a robust FeN4 coordination.140 The surfactant, cetyltrimethylammonium bromide (CTAB), used in the synthesis has two functions: modulating the crystallinity of the MOF precursor and concurrently coordinating with the metal sites on its surface to form a surfactant shell. Additionally, the pyrolysis of the CTAB layer results in the formation of a nitrogen-doped carbon shell, effectively preventing the collapse of the MOF framework (Fig. 4(b1)). Simultaneously, CTAB acted as an additional carbon and nitrogen source during the pyrolysis process, stabilizing single Fe atom sites and reducing their aggregation. The resulting Fe SACs/NC with Fe loading up to 8.5 wt% presented the best catalytic activity for H2O2 activation (Fig. 4(b2)). Trimethylolpropane and ciprofloxacin were completely degraded within 30 min in this Fenton-like process catalyzed by Fe SACs/NC, while the other contaminants attained 100% removal within 60 min. Moreover, Fe SACs/NC exhibited appreciable stability in the heterogeneous Fenton-like reaction, and the dominant active sites of the FeN4 coordination structure were determined by the characterization of EXAFS. Fig. 4(b3) showed the DFT calculation on Gibbs free energy evolution (ΔG) of the FeN4 and Fe3N sites involved in H2O2 adsorption and activation. The ΔG value of Fe SAC/NC with FeN4 sites (−3.11 eV) was lower than that of Fe NP/NC with Fe3N sites (−2.76 eV) during the conversion of OOH* to OH*, revealing more facile cleavage of H2O2 catalyzed by Fe SAC/NC to form ·OH. This result further clarified the remarkable catalytic activity of FeN4 sites in facilitating H2O2 activation. However, the MN4 coordination configuration of MOF-supported SACs is not rigid. For example, Zhan et al. developed a Co-SA catalyst with a variable Co–N4 configuration by the pyrolysis of Zn and Co co-doped MOFs (Fig. 4(c1)).141 The atomically dispersed Co sites as well as the coordination environments were characterized by HAADF-STEM and EXAFS, respectively. Two possible CoN4 configurations were proposed: a traditional CoN4 configuration embedded in an intact graphite layer, and a CoN2+2 bridged between two adjacent graphite edges. Theoretical estimation of the Co–N bond length (1.88 Å) within the CoN2+2 site closely approximated the value of 1.89 Å as determined by EXAFS analysis. The DFT results further showed that the CoN2+2 site was more energy favorable for PMS adsorption compared to the CoN4 site (Fig. 4(c2)). These results conclusively corroborated the vital role of the CoN2+2 configuration in Co-SA for PMS-based Fenton-like reactions. The above examples illustrate that different anchoring sites of MOF-based supports for a single metal atom lead to their variable coordination configurations, thus affecting the catalytic activity and stability of SACs.
 | ||
| Fig. 4 (a) The typical configuration of MOFs and its derived carbon-based SACs.60 (b1) Preparation of Fe-SAC/NC. (b2) Removal of different organic contaminants during the use of the Fe-SAC/NC-H2O2 oxidation system. (b3) Reaction pathways for the adsorption and activation of H2O2 at FeN4 and Fe3N sites investigated by DFT. Reproduced with permission.140 (c1) Synthesis scheme of Co-SA. (c2) Comparative DFT calculation results of PMS adsorption on CoN2+2 and CoN4 surface, respectively.141 Figures have been adapted from ref. 60, 140 and 141 with permissions from Elsevier B.V., Copyright 2023, Elsevier B.V., Copyright 2022, and Wiley-VCH, Copyright 2020, respectively. | ||
In addition to MOFs, 2D layered g-C3N4 is commonly employed as a support for SACs, because it has uniform distribution of vacancies, which provides abundant nitrogen coordination sites for stabilizing single-metal atoms.142 Moreover, g-C3N4 exhibits excellent chemical stability and cost-effectiveness, further enhancing its suitability as a support material for SACs.143,144 Currently, a novel strategy has been developed utilizing various nitrogen-rich carbon organic small molecules (such as urea, melamine, and dicyandiamide) to self-assemble with metal ions, followed by a one-step pyrolysis to obtain g-C3N4-supported SACs applied in Fenton-like reactions.45,65,140,145 In comparison with metal oxides, this catalyst exhibits an expanded applicable pH range and suppressed metal leaching during the Fenton-like reaction. Cui et al. successfully incorporated single-atom Cu into g-C3N4 by a one-pot pyrolysis using Cu(NO3)2 and melamine as the precursors.146 Cu–C3N4 demonstrated remarkable catalytic activity in the generation of ·OH via activating H2O2 at pH = 7.0, resulting in the complete decomposition of RhB within 5 minutes, exceeding the performance of conventional Cu2O and CuO catalysts. Furthermore, the stability of Cu-C3N4 was assessed by immobilizing it on a filter composed of carbon fibers. The filter maintained efficient activity for dye removal over extended periods, with acceptable Cu leaching (0.1 ppm). Furthermore, for carbon-based SACs with M–N coordination, compared to other carbon supports, g-C3N4 exhibits an extremely high N/C ratio (57%) in its bulk structure, facilitating the increase of metal loading density.100 Furthermore, its thermal instability allows for facile introduction of abundant carbon–nitrogen vacancies in g-C3N4, typically enabling single metal atoms coordinated with these vacancies to achieve high coordination numbers (>4).147 Zhu et al. synthesized Fe-based SACs using g-C3N4 as a support, featuring FeN5 coordination (CNFe2-0.6) with a high Fe loading content (16.64 wt%) (Fig. 5(a1)).116 The CNFe2-0.6 can rapidly activate PMS through a pathway of surface contact oxidation, resulting in the rapid degradation of sulfamethoxazole (SMX) (Fig. 5(a2)). The degradation rate of SMX accelerated with the increase of Fe loading (MFe), but Fe agglomeration when MFe exceeded 16.64 wt% decreased its catalytic performance (Fig. 5(a3)). DFT calculations revealed the thermodynamic preference of FeN5 sites (−1.40 eV) over FeN3 sites (−0.37 eV) for electron acquisition from contaminants. Moreover, the energy required for –SO4H desorption from the FeN5 site is lower than that from the FeN3 site, facilitating catalyst regeneration (Fig. 5(a4) and (a5)).
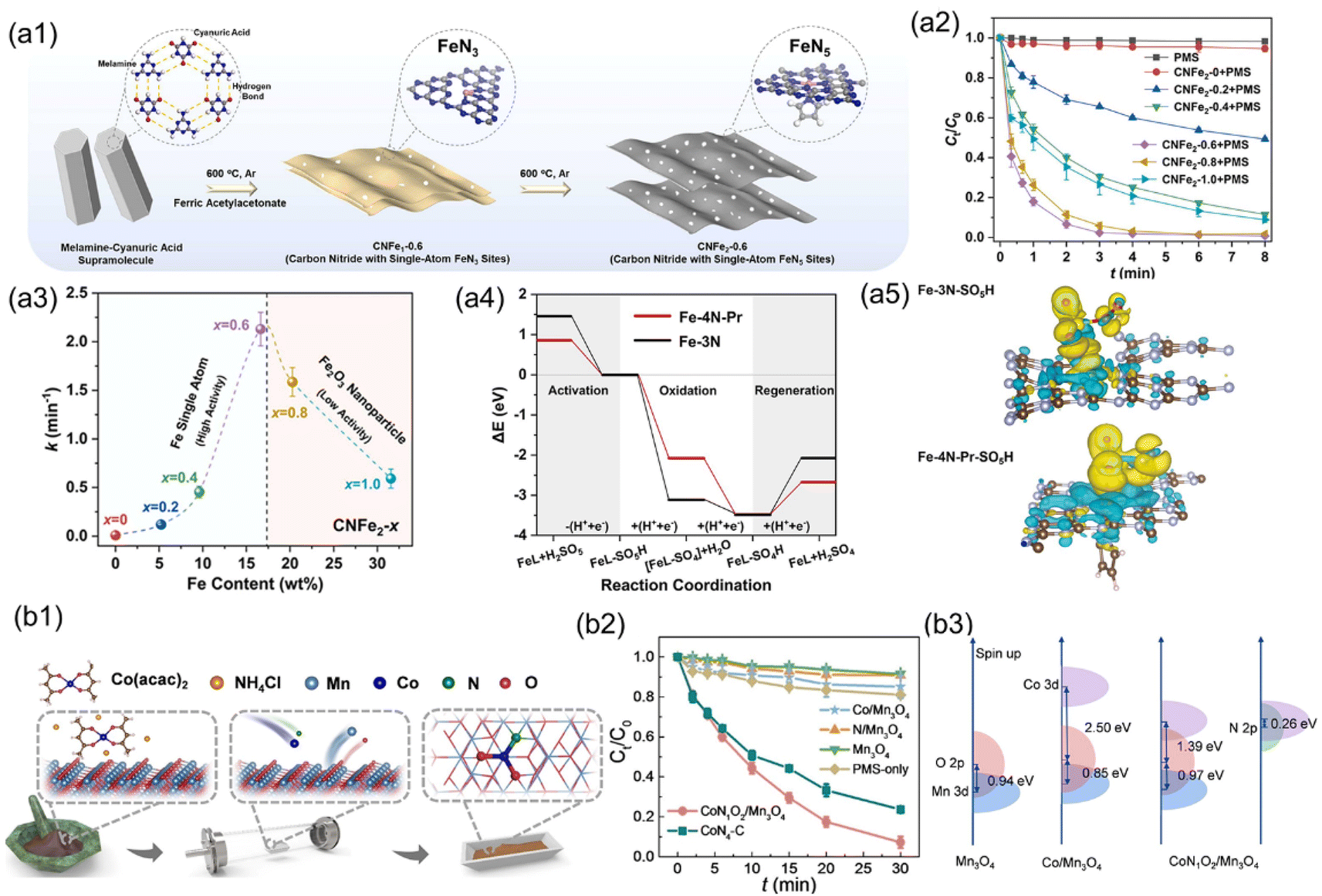 | ||
| Fig. 5 (a1) The synthetic procedures for CNFe1-0.6 and CNFe2-0.6. (a2) SMZ degradation during PMS activation over different CNFe2−x (x = 0–1.0) catalysts. (a3) The correlation of degradation rate constants (k) of SMZ with Fe content in CNFe2−x. (a4) Energy diagrams illustrating PMS activation on Fe-4N-Pr and Fe-3N. (a5) Graphs depicting charge density for Fe-3N-SO5H and Fe-4N-Pr-SO5H species.116 (b1) The synthesis of CoN1O2/Mn3O4. (b2) SMX degradation in the PMS system catalyzed by different catalysts. (b3) Illustration of Mn 3d-O/N 2p overlap for CoN1O2/Mn3O4.47 Figures have been adapted from ref. 116 and 47 with permissions from Elsevier B.V., Copyright 2022, Elsevier B.V., and Wiley-VCH, Copyright 2023, respectively. | ||
In summary, the main advantage of using carbon substrates as carriers for the immobilization of single metal atoms is the broadly available and low-cost carbon precursors (e.g., biomass). In addition, metal atoms can coordinate with heteroatoms such as N, C, O, S, and B to form strong metal–support interactions, over the carbon substrates. The doped metal atoms can alter the electronic properties of the carbon substrate, enhance the adsorption of peroxide molecules, and induce tunable catalytic pathways in Fenton-like reactions. The distinct mechanisms triggered by carbon-based SACs are likely closely related to their structural characteristics, which are determined by the types of single-atom metal, substrates, and local coordination environments. These aspects are discussed in detail in the subsequent sections.
2.2 Metal oxide-based SACs
The inherent uncontrollable pyrolysis process of carbon-based supports often results in uncertain metal coordination within carbon-based SACs.41,148 In recent years, several metal oxides have been employed as the supports for heterogeneous SAC catalysts for Fenton-like reactions (Table 1).As metal oxides with precise crystalline structures offer advantages over carbon-based supports, isolated metal sites with well-defined coordination can be fabricated.47,137 For instance, Li et al.47 employed spinel Mn3O4 as the support to anchoring Co single atoms (CoN1O2/Mn3O4) via a reverse atom-trapping strategy (Fig. 5(b1)). The CoN1O2/Mn3O4 containing 0.8 wt% Co exhibited superior performance for PMS activation, achieving complete SMX removal within 30 min (Fig. 5(b2)). The projected density of states (PDOS) for CoN1O2/Mn3O4 indicated that the co-coordination effects of nitrogen and oxygen with the cobalt center led to a notable decrease (1.39 eV) in the energy difference between the Co d- and O p-band, thereby enhancing the hybridization of Co–O interactions (Fig. 5(b3)). Furthermore, the introduction of nitrogen into CoN1O2 moieties promoted the electronic delocalization of cobalt sites, restraining the filling of Co 3d orbitals, thus facilitating PMS adsorption and high valent Co(IV)O formation. Fusion and aggregation of single metal atoms are common occurrences in synthesis and catalysis. The Fenton-like catalytic performance of metal oxide-based SACs can be enhanced by the surface morphology of metal oxides to increase the exposure of active sites, thus facilitating electron transfer. Xie et al. employed a solvothermal-assisted spontaneous exfoliation method to fabricate Au atoms supported on 2D VO2(B) nanobelts (Au/VO2) and investigated their efficacy in degrading organic pollutants using PDS activation.135 Due to the similar covalent radii of Au (0.134 nm) and V (0.122 nm) atoms, Au atoms are easily immobilized on the surface of the VO2(B) nanobelt. The 2D nanobelt architecture enhances the exposure range of Au, thus increasing the number of active sites available for S2O82− absorption. Besides, the strong “electronic metal–support interactions” induced the positively charged Au atoms in Au/VO2, thus lowering the energy barrier for the decomposition of S2O82− to SO4˙−. However, the metal oxide-based SACs applied in Fenton-like reactions are relatively few. Further investigation is particularly needed on the structural characteristics of metal oxides, such as surface defects, crystalline phase, coordination, and their impact on the Fenton-like activity and reaction mechanisms.
2.3 Synthesis strategies of SACs applied in Fenton-like reactions
The synthesis methods of SACs for Fenton-like reactions have advanced unprecedentedly. Diverse synthesis strategies, including pyrolysis, solvothermal method, ball milling and chemical reduction, have been reported for fabricating highly efficient SACs. Herein, we focus on the synthetic methods with an emphasis on regulating the coordination environment of SACs.Pyrolysis is the most popular method for the synthesis of SACs, especially carbon-based SACs. The pyrolysis temperature is usually higher than 700 °C.59 Adjusting the pyrolysis temperature can change the spin and oxidation states of the isolated metal sites, thus impacting the catalytic activity and selectivity of SACs. For instance, Zhang et al. demonstrated spin state-dependent performance for PMS activation in organic pollutant oxidation by adjusting the pyrolysis temperature of Fe SACs (Fig. 6a and b).86 The coordination number of the isolated metal center is a valid descriptor reflecting the structure–activity relationship of the SACs, since the coordination number is very sensitive to the pyrolysis temperature, which determines the structure of SACs.149 The coordination number of SACs generally decreases with the increase of the pyrolysis temperature. Wu et al. constructed a set of Fe SACs with different coordination numbers (FeNx, x = 2, 3, 4) by controlling the pyrolysis temperature from 900 °C to 1100 °C (Fig. 6c).150 In particular, the Fenton-like catalytic performance of FeNx increased with the increased coordination numbers. Furthermore, various heteroatoms including N, O, C, P, B, Cl, etc., can be coordinated with the isolated metal sites and tune the electronic properties of the metal center and supports, which can be modified by adding regulators or precursors containing different hetero atoms during pyrolysis.151,152 Li et al. designed Co SACs with a Co-P1N3 moiety through in situ phosphorylation of triphenylphosphine encapsulated within Co MOFs, while traditional Co-N4 SACs were obtained by the direct pyrolysis of Co MOFs (Fig. 6d).153 However, most precursors tend to lose their original structures during high-temperature pyrolysis, resulting in a tendency for the metal atoms to rebuild into the most preferred coordination structure (i.e., M–Nx).154 Due to the uncontrollable interaction between the metal center and coordination atoms, the high-temperature pyrolysis strategy usually produces multiple active sites in the obtained SACs.155,156 Therefore, there is an urgent need for pyrolysis methods to precisely regulate the coordination environments of the metal centers.
 | ||
| Fig. 6 (a) The preparation of FeNx-C catalysts (Fe SACs); (b) the proposed mechanism of FeNx-C/PMS system;86 Copyright 2021 American Chemical Society. (c) The synthesis strategy of FeNx catalysts based on pyrolysis temperature and the relationship between Fe–N coordination number and the intrinsic reactivity;150 Copyright © 2023 American Chemical Society. (d) The fabrication process of Co-SA/P catalysts;153 Copyright © 2020 American Chemical Society. (e) Schematic of the synthesis process for Ni SACs;161 Copyright © 2020 American Chemical Society. (f) The preparation strategy of Pt/f-MWCNTs;162 figures have been adapted from ref. 86, 150, 153, 161 and 162 with permissions from the American Chemical Society, Copyright 2022, American Chemical Society, Copyright 2023, American Chemical Society, Copyright 2020, American Chemical Society, Copyright 2020 and Elsevier B.V. Copyright 2019, respectively. | ||
Compared to the pyrolysis method, the low-temperature synthesis strategy can better regulate the coordination structure of SACs by preserving the coordination characteristics of precursors.59,157 This approach is promising for constructing SACs with well-defined coordination environments. Typical low-temperature synthesis methods, such as electrodeposition and photodeposition, finely tune the coordination environment of SACs by modifying the properties of surface functional groups.158–160 For example, Qiu et al. developed a one-step electrochemical synthesis method for large-scale production of atomically dispersed Ni sites (Fig. 6e). In this process, highly electronegative oxygen defects were formed on the graphite foil surface, anchoring Ni2+ ions from the electrolyte by forming Ni–O6 coordination.161 Similarly, Lin et al. used photodeposition to construct Pt SACs on CNTs, with Pt atoms anchored by four oxygen atoms from surface functional groups (Fig. 6f).162 The study on low-temperature synthesis of SACs is still in its early stages. Combining the advantages of both methods could thus lead to the development of SACs with customizable and well-defined coordination structures for efficient Fenton-like reactions.
3. Mechanisms of SAC-based Fenton-like reactions
The catalytic performance of SACs depends on the loading amount and types of isolated metal center, as well as surrounding electronic and chemical factors (such as the coordination environment). With the rapid advancement of advanced characterization instruments and theoretical calculations, the intuitive definition of catalytic sites for individual metal atoms and the simulation of catalytic reaction mechanisms have become clearer.163,164 This clarification provides insights into the chemical states and coordination environments of catalytic sites involved in Fenton-like oxidation, along with their engagements with oxidants (e.g., H2O2, PMS, and PDS) or target pollutants, electron-transfer process, and the formation and evolution of ROS.52,165 The activation mechanisms of peroxides by SACs in Fenton-like reactions have been extensively studied, yielding significant insights into their intrinsic activation processes.11,166 To date, various radical (e.g. ·OH and SO4˙−) and nonradical pathways (e.g.1O2, high-valent metal-oxo (HVMO) species, and direct electron transfer) are considered as the primary activation mechanisms, which will be discussed in detail in this section.3.1 Radical pathways and the associated active sites
The radical based-catalytic mechanism in Fenton-like reaction systems has been extensively studied and recognized, owing to the highly reactive radicals, such as ·OH and SO4˙−, serving as efficient ROS for mineralizing refractory ECs.167 The generation of highly reactive ·OH primarily depends on the cleavage of peroxides adsorbed on the active sites of the Fenton-like catalyst surface. Recently, Yin et al. reported the efficient catalytic activation of H2O2 by using a single-atom Fe catalyst (SAFe-SBA) for the removal of p-hydroxybenzoic acid (HBA).14 In comparison to the catalysts containing clustered Fe sites, SAFe-SBA exhibited superior activity (Fig. 7a). 1.0SAFe-SBA with a lower Fe loading of 1.0 mM Fe per g showed optimal catalytic activity, achieving 100% removal of HBA within 120 min. Through the radical quenching tests and electron paramagnetic resonance (EPR) analysis (Fig. 7b), the potential catalytic reaction mechanism of SAFe-SBA/H2O2 (Fig. 7c) as described by eqn (1)–(3) is proposed.| Si−O−Fe(III) + H2O2 → Si−O−Fe(III)−H2O2 → Si−O−Fe(II) + HO˙2 | (1) |
| Si–O–Fe(II) + H2O2 → Si–O–Fe(III) + 2·OH | (2) |
| HBA + ·OH → intermediates → CO2 + H2O E0 = 1.9–2.7 V | (3) |
| HSO−5 + 2H+ + 2e− → HSO−4 + H2O E0 = 1.81 V | (4) |
| S2O2−8 + 2H+ + 2e− ↔ 2HSO−4E0 = 2.08 V | (5) |
| SO4˙− + e− ↔ SO2−4E0 = 2.5–3.1 V | (6) |
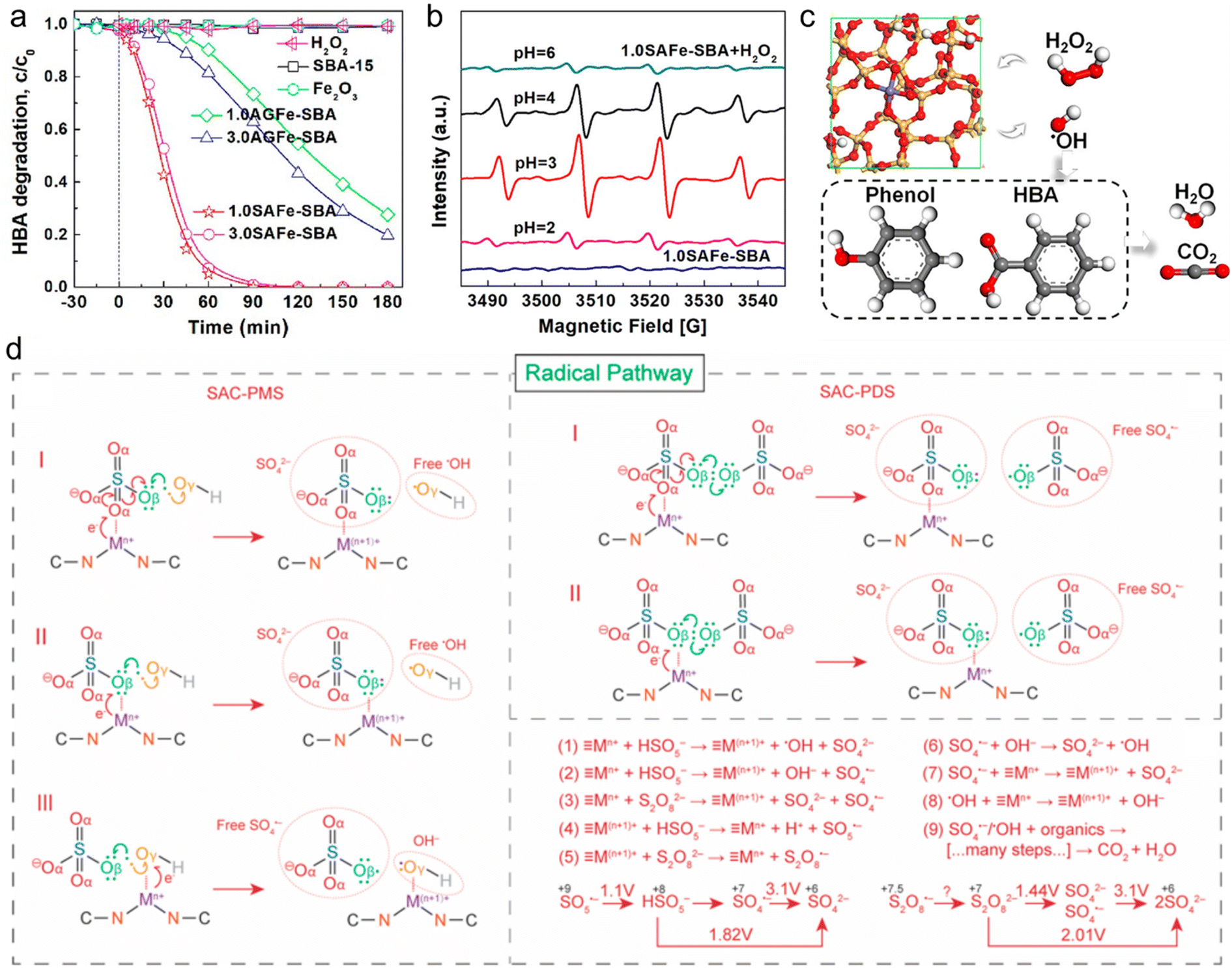 | ||
| Fig. 7 (a) Adsorption and oxidation of HBA by H2O2 catalyzed activation on different catalysts. (b) EPR spectra for Fenton-like reactions catalyzed by 1.0SAFe-SBA under different pH conditions. (c) Proposed mechanism for H2O2 activation on SAFe-SBA surface.14 (d) The radical generation pathway and reaction steps of the persulfate-based Fenton-like process catalyzed by SACs.170 Figures have been adapted from ref. 14 and 170 with permissions from the American Chemical Society, Copyright 2019, and American Chemical Society, Copyright 2022, respectively. | ||
In addition, SO4˙− is predominantly generated from two types of persulfate oxidants (PDS and PMS), which can cleave O–O bonds through either energy transfer or reductive electron transfer (i.e., Fenton-like reactions) to oxidize target pollutants in wastewater (eqn (4)–(6)).11,168,169 During the Fenton-like reaction, single metal atoms (≡Mn+) serve as the catalytic active sites, binding to persulfate and undergoing oxidation to form ≡M(n+1)+ through one-electron transfer, thereby generating reactive radicals (SO4˙− and ·OH). Subsequently, upon detachment of surface-bound SO42− and OH−, ≡M(n+1)+ can be further reduced to ≡Mn+ by persulfate, thus completing the redox cycle (Fig. 7d).170,171 However, this reduction process remains speculative and lacks experimental validation. Due to the oxidation potential of PMS (HSO5−/SO5˙−) at E0 = 1.1 V, ≡Mn+/≡M(n+1)+ sites such as Mn3+/Mn2+ (E0 = 1.51 V), Co3+/Co2+ (E0 = 1.82 V), and Ag2+/Ag+ (E0 = 1.98 V) are thermodynamically favorable for PMS oxidation.172 The E0 value for S2O8˙−/S2O82− is currently unavailable in the literature, and more efforts should be taken to understand PDS oxidation by ≡M(n+1)+. Different types of oxidants significantly affect the catalytic activity of Fenton-like catalysts and the generation of ROS in the reaction system, thereby influencing the degradation of refractory pollutants. Due to the asymmetric structure and longer O–O bond (IO–O = 1.326 Å), PMS can easily be activated than PDS (IO–O = 1.322 Å).52,173 Thus, PMS emerges as a highly promising reagent for wastewater oxidation remediation due to its mildness, ease of transport, and storage. The efficacy of SO4˙− generated in PMS-based Fenton-like reactions stems from its elevated redox potential (2.5–3.1 V) and prolonged half-life (30–40 μs) relative to ·OH (1.8–2.7 V, 20 ns).11 Additionally, the reactivity of SO4˙− towards organic compounds is essentially independent of pH.52
3.2 Nonradical pathways and the associated active sites
According to an analysis of the literature about SAC-based Fenton-like systems, nonradical pathways with high selectivity accounted for 60.0%, demonstrating their dominance in this field (Fig. 3b). The single-atom metal or non-metal active sites on carbon supports in SACs could involve in Fenton-like processes to generate the nonradical species. Additionally, among the different reaction pathways (Fig. 3b), singlet oxygen (43.1%) was frequently identified as the primary ROS oxidizing target ECs in nonradical-based SACs/peroxide systems.Singlet oxygen (1O2) oxidation is a frequently observed nonradical pathway in SAC-based Fenton-like systems.40,174,175 Taking PMS as the example, we then proceeded to discuss the formation pathway of 1O2 in SACs/PMS systems and the associated active sites (Fig. 8). Recent studies have utilized DFT calculations to explain the 1O2 generation pathways on single-atom metal sites (≡Mn+).83,100,176 PMS initially adsorbs onto the ≡Mn+ site, undergoing decay to form a metal–O complex structure (≡M(n+2)+O), which serves as a crucial intermediate for the 1O2 formation.177,178 However, the precise configuration of ≡M(n+2)+O remains unclear and requires further experimental and theoretical validation. It is hypothesized that the role of ≡M(n+2)+O is similar to ketone groups (R2CO) in carbon materials, further catalyzing the decay of PMS with metal–dioxirane intermediates to generate 1O2.179–181 Moreover, in comparison to metal nanoparticles, SACs are more prone to react with peroxides to form a structure like ≡M(n+2)+O. If the above hypothesis is right, this mechanism could explain why 1O2 is reported to dominate in SAC-based Fenton-like systems (Fig. 3b).39,100,165 As illustrated in Fig. 8, besides R2CO, other non-metal sites (e.g.+C–N) on carbon-supported SACs can also facilitate PMS activation to form O2˙−, which serves as an intermediate product subsequently transformed into 1O2.182 Particularly, electron-rich heteroatoms such as nitrogen (N) can increase the charge density of adjacent positive carbon atoms, which accelerate PMS decay to produce O2˙− by weakening the O–O bond.183,184 Furthermore, the alteration in charge distribution due to N modification can prompt the electron loss from PMS to the positive carbon atoms, leading to the generation of SO5˙−, and subsequently produce 1O2viaeqn (7).185 In addition, lattice oxygen (OL) or oxygen vacancy (OV) sites in metal oxide-based SACs can also promote PMS activation to generate 1O2.186
| SO5˙− + SO5˙− → 2SO2−4 + 1O2 | (7) |
 | ||
| Fig. 8 Different nonradical pathways of SAC-based Fenton-like reactions. | ||
Direct electron transfer serves as a nonradical pathway in the SAC-based Fenton-like systems.111,187 Mechanisms based on direct electron transfer for persulfate activation have been proposed for both carbon-based materials and other nanocatalysts.25,188,189 Similarly, in SAC-based Fenton-like processes, electrons are transferred from target contaminants (electron donors) to active sites (metal sites or non-metal sites) on the carbon supports of SACs, and then to persulfate (electron acceptors), achieving the oxidation of pollutants (Fig. 8).41,114 In contrast to the radical, persulfate acquires two electrons from pollutants to produce sulfate ions in the nonradical mechanism, where metal or non-metal sites merely act as mediators for electron transfer. DFT studies have investigated the charge distribution and electron density at single atom sites and surrounding the metal–support junction.117,190 For carbon-supported SACs, some studies have demonstrated that the total density of states (TDOS) near the Fermi level of single-atom metal sites was higher than that of adjacent C and N atoms, due to the contribution of metal d orbitals (e.g., Co, Fe, Mn and Cu).45,191,192 The increased TDOS reduced the interface barrier between the metal and support, consequently triggering more free electron accumulation at the metal sites, facilitating charge transfer between the metal and support. This also implied a higher charge accumulation between single-atom metal sites and their adjacent coordinating atoms, promoting persulfate reduction.111,193 Previous DFT analysis also indicated that the Fermi level of SACs supported on carbon was more negative compared to the original carbon support, approaching the conduction band and thus acting as a better electron donor,114,194 which could help to enhance the Fenton-like activity of SACs and promote the decomposition of peroxides to produce active oxygen species for EC oxidation.
The HVMO pathway has been systematically investigated in SAC-based Fenton-like reactions.168,170 Compared with SO4˙− and ·OH, the HVMO species Fe(IV)-oxo (E0 = 2.0 V) is a milder oxidant, but its lifetime can last up to 0.1 s, becoming the suitable ROS for oxidizing target ECs in the complicated water matrix.195,196 The HVMO species (≡M(n+2)+O), such as CoIVO,47,49 FeVO,72 FeIVO,197,198 CuIIIO,46 and MnIVO,45 serve as crucial ROS in Fenton-like processes and have been identified as the major contributors in certain SAC systems. ≡M(n+2)+O could be formed by transferring two electrons from low-valent metal single-atom sites (≡Mn+) to peroxide (Fig. 8), which is associated with the decay pathway of peroxides. Current research only offers some experimental clues, but without detailed pathways regarding the arrangement of peroxide molecules and the formation of ≡M(n+2)+O. DFT studies suggested that the dissolved Co2+ ion interacted with PMS (HSO5−) through type III configurations, followed by O–O bond cleavage and deprotonation to generate CoIVO2+.199,200 The formation mechanism of HVMO in SACs may share similarities with the aforementioned mechanism, but further validation is required. Methylphenyl sulfoxide (PMSO) is commonly employed to elucidate the role of ≡M(n+2)+O, which can be highly selectively oxidized into PMSO2 by HVMO.200 Furthermore, the specific oxygen atom exchange between HVMO and H2O enables verification of ≡M(n+2)+O by using 18O isotope labeling techniques.47,200 However, it is widely recognized that the reaction kinetics of HVMO species with common organic contaminants is 102–104 M−1 s−1, which is lower than that of 1O2 (102–108 M−1 s−1) and radicals (106–1011 M−1 s−1),195 raising concerns about the effectiveness of HVMO species.
Although the mild redox potential of the nonradical pathway leads to the lower mineralization efficiency of organic matter compared to the highly oxidative radical system, its high selectivity can trigger polymerization reactions to generate value-added polymer products and reduce CO2 emissions.201,202 This selectivity also facilitates the effective oxidation of target organics, avoiding interference with background matrices and preventing the formation of halogenated disinfection by-products, thus showing unique advantages in treating actual wastewater.170 In addition, the redox potential of the nonradical pathway can be modulated by adjusting the structure, composition and physicochemical properties of the catalyst.203 The degradation efficiency of organic matter in nonradical pathways is determined by the activity, type, concentration, and reaction time of reactive species.204 In contrast, the radical pathway exhibits non-selectivity for most organic pollutants, are influenced by the water matrix, and may produce halogenated by-products. Thus, evaluating the properties and formation of nonradical and radical pathways can fill the knowledge gap and aid to establish the structure–activity–mechanism relationship between SACs and activation pathways, providing guidance for designing highly efficient SACs and effectively regulating activation pathways in Fenton-like systems.
4. Crucial factors for the performance of SACs
The types of ROS generated in SAC-based AOPs is primarily governed by the active sites (Table 1). Indeed, various oxidation pathways were elicited during Fenton-like reactions across different metal and non-metal active sites in SACs, stemming from the difference in surface adsorption energy, electronic properties, and charge transfer capabilities. The radical pathways (such as SO4˙− and ·OH) are typically formed by the cleavage of the O–O bond in peroxides. However, electron transfer and hydrogen abstraction reactions influence the generation of radicals. Two-electron transfer at single-atom metal sites may lead to the formation of HVMO. Hydroxyl groups on the carbonaceous surfaces in SACs can facilitate PMS adsorption, favoring the formation of surface-active complexes. Defects affect the electron distribution on the catalyst surface, generating active sites and promoting the preferential cleavage of O–H bonds in PMS to produce SO5˙− intermediates, thus generating 1O2. In summary, the ease of gaining or losing electrons at active sites in SACs influences the activation pathways of peroxides. In this section, the metal types, coordination environments and metallic electron spin state are discussed as the crucial factors for ROS generation in the Fenton-like catalysis (Fig. 9).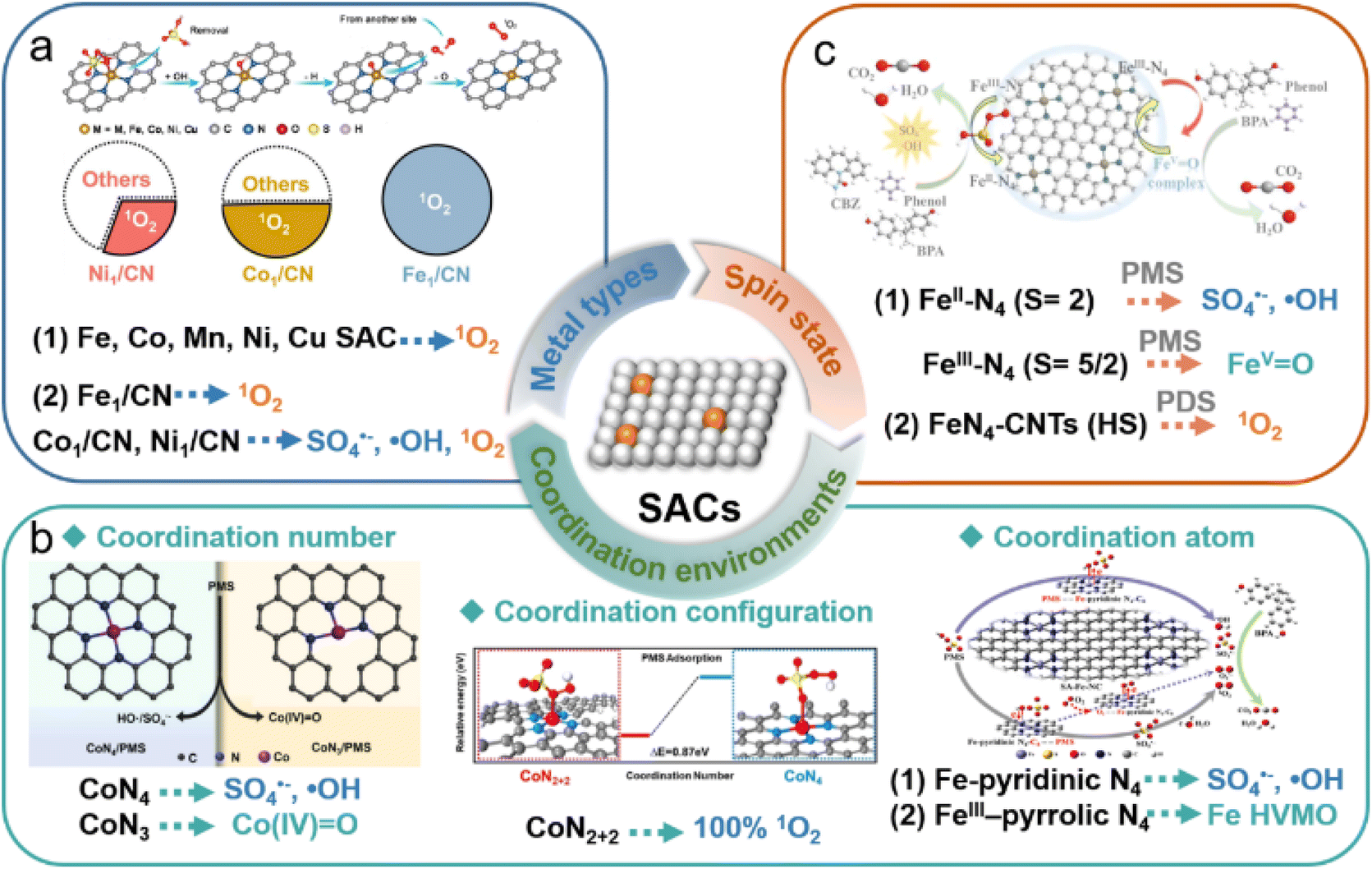 | ||
| Fig. 9 Effects of (a) metal types,100,176 (b) coordination environments,86,107 and (c) metallic spin states81,141 on the generated oxidative species in the SAC-based Fenton-like process. Figures have been adapted from ref. 81, 86, 100, 107, 141 and 176 with permissions from Elsevier B.V., Copyright 2023, Elsevier B.V., Copyright 2023, Wiley-VCH, Copyright 2021, Wiley-VCH, Copyright 2021, American Chemical Society, Copyright 2022, and Wiley-VCH, Copyright 2021, respectively. | ||
4.1 Metal types
Transition metals, such as Co, Fe, Mn, Cu, and Ni are the most commonly utilized metal active sites for persulfate activation. Due to the distinct electronic structures of these transition metal atoms, their interactions with persulfate molecules, as well as the catalytic activities and reactive species, are significantly different (Fig. 9a). In previous studies, we examined the intrinsic differences of Co, Fe, Mn, and Ni sites in PMS activation by constructing a series of M–N–C SACs with similar coordination configurations.41 In metal-based Fenton-like reactions, the eg orbitals of transition metals typically participate in σ bonding with oxygen-containing species through overlap with O 2p orbitals, thereby influencing the bonding strength and electron transfer rate between the metal center and oxidants.205 The electron distribution of the eg orbital in the transition metals within M–N–C elucidated the correlation between the electron spin of metal sites and Fenton-like catalytic activity, providing profound insights into the mechanism of PMS activation by metal sites.The oxidation pathways of Fenton-like reactions are highly dependent on metal types in the SACs. Zhang et al. systematically investigated the PMS activation pathways using Co, Ni, and Fe SACs with similar configurations (labeled M1/CN).100 The quantitative experiments, trapping tests, and EPR analysis demonstrated that Fe1/CN efficiently activated PMS to produce 100% 1O2. However, the selectivities of Co1/CN and Ni1/CN for generating 1O2 were 49% and 27%, respectively. DFT calculations confirmed that the Fe site of Fe1/CN served as the adsorption site for PMS, promoting PMS oxidation to generate SO5˙−, which decays to produce 1O2. In contrast, the N sites of Co1/CN and Ni1/CN adsorbed PMS, facilitating PMS reduction to produce SO4˙− and ·OH. Recently, Cu- and Fe SACs with uniform MN4 coordination were fabricated to evaluate the triggered versatile pathways of PMS activation.206 The results showed that high-valent Fe-oxo species contributed to 74.5% of organic oxidation in the Fe-SAC/PMS system, while radicals (SO4˙− and ·OH) predominated (58.3%) in the Cu-SAC/PMS system. The difference in their catalytic performances and generated reactive species was essentially attributed to variations in the spin state of the metal sites (FeN4 and CuN4) during PMS adsorption and the cleavage pathway of O–O in the adsorbed PMS.
4.2 Coordination environments
The coordination environments of isolated metal atoms in SACs are usually considered as the vital factors for tuning the inherent catalytic efficiency of Fenton-like reactions. As a result of variations in the coordination environment, the electronic structure of single atoms can be adjusted, thereby modifying the affinity of the substrate towards metal sites and enhancing catalytic efficiency.207 Notably, these changes in coordination environments are anticipated to influence PMS activation pathways by regulating electron transfer between PMS and single atoms, facilitating the targeted production of specific ROS. The coordination numbers, coordination atoms, and coordination configurations are the key influencing factors of the coordination environments of SACs (Fig. 9b).In principle, with a decrease in coordination number (n < 4), SACs are expected to feature more unsaturated metal sites MNx (x = 2, 3), characterized by reduced steric hindrance,208 facilitating interactions with peroxides to form reaction intermediates, consequently enhancing their catalytic performance. For example, Liang et al. demonstrated that CoSA-N3-C, featuring Co–N3 coordination configurations, exhibited higher activity than the Co-SACs with Co–N4 coordination in activating PDS for BPA degradation.48 This enhancement was attributed to the heightened electron density at single Co atoms achieved through lower coordination number engineering, bolstering the interaction between Co sites with PDS, thus facilitating the generation of active radicals. Furthermore, unsaturated Co–N3 sites have been shown to possess high spin states, exhibiting enhanced electron-donating capabilities and electron flow during PMS activation, which triggered the generation of Co(IV)-oxo species. In contrast, the saturated CoN4 sites with low spin states were inclined towards radical formation.107
Despite the high electronegativity of N, the catalytic activity of the common MN4 sites in SACs is limited due to their symmetric electron distribution. SACs can improve catalytic performance in Fenton-like reactions by doping with heteroatoms (e.g., O, S, P, B).47,174,209 Substituting N coordination atoms in SACs with other atoms alters the electronic structure of the metal center, thereby optimizing the adsorption of intermediates by enhancing d-band delocalization or modifying charge density differences. Some reported SACs include W-N1O3,210 Co-N3S1,80 Co-N3O1,209 and Co-SiN3.89 Wang et al. observed that PMS exhibited excellent adsorption and activation properties over cobalt-based SACs with the Co–N3O1 configuration.209 Compared to the Co–N4 moiety, SACs featuring the Co–N3O1 moiety demonstrated significantly improved performance in ciprofloxacin (CIP) degradation and enhanced selectivity in PMS activation for 1O2 generation, confirming the role of oxygen atom substitution in the coordination environment of the Co single atoms. DFT calculations were used to investigate the electronic structure at Co sites modified by oxygen atoms, which facilitated PMS adsorption and *O formation, resulting in rapid and selective 1O2 generation.
Besides the coordination number and coordination atoms, coordination configurations of the metal atoms in SACs lead to diverse Fenton-like activity and activation pathways.35,141,209 Mi et al. achieved high selectivity in PMS activation for 1O2 production by employing Co SACs featuring distinctive Co–N2+2 configurations.141 EXAFS spectra and theoretical predictions confirmed the Co–N2+2 configuration. Compared to the traditional Co–N4, PMS adsorption was more favorable at Co–N2+2 sites due to the shorter Co–N bonds, which enhanced electron transfer from the carbon substrate, increasing the electropositivity of Co atoms. During PMS activation, PMS was more readily oxidized to SO5˙− at weakly positively charged Co single-atom sites compared to strongly positively charged Co nanoparticles, leading to the generation of 1O2 with low activation energy.
4.3 Metallic electron spin state
In SAC-based Fenton-like reactions, the regulation of catalytic activity by transition metal types and their coordination environment essentially involves modulating the inherent electronic properties of transition metal sites (oxidation or spin states).41,107 Therefore, the metallic spin state is a key factor in revealing the catalytic performance of SACs (Fig. 9c). In addition to regulating the spin states of metal sites through different coordination environments, transition metal-based SACs with the same coordination configuration can also modulate the metallic spin state with the oxidation state to achieve diverse reactive species-dominated Fenton-like reactions. Zhang et al. elucidated the spin-state-dependent PMS activation pathways catalyzed by Fe–Nx–C catalysts with the FeN4 configuration.86 The spin states of FeII and FeIII in FeN4 sites have been verified to result in different catalytic pathways. DFT calculation revealed that both high-spin FeIIN4 and FeIIIN4 species exhibited significant activity efficiency in Fenton-like catalysis. FeIIN4 (S = 2) with a high-spin state tended to produce ·OH and SO4˙−via single-electron transfer during PMS activation, the FeIIIN4 with (S = 5/2) is indeed high-spin state was more inclined to generate FeIV-oxo species through a two-electron transfer. This suggested that the oxidation state of metal atomic centers would affect their electron spin structure, regulating the coupling of oxygen-containing adsorbents (such as PMS) on SACs and the transfer of spin-oriented electrons, thus triggering different catalytic mechanisms for the oxidation of organic pollutants. Li et al. proved that FeN4 with low or medium spin states functioned as the primary sites for PMS activation, while FeN4 sites with high-spin states played a crucial role in 1O2 production.211 This result further validated that the spin-electronic structure of metal atoms played a vital role in the adsorption configuration of PMS on SACs, thus modulating the selective production of ROS. Furthermore, different metal sites equipped with the same coordination configuration (FeN4 and CuN4) could induce versatile reaction pathways for activating PMS. The high-spin FeN4 and CuN4 separately triggered the production of high-valent Fe-oxo species and ·OH/SO4˙−via efficient PMS activation,206 clarifying that PMS adsorption and the breaking of O–O bonds were affected by the metal types and their spin states.Overall, the various catalytic mechanisms of SAC-based Fenton-like systems depend not only on the type of metal atoms, coordination environments, and metal–electronic properties, but also on the adsorption configuration of peroxides on SACs.33,170 The factors of metal types and coordination environments would affect the spin-electronic structure of metal sites, tuning the coupling of oxygen-containing adsorbents on metal active sites, thus resulting in the versatile catalytic activity and mechanisms.41,206,212 Additionally, as shown in Table 1, these different catalytic mechanisms mainly occurred in SACs/persulfate (PS) systems due to the different types of oxygen in the PS molecules (Fig. 10), resulting in the selectivity of the generated ROS.33 These distinct adsorption configurations could influence the electron transfer and binding strength between PS (or its intermediates) and SACs. For instance, the electron transfer from the single-atom site to the oxygen atom in the –SO4 (Oα/Oβ) or –OH (Oγ) group of PS could produce different types of ROS (Fig. 7d).170 Therefore, the metal species, local coordination environment and electronic properties of SACs determine the molecular arrangement of PS on SACs, resulting in the production of different ROS. Although previous experimental observations and theoretical calculations provide some clues, further in-depth mechanistic studies are still needed.
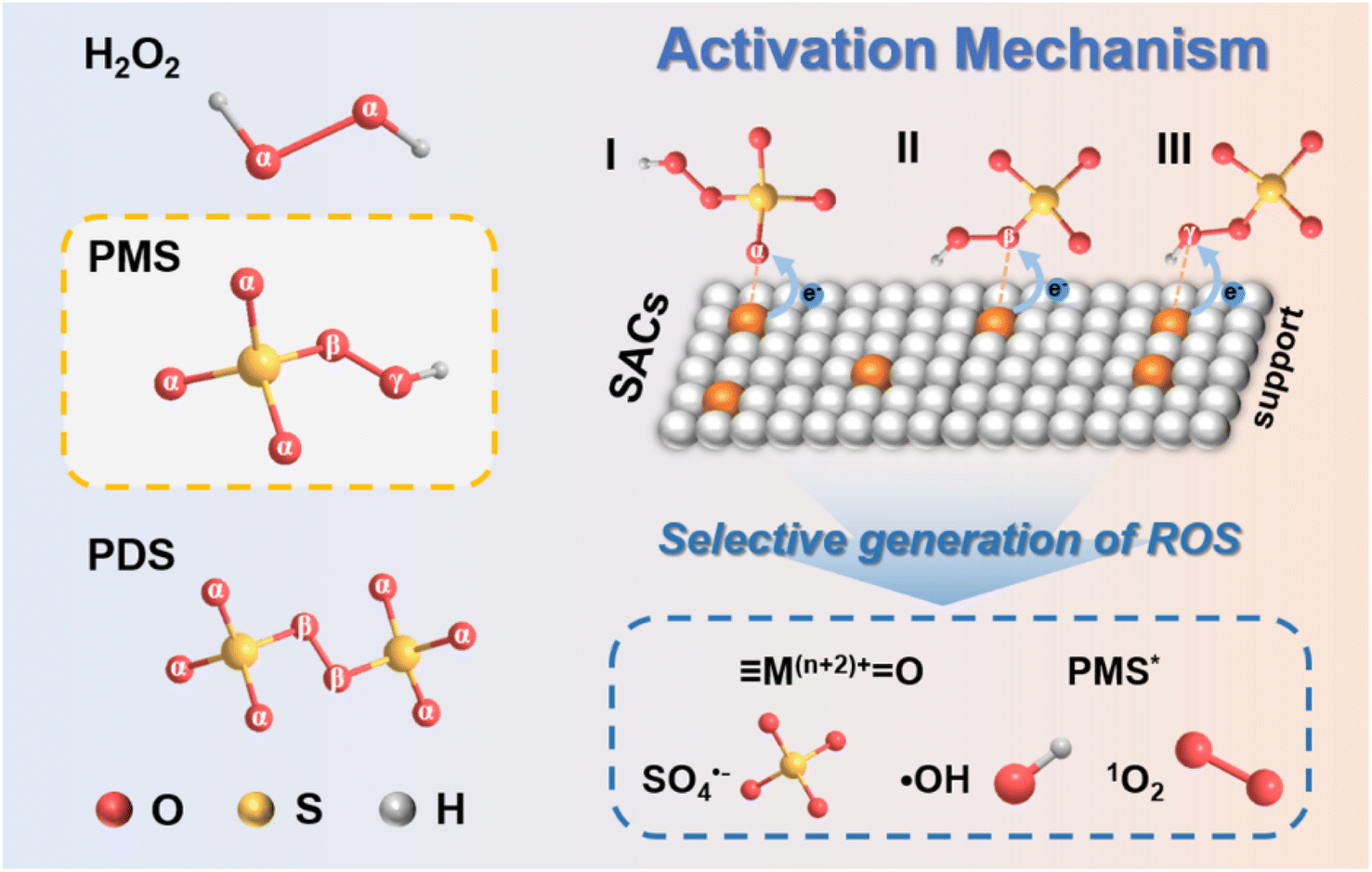 | ||
| Fig. 10 The adsorption configuration of peroxides on SACs depends on their coordination with different types of O in peroxides. | ||
5. Summary and perspectives
Significant progress has been made in the study of heterogeneous catalysts for Fenton-like reactions. The performance and catalytic mechanisms of these catalysts are highly sensitive to the type, size, and structure of the metal active centers. The development of SACs has facilitated in-depth exploration of structure–performance relationships, laying the foundation for the development of more advanced heterogeneous catalysts. This review summarized the current SACs with different metal types and supports (such as carbon and metal oxides) applied in Fenton-like processes for organic removal, discussed the catalytic mechanisms induced by SACs and their associated active sites, and then emphasized the influence of central metal site types, coordination configuration and numbers, as well as spin electronic structures on the catalytic activity of SACs and mechanisms of Fenton-like reactions. These studies highlight a fundamental understanding of the relationship between the electronic structure of SACs and their Fenton-like activity.Although SACs show great potential as efficient heterogeneous Fenton-like catalysts, they still face significant challenges in both laboratory studies and practical applications in wastewater remediation. Systematic validation of these barriers and opportunities is needed to achieve a more robust SAC design and to gain insights into the underlying catalytic mechanisms. These challenges and opportunities are summarized below:
(1) Design of highly active SACs. Fine synthesis of single-atom catalysts is difficult. Controlling the synthesis of SACs with desirable well-defined coordination structures and high-density metal centers is crucial for accurately correlating the coordination configuration with their catalytic performances, especially when multiple potential active sites trigger diverse oxidation pathways for target pollutant degradation. Such controllable synthetic approaches are essential for understanding the structure–activity relationship, and enabling the design of highly active materials on demand. Furthermore, scalable production of SACs based on advanced manufacturing techniques urgently needs to be explored.
(2) Catalytic mechanistic validation. The catalytic pathways of SACs in Fenton-like systems remain uncertain, with several key scientific issues requiring further investigation. (i) In the radical pathway, the reduction kinetics from M(n+1)+ to Mn+ are limited, which decreases the cyclic activity of SACs. A critical challenge is to achieve rapid redox cycling of M(n+1)+/Mn+ in the radical pathway. (ii) In the non-radical pathway (excluding the high-valent metal pathway), identifying active sites compatible with 1O2 and ETP is essential. (iii) How different oxidation states of metal sites selectively induce the high-valent metal pathway needs to be determined. Addressing these issues will enhance our understanding of the various catalytic mechanisms in Fenton-like reactions, aiding the design of SACs with optimized kinetics and selectivity. Additionally, the physicochemical properties of SACs may change significantly under different water conditions (e.g., coexisting anions, pH, and organic matter), potentially triggering different reaction pathways in Fenton-like systems. These variations also depend on the synthesis methods, metal loading, and coordination environment of the SACs. Therefore, to comprehensively understand the role of SACs in Fenton-like processes, an extensive and systematic investigation considering these factors is necessary.
(3) Theoretical simulation. Understanding the generation of ROS and the influencing factors is crucial for designing better SACs in Fenton-like reactions. Current theoretical simulations mainly focus on thermodynamic properties (e.g., the adsorption energies (Eads), O–O bond elongation of peroxide, charge accumulation around the single atom site). However, DFT is computationally expensive to calculate these parameters for each SAC on different supports, considering variations in water quality parameters. A more viable alternative is the rational design of heterogeneous SACs using machine learning and high-throughput simulations.
(4) Costs and commercial applications. To transition SACs from lab studies to commercial use, researchers are suggested to focus on rational designs with controllable metal loading, valence states, and coordination configuration. This will facilitate their implementation in wastewater treatment plants. SACs with utmost atom utilization efficiency are still in early development stages, requiring cost and lifecycle assessments for environmental applications. The key advantages of SACs applied for wastewater treatment are their stability and reusability after reaction. Although metal leaching in SAC-based Fenton-like reactions is significantly lower, its further reduction is needed to meet the stringent future guidelines, which may be achieved by enhancing the stability of SACs through better interactions with their supports.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Conceptualization, validation, funding acquisition and writing-original draft: J. Miao. Conceptualization and investigation: Y. Jiang. Investigation and writing – review & editing: X. Wang, X. Li and Y. Zhu. Writing – review & editing: Z. Shao. Funding acquisition, writing and supervision – review & editing: M. Long.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from the National Natural Science Foundation of China (nos. 22106104, 52070218 and 22376138) and the State Key Laboratory of Materials-Oriented Chemical Engineering (No. SKL-MCE-23B10) are acknowledged.References
- J. P. R. Sorensen, D. J. Lapworth, D. C. W. Nkhuwa, M. E. Stuart, D. C. Gooddy, R. A. Bell, M. Chirwa, J. Kabika, M. Liemisa, M. Chibesa and S. Pedley, Water Res., 2015, 72, 51–63 Search PubMed.
- S. Khan, M. Naushad, M. Govarthanan, J. Iqbal and S. M. Alfadul, Environ. Res., 2022, 207, 112609 Search PubMed.
- Z. Zhang, Q. Xiao, X. Du, T. Xue, Z. Yan, Z. Liu, H. Zhang and T. Qi, J. Alloys Compd., 2022, 897, 162742 Search PubMed.
- X. Wang, C. Xu, Y. Zhu, C. Zhou, Y. Yang, J. Miao, W. Zhou and Z. Shao, Surf. Interfaces, 2024, 44, 103820 Search PubMed.
- S. Lim, J. L. Shi, U. von Gunten and D. L. McCurry, Water Res., 2022, 213, 118053 Search PubMed.
- K. Maduna, N. Kumar, A. Aho, J. Wärnå, S. Zrnčević and D. Y. Murzin, ACS Omega, 2018, 3, 7247–7260 Search PubMed.
- M. Y. Kilic, W. H. Abdelraheem, X. He, K. Kestioglu and D. D. Dionysiou, J. Hazard. Mater., 2019, 367, 734–742 Search PubMed.
- M. Munoz, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2015, 176–177, 249–265 Search PubMed.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 Search PubMed.
- D. Meyerstein, Nat. Rev. Chem., 2021, 5, 595–597 Search PubMed.
- J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 Search PubMed.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Water Res., 2018, 139, 118–131 Search PubMed.
- M. P. Rayaroth, C. T. Aravindakumar, N. S. Shah and G. Boczkaj, Chem. Eng. J., 2022, 430, 133002 Search PubMed.
- Y. Yin, L. Shi, W. Li, X. Li, H. Wu, Z. Ao, W. Tian, S. Liu, S. Wang and H. Sun, Environ. Sci. Technol., 2019, 53, 11391–11400 Search PubMed.
- J. Miao, Y. Zhu, Y. Wei, X. Wen, Z. Shao, B. Zhou, C. Wu and M. Long, J. Hazard. Mater., 2024, 465, 133344 Search PubMed.
- H. Dong, Z. Qiang, J. Hu and C. Sans, Chem. Eng. J., 2017, 316, 288–295 Search PubMed.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000–1007 Search PubMed.
- S. Xiao, M. Cheng, H. Zhong, Z. Liu, Y. Liu, X. Yang and Q. Liang, Chem. Eng. J., 2020, 384, 123265 Search PubMed.
- S. Sun, S. Song, S. Yang, Y.-L. He, Y. Shi, P. Zhou, Z.-k. Xiong, Y. Liu, H. Zhang, Y. Du, C.-S. He and B. Lai, Chin. Chem. Lett., 2024, 35, 109242 Search PubMed.
- P. Hu and M. Long, Appl. Catal., B, 2016, 181, 103–117 Search PubMed.
- B. Liu, B. Huang, Z. Wang, L. Tang, C. Ji, C. Zhao, L. Feng and Y. Feng, J. Environ. Chem. Eng., 2023, 11, 109586 Search PubMed.
- M. Huang, T. Zhou, X. Wu and J. Mao, Water Res., 2017, 119, 47–56 Search PubMed.
- J. Feng, X. Hu and P. L. Yue, Environ. Sci. Technol., 2004, 38, 5773–5778 Search PubMed.
- S. Zhang, M. Sun, T. Hedtke, A. Deshmukh, X. Zhou, S. Weon, M. Elimelech and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 10868–10875 Search PubMed.
- Y.-Y. Ahn, H. Bae, H.-I. Kim, S.-H. Kim, J.-H. Kim, S.-G. Lee and J. Lee, Appl. Catal., B, 2019, 241, 561–569 Search PubMed.
- Z. Shen, Y. Zhou, Y. Guo, J. Zhao, J. Song, Y. Xie, Y. Ling and W. Zhang, Chin. Chem. Lett., 2021, 32, 2524–2528 Search PubMed.
- L. Xu and L. Liu, Appl. Catal., B, 2022, 304, 120953 Search PubMed.
- X. Qiu, S. Yang, M. Dzakpasu, X. Li, D. Ding, P. Jin, R. Chen, Q. Zhang and X. C. Wang, Chem. Eng. J., 2019, 372, 605–615 Search PubMed.
- H. Zhou and Y. Wu, Sci. Bull., 2023, 68, 465–468 Search PubMed.
- Y. Shang, X. Xu, B. Gao, S. Wang and X. Duan, Chem. Soc. Rev., 2021, 50, 5281–5322 Search PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 Search PubMed.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 Search PubMed.
- J. Guo, B. Gao, Q. Li, S. Wang, Y. Shang, X. Duan and X. Xu, Adv. Mater., 2024, 2403965 Search PubMed.
- Q.-Y. Wu, Z.-W. Yang, Z.-W. Wang and W.-L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219923120 Search PubMed.
- X. Zhou, M.-K. Ke, G.-X. Huang, C. Chen, W. Chen, K. Liang, Y. Qu, J. Yang, Y. Wang, F. Li, H.-Q. Yu and Y. Wu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119492119 Search PubMed.
- Y. Chen, Y. Yang, N. Ren and X. Duan, Curr. Opin. Chem. Eng., 2023, 41, 100942 Search PubMed.
- L.-H. Xu, W. Liu and K. Liu, Adv. Funct. Mater., 2023, 33, 2304468 Search PubMed.
- P. Cao, X. Quan, K. Zhao, S. Chen, H. Yu and Y. Su, Environ. Sci. Technol., 2020, 54, 12662–12672 Search PubMed.
- B. Huang, Z. Wu, H. Zhou, J. Li, C. Zhou, Z. Xiong, Z. Pan, G. Yao and B. Lai, J. Hazard. Mater., 2021, 412, 125253 Search PubMed.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang, J. Gao, J. Wang, Y. Huang, T. Zhang and B. Liu, J. Am. Chem. Soc., 2018, 140, 12469–12475 Search PubMed.
- J. Miao, Y. Zhu, J. Lang, J. Zhang, S. Cheng, B. Zhou, L. Zhang, P. J. J. Alvarez and M. Long, ACS Catal., 2021, 11, 9569–9577 Search PubMed.
- F. Chen, X.-L. Wu, C. Shi, H. Lin, J. Chen, Y. Shi, S. Wang and X. Duan, Adv. Funct. Mater., 2021, 31, 2007877 Search PubMed.
- T. Wang, J. Zhou, W. Wang, Y. Zhu and J. Niu, Chin. Chem. Lett., 2022, 33, 2121–2124 Search PubMed.
- L.-C. Wang, L.-C. Chang, W.-Q. Chen, Y.-H. Chien, P.-Y. Chang, C.-W. Pao, Y.-F. Liu, H.-S. Sheu, W.-P. Su, C.-H. Yeh and C.-S. Yeh, Nat. Commun., 2022, 13, 7772 Search PubMed.
- J. Miao, J. Song, J. Lang, Y. Zhu, J. Dai, Y. Wei, M. Long, Z. Shao, B. Zhou, P. J. J. Alvarez and L. Zhang, Environ. Sci. Technol., 2023, 57, 4266–4275 Search PubMed.
- F. Li, Z. Lu, T. Li, P. Zhang and C. Hu, Environ. Sci. Technol., 2022, 56, 8765–8775 Search PubMed.
- X. Li, X. Wen, J. Lang, Y. Wei, J. Miao, X. Zhang, B. Zhou, M. Long, P. J. J. Alvarez and L. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303267 Search PubMed.
- X. Liang, D. Wang, Z. Zhao, T. Li, Z. Chen, Y. Gao and C. Hu, Appl. Catal., B, 2022, 303, 120877 Search PubMed.
- J. Song, N. Hou, X. Liu, M. Antonietti, P. Zhang, R. Ding, L. Song, Y. Wang and Y. Mu, Adv. Mater., 2023, 35, 2209552 Search PubMed.
- Y. Li, J. Hu, Y. Zou, L. Lin, H. Liang, H. Lei, B. Li and X.-y. Li, Chem. Eng. J., 2023, 453, 139890 Search PubMed.
- K. Yin, R. Wu, Y. Shang, D. Chen, Z. Wu, X. Wang, B. Gao and X. Xu, Appl. Catal., B, 2023, 329, 122558 Search PubMed.
- L. Hao, C. Guo, Z. Hu, R. Guo, X. Liu, C. Liu and Y. Tian, Nanoscale, 2022, 14, 13861–13889 Search PubMed.
- C. Zhai, Y. Chen, X. Huang, A. B. Isaev and M. Zhu, Environ. Funct. Mater., 2022, 1, 219–229 Search PubMed.
- Y. Zeng, E. Almatrafi, W. Xia, B. Song, W. Xiong, M. Cheng, Z. Wang, Y. Liang, G. Zeng and C. Zhou, Coord. Chem. Rev., 2023, 475, 214874 Search PubMed.
- C. Chen, L. Liu, Y. Li, L. Zhou and Y. Lan, J. Hazard. Mater., 2020, 407, 124559 Search PubMed.
- W. Ma, X. Ren, J. Li, S. Wang, X. Wei, N. Wang and Y. Du, Small, 2023, 2308957 Search PubMed.
- Y. Chen, H. Kang, M. Cheng, G. Zhang, G. Wang, H. Liu, W. Xiao, Y. Liu and H. Jiang, Adv. Funct. Mater., 2023, 2309223 Search PubMed.
- Z. Qi, Y. Zhou, R. Guan, Y. Fu and J.-B. Baek, Adv. Mater., 2023, 35, 2210575 Search PubMed.
- B. Han, Y. Luo, Y. Lin, B. Weng, D. Xia, Y. Zhou, C. Guan, Z. Wang, X. Wei and J. Jiang, Chem. Eng. J., 2022, 447, 137551 Search PubMed.
- W. Qu, C. Chen, Z. Tang, H. Wen, L. Hu, D. Xia, S. Tian, H. Zhao, C. He and D. Shu, Coord. Chem. Rev., 2023, 474, 217855 Search PubMed.
- S. P, J. John, T. P. D. Rajan, G. M. Anilkumar, T. Yamaguchi, S. C. Pillai and U. S. Hareesh, J. Mater. Chem. A, 2023, 11, 8599–8646 Search PubMed.
- F. Chen, X. L. Wu, C. Shi, H. Lin, J. Chen, Y. Shi, S. Wang and X. Duan, Adv. Funct. Mater., 2021, 31, 2007877 Search PubMed.
- B. Shi, H. Li, X. Fu, C. Zhao, M. Li, M. Liu, W. Yan and H. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 53767–53776 Search PubMed.
- Q. Meng, H. Hu, D. Hao, S. Yuan, J. He, X. Yu, Z. Xie, K. Wang, Y. Tang, K. Zhao and C. Xu, J. Environ. Chem. Eng., 2022, 10, 108843 Search PubMed.
- X. Song, Y. Shi, Z. Wu, B. Huang, X. Wang, H. Zhang, P. Zhou, W. Liu, Z. Pan, Z. Xiong and B. Lai, Appl. Catal., B, 2024, 340, 123240 Search PubMed.
- S. Liu, D. Liu, Y. Sun, P. Xiao, H. Lin, J. Chen, X.-L. Wu, X. Duan and S. Wang, Appl. Catal., B, 2022, 310, 121327 Search PubMed.
- H. Fu, J. Wei, G. Chen, M. Xu, J. Liu, J. Zhang, K. Li, Q. Xu, Y. Zou, W.-x. Zhang, S. Xi, X. Chen, S. Li and L. Ling, Appl. Catal., B, 2023, 321, 122012 Search PubMed.
- M. Li, L. Hu, Y. Yuan, M. Li, C. Huang, X. Hu, J. Deng, Y. Xie, P. Wang and H. Jiang, Sep. Purif. Technol., 2024, 332, 125823 Search PubMed.
- Y. Shang, X. Liu, Y. Li, Y. Gao, B. Gao, X. Xu and Q. Yue, Chem. Eng. J., 2022, 446, 137120 Search PubMed.
- J. Yuan, Y. Han, K. Sathiyan, V. K. Sharma, A. H. Mokarizadeh, M. Tsige, J. Jiang and X. Ma, RSC Sustainability, 2023, 1, 2296–2304 Search PubMed.
- H. Zhang, G. Xu and Y. Yu, Environ. Pollut., 2023, 333, 121983 Search PubMed.
- N. Jiang, H. Xu, L. Wang, J. Jiang and T. Zhang, Environ. Sci. Technol., 2020, 54, 14057–14065 Search PubMed.
- S. Zuo, X. Jin, X. Wang, Y. Lu, Q. Zhu, J. Wang, W. Liu, Y. Du and J. Wang, Appl. Catal., B, 2021, 282, 119551 Search PubMed.
- H. Qin, X. Liu, X. Liu, H. Zhao and S. Mao, Nano-Micro Lett., 2023, 15, 193 Search PubMed.
- Z. Zhao, P. Zhang, H. Tan, X. Liang, T. Li, Y. Gao and C. Hu, Small, 2023, 19, 2205583 Search PubMed.
- Z. Du, J. Qin, K. Zhang, L. Jia, K. Tian, J. Zhang and H. Xie, Appl. Surf. Sci., 2022, 591, 153124 Search PubMed.
- C. Zhou, Y. Liang, W. Xia, E. Almatrafi, B. Song, Z. Wang, Y. Zeng, Y. Yang, Y. Shang, C. Wang and G. Zeng, J. Hazard. Mater., 2023, 441, 129871 Search PubMed.
- R. Guo, Z. Bi, B. Xi, C. Guo, H. Zhang, N. Lv, G. Hu and J. Xu, Chem. Eng. J., 2024, 481, 1486229 Search PubMed.
- Z. Zhao, H. Tan, P. Zhang, X. Liang, T. Li, Y. Gao and C. Hu, Angew. Chem., Int. Ed., 2023, 62, e202219178 Search PubMed.
- P. Cui, Q. Yang, C. Liu, Y. Wang, G. Fang, D. D. Dionysiou, T. Wu, Y. Zhou, J. Ren, H. Hou and Y. Wang, ACS ES&T Eng., 2021, 1, 1460–1469 Search PubMed.
- Y. Gao, Y. Zhu, Z. Chen and C. Hu, ACS ES&T Eng., 2020, 1, 76–85 Search PubMed.
- X. Liu, Y. Pei, M. Cao, H. Yang and Y. Li, Chem. Eng. J., 2022, 450, 138194 Search PubMed.
- X. Zhao, X. Li, Z. Zhu, W. Hu, H. Zhang, J. Xu, X. Hu, Y. Zhou, M. Xu, H. Zhang and G. Hu, Appl. Catal., B, 2022, 300, 120759 Search PubMed.
- H. Xu, N. Jiang, D. Wang, L. Wang, Y. Song, Z. Chen, J. Ma and T. Zhang, Appl. Catal., B, 2020, 263, 118350 Search PubMed.
- J. Yang, P. Li, X. Duan, D. Zeng, Z. Ma, S. An, L. Dong, W. Cen and Y. He, J. Hazard. Mater., 2022, 430, 128463 Search PubMed.
- B. Zhang, X. Li, K. Akiyama, P. A. Bingham and S. Kubuki, Environ. Sci. Technol., 2022, 56, 1321–1330 Search PubMed.
- H. Zhang, G. Xu and Y. Yu, Chem. Eng. J., 2023, 476, 146721 Search PubMed.
- Y. Zou, J. Hu, B. Li, L. Lin, Y. Li, F. Liu and X.-y. Li, Appl. Catal., B, 2022, 312, 121408 Search PubMed.
- X. Dong, Z. Chen, A. Tang, D. D. Dionysiou and H. Yang, Adv. Funct. Mater., 2022, 32, 2111565 Search PubMed.
- C. Gu, Y. Zhang, P. He, J. Zhu and M. Gan, Environ. Sci.: Nano, 2022, 9, 3551–3561 Search PubMed.
- B. Huang, X. Ren, J. Zhao, Z. Wu, X. Wang, X. Song, X. Li, B. Liu, Z. Xiong and B. Lai, Environ. Sci. Technol., 2023, 57, 14071–14081 Search PubMed.
- Z. Wang, Y. Wang, W. Wang, D. Wu, Q. Wu and H. Hu, Appl. Catal., B, 2023, 324, 122248 Search PubMed.
- H.-Q. Zhao, J.-S. Song, P. Lu and Y. Mu, Chem. Eng. J., 2023, 456, 141045 Search PubMed.
- G. Liao, X. Qing, P. Xu, L. Li, P. Lu, W. Chen and D. Xia, Chem. Eng. J., 2022, 427, 132027 Search PubMed.
- X. Zhao, X. Jia, H. Li, H. Zhang, X. Zhou, Y. Zhou, H. Wang, L. Yin, T. Wågberg and G. Hu, Chem. Eng. J., 2022, 450, 138098 Search PubMed.
- W. Chen, X. Li, X. Wei, G. Liao, J. Wang and L. Li, Sci. Total Environ., 2023, 858, 160097 Search PubMed.
- M. Yang, K. Wu, S. Sun, J. Duan, X. Liu, J. Cui, S. Liang and Y. Ren, ACS Catal., 2022, 13, 681–691 Search PubMed.
- L. Yang, H. Yang, S. Yin, X. Wang, M. Xu, G. Lu, Z. Liu and H. Sun, Small, 2022, 18, e2104941 Search PubMed.
- T. Zeng, X. Tang, Z. Huang, H. Chen, S. Jin, F. Dong, J. He, S. Song and H. Zhang, Environ. Sci. Technol., 2023, 57, 20929–20940 Search PubMed.
- L.-S. Zhang, X.-H. Jiang, Z.-A. Zhong, L. Tian, Q. Sun, Y.-T. Cui, X. Lu, J.-P. Zou and S.-L. Luo, Angew. Chem., Int. Ed., 2021, 60, 21751–21755 Search PubMed.
- X. Zhang, C. Li, X. Wang, S. Yang, Y. Tan, F. Yuan, S. Zheng, D. D. Dionysiou and Z. Sun, Small, 2022, 18, e2204793 Search PubMed.
- S. Ma, S. Tang, T. Zhang, W. Jin, H. Zhu, Y. Zhao and D. D. Dionysiou, Chem. Eng. J., 2023, 474, 145488 Search PubMed.
- S. Wu, H. Liu, C. Yang, X. Li, Y. Lin, K. Yin, J. Sun, Q. Teng, C. Du and Y. Zhong, Chem. Eng. J., 2020, 392, 123683 Search PubMed.
- W. Tang, H. Zhang, X. Yang, Z. Dai, Y. Sun, H. Liu, Z. Hu and X. Zheng, Appl. Catal., B, 2023, 320, 121952 Search PubMed.
- X. Peng, J. Wu, Z. Zhao, X. Wang, H. Dai, Y. Wei, G. Xu and F. Hu, Chem. Eng. J., 2022, 429, 132245 Search PubMed.
- W. Miao, Y. Liu, D. Wang, N. Du, Z. Ye, Y. Hou, S. Mao and K. Ostrikov, Chem. Eng. J., 2021, 423, 130250 Search PubMed.
- J. Song, N. Hou, X. Liu, M. Antonietti, Y. Wang and Y. Mu, Appl. Catal., B, 2023, 325, 122368 Search PubMed.
- H. C. Zhang, P. X. Cui, D. H. Xie, Y. J. Wang, P. Wang and G. P. Sheng, Adv. Sci., 2023, 10, e2205681 Search PubMed.
- C. Wang, X. Wang, H. Wang, L. Zhang, Y. Wang, C.-L. Dong, Y.-C. Huang, P. Guo, R. Cai, S. J. Haigh, X. Yang, Y. Sun and D. Yang, J. Hazard. Mater., 2023, 455, 131622 Search PubMed.
- H. Zhang, L. Lyu, Q. Fang, C. Hu, S. Zhan and T. Li, Appl. Catal., B, 2021, 286, 119912 Search PubMed.
- Y. Qi, J. Li, Y. Zhang, Q. Cao, Y. Si, Z. Wu, M. Akram and X. Xu, Appl. Catal., B, 2021, 286, 119910 Search PubMed.
- C. Zhu, Y. Nie, S. Zhao, Z. Fan, F. Liu and A. Li, Appl. Catal., B, 2022, 305, 121057 Search PubMed.
- M. Xie, M. Yao, S. Zhang, L. Kong, L. Zhao, J. Zhan and R.-S. Zhao, Sep. Purif. Technol., 2023, 304, 122398 Search PubMed.
- J. Pan, B. Gao, P. Duan, K. Guo, M. Akram, X. Xu, Q. Yue and Y. Gao, J. Mater. Chem. A, 2021, 9, 11604–11613 Search PubMed.
- T. Chen, Z. Zhu, X. Shen, H. Zhang, Y. Qiu and D. Yin, Chem. Eng. J., 2022, 450, 138469 Search PubMed.
- C. Zhu, Y. Nie, F. Cun, Y. Wang, Z. Tian and F. Liu, Appl. Catal., B, 2022, 319, 121900 Search PubMed.
- P. Duan, J. Pan, W. Du, Q. Yue, B. Gao and X. Xu, Appl. Catal., B, 2021, 299, 120714 Search PubMed.
- P. Duan, M. Li, X. Xu, Q. Yue, Y. Gao and B. Gao, Appl. Catal., B, 2024, 344, 123679 Search PubMed.
- J. Li, S. Zhao, C. Li, S. Kawi, K. Wang, J. Huang and S. Liu, J. Colloid Interface Sci., 2024, 656, 58–67 Search PubMed.
- Y. Xiao, J. Hu, X.-y. Li, Y. Zou, Y. Li, L. Lin and B. Li, Chem. Eng. J., 2023, 474, 145973 Search PubMed.
- Y. Chai, H. Dai, X. Duan, Z. Sun, F. Hu, J. Qian and X. Peng, Appl. Catal., B, 2024, 341, 123289 Search PubMed.
- Y. Li, T. Yang, S. Qiu, W. Lin, J. Yan, S. Fan and Q. Zhou, Chem. Eng. J., 2020, 389, 124382 Search PubMed.
- Y. Xiong, H. Li, C. Liu, L. Zheng, C. Liu, J. O. Wang, S. Liu, Y. Han, L. Gu, J. Qian and D. Wang, Adv. Mater., 2022, 34, e2110653 Search PubMed.
- Q. Zhou, N. Lv, J. Wang, H. Shi and J. Song, Appl. Surf. Sci., 2023, 640, 158333 Search PubMed.
- Z. Huang, H. Yu, L. Wang, M. Wang, X. Liu, D. Shen, S. Shen, S. Ren, T. Lin and S. Lei, Sep. Purif. Technol., 2023, 305, 122402 Search PubMed.
- M. Li, J. Chen, W. Wu, S. Wu, L. Xu and S. Dong, Nano Res., 2023, 16, 4678–4684 Search PubMed.
- Z. Guo, Y. Xie, J. Xiao, Z. J. Zhao, Y. Wang, Z. Xu, Y. Zhang, L. Yin, H. Cao and J. Gong, J. Am. Chem. Soc., 2019, 141, 12005–12010 Search PubMed.
- A. Chen, J. Xiao, X. Kong, L. Chen, C. Li, Y. Wei, Q. Du, W. Sun and J. Zhang, Sep. Purif. Technol., 2023, 314, 123525 Search PubMed.
- C. Yang, S. Shang, Y. Fan, K. Shih, X.-y. Li and L. Lin, Appl. Catal., B, 2023, 325, 122344 Search PubMed.
- J. Li, S. Zhao, S.-Z. Yang, S. Wang, H. Sun, S. P. Jiang, B. Johannessen and S. Liu, J. Mater. Chem. A, 2021, 9, 3029–3035 Search PubMed.
- L. Wang, X. Guo, Y. Chen, S. Ai and H. Ding, Appl. Surf. Sci., 2019, 467, 954–962 Search PubMed.
- Y. Liu, W. Miao, Y. Feng, X. Fang, Q. Li, N. Du, D. Wang and S. Mao, J. Hazard. Mater., 2021, 403, 123691 Search PubMed.
- J. Yang, D. Zeng, Q. Zhang, R. Cui, M. Hassan, L. Dong, J. Li and Y. He, Appl. Catal., B, 2020, 279, 119363 Search PubMed.
- X. Dai, S. Adomeit, J. Rabeah, C. Kreyenschulte, A. Bruckner, H. Wang and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 5251–5255 Search PubMed.
- M. Xie, F. Dai, J. Li, X. Dang, J. Guo, W. Lv, Z. Zhang and X. Lu, Angew. Chem., Int. Ed., 2021, 60, 14370–14375 Search PubMed.
- A. A. Ioannidi, G. Bampos, M. Antonopoulou, P. Oulego, D. Mantzavinos and Z. Frontistis, J. Environ. Chem. Eng., 2024, 12, 111851 Search PubMed.
- X. Mi, H. Zhong, H. Zhang, S. Xu, Y. Li, H. Wang, S. Zhan and J. C. Crittenden, Environ. Sci. Technol., 2022, 56, 2637–2646 Search PubMed.
- H. Zhao, Y. Liu, D. Wu, H. Yu, X. Zhang, H. Wang, X. Shang and M. Lv, Environ. Pollut., 2023, 335, 122298 Search PubMed.
- D. Ouyang, Y. Chen, J. Yan, L. Qian, L. Han and M. Chen, Chem. Eng. J., 2019, 370, 614–624 Search PubMed.
- P. Xia, Z. Ye, L. Zhao, Q. Xue, S. Lanzalaco, Q. He, X. Qi and I. Sirés, Appl. Catal., B, 2023, 322, 122116 Search PubMed.
- X. Mi, P. Wang, S. Xu, L. Su, H. Zhong, H. Wang, Y. Li and S. Zhan, Angew. Chem., Int. Ed., 2021, 60, 4588–4593 Search PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 Search PubMed.
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Small Methods, 2019, 3, 1800368 Search PubMed.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, N. López, S. M. Collins, R. K. Leary, P. A. Midgley, R. Hauert and J. Pérez-Ramírez, Natl. Sci. Rev., 2018, 5, 642–652 Search PubMed.
- Y. Xu, M. Guo, C. Ge, P. Zhang, W. Xu, L. Zhang, S. Zhou and J. Liao, Appl. Surf. Sci., 2023, 640, 158290 Search PubMed.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li, S. Dai, G. Chen, K. Wang, H. Wang, J. K. Chen, W. Mitch and Y. Cui, Nat. Sustain., 2021, 4, 233–241 Search PubMed.
- L. Su, P. Wang, X. Ma, J. Wang and S. Zhan, Angew. Chem., Int. Ed., 2021, 60, 21261–21266 Search PubMed.
- Y. Xiang, H. Liu, E. Zhu, K. Yang, D. Yuan, T. Jiao, Q. Zhang and S. Tang, Sep. Purif. Technol., 2022, 295, 121293 Search PubMed.
- C. Choi, S. Yoon and Y. Jung, Chem. Sci., 2021, 12, 3551–3557 Search PubMed.
- Z. Wu, B. Huang, X. Wang, C.-S. He, Y. Liu, Y. Du, W. Liu, Z. Xiong and B. Lai, Environ. Sci. Technol., 2023, 57, 14046–14057 Search PubMed.
- Z. Chen, A. Huang, K. Yu, T. Cui, Z. Zhuang, S. Liu, J. Li, R. Tu, K. Sun, X. Tan, J. Zhang, D. Liu, Y. Zhang, P. Jiang, Y. Pan, C. Chen, Q. Peng and Y. Li, Energy Environ. Sci., 2021, 14, 3430–3437 Search PubMed.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 Search PubMed.
- J. Wan, Z. Zhao, H. Shang, B. Peng, W. Chen, J. Pei, L. Zheng, J. Dong, R. Cao, R. Sarangi, Z. Jiang, D. Zhou, Z. Zhuang, J. Zhang, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 8431–8439 Search PubMed.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 Search PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 Search PubMed.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 Search PubMed.
- Y.-K. Lv, K. Wang, W.-Y. Sun, P. Peng and S.-Q. Zang, Adv. Sci., 2023, 10, 2304656 Search PubMed.
- B. Yan, H. Song and G. Yang, Chem. Eng. J., 2022, 427, 131795 Search PubMed.
- H. Zhang, W. Tian, X. Duan, H. Sun, Y. Huang, Y. Fang and S. Wang, Chin. J. Catal., 2022, 43, 2301–2315 Search PubMed.
- Y. Shi, C. Lee, X. Tan, L. Yang, Q. Zhu, X. Loh, J. Xu and Q. Yan, Small Struct., 2022, 3, 2100185 Search PubMed.
- Z. Liu, S. Li, J. Yang, X. Tan, C. Yu, C. Zhao, X. Han, H. Huang, G. Wan, Y. Liu, K. Tschulik and J. Qiu, ACS Nano, 2020, 14, 11662–11669 Search PubMed.
- J. Ji, Y. Zhang, L. Tang, C. Liu, X. Gao, M. Sun, J. Zheng, M. Ling, C. Liang and Z. Lin, Nano Energy, 2019, 63, 103849 Search PubMed.
- Y. Shang, X. Xu, B. Gao, S. Wang and X. Duan, Chem. Soc. Rev., 2021, 50, 5281–5322 Search PubMed.
- C. Rivera-Cárcamo and P. Serp, ChemCatChem, 2018, 10, 5058–5091 Search PubMed.
- J. Wang, S. Bi, Y. Zhang, Y. Shen, L. Lu, X. Duan, X. Tan and S. Wang, Curr. Opin. Chem. Eng., 2023, 41, 100931 Search PubMed.
- Y. Ding, X. Wang, L. Fu, X. Peng, C. Pan, Q. Mao, C. Wang and J. Yan, Sci. Total Environ., 2021, 765, 142794 Search PubMed.
- M. B. Gawande, P. Fornasiero and R. Zboril, ACS Catal., 2020, 10, 2231–2259 Search PubMed.
- V. K. Sharma, X. Ma and R. Zboril, Chem. Soc. Rev., 2023, 52, 7673–7686 Search PubMed.
- I. A. Ike, K. G. Linden, J. D. Orbell and M. Duke, Chem. Eng. J., 2018, 338, 651–669 Search PubMed.
- X. Wu and J.-H. Kim, ACS ES&T Eng., 2022, 2, 1776–1796 Search PubMed.
- Z. Huang, H. Bao, Y. Yao, W. Lu and W. Chen, Appl. Catal., B, 2014, 154–155, 36–43 Search PubMed.
- T. N. Das, R. E. Huie and P. Neta, J. Phys. Chem. A, 1999, 103, 3581–3588 Search PubMed.
- S. Liu, Z. Zhang, F. Huang, Y. Liu, L. Feng, J. Jiang, L. Zhang, F. Qi and C. Liu, Appl. Catal., B, 2021, 286, 119921 Search PubMed.
- Y. Long, Z. Cao, W. Wu, W. Liu, P. Yang, X. Zhan, R. Chen, D. Liu and W. Huang, Appl. Catal., B, 2024, 344, 123643 Search PubMed.
- B. Sheng, C. Deng, Y. Li, S. Xie, Z. Wang, H. Sheng and J. Zhao, ACS Catal., 2022, 12, 14679–14688 Search PubMed.
- Y. Gao, T. Wu, C. Yang, C. Ma, Z. Zhao, Z. Wu, S. Cao, W. Geng, Y. Wang, Y. Yao, Y. Zhang and C. Cheng, Angew. Chem., Int. Ed., 2021, 60, 22513–22521 Search PubMed.
- Y. You, Org. Biomol. Chem., 2018, 16, 4044–4060 Search PubMed.
- C. Liang and H.-W. Su, Ind. Eng. Chem. Res., 2009, 48, 5558–5562 Search PubMed.
- J. O. Edwards, R. H. Pater, R. Curclf and F. D. Furia, Photochem. Photobiol., 1979, 30, 63–70 Search PubMed.
- A. Lange and H.-D. Brauer, J. Chem. Soc., Perkin Trans. 2, 1996, 5, 805–811 Search PubMed.
- R. E. Montgomery, J. Am. Chem. Soc., 1974, 96, 7820–7821 Search PubMed.
- C. Chen, T. Ma, Y. Shang, B. Gao, B. Jin, H. Dan, Q. Li, Q. Yue, Y. Li, Y. Wang and X. Xu, Appl. Catal., B, 2019, 250, 382–395 Search PubMed.
- S. Liu, C. Lai, B. Li, X. Liu, X. Zhou, C. Zhang, L. Qin, L. Li, M. Zhang, H. Yi, Y. Fu, H. Yan and L. Chen, Chem. Eng. J., 2022, 427, 131655 Search PubMed.
- N. Li, J. Ye, H. Dai, P. Shao, L. Liang, L. Kong, B. Yan, G. Chen and X. Duan, Water Res., 2023, 235, 119926 Search PubMed.
- Y. Gao, Z. Chen, Y. Zhu, T. Li and C. Hu, Environ. Sci. Technol., 2020, 54, 1232–1241 Search PubMed.
- Y. Liu, H. Guo, Y. Zhang, W. Tang, X. Cheng and W. Li, Chem. Eng. J., 2018, 343, 128–137 Search PubMed.
- T. Yang, S. Fan, Y. Li and Q. Zhou, Chem. Eng. J., 2021, 419, 129590 Search PubMed.
- W. Ren, L. Xiong, G. Nie, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2020, 54, 1267–1275 Search PubMed.
- X. Duan, H. Sun, Z. Ao, L. Zhou, G. Wang and S. Wang, Carbon, 2016, 107, 371–378 Search PubMed.
- Y. Li, T. Kong and S. Shen, Small, 2019, 15, 1900772 Search PubMed.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 Search PubMed.
- P. Su, W. Pei, X. Wang, Y. Ma, Q. Jiang, J. Liang, S. Zhou, J. Zhao, J. Liu and G. Q. Lu, Angew. Chem., Int. Ed., 2021, 60, 16044–16050 Search PubMed.
- Z. Li, K. Li, S. Ma, B. Dang, Y. Li, H. Fu, J. Du and Q. Meng, J. Colloid Interface Sci., 2021, 582, 598–609 Search PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 Search PubMed.
- Z. Wang, J. Jiang, S. Pang, Y. Zhou, C. Guan, Y. Gao, J. Li, Y. Yang, W. Qiu and C. Jiang, Environ. Sci. Technol., 2018, 52, 11276–11284 Search PubMed.
- Z. Wang, W. Qiu, S. Y. Pang, Q. Guo, C. Guan and J. Jiang, Environ. Sci. Technol., 2022, 56, 1492–1509 Search PubMed.
- C. Cheng, W. Ren, F. Miao, X. Chen, X. Chen and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218510 Search PubMed.
- J. Cui, S. Shao, J. Gao, Z. Yang, L. Li, S. Zeng, K. Wang, J. Cui, Y. Zhao and C. Hu, ACS ES&T Water, 2022, 2, 2698–2705 Search PubMed.
- H. Li, Z. Zhao, J. Qian and B. Pan, Environ. Sci. Technol., 2021, 55, 6397–6406 Search PubMed.
- Y. Zong, H. Zhang, X. Zhang, W. Liu, L. Xu and D. Wu, Appl. Catal., B, 2022, 300, 120722 Search PubMed.
- X. Zhang, J. Tang, L. Wang, C. Wang, L. Chen, X. Chen, J. Qian and B. Pan, Nat. Commun., 2024, 15, 917 Search PubMed.
- M. Li, H. Li, C. Ling, H. Shang, H. Wang, S. Zhao, C. Liang, C. Mao, F. Guo, B. Zhou, Z. Ai and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2304562120 Search PubMed.
- W. Ren, C. Cheng, P. Shao, X. Luo, H. Zhang, S. Wang and X. Duan, Environ. Sci. Technol., 2022, 56, 78–97 Search PubMed.
- Z.-H. Xie, C.-S. He, H.-Y. Zhou, L.-L. Li, Y. Liu, Y. Du, W. Liu, Y. Mu and B. Lai, Environ. Sci. Technol., 2022, 56, 8784–8795 Search PubMed.
- Z.-Y. Guo, C.-X. Li, M. Gao, X. Han, Y.-J. Zhang, W.-J. Zhang and W.-W. Li, Angew. Chem., Int. Ed., 2021, 60, 274–280 Search PubMed.
- M. Yang, R. Wu, S. Cao, Y. Li, S. Huo, W. Wang, Z. Hu and X. Xu, Chem. Eng. J., 2023, 451, 138606 Search PubMed.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 Search PubMed.
- F. Calle-Vallejo, D. Loffreda, M. T. M. Koper and P. Sautet, Nat. Chem., 2015, 7, 403–410 Search PubMed.
- Z. Wang, E. Almatrafi, H. Wang, H. Qin, W. Wang, L. Du, S. Chen, G. Zeng and P. Xu, Angew. Chem., Int. Ed., 2022, 61, e202202338 Search PubMed.
- Y. Gu, T. Xu, X. Chen, W. Chen and W. Lu, Chem. Eng. J., 2022, 427, 131973 Search PubMed.
- M. Li, Z. Li, X. Yu, Y. Wu, C. Mo, M. Luo, L. Li, S. Zhou, Q. Liu, N. Wang, K. Lun Yeung and S. Chen, Chem. Eng. J., 2022, 431, 133339 Search PubMed.
- Y. Zhao, S. Chen, H. Qie, S. Zhu, C. Zhang, X. Li, W. Wang, J. Ma and Z. Sun, Water Res., 2023, 236, 119957 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |