
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of the anion exchange between I− and NCS− coordinated to Co2+-centered tetrahedra in the organic–inorganic hybrid halides†
Takuya
Ohmi‡
 a,
Ryuju
Ito‡
a,
Sergey A.
Nikolaev
a,
Kotaro
Fujii
b,
Iain W. H.
Oswald
c,
Koki
Matsushima
ad,
Yuiga
Nakamura
e,
James R.
Neilson
c,
Masatomo
Yashima
b,
Masaki
Azuma
fag and
Takafumi
Yamamoto
*da
a,
Ryuju
Ito‡
a,
Sergey A.
Nikolaev
a,
Kotaro
Fujii
b,
Iain W. H.
Oswald
c,
Koki
Matsushima
ad,
Yuiga
Nakamura
e,
James R.
Neilson
c,
Masatomo
Yashima
b,
Masaki
Azuma
fag and
Takafumi
Yamamoto
*da
aMaterials and Structures Laboratory, Institute of Integrated Research, Institute of Science Tokyo, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8501, Japan. E-mail: yama@kuchem.kyoto-u.ac.jp
bDepartment of Chemistry, School of Science, Institute of Science Tokyo, 2-12-1-W4-17 Ookayama, Meguro-ku, Tokyo 152-8551, Japan
cDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523-1872, USA
dDepartment of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa Oiwake-cho, Sakyoku, Kyoto 606-8502, Japan
eJapan Synchrotron Radiation Research Institute (JASRI), SPring-8, 1-1-1, Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
fResearch Center for Autonomous Systems Materialogy (ASMat), Institute of Integrated Research, Institute of Science Tokyo, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8501, Japan
gKanagawa Institute of Industrial Science and Technology, 705-1 Shimoimaizumi, Ebina 243-0435, Japan
First published on 25th June 2025
Abstract
Mixed-anion compounds provide a unique playground for local coordination engineering due to their heteroleptic coordination around cations. Here, we successfully synthesized novel thiocyanate-containing organic–inorganic hybrid halides (CH3NH3)2Co(NCS)4 and (CH3NH3)3CoI4(NCS) with isolated Co(NCS)4 tetrahedra and mixed-anion CoI3(NCS) tetrahedra, respectively. Together with (CH3NH3)2CoI4 with CoI4 tetrahedra, they provide different types of anion-coordination around Co2+ ions. We compare the magnetic properties of these compounds and discuss the effects of anion exchange between I− and NCS− using density functional theory calculations.
Introduction
Mixed-anion systems, where multiple anions are contained in a single compound, have received increasing attention due to their fundamental and industrial importance.1–3 Having crystal structures with the heteroleptic coordination around cations, such compounds hold the potential to exhibit unique properties that cannot be realized in single-anion compounds. For example, using noncentrosymmetric units with a mixed anion coordination, a methodology for novel design of the polar structures was demonstrated in oxyfluoride CuVOF4(H2O)7.4 Oxyselenide BaFe2Se2O with trans-coordinated FeSe4O2 octahedra was reported to have a noncollinear magnetic order with double-k vectors that originates from spin–orbit coupling induced by the heteroleptic coordination around the iron.5 SrVO2H with trans-coordinated VO4H2 octahedra exhibits a quasi-2D electronic structure due to the π-blocking nature of H−.6,7Recently, organic–inorganic hybrid compounds with molecular cations have also attracted a great deal of interest in the field of advanced functional materials. For example, hybrid lead halide perovskites, such as CH3NH3PbI3 (CH3NH3+ = methylammonium ion), are emerging as solar cell materials.8 Hybrid halides can also accommodate molecular anions, so-called pseudo-halide ions, such as thiocyanate (NCS−) and formate ion (HCOO−).9–17 The substitution of anionic ligands in hybrid compounds provides diverse coordination environments for exploring physical properties.
Thiocyanate ions are widely studied as ligands for hybrid compounds such as molecular organic flameworks because of their versatility in bonding with metals.18–23 Being an anisotropic molecule, a thiocyanate ion can provide various coordination motifs, as reported in (CH3NH3)2PbI2(SCN)2 with M-SCN motif,9,10,17 [BzPy]2Co(NCS)4 (BzPy = benzyl pyridinium) and (C4H12N)2Co(NCS)4 with M-NCS motif,19,24 and M(NCS)2(NCMe)2 (M = Cr, Mn) with M-NCS-M motif.18 Furthermore, there have been reported thiocyanate-based hybrid compounds with mixed-ligands, such as Hg3CdCl2(SCN)6 and Hg4CdBr4(SCN)6.10,17,20,21,25–27 Another example HgCo(NCS)4 with isolated Co(NCS)4 tetrahedra has long been studied as a magnetic susceptibility standard.28–33 Some researchers also reported the magnetic properties of the complexes containing Co(NCS)4 tetrahedra and organic cations.19,34–38 However, the effect of the mixed-ligand coordination in the Co2+-centered tetrahedra on magnetism has not yet been revealed.
Herein, we synthesized two new cobalt-based hybrid compounds with structural motifs comprised of the tetrahedrally coordinated Co2+ ions: (CH3NH3)3CoI4(NCS) with the mixed-anion CoI3(NCS) tetrahedra and (CH3NH3)2Co(NCS)4 with isotropic Co(NCS)4 tetrahedra. Including (CH3NH3)2CoI4 with CoI4 tetrahedra,39 we consider a series of three compounds and demonstrate how the anionic coordination of Co2+ ions affects their magnetic properties using magnetization measurements and first-principles calculations.
Experimental and computational
A powder sample of (CH3NH3)2Co(NCS)4 was prepared using the solvent evaporation method. A stoichiometric amount of solid CH3NH3SCN (>97.0%, Tokyo Chemical Industry Co., Ltd) and Co(NCS)2 (96%, Sigma-Aldrich) were dissolved with 2-methoxyethanol solvent (>99.0%, Kanto Chemical Co., Inc.). The solvent was evaporated in air at RT, and dark blue (CH3NH3)2Co(NCS)4 crystals were precipitated. The product was filtered and dried under reduced pressure overnight. The crystals were finely ground with agate mortar. Powder samples of (CH3NH3)3CoI4(NCS), and (CH3NH3)2CoI4 were synthesized by using solid-state reactions. A stoichiometric amount of CH3NH3I (>97.0% Tokyo Chemical Industry Co., Ltd), CoI2 (99.5%, Alfa Aesar), and Co(NCS)2 were mixed and pelletized in an N2-filled glovebox. Then, the pellets were sealed in an evacuated Pyrex tube and heated at 120 °C for 36 hours. Since powder samples of (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4 showed gradual decomposition into their precursors in the air over several days at RT, the samples were kept in an N2-filled glove box.(CH3NH3)2Co(NCS)4 single crystals were obtained in the same way as (CH3NH3)2Co(NCS)4 powder. 0.254 g of CH3NH3(SCN) and 0.246 g of Co(NCS)2 were combined with 10 ml of 2-methoxyethanol. After evaporating the solution for a few days, the blue-colored crystals could be found in the residual solvent. Single crystals of (CH3NH3)3CoI4(NCS) were synthesized by a solid-state reaction. The pellet in the sealed tube was heated at 140 °C and slowly cooled to room temperature for 24 hours.
Powder X-ray diffraction (XRD) data were collected with the Malvern Panalytical Aeris Benchtop XRD System using Cu Kα radiation. The measurement was performed in air at RT. All measurement processes were completed within 10 minutes to minimize degradation. Rietveld refinements of the obtained patterns were performed with the TOPAS v6 software. The crystal structures are visualized using the VESTA 3.40
Single crystal XRD data of (CH3NH3)2Co(NCS)4 was collected at the beamline BL02B1 of SPring-8, the Japan Synchrotron Radiation Research Institute (JASRI). The wavelength of an incident X-ray beam was 0.4132 Å. The selected crystals were covered with Fluorolube oil (Sigma-Aldrich), and mounted onto a goniometer under the cold N2 gas stream. The diffraction was collected at 100 K. The structures were solved by the intrinsic phase methods (SHELXT 2014/5),41,42 and refined by full-matrix least-squares methods with SHELXL (2018/3)43 on Olex2 software (ver. 1.5).44 To compensate for the residual electron density observed around the Co2+ ion in (CH3NH3)2Co(NCS)4, anharmonic displacement parameters were employed during the refinement.45 Due to the highly disordered Methylammonium ions, the data were treated by the SQUEEZE program in PLATON.46 Structural details of the refinement, crystallographic parameters, and atomic positions can be found in Tables S1–S3 in ESI.† Single crystal XRD data of (CH3NH3)3CoI4(NCS) was collected at RT using a Bruker D8 Quest ECO diffractometer equipped with a microfocus Mo Kα radiation source and Photon 50 CMOS half-plate detector. Single Bruker SAINT was used for the integration and scaling of collected data and the SADABS was used for the absorption correction.47 Structural details of the refinement, crystallographic parameters, bond lengths, bond angles, and experimental conditions are listed in Tables S4–S6 in ESI.†
Diffuse Reflectance spectra were measured using a JASCO V-670 UV-visible spectrophotometer. Each sample was filled into a round holder with a quartz glass and BaSO4. The spectra were collected from λ = 200 to 1200 nm. These measurements were carried out using an integration sphere at RT in air.
Magnetic susceptibility was measured on a Quantum Design MPMS SQUID magnetometer equipped with RSO option, in an applied magnetic field of 1000 Oe from 4 K to 300 K. Powder samples of ∼20 mg sealed in plastic wrap were used for the measurement. Note that (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4 can decompose into precursors after a few hours in air (Fig. S1†). Thus, the samples were immediately sealed in plastic wrap after taking them out from the glove box. Subsequently, it was immediately transferred to the MPMS chamber.
First-principles calculations were performed within the projector-augmented plane-wave formalism48 using the generalized gradient approximation (GGA, Perdew–Burke–Ernzerhof)49 for the exchange–correlation potential as implemented in the Vienna Ab initio Simulation Package (VASP).50 Non-magnetic calculations were performed without spin–orbit coupling using the experimental crystal structures. Calculations are carried out with an energy cut-off set to 500 eV and the energy convergence criterion of 10−8 eV. The Brillouin zone was sampled by a 6 × 6 × 6, 4 × 4 × 3, and 4 × 4 × 2 k-point mesh51 for (CH3NH3)2Co(NCS)4, (CH3NH3)3CoI4(NCS), and (CH3NH3)2CoI4, respectively. Since the methylammonium cations in (CH3NH3)2Co(NCS)4 are allowed to rotate at given positions (schematically, the 6b Wyckoff positions), we considered two structures of (CH3NH3)2Co(NCS)4: the one with fixed disorder where positions and orientations of methylammonium cations are chosen arbitrarily (4 out of 6b positions are occupied to preserve the charge neutrality), and the one without methylammonium cations by imposing the total charge of 4e− in the unit cell (that corresponds to removing four methylammonium cations from the unit cell). Crystal-field splitting of the Co 3d states is calculated in the basis of Wannier functions, as implemented in the Wannier 90 package.52
Parameters of the electronic model were calculated for a single [CoX4]2− complex with X = I and NCS, assuming to have a perfect tetrahedral coordination. The corresponding bond lengths are adopted from the experimental structures (for X = I, we take the average Co–I bond length of 2.604 Å). Each tetrahedron is placed in a cubic box with a side length of 40 Å, where the total charge of 2e− is imposed. Electronic structure calculations are performed in VASP as outlined above and Quantum-ESPRESSO using norm-conserving pseudopotentials with the energy cut-off of 100 Ry.53 The one-electron part of the model including crystal field and spin–orbit coupling is obtained from electronic structure calculations with spin–orbit coupling using the Wannier functions, which are constructed by projecting the states near the Fermi level onto atomic d orbitals. The on-site Coulomb interactions are calculated from electronic structure calculations without spin–orbit coupling using constrained random phase approximation in the basis of Wannier functions,54 as implemented in the RESPACK package.55,56 Calculations of the polarization function, which accounts for the screening effects, are carried out with a large number of unoccupied states (∼100).
Results and discussion
Obtained (CH3NH3)2Co(NCS)4 single crystals are block-shaped with 10–200 μm3 size and have a translucent blue color. The structure analysis of single crystal synchrotron XRD data at 100 K revealed that the compound has a cubic structure (space group: I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m) with a = 9.63590(10) Å. The finalized structural refinement converged to R1 = 5.17%, and the crystal structure was determined except for the methylammonium ions. The obtained crystal structure is shown in Fig. 1a. Details of crystallographic information are shown in the ESI (Table S1–S3†). The methylammonium ions are disordered in the voids of the crystal lattice even though measured at 100 K. Thus, we executed the SQUEEZE procedure to converge the refinement.46 Note that there are six interstitial cation sites (three for face-centered positions and three for edge-centered positions; Fig. 1a) in a single unit lattice: the four sites are occupied by methylammonium ions, causing a significant site disorder. The electron density squeezed from the procedure was 72 electrons, which agrees with the number of electrons in the four methylammonium ions. This suggests that no solvent molecules are intercalated in the lattice.
3m) with a = 9.63590(10) Å. The finalized structural refinement converged to R1 = 5.17%, and the crystal structure was determined except for the methylammonium ions. The obtained crystal structure is shown in Fig. 1a. Details of crystallographic information are shown in the ESI (Table S1–S3†). The methylammonium ions are disordered in the voids of the crystal lattice even though measured at 100 K. Thus, we executed the SQUEEZE procedure to converge the refinement.46 Note that there are six interstitial cation sites (three for face-centered positions and three for edge-centered positions; Fig. 1a) in a single unit lattice: the four sites are occupied by methylammonium ions, causing a significant site disorder. The electron density squeezed from the procedure was 72 electrons, which agrees with the number of electrons in the four methylammonium ions. This suggests that no solvent molecules are intercalated in the lattice.
 | ||
| Fig. 1 Crystal structures of (a) (CH3NH3)2Co(NCS)4, (b) (CH3NH3)3CoI4(NCS), and (c) (CH3NH3)2CoI4.39 The solid black line shows the unit cell. Disordered methylammonium ions are not shown in (a), instead, interstitial voids that can be potentially occupied by methylammonium ions are indicated by gray circles. Local structures around Co2+ ions (d–f). | ||
We also synthesized (CH3NH3)3CoI4(NCS) single crystals by a solid-state reaction of a stoichiometric mixture of the precursors with slow cooling. The obtained crystals are translucent light green rectangular prisms. The single crystal X-ray analysis of the data at 300 K revealed that the compound has an orthorhombic structure (space group: P212121) with a = 10.4690(9) Å, b = 11.5048(9) Å, and c = 15.7129(12) Å (see the details in Tables 4–6 of ESI†). Fig. 1b shows the structure of (CH3NH3)3CoI4(NCS), where the interstitial space between the isolated CoI3(NCS) tetrahedra is occupied by CH3NH3+ and I− ions.
Together with (CH3NH3)2CoI4,39 crystal structures of the newly synthesized (CH3NH3)3CoI4(NCS) and (CH3NH3)2Co(NCS)4 compounds are shown in Fig. 1a–c. Compared with these three compounds, anion species coordinated to Co2+-centered tetrahedra exchanges from I− to NCS− (Fig. 1d–f). Among them, (CH3NH3)3CoI4(NCS) has a mixed-anion coordination with one NCS− and three I− ligands. Although there have been some reports of mixed-anion transition metal compounds incorporating both NCS− and halide ligands,21,25,26 (CH3NH3)3CoI4(NCS) represents the first example of a tetrahedral unit composed of three I− and one NCS− ligands to the best of our knowledge. This distinct coordination environment serves as a useful platform for systematic studies of mixed-anion tetrahedral coordination.
The body color of the powder samples changes from blue to green with the anion substitution from I to SCN (Fig. S2†). Diffuse reflectance spectra of the powder samples show an energy shift in the absorption bands corresponding to the d–d transition of Co2+, due to the change in the crystal-field splitting caused by anion substitution (Fig. S3†).
To investigate the effect of the local coordination, we analyzed the magnetic properties using powder samples. The powder sample of (CH3NH3)2Co(NCS)4 was prepared by fine grounding the crystals prepared with the solution method (Fig. S2a†). Powder samples of (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4 were synthesized by a solid-state reaction (Fig. S2b and c†). The XRD patterns of the powder samples are shown in Fig. 2. The simulation patterns match with the calculated patterns well. The lattice parameter obtained by Le Bail analysis is a = 9.7723(3) Å for (CH3NH3)2Co(NCS)4 (Fig. S4†). (CH3NH3)2Co(NCS)4 did not show any structural phase transition in the range of 100 K–300 K (Fig. S5†). (CH3NH3)2Co(NCS)4 are phase pure, while (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4 contain a tiny amount of CH3NH3I impurity. To estimate the phase fractions of the powder samples, we performed Rietveld analysis of the XRD patterns (Fig. S6†), and found that the phase fraction of the CH3NH3I impurity was 0.57 mol% for (CH3NH3)3CoI4(NCS) and 0.95 mol% for (CH3NH3)2CoI4. Thus, the contribution of impurities to the magnetic properties can be regarded as almost negligible.
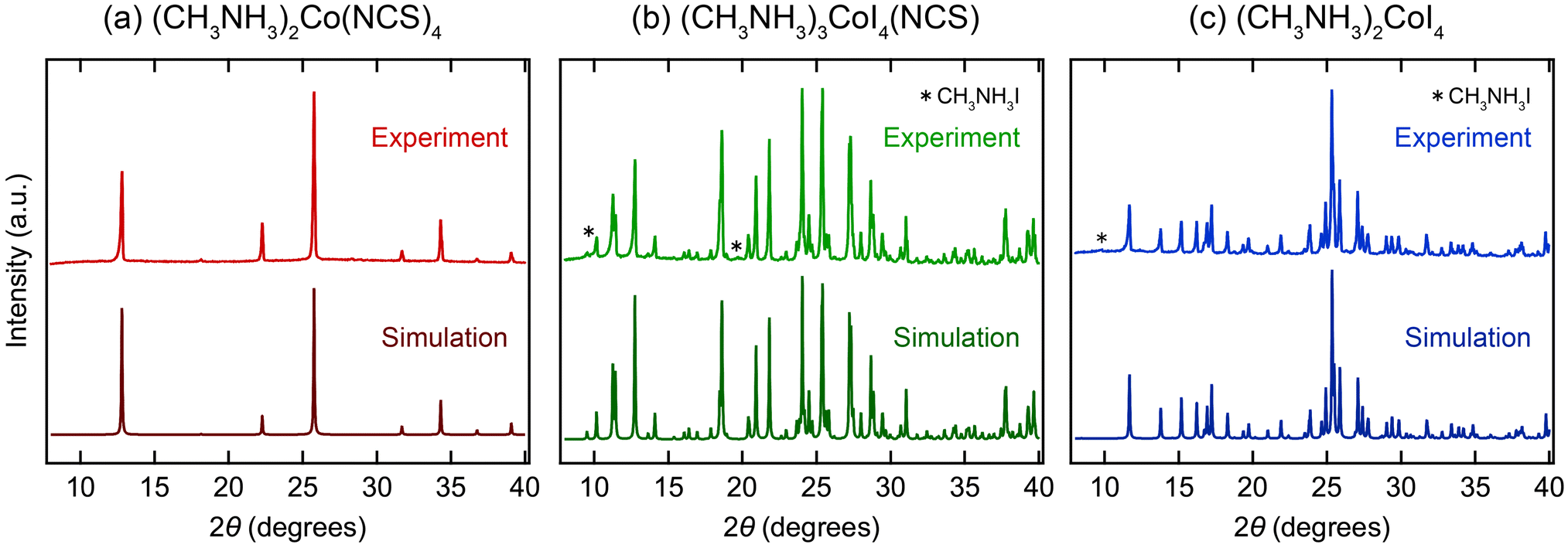 | ||
| Fig. 2 XRD patterns of (a) (CH3NH3)2Co(NCS)4, (b) (CH3NH3)3CoI4(NCS), and (c) (CH3NH3)2CoI4. The upper and lower patterns show the experimental and calculated ones, respectively. * indicates the diffraction from the impurity of CH3NH3I. Simulated patterns of (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4 are from the structure obtained from single XRD analysis and ref. 39, respectively. Note that the simulated pattern of (CH3NH3)2Co(NCS)4 is calculated from a hypothetical structure (Fig. S7†), modified from the structure obtained by single crystal XRD analysis. | ||
Fig. 3a shows the temperature dependence of magnetic susceptibilities in all three samples. With decreasing temperatures down to 4 K, the susceptibility gradually increases, suggesting that all three compounds are paramagnetic above 4 K. The temperature dependence of the inverse susceptibility can be fitted using the Curie–Weiss equation χ−1 = (T − ΘW)/(C + χ0 (T − ΘW)), where C, χ0, and ΘW stand for the Curie constant, temperature independent susceptibility, and the Weiss temperature, respectively.
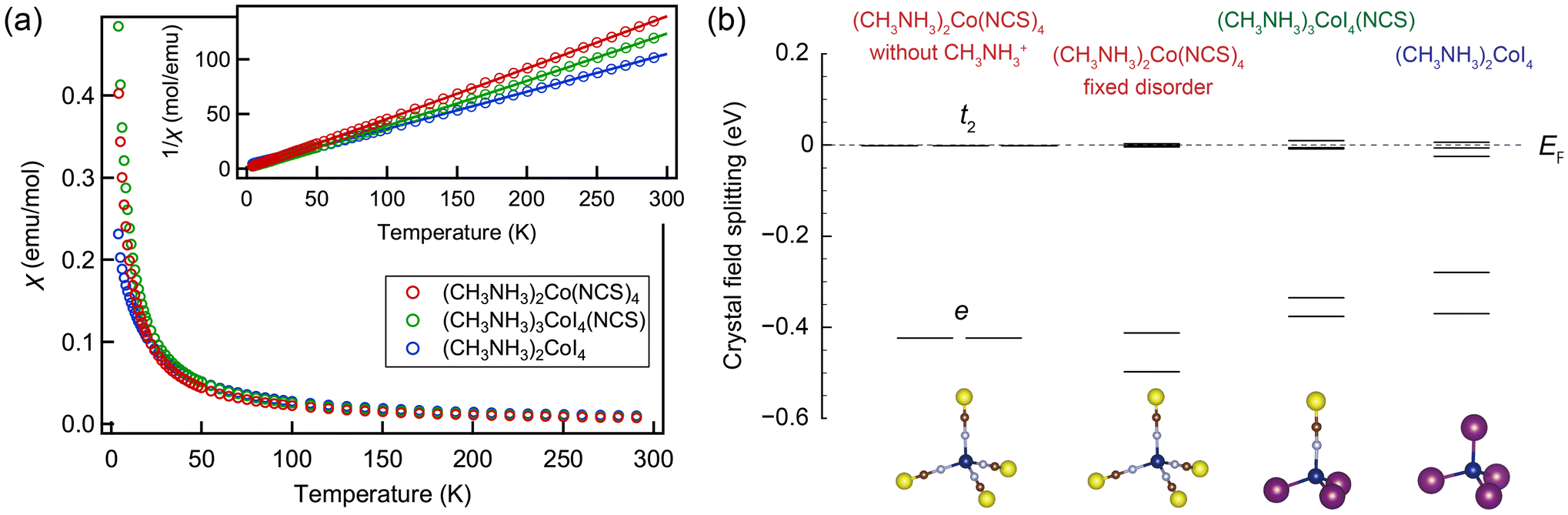 | ||
| Fig. 3 (a) Temperature dependence of magnetic susceptibilities for (CH3NH3)2Co(NCS)4, (CH3NH3)3CoI4(NCS), and (CH3NH3)2CoI4. Inset shows temperature dependence of the inverse susceptibilities and Curie–Weiss fitting curves. (b) The crystal-field splitting of the Co 3d states calculated from a non-magnetic state in (CH3NH3)2CoI4, (CH3NH3)3CoI4(NCS), and (CH3NH3)2Co(NCS)4. The energy states are shown with respect to the Fermi level. | ||
The resulting parameters are summarized in Table 1. Here, the effective magnetic moment is given by  , where the orbital contribution is pushed into the g-factor. One can see that the Curie constant increases in a series from (CH3NH3)2Co(NCS)4 to (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4. Similarly, the effective magnetic moment μeff and the g-factor become larger with the introduction of iodide ions. In fact, g = 2.54 of (CH3NH3)2CoI4 is consistent with 2.59 reported in (C9H5N)2CoI4 with CoI4 tetrahedra.57,58 Likewise, g = 2.20 estimated for (CH3NH3)2Co(NCS)4 is also in good agreement with previous results for similar compounds containing Co(NCS)4 complexes.18,19,28,33 Notably, both μeff and g are found to exceed their spin-only values, 3.87μB and 2, respectively, which can be nominally assigned to a high-spin state (S = 3/2) of the tetrahedrally coordinated Co2+ ion, thus implying the existence of the orbital contribution that becomes more pronounced as NCS− is substituted by I−.
, where the orbital contribution is pushed into the g-factor. One can see that the Curie constant increases in a series from (CH3NH3)2Co(NCS)4 to (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4. Similarly, the effective magnetic moment μeff and the g-factor become larger with the introduction of iodide ions. In fact, g = 2.54 of (CH3NH3)2CoI4 is consistent with 2.59 reported in (C9H5N)2CoI4 with CoI4 tetrahedra.57,58 Likewise, g = 2.20 estimated for (CH3NH3)2Co(NCS)4 is also in good agreement with previous results for similar compounds containing Co(NCS)4 complexes.18,19,28,33 Notably, both μeff and g are found to exceed their spin-only values, 3.87μB and 2, respectively, which can be nominally assigned to a high-spin state (S = 3/2) of the tetrahedrally coordinated Co2+ ion, thus implying the existence of the orbital contribution that becomes more pronounced as NCS− is substituted by I−.

| (CH3NH3)2Co(NCS)4 | (CH3NH3)3CoI4(NCS) | (CH3NH3)2CoI4 | |
|---|---|---|---|
| C (emu K mol−1) | 2.26(4) | 2.64(2) | 3.03(6) |
| Θ W (K) | −1.20(8) | −1.15(4) | −9.21(9) |
| χ 0 (emu mol−1) | −0.00032(1) | −0.000693(9) | −0.00027(2) |
| exp. μeff (μB) | 4.26 | 4.59 | 4.92 |
| g | 2.20 | 2.37 | 2.54 |
Indeed, previous studies showed that the effective magnetic moments of the tetrahedrally coordinated [CoX4]2− complexes (X = Cl, Br, and I) can have an orbital contribution due to spin–orbit coupling.56 For the g-factor, this contribution can be estimated using perturbation theory:57,58
| g = 2.00 − 8λ′/Δ | (1) |
The effects of the local environment on the magnetic properties of [CoX4]2− complexes are further analyzed using electronic structure calculations. To this end, we start by considering a single Co2+ ion in perfect tetrahedral coordination whose electronic properties can be described by the following effective model:
 | (2) |
 are in the range of ∼0.03–0.06 and ∼0.02–0.04 eV for X = I and NCS, respectively). Calculations for Co(NCS)4 and CoI4 tetrahedra yield the g-factors of 2.26 and 2.58, respectively, in good agreement with experimental estimates.
are in the range of ∼0.03–0.06 and ∼0.02–0.04 eV for X = I and NCS, respectively). Calculations for Co(NCS)4 and CoI4 tetrahedra yield the g-factors of 2.26 and 2.58, respectively, in good agreement with experimental estimates.
Similar trends can be seen in the real structures, where CoX4 tetrahedra appear to be distorted. The crystal-field splitting of the Co 3d states in all three compounds obtained is presented in Fig. 3b. While the Co 3d levels in the tetrahedral environment can be schematically grouped into the e and t2 levels, the exact splitting is obtained only for (CH3NH3)2Co(NCS)4 without the methylammonium cations, where the Co(NCS)4 tetrahedra possess the Td point group symmetry. By fixing the positions of the methylammonium cations, the symmetry of (CH3NH3)2Co(NCS)4 (see Fig. S9†) is lowered, resulting in a further splitting of the Co 3d states. The e and t2 levels are also split in (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4, where the corresponding tetrahedra are distorted, and the symmetry is lowered to the D2 and D2h point groups, respectively. This symmetry-lowering results in further splitting within the Co 3d levels (Fig. 3b). However, these distortions remain moderate, and thus the tetrahedral character (splitting to t2 and e) still dominates the crystal-field splitting. Overall, our calculations reveal that the crystal-field splitting of the Co 3d states steadily decreases from (CH3NH3)2Co(NCS)4 to (CH3NH3)3CoI4(NCS) and (CH3NH3)2CoI4, which is qualitatively consistent with the observed changes in magnetic properties.
Conclusions
We reported new thiocyanate-containing organic–inorganic hybrid halides (CH3NH3)2Co(NCS)4 and (CH3NH3)3CoI4(NCS) with isolated Co(NCS)4 and CoI3(NCS) tetrahedra, respectively. Together with (CH3NH3)2CoI4, they have different types of anion-coordination around Co2+. We revealed that the effective magnetic moment deviates from the spin-only value and the g-factor increases when I− is introduced. The results of first-principles calculations showed that these changes can be ascribed to the enhanced orbital contribution due to the presence of I ions.Author contributions
TO and RI contributed equally to this work. TY designed the research. TO, RI, KM, and TY synthesized the samples. TO and RI performed powder XRD and magnetic measurements. KM conducted the DRS measurements. RI, KF, and IWHO performed laboratory single-crystal XRD measurements. TO and YN performed synchrotron single-crystal XRD measurements. TO, KF, and IWHO carried out the single-crystal structure analysis. SAN conducted the DFT calculations. MY, JRN, and MA contributed to discussions, validation, and resources. RI and SAN wrote the initial version of the manuscript, and TO and TY reviewed and edited the final version, with comments from all authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2365182 (for (CH3NH3)2Co(NCS)4) and CCDC 2325726 (for (CH3NH3)3CoI4(NCS)†) contain the supplementary crystallographic data for this paper.Acknowledgements
We thank Dr Shiro Funahashi at the National Institute for Materials Science for fruitful discussions on single-crystal XRD analysis. We also thank Dr Chishiro Michioka at Kyoto University for fruitful discussions on magnetism. Grant-in-Aid supported this work for Transformative Research Areas (A) “Supra-ceramics” (JSPS KAKENHI Grant Numbers JP22H05147, and JP23H04618), JSPS KAKENHI Grant Number JP22H01767. This work was also supported by Design and Engineering by Joint Inverse Innovation for Materials Architecture (DEJI2MA), MEXT. TY was supported by Tokyo Tech Challenging Research Award. TO was supported by the Japan Society for the Promotion of Science for Young Scientists (JP22J20672) and the MEXT Project of the Tokyo Tech Academy for Convergence of Materials and Informatics (TAC-MI). The synchrotron radiation experiments were performed at the BL02B1 and BL02B2 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2024A1508, 2022A1191).References
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, e1805295 CrossRef PubMed.
- K. Maeda, F. Takeiri, G. Kobayashi, S. Matsuishi, H. Ogino, S. Ida, T. Mori, Y. Uchimoto, S. Tanabe, T. Hasegawa, N. Imanaka and H. Kageyama, Bull. Chem. Soc. Jpn., 2022, 95, 26–37 CrossRef.
- M. D. Donakowski, R. Gautier, J. Yeon, D. T. Moore, J. C. Nino, P. S. Halasyamani and K. R. Poeppelmeier, J. Am. Chem. Soc., 2012, 134, 7679–7689 CrossRef PubMed.
- F. Takeiri, Y. Matsumoto, T. Yamamoto, N. Hayashi, Z. Li, T. Tohyama, C. Tassel, C. Ritter, Y. Narumi, M. Hagiwara and H. Kageyama, Phys. Rev. B, 2016, 94, 184426 CrossRef.
- J. Bang, S. Matsuishi, H. Hiraka, F. Fujisaki, T. Otomo, S. Maki, J.-I. Yamaura, R. Kumai, Y. Murakami and H. Hosono, J. Am. Chem. Soc., 2014, 136, 7221–7224 CrossRef.
- T. Yamamoto, D. Zeng, T. Kawakami, V. Arcisauskaite, K. Yata, M. A. Patino, N. Izumo, J. E. McGrady, H. Kageyama and M. A. Hayward, Nat. Commun., 2017, 8, 1217 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- M. Daub and H. Hillebrecht, Angew. Chem., Int. Ed., 2015, 54, 11016–11017 CrossRef PubMed.
- T. Yamamoto, I. W. H. Oswald, C. N. Savory, T. Ohmi, A. A. Koegel, D. O. Scanlon, H. Kageyama and J. R. Neilson, Inorg. Chem., 2020, 59, 17379–17384 CrossRef.
- T. Ohmi, I. W. H. Oswald, J. R. Neilson, N. Roth, S. Nishioka, K. Maeda, K. Fujii, M. Yashima, M. Azuma and T. Yamamoto, J. Am. Chem. Soc., 2023, 145, 19759–19767 CrossRef PubMed.
- T. Ohmi, J. R. Neilson, W. Taniguchi, T. Fukui, T. Nagase, Y. Haruta, M. I. Saidaminov, T. Fukushima, M. Azuma and T. Yamamoto, ACS Mater. Lett., 2024, 6, 1913–1919 CrossRef.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef.
- W. Hui, L. Chao, H. Lu, F. Xia, Q. Wei, Z. Su, T. Niu, L. Tao, B. Du, D. Li, Y. Wang, H. Dong, S. Zuo, B. Li, W. Shi, X. Ran, P. Li, H. Zhang, Z. Wu, C. Ran, L. Song, G. Xing, X. Gao, J. Zhang, Y. Xia, Y. Chen and W. Huang, Science, 2021, 371, 1359–1364 CrossRef.
- M. J. Cliffe, E. N. Keyzer, M. T. Dunstan, S. Ahmad, M. F. L. De Volder, F. Deschler, A. J. Morris and C. P. Grey, Chem. Sci., 2019, 10, 793–801 RSC.
- J. Y. Lee, S. Ling, S. P. Argent, M. S. Senn, L. Cañadillas-Delgado and M. J. Cliffe, Chem. Sci., 2021, 12, 3516–3525 RSC.
- T. Ohmi, T. Miura, K. Shigematsu, A. A. Koegel, B. S. Newell, J. R. Neilson, T. Ikoma, M. Azuma and T. Yamamoto, CrystEngComm, 2022, 24, 5428–5434 RSC.
- E. Shurdha, C. E. Moore, A. L. Rheingold, S. H. Lapidus, P. W. Stephens, A. M. Arif and J. S. Miller, Inorg. Chem., 2013, 52, 10583–10594 CrossRef.
- Z. Zhang, J. Xu, S. Yan, Y. Chen, Y. Wang, Z. Chen and C. Ni, Crystals, 2017, 7, 92 CrossRef.
- D. Li, Q. Zhang, X. Wang, S. Li, H. Zhou, J. Wu and Y. Tian, Dyes Pigm., 2015, 120, 175–183 CrossRef.
- A. Mosset, M. Bagieu-Beucher, A. Lecchi, R. Masse and J. Zaccaro, Solid State Sci., 2002, 4, 827–834 CrossRef.
- S. E.-D. H. Etaiw, M. M. El-bendary, A. E.-A. S. Fouda and M. M. Maher, Prot. Met. Phys. Chem. Surf., 2017, 53, 937–949 CrossRef.
- M. J. Cliffe, Inorg. Chem., 2024, 63, 13137–13156 CrossRef.
- N. M. Byrne, M. H. Schofield, A. D. Nicholas and C. L. Cahill, Dalton Trans., 2021, 50, 9158–9172 RSC.
- S. S. Singh, R. S. Prasad, S. Upreti, N. K. Jha and A. Ramanan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1014–m1015 CrossRef.
- R. S. Prasad, S. S. Singh, S. Upreti, N. K. Jha and A. Ramanan, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2006, 62, m2990–m2991 CrossRef.
- E. V. Peresypkina, A. V. Virovets, J. V. Akhmerkina and L. B. Serezhkina, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m355–m356 CrossRef PubMed.
- D. Nelson and L. W. Ter Haar, Inorg. Chem., 1993, 32, 182–188 CrossRef.
- R. Boča, J. Titiš, C. Rajnák and J. Krzystek, Dalton Trans., 2021, 50, 3468–3472 RSC.
- F. A. Cotton, D. M. L. Goodgame, M. Goodgame and A. Sacco, J. Am. Chem. Soc., 1961, 83, 4157–4161 CrossRef.
- J.-C. G. Bünzli, Inorg. Chim. Acta, 1979, 36, L413–L414 CrossRef.
- D. B. Brown, V. H. Crawford, J. W. Hall and W. E. Hatfield, J. Phys. Chem., 1977, 81, 1303–1306 CrossRef.
- C. J. O’Connor, E. Sinn, E. J. Cukauskas and B. S. Deaver Jr., Inorg. Chim. Acta, 1979, 32, 29–32 CrossRef.
- H.-Q. Ye, Y.-Y. Li, R.-K. Huang, X.-P. Liu, W.-Q. Chen, J.-R. Zhou, L.-M. Yang and C.-L. Ni, J. Struct. Chem., 2014, 55, 691–696 CrossRef.
- H.-Q. Ye, L.-J. Su, X.-X. Chen, X. Liao, Q.-T. Liu, X.-Y. Wu, J.-R. Zhou, L.-M. Yang and C.-L. Ni, Synth. Met., 2015, 199, 232–240 CrossRef.
- W.-Q. Chen, L.-J. Su, X.-Q. Cai, J.-J. Yang, Y.-L. Qian, X.-P. Liu, L.-M. Yang, J.-R. Zhou and C.-L. Ni, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2014, 44, 980–985 CrossRef.
- X. Chen, S.-L. Dai, Z.-P. Cheng, L.-B. Liang, S. Han, J.-F. Liu, J.-R. Zhou, L.-M. Yang and C.-L. Ni, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 987–993 CrossRef.
- X. Chen, W.-Q. Chen, S. Han, J.-F. Liu, J.-R. Zhou, L.-L. Yu, L.-M. Yang, C.-L. Ni and X.-L. Hu, J. Mol. Struct., 2010, 984, 164–169 CrossRef.
- M. Daub, R. Stroh and H. Hillebrecht, Z. Anorg. Allg. Chem., 2016, 642, 268–274 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- B. T. M. Willis, Acta Crystallogr., Sect. A: Found. Crystallogr., 1969, 25, 277–300 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS.
- J. D. Pack and H. J. Monkhorst, Phys. Rev., 1977, 16, 1748–1749 Search PubMed.
- A. I. Liechtenstein, V. I. Anisimov VI and J. Zaanen, Phys. Rev. B, 1995, 52, R5467–R5470 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef.
- F. Aryasetiawan, M. Imada, A. Georges, G. Kotliar, S. Biermann and A. I. Lichtenstein, Phys. Rev. B, 2004, 70, 195104 CrossRef.
- K. Nakamura, Y. Nohara, Y. Yosimoto and Y. Nomura, Phys. Rev. B, 2016, 93, 085124 CrossRef.
- K. Nakamura, Y. Yoshimoto, Y. Nomura, T. Tadano, M. Kawamura, T. Kosugi, K. Yoshimi, T. Misawa and Y. Motoyama, Comput. Phys. Commun., 2021, 261, 107781 CrossRef CAS.
- R. H. Holm and F. A. Cotton, J. Chem. Phys., 1959, 31, 788–792 CrossRef CAS.
- R. Schlapp and W. G. Penney, Phys. Rev., 1932, 42, 666–686 CrossRef CAS.
- J. C. Slater, Quantum theory of atomic structure: V. 1, McGraw-Hill, New York, NY, 1960 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Photographs of the powder samples, crystallographic data, additional powder XRD data, diffuse-reflectance spectra, additional DFT results. CCDC 2365182 and 2325726. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00924c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |