
In situ analysis of a boron-based catalytic electrode with trace platinum for efficient hydrogen evolution in a wide pH range†
Xunwei
Ma‡
ab,
Yifan
Zhang‡
a,
Liugang
Wu
a,
Zijun
Huang
a,
Jiyuan
Yang
d,
Chunguang
Chen
a,
Shengwei
Deng
e,
Lincai
Wang
 *b,
Jian
Chen
*c and
Weiju
Hao
*a
*b,
Jian
Chen
*c and
Weiju
Hao
*a
aUniversity of Shanghai for Science and Technology, Shanghai 200093, P. R. China. E-mail: wjhao@usst.edu.cn
bSchool of Resources and Environmental Engineering, Shanghai Polytechnic University, Shanghai 201209, P. R. China. E-mail: lcwang@sspu.edu.cn
cDepartment of Orthopedics, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, Shanghai Jiaotong University, Shanghai 200080, China. E-mail: chenjianpumch@163.com
dDepartment of Materials Science and Engineering, National University of Singapore, Singapore
eCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, Zhejiang 310014, P. R. China
First published on 6th November 2024
Abstract
The development of highly active and cost-effective catalytic electrodes that function effectively across a wide range of pH values is one of the challenges to achieving efficient and stable hydrogen production via electrolytic water. This work constructs a self-supported catalytic electrode (Pt-NiB@NF) by growing boron-based catalytic materials in situ on nickel foam (NF) through mild electroless plating and then rapidly “decorating” trace amounts of platinum (Pt) on the precursor surface via electrodeposition. Decorating with trace amounts of Pt (0.58 wt%) achieves a 3.5-fold enhancement in the performance of NiB@NF. Pt-NiB@NF exhibits low hydrogen evolution reaction (HER) overpotentials of 70 mV and 12 mV at a current density of 100 mA cm−2 in neutral high-salt media and alkaline environments, respectively. Meanwhile, Pt-NiB@NF demonstrates long-term stability at industrial-scale current densities, maintaining for 120 hours at 100 mA cm−2 in neutral high-salt media and for 1200 hours at 500 mA cm−2 in alkaline electrolyte. The strategy of mild electroless plating and rapid electroplating realizes large-area electrode preparation for assembling a proton exchange membrane electrolyzer, more promising for industry-grade hydrogen production via water splitting. This work provides an optimized solution for the commercialization and large-scale production of high-performance Pt-based electrodes through a simple preparation strategy.
Introduction
Hydrogen (H2) has the potential to replace traditional energy sources due to its high energy density, renewability, and clean combustion byproducts.1,2 Water electrolysis can use electricity generated from intermittent energy sources such as solar or wind energy to achieve green hydrogen production.3,4 The hydrogen evolution reaction (HER) is an important half-reaction in water splitting by electrochemically reducing H+/H2O to H2, and it can occur in acidic, alkaline, and neutral media.5,6 However, the reaction is more challenging under alkaline and neutral conditions due to the additional water dissociation steps required, leading to slower reaction kinetics.7,8 Especially, current electrocatalysts rarely exhibit high performance in neutral media.9,10 It may provide a key technology for direct seawater or high-salinity water splitting if efficient neutral water splitting can be achieved.11 Therefore, designing catalytic electrodes with high catalytic activity, stability, and low cost in neutral or alkaline media holds great promise and challenges.Transition metal borides (TMBs) are potential materials for HER catalytic electrodes because of their adjustable structure, high selectivity, good conductivity, excellent corrosion resistance and low cost.12 For example, Wang et al. used pulsed laser deposition-assisted synthesis of MoB–Ni3B for hydrogen production by alkaline (1.0 M KOH) and acidic (0.5 M H2SO4) water splitting to reach a current density of 10 mA cm−2 with overpotentials of 35 mV and 57 mV, respectively.13 Although boron-based materials showed promising potential in promoting water splitting, the problems of complex synthesis processes, high overpotentials and poor long-term stability faced under industrial-scale conditions need to be further addressed.14 Studies showed that surface or interfacial engineering can alter the catalytic electrode active sites and modulate the adsorption behaviour of reaction intermediates to achieve enhanced catalytic activity.15 In particular, designing metal–carrier interactions can effectively modulate the electronic structure and enhance the kinetics of the HER.16,17 Lau et al. constructed built-in electric field (BEF) metal-supported interfacial electrocatalysts Pt@CoOx, revealing that a strong BEF induced by the work function (ΔΦ) optimizes the hydrogen adsorption free energy (ΔGH*) and hydroxide adsorption free energy (ΔGOH*), thereby promoting neutral HER kinetics.18 Qiu and Cheng et al. fabricated a RuIr@BCN catalytic electrode by loading iridium (Ir) and ruthenium (Ru) nanoalloys onto boron/nitrogen co-doped graphite nanotubes (BCNs) through pyrolysis, which exhibited excellent alkaline HER activity with an overpotential of 23.6 mV at a current density of 10 mA cm−2.19 Platinum group metals (PGMs) such as platinum (Pt), iridium (Ir), and ruthenium (Ru) exhibit excellent catalytic performance.20–22 Constructing metal–carrier heterostructures of PGM-TMB opens new possibilities to enhance the catalytic activity and industrial-grade stability of the HER. However, the scarcity and high cost of PGMs have hindered their large-scale application.23 Meanwhile, achieving the real utilization of catalytic electrodes remains a challenge with the core issue being the design of composite advantage electrodes that combine “High stability, Durability, and Cost-effectiveness”. Therefore, to ensure the catalytic electrode's ultra-high activity and stability, efforts should be made to maximize the utilization efficiency of platinum group metals to reduce the content of platinum group metals in the catalytic electrode.
Furthermore, catalytic electrodes that can efficiently catalyze water splitting in a wide range of pH media are more promising for industrialization and commercialization. In particular, the efficient and stable neutral salt water splitting is beneficial for: (1) directly splitting seawater and high-salt wastewater, or (2) coupling HER withthe chlor-alkali process, which facilitates green hydrogen production in multiple scenarios and demonstrates greater commercial value.24,25 However, seawater and high-concentration salt water contain a large number of chloride ions (Cl−), and when the electrode materials are exposed to these electrolytes for a long time, the chloride ions will combine with the metal sites and lead to corrosion of the electrode materials.26 Moreover, with the increase of salt concentration in the electrolyte, the catalytic activity will be further reduced.27 Researchers devised methods such as site-specific adsorption as well as electrostatic shielding to reduce the effect of Cl− on water splitting.28 Shen et al. showed that the oxide (CeO2)−anion (B(OH)4−) dual-layer was effective in resisting the invasion of Cl− (0.5 M).29 The boron component on the surface of TMBs is easily oxidized to produce a borate anion layer when exposed to air, providing new possibilities for electrostatic repulsion of Cl− by TMB electrode materials.30 The challenge of directly splitting highly concentrated salt water with complex and corrosive compositions for hydrogen production lies in the preparation of cost-effective, highly active, and durable electrode materials through easy and controllable preparation strategies.
Based on the above considerations, this work used a mild electroless plating method to deposit a nickel–boron (Ni–B) catalytic material in situ on nickel foam (NF), and then quickly “decorated” it with trace amounts of Pt through electroplating, to construct efficient and stable Pt-NiB@NF self-supported catalytic electrodes. This strategy had the following advantages: (1) the nickel foam (NF) as the substrate provided a high surface area and good electron transfer ability for the catalytic electrode; (2) decorating Ni–B with Pt adjusted the electronic structure, providing excellent corrosion resistance and stability; (3) the mild electroless plating coupled with the electroplating process provided favorable conditions for preparing industrial-grade and practical electrodes. The Pt-NiB@NF catalyst achieved a HER current density of 10 mA cm−2 with only 12 mV overpotential in an alkaline environment (1.0 M KOH). Moreover, it could be durable for over 1200 hours at a high current density of 500 mA cm−2 without significant performance degradation. The catalyst reached a HER current density of 10 mA cm−2 with only 70 mV overpotential in a neutral high-salt environment (1.0 M NaCl). It also demonstrated stable operation for over 120 hours at a high current density of 100 mA cm−2. Furthermore, the large area Pt-NiB@NF electrode was applied in a proton exchange membrane electrolyzer cell (PEMEC), where it could achieve efficient hydrogen production (approximately 130 mL min−1) in both neutral and alkaline environments. This work provides a reference for designing efficient, stable, and economical HER catalytic electrodes and offers strategies for the commercialization and scaling up of high-performance Pt-based catalytic electrodes.
Results and discussion
Characterization of catalytic electrodes
To construct an efficient, economical, and stable self-supported catalytic electrode, a Pt-NiB@NF electrode was prepared using a mild electroless plating and electroplating method in this work (Scheme 1). Initially, NF (1.0 cm × 0.5 cm) was immersed in a 2.0 M HCl solution for 30 minutes for activation. The activated NF was then immersed in the Ni–B electroless plating solution and reacted for different durations (1 h, 3 h, 5 h, and 7 h) at 298 K. Scanning electron microscopy (SEM) analysis results revealed distinct surface morphologies on the electrode as the electroless plating duration increased. After 1 h of electroless plating, the loading amount was low (2.2 mg cm−2) with a few nanoparticles attached to the NF. Following 3 h of electroless plating, the loading amount increased to 6.2 mg cm−2 and the electrode surface formed a regular, dense particle structure, with the holes and defects on the substrate material almost disappearing. With further increase in the electroless plating time, the loading amounts at 5 h and 7 h were 8.9 mg cm−2 and 11.3 mg cm−2, respectively, leading to the collapse of the catalytic material and accumulation of nano-materials (Fig. S1 and S2†). The catalytic activity of the NiB@NF-x h electrodes was investigated by testing the linear sweep voltammetry (LSV) polarization curves and electrochemical impedance spectroscopy (EIS) plots in 1.0 M KOH. The results showed that the HER performance of NiB@NF-3 h (η10 = 24 mV) was the highest, with the smallest electrochemical impedance (Rct = 0.51 Ω), outperforming those of NiB@NF-1 h (η10 = 75 mV and Rct = 1.26 Ω), NiB@NF-5 h (η10 = 55 mV and Rct = 0.6 Ω), and NiB@NF-7 h (η10 = 40 mV and Rct = 0.64 Ω) (Fig. S3 and S4†). To further enhance the catalytic activity, trace amounts of Pt were deposited on NiB@NF-3 h by electrodeposition. The NiB@NF surface was loaded with a small amount of Pt with 0.21 mg cm−2 after 1 min of plating. The loading amount reached 0.67 mg cm−2 when the electroplating time was 5 min and the Pt component was distributed in an orderly manner on NiB@NF, with protruding spherical structures effectively providing more specific surface area and abundant active sites. Upon electroplating for 10 min, a large amount of Pt accumulated on NiB@NF, leading to some fractures, and the loading amount was 1.07 mg cm−2 (Fig. S5 and S6†). The electrochemical test results indicated that the HER performance of Pt-NiB@NF-5 min (η10 = 12 mV and Rct = 0.27 Ω) was better than that of Pt-NiB@NF-10 min (η10 = 14 mV and Rct = 0.45 Ω) and Pt-NiB@NF-1 min (η10 = 16 mV and Rct = 0.73 Ω) (Fig. S7 and S8†).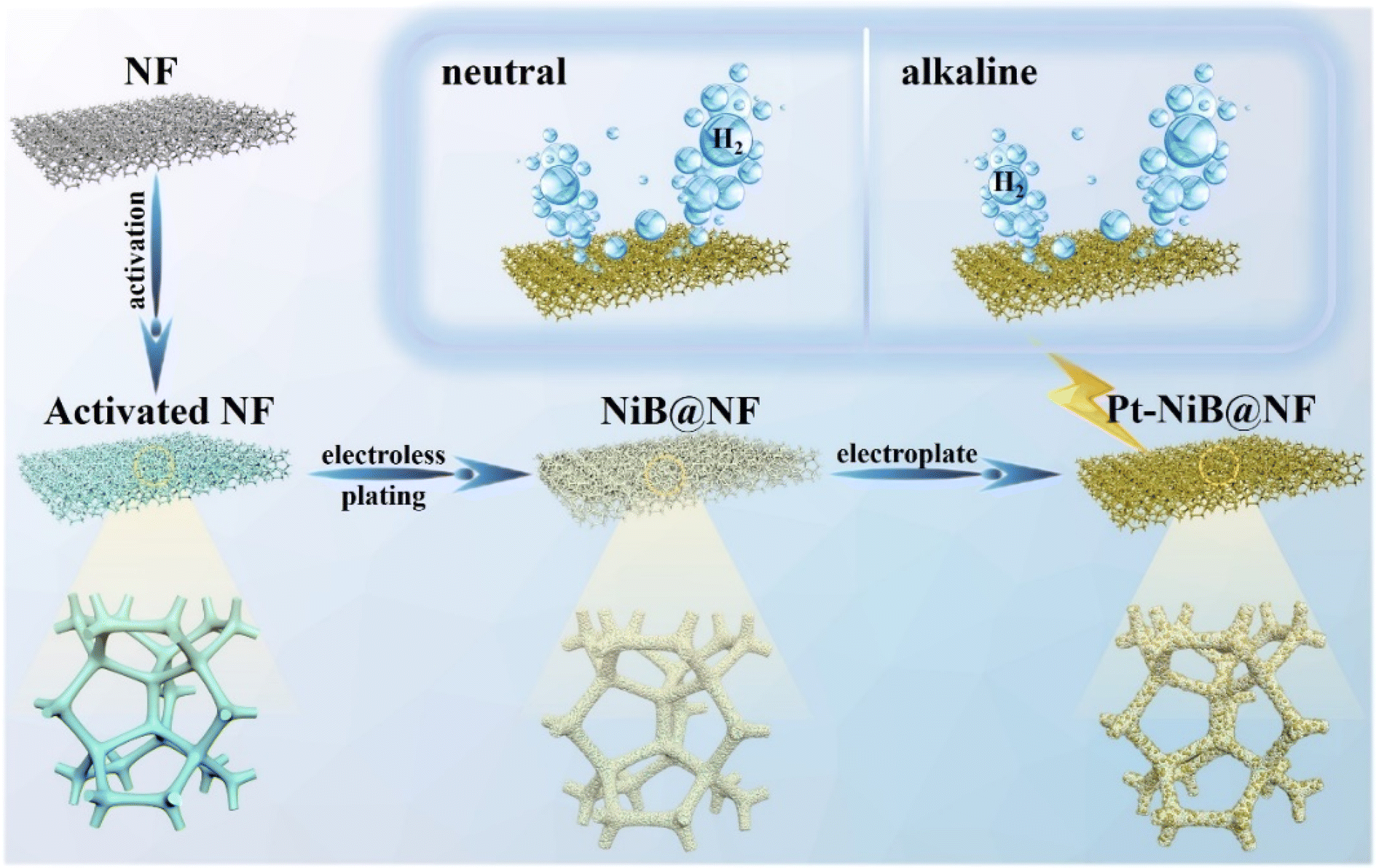 | ||
| Scheme 1 The optimal synthesis route of the Pt-NiB@NF electrode for the HER in both neutral and alkaline systems. | ||
The NiB@NF and Pt-NiB@NF electrodes were characterized using X-ray diffraction (XRD) (Fig. 1a). Three distinct diffraction peaks were observed at 44.7°, 52.1°, and 76.8° for both samples, corresponding to the (111), (200), and (220) crystal planes of face-centered cubic nickel foam (NF) metal (PDF#03-1051).26,31 A diffraction peak at 27.8° was observed, attributed to the (310) crystal plane of B2O3 (PDF#29-0236).32 Due to the low Pt content and highly dispersed Pt atoms, the Pt material might exist in an amorphous or weakly crystalline state, and hence no diffraction peaks corresponding to Pt compounds were observed in the XRD pattern.33 The SEM image of NiB@NF showed that dense polyhedral particles grew uniformly on the NF substrate (Fig. 1b). The porous structure provided abundant sites for Pt attachment. Compared to NiB@NF, the surface of Pt-NiB@NF was uniformly loaded with a large number of spherical particles, providing more active sites for the catalytic process (Fig. 1c). Furthermore, SEM elemental mapping confirmed the uniform distribution of Pt, Ni, B, and O elements on the surface of Pt-NiB@NF (Fig. 1d). Inductively coupled plasma optical emission spectroscopy (ICP-OES) confirmed that the mass fractions of Ni, B, and Pt in Pt-NiB@NF were 95.31%, 1.66%, and 0.58%, while the mass fractions of Ni and B in NiB@NF were 97.80% and 1.84%, respectively (Table S1†). To further study the crystalline state of the synthesized material, the surface catalytic layer was stripped from Pt-NiB@NF through ultrasonic treatment for transmission electron microscopy (TEM) analysis. As shown in Fig. 1e, particles with a diameter of 5–10 nm were uniformly attached to the surface of nanospheres with a diameter of 200–300 nm. High-resolution transmission electron microscopy (HRTEM) revealed that the surface of Pt–NiB contains many amorphous/weakly crystalline regions (Fig. 1f). In certain areas, lattice fringes with a spacing of 0.202 nm, corresponding to the (111) plane of metallic Ni, were observed (Fig. 1f1 and 2). Meanwhile, lattice fringes with a spacing of 0.226 nm were also observed dispersed at the edges of these amorphous/weakly crystalline regions, which can be attributed to the (111) plane of Pt (Fig. 1f3 and 4). It indicated that the surface-deposited Pt species are loaded onto the NiB substrate in the form of clusters or nanoparticles. Similar results were obtained after multiple repeated experiments (Fig. S9†). Additionally, the selected area electron diffraction (SAED) pattern further confirmed the crystal structure of Pt-NiB@NF, with diffraction rings matching the (111) plane of Pt and (200) and (220) planes of Ni (Fig. S10†), consistent with the results of XRD. These results indicate that the NiB substrate mainly existed in an amorphous/weakly crystalline form. This was because electroless deposition was a self-growth process, and the low temperature prevented the formation of crystalline nuclei, leading to a disordered amorphous/weakly crystalline structure, which was also reported in the literature.29,34 The electrodeposited Pt species were anchored on these amorphous/weakly crystalline NiB substrates in the form of clusters or nanoparticles.
 | ||
| Fig. 1 (a) XRD patterns of the Pt-NiB@NF and NiB@NF electrodes. SEM images of the (b) NiB@NF and (c) Pt-NiB@NF electrodes. (d) SEM image and EDS mapping of Pt-NiB@NF showing uniform distribution of Pt, Ni, O, and B. (e) TEM and (f) HRTEM images of Pt–NiB. (g) Image of the 5.0 cm × 5.0 cm Pt-NiB@NF electrode. (h and i) Dynamic droplet contact hysteresis on NiB@NF and Pt-NiB@NF electrodes. | ||
To further analyze the surface valence states of NiB@NF and Pt-NiB@NF electrodes, X-ray photoelectron spectroscopy (XPS) was conducted. The XPS survey spectrum of Pt-NiB@NF confirmed the presence of Ni, B, O, and Pt elements (Fig. S11†). The Pt 4f spectrum of Pt-NiB@NF exhibited two main peaks at 71.9 eV and 75.1 eV, corresponding to Pt 4f7/2 and Pt 4f5/2, respectively (Fig. S12a†).35 A peak appearing at 68.49 eV overlapped partially with the binding energy peak of Pt 4f, and this peak corresponded to Ni 3p.36 In the Ni 2p spectrum of Pt-NiB@NF, the peaks located at 853.2, 855.8/873.3, 857.8/876.1, and 861.9/879.8 eV can be attributed to Ni–B bonds (Niδ), divalent Ni species (Ni2+), trivalent Ni species (Ni3+), and satellite peaks (Fig. S12b†).37,38 Compared to the Ni 2p spectrum of NiB@NF, the peaks of oxidized Ni species undergo a positive shift, while the peak of Niδ shifted positively by 0.1 eV. It indicated that the introduction of Pt results in a reduction of electron density around the Ni atoms. The peak at 192.2 eV in the B 1s spectrum was attributed to the B–O bonding, indicating that the surface boron on the electrode had been oxidized due to exposure to air (Fig. S12c†).39,40 Further observation of the O 1s spectrum of Pt–NiB shows that the peaks of surface-adsorbed water (OH2O), hydroxide (OOH), and lattice oxygen (OLattice) are located at 532.7, 531.6, and 530.9 eV, respectively (Fig. S12d†).41 Compared to the peak ratio in the O 1s spectrum of NiB@NF (OLattice/OOH = 0.6/1), that in the figure of Pt-NiB@NF increased to 0.74/1, further indicating that the introduction of Pt leads to the formation of more high-valence oxides on the surface of the catalytic material. These characterization results demonstrated the successful deposition of Pt on NiB@NF and the modulation of the intrinsic electronic structure.
The surface wettability of the NiB@NF and Pt-NiB@NF electrodes was studied by analysing the dynamic contact angle hysteresis through liquid droplet contact imaging. Fig. 1h and i show that both NiB@NF and Pt-NiB@NF exhibited good hydrophilicity. Specifically, Pt-NiB@NF achieved complete droplet penetration in just 118 ms, which was faster than NiB@NF (237 ms). Due to the large number of gas bubbles generated during the HER process at high current densities, the catalyst surface would be covered, increasing the charge and proton transfer resistance at the interface, which inhibited HER activity. Catalysts with superhydrophilicity contributed to the kinetics of the HER.42 On the other hand, an uneven, porous surface utilized the capillary effect to transport liquid electrolyte to the surface, enhancing hydrophilicity and reducing the adhesion of gas bubbles.43
HER electrocatalytic performance
Enabling catalytic electrodes to catalyze hydrogen production in neutral or wide pH systems, the raw materials for water electrolysis were not only expanded but also the application scenarios were broadened. With the exceptional structural morphology of the catalytic electrodes and the incorporation of trace amounts of the precious metal Pt, the electrodes exhibited high hydrogen evolution reaction (HER) activity in neutral high-salt environments (1.0 M NaCl) and alkaline environments (1.0 M KOH). HER testing was conducted on the catalytic electrode with a three-electrode system at 298 K at a scan rate of 5 mV s−1. The HER activity of the NiB@NF electrode was significantly enhanced after a small amount of Pt was loaded. The LSV curves without iR-compensation showed that the overpotential of Pt-NiB@NF was only 70 mV at a current density of 10 mA cm−2, outperforming bare NF (η10 = 530 mV) and NiB@NF (η10 = 129 mV), and significantly lower than that of a commercial Pt electrode (η10 = 251 mV) (Fig. 2a). Compared with recently reported HER catalytic materials as shown in Table S2†, Pt-NiB@NF exhibited significant advantages. The Tafel slope was an important parameter to study the reaction pathway and kinetics of electrocatalysts. The Tafel slope of Pt-NiB@NF was 72.5 mV dec−1, much lower than that of the commercial Pt foil electrode (124.7 mV dec−1), bare NF (183.8 mV dec−1), and NiB@NF (103.5 mV dec−1) (Fig. 2b), indicating faster electrochemical kinetics through the Volmer–Heyrovsky mechanism. To study the charge transfer resistance of catalytic electrodes during the hydrogen evolution process, electrochemical impedance analysis was conducted, as shown in Fig. 2c. Rs represents the solution resistance (Ω), CPE represents a capacitive element with a constant phase angle, and Rct represents the diffusion resistance of electrolyte ions within the electrode pores (Ω). The fitting results revealed the resistance (Rct) values of the catalytic electrodes in the following order: bare NF (25.28 Ω) > Pt foil (22.87 Ω) > NiB@NF (1.21 Ω) > Pt-NiB@NF (0.86 Ω). The low resistance of Pt-NiB@NF indicated that it exhibited fast electron transfer kinetics at the interface between the electrolyte and the electrocatalyst. | ||
| Fig. 2 HER performance in the 1.0 M NaCl solution. (a) LSV curves of bare NF, commercial Pt foil, NiB@NF, and Pt-NiB@NF with a sweep rate of 5 mV s−1 without infrared correction. (b) Tafel slopes and (c) EIS plots of bare NF, Pt foil, NiB@NF, and Pt-NiB@NF. (d) The calculated Cdl values, (e) j/Cdl curves, (f) mass-normalized LSV curves, and (g) TOF values of NiB@NF and Pt-NiB@NF. (h) Long-term stability of Pt-NiB@NF at 100 mA cm−2 in the 1.0 M NaCl solution. | ||
The electrochemically active surface area (ECSA) was an important indicator for evaluating the catalytic performance of electrodes. In this work, cyclic voltammetry was conducted in the non-faradaic region (0.09–0.15 V vs. RHE) at different scans to obtain the double-layer capacitance (Cdl) for evaluating the ECSA (Fig. S13†). The Cdl value of the Pt-NiB@NF electrode (756.0 mF cm−2) was significantly higher than that of the NiB@NF electrode (708.6 mF cm−2) (Fig. 2d), indicating that the trace loading of Pt played a positive role in enhancing the catalytic performance of the electrode. To eliminate the influence of the electrochemically active surface area on catalytic activity, the polarization curves were normalized by Cdl. As shown in Fig. 2e, Pt-NiB@NF exhibited a higher j/Cdl value compared to NiB@NF at the same overpotential, indicating that Pt-NiB@NF possessed higher catalytic activity. To eliminate the impact of the loading amount on catalytic activity, the normalization of polarization curves by the loading amount was conducted (Fig. 2f), confirming the high HER catalytic activity of Pt-NiB@NF. By calculating the turnover frequency (TOF) of each surface site of the catalyst for H2 conversion, the intrinsic activity of all catalysts was explored. As shown in Fig. 2g, the TOF of Pt-NiB@NF was 3.46 s−1, which was 2.4 times higher than that of NiB@NF (1.45 s−1) at an overpotential of 350 mV, indicating that Pt-NiB@NF had higher intrinsic activity. The results demonstrated that the doping of Pt provided more active sites for NiB@NF, thereby enhancing the HER catalytic activity.
The long-term stability under corrosive conditions was an important indicator for industrial applications. The hydrolysis system with Pt-NiB@NF as both the cathode and the anode was continuously stabilized in a neutral high-salt medium (1.0 M NaCl) at a high current density of 100 mA cm−2 for more than 120 h with only a 9.17% increase in potential (Fig. 2h). The LSV curve for Pt-NiB@NF showed minimal changes after 120 hours of catalytic electrolysis (Fig. 2h, inset). The high catalytic activity exhibited in a neutral high-salt medium and the long-term stability at high current densities hold promise for industrial applications of Pt-NiB@NF in areas such as seawater electrolysis, high-salinity lake water electrolysis, and co-electrolysis of high-salt wastewater for hydrogen production.
The catalytic electrode was further tested for catalysis in 1.0 M KOH to expand its applicability. Fig. 3a shows the LSV curve of the electrode in 1.0 M KOH. Pt-NiB@NF still exhibited the best HER performance (η10 = 12 mV), far surpassing the commercial Pt foil (η10 = 57 mV), NiB@NF (η10 = 24 mV), and bare NF (η10 = 237 mV), respectively. Compared to recently reported HER catalytic materials, Pt-NiB@NF demonstrated significant advantages under alkaline conditions (Table S3†). The Tafel slope of Pt-NiB@NF was 48.6 mV dec−1, indicating that it corresponded to the “Volmer–Heyrovsky” catalytic process in alkaline HER.44 The Tafel slope of Pt-NiB@NF was significantly lower than that of the commercial Pt foil electrode (73.7 mV dec−1), NiB@NF (90.7 mV dec−1), and bare NF (136.4 mV dec−1) (Fig. 3b). To further confirm the rapid HER kinetics of Pt-NiB@NF, the charge transfer rate of the electrode was explored through Nyquist plots (Fig. 3c). The charge transfer resistance (Rct) of Pt-NiB@NF was 0.27 Ω, significantly lower than that of Pt foil (6.43 Ω) and NiB@NF (0.51 Ω), indicating that Pt-NiB@NF had a faster charge transfer rate.
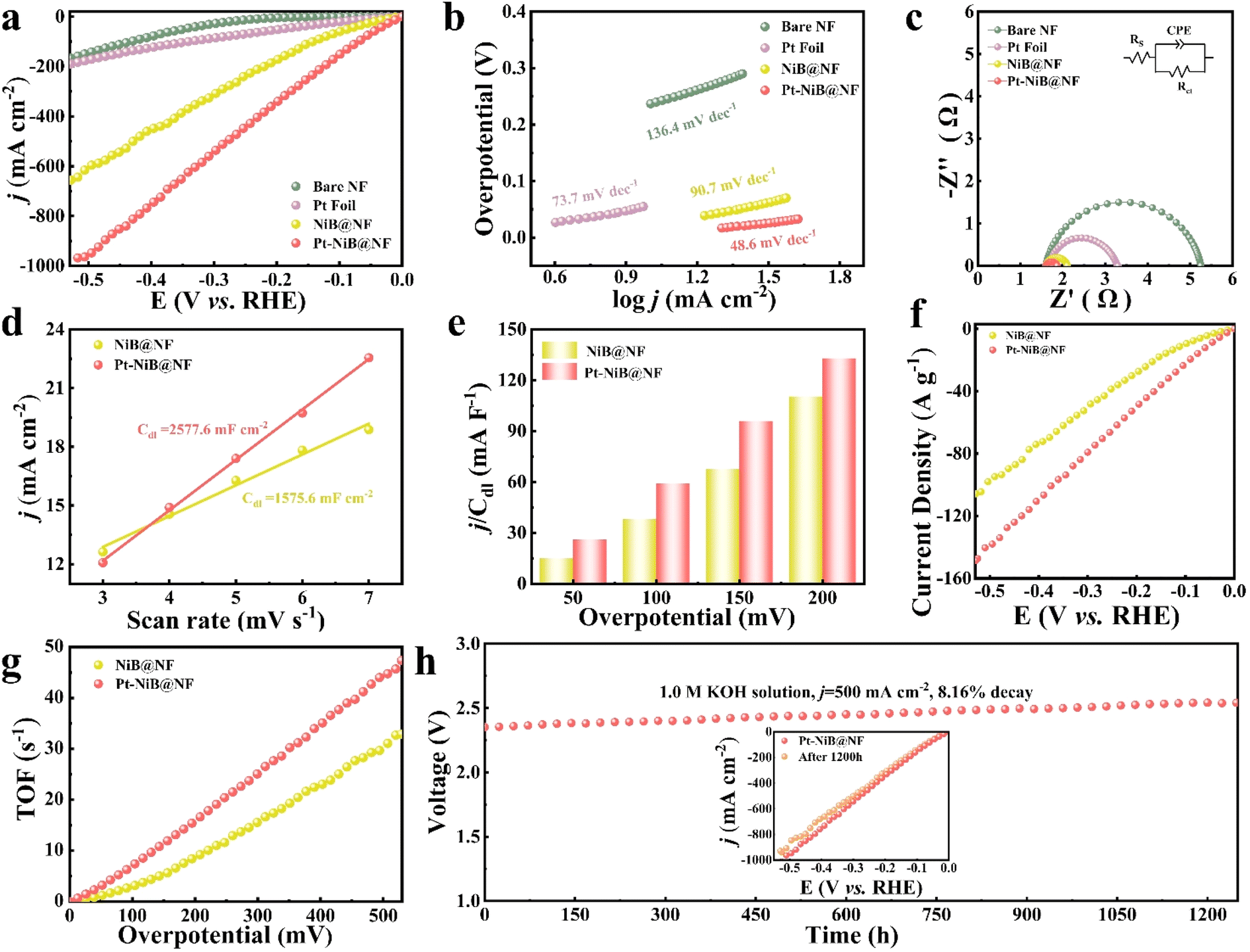 | ||
| Fig. 3 HER performance in the 1.0 M KOH solution. (a) LSV curves of bare NF, commercial Pt foil, NiB@NF, and Pt-NiB@NF at a sweep rate of 5 mV s−1 without infrared correction. (b) Tafel slopes and (c) EIS plots of bare NF, Pt foil, NiB@NF, and Pt-NiB@NF. (d) The calculated Cdl values, (e) j/Cdl, (f) mass-normalized LSV curves, and (g) TOF values of NiB@NF and Pt-NiB@NF. (h) Long-term stability of Pt-NiB@NF at 500 mA cm−2 in the 1.0 M KOH solution. | ||
The Cdl value of Pt-NiB@NF was 2577.6 mF cm−2, superior to that of NiB@NF (1575.6 mF cm−2) (Fig. 3d), indicating that the loading of Pt enhanced the electrochemically active surface area (ECSA) of the electrode (Fig. S14†). The j/Cdl value of Pt-NiB@NF consistently remained higher than that of NiB@NF (Fig. 3e), suggesting that after excluding the influence of the electrochemically active surface area on catalytic activity, Pt-NiB@NF still exhibited higher activity. Fig. 3f reflects the catalytic performance enhancement per unit loading amount after decorating NiB@NF with Pt, demonstrating that Pt-NiB@NF exhibited higher intrinsic catalytic activity towards the HER. The TOF value of Pt-NiB@NF consistently remained higher than that of NiB@NF (Fig. 3g), showing that the doping of Pt significantly improved the electrode's intrinsic activity. More importantly, Pt-NiB@NF exhibited extremely high stability, enduring electrolysis for over 1200 hours at an industrial-level current density of 500 mA cm−2, with only an 8.16% increase in the potential (Fig. 3h), showing virtually unchanged performance (Fig. 3h, inset).
Mechanism analysis of the hydrogen evolution process
Based on the excellent performance, stability, and cost-effectiveness of the Pt-NiB@NF catalytic electrode, detailed characterization of the composition, structure, and changes involved in the catalytic process was conducted. The XRD patterns of Pt-NiB@NF samples after HER stability testing in a neutral high-salt medium and alkaline medium (referred to as neutral-HER and alkaline-HER) are shown in Fig. S15.† The peaks at 44.7°, 52.1°, and 76.8° corresponded to the (111), (200), and (220) crystal planes of nickel, respectively. The peak at 27.8° corresponded to the (310) crystal plane of B2O3. Compared to the initial sample, no new peaks were observed, indicating that after stability testing through the hydrogen evolution reaction (HER) in 1.0 M NaCl and 1.0 M KOH, the Pt-NiB@NF catalytic electrode did not undergo phase transformation. The result suggested that Pt-NiB@NF demonstrated good structural stability. Moreover, the SEM results after long-term stability testing revealed that the Pt-NiB@NF electrode retained its three-dimensional reticular structure, with the surface covered by dense spherical particles, and no obvious morphological changes were observed (Fig. S16 and S17†).The FT-IR spectrum elucidated the chemical structure of Pt-NiB@NF after HER stability testing (Fig. S18†). There were two peaks at 1632/3451 cm−1 corresponding to the bending and stretching vibrations of adsorbed H2O molecules, formed by absorbed water or metal hydroxides.45 The peak at 1092 cm−1 was attributed to the stretching vibration of B–O bonds. Compared to the original Pt-NiB@NF, the M–O stretching vibration at 672 cm−1 was not prominently observed in the neutral-post and alkaline-post samples. Instead, a stretching vibration peak belonging to Pt–O–Ni appeared at 745 cm−1.46 This indicated that during the HER catalytic reaction, the catalytic material might have undergone surface restructuring, resulting in the formation of a hydroxide coverage layer.
The surface composition and chemical states of Pt-NiB@NF after HER stability testing were examined by XPS. As shown in Fig. S19,† in the XPS spectra of neutral-post and alkaline-post samples, signals for Pt, Ni, B, and O elements were observed, consistent with the initial sample. In the Pt 4f spectra of the neutral/alkaline-post samples (Fig. 4a), peaks for Pt 4f7/2 and Pt 4f5/2 were observed at approximately 71.3/71.3 eV and 74.9/74.6 eV, respectively. Compared to the initial sample, the peaks of Pt 4f7/2 and Pt 4f5/2 in the neutral/alkaline-post figures shifted to lower binding energies. It may be attributed to the surface reconfiguration of the NiB support during the HER process, which interacts with the Pt metal layer resulting in electron transfer.47 Further observation of the Ni 2p spectrum in neutral-post sample reveals that Ni3+ 2p3/2, Ni2+ 2p3/2, Ni3+ 2p1/2, and Ni2+ 2p1/2 shift towards lower binding energies by 1.1, 0.5, 0.5, and 0.4 eV, respectively (Fig. 4b). Meanwhile, the peak area ratio of Ni3+/Ni2+ changes from 0.9/1.5 to 0.8/1.4. In the Ni 2p spectrum of the alkaline-HER sample, the peaks of Ni3+ 2p3/2, Ni2+ 2p3/2, Ni3+ 2p1/2, and Ni2+ 2p1/2 are located at 874.7, 872.7, 857.2, and 855.3 eV, respectively. Compared to the initial sample, the peaks of the high-valence Ni species also show a negative shift, and the peak area ratio of Ni3+/Ni2+ decreases to 0.7/1.4. This is due to the reduction of high-valence nickel species on the surface of Pt-NiB@NF to a lower valence state, resulting in the formation of nickel-based (hydroxy) oxides.48 This suggested the possible surface reconstruction of Ni(OH)2 on the samples, which might be the true active species in the HER process.49 In the B 1s spectra (Fig. 4c), the peak intensities of B–O at 192.17 eV and 192.13 eV in the neutral/alkaline-post samples, respectively, showed a sharp decrease after the HER.50 This may be due to the leaching of borate anionic elements from the surface into the electrolyte.29 The significant decrease in the intensity of the B–O peak indicated that the boron-modified NF with abundant defects could confine Ni in the Ni2+ states. This provided ample water adsorption/dissociation sites and contributed to modulating the electronic structures of Pt and Ni.51 The O 1s spectra revealed characteristic peaks of surface-adsorbed water (OH2O), hydroxide (OOH), and lattice oxygen (OLattice) at 532.3/532.1 eV, 531.0/530.8 eV, and 528.7/529.3 eV in the neutral/alkaline-post samples (Fig. 4d). Compared to the initial sample, the peak area ratios (OLattice/OOH) of hydroxide (OOH) and lattice oxygen (OLattice) for neutral-post and alkaline-post samples decreased from 0.74/1 to 0.13/1 and 0.3/1, respectively. The significant decrease in the OLattice peak ratio and the negative shift in binding energy may be related to the partial substitution of anions in metal oxides,52 possibly due to the generation of Ni(OH)2 from the reduction of high-valent nickel species during the HER process. Additionally, the negative shift of the OOH peaks may be due to changes in surface OH− adsorption sites after the catalyst material undergoes surface reconstruction, leading to a decrease in binding energy.
 | ||
| Fig. 4 XPS spectra of (a) Pt 4f, (b) Ni 2p, (c) B 1s and (d) O 1s for Pt-NiB@NF and post-HER Pt-NiB@NF in 1.0 M KOH and 1.0 M NaCl. (e) Operando Raman spectra of Pt-NiB@NF at various potentials. (f) Schematic illustration showing the HER mechanism of Pt-NiB@NF in 1.0 M NaCl and 1.0 M KOH. | ||
The in situ Raman spectroscopic characterization elucidated the surface reconstruction components of Pt-NiB@NF during the HER (Fig. 4e). The initial sample exhibited peaks at approximately 545 cm−1 and 816 cm−1, attributed to oxidized Ni and B species, respectively.53 Previous studies have shown that the anions on the electrode surface can electrostatically repel Cl− and prevent corrosion of the electrode material.26,28,29,54 Therefore, the negatively charged surface of these surface borate anions with better corrosion resistance in high-salt (1 M NaCl) electrolytes mainly originates from this electrostatic shielding effect. Additionally, upon applying a potential of −0.10 V vs. RHE in 1.0 M NaCl electrolyte, a novel, broadened Raman peak emerged near 470 cm−1, whose intensity increased with increasingly negative potentials, indicative of Ni(OH)2 species.55 Furthermore, upon imposition of a −1.0 V reduction potential, a band at 1627 cm−1 was discerned, corresponding to the H–O–H bending mode (δH–O–H) of interfacial water.56,57 These findings underscore the formation of Ni(OH)2 species on the catalyst surface during the HER process. The surface reconstruction of the NiB support leads to changes in the electronic structure. As the support for Pt, NiB acted as the “environment” for the surface Pt metal.58 Therefore, the metal–support interaction further induced electron transfer between the surface Pt metal and the support. This redistribution of charges influences the adsorption/desorption of intermediate species such as H* or OH*, thereby enhancing the HER kinetics. The specific catalytic mechanism of Pt-NiB@NF in neutral and alkaline solutions during the HER process is illustrated as Fig. 4f. During the HER, water molecules acquired an electron on the electrode surface and dissociated into OH− ions and Hads (hydrogen adsorption). The Hads reacted with water molecules and electrons to form OH− ions and hydrogen molecules (H2). The H2 accumulated on the cathode surface until it reached saturation, at which point it desorbed from the electrode surface and was released into the gas phase. Pt/Ni(OH)2/NiB served as the true active species during the HER process.
Based on the analysis above, the exceptional HER of the Pt-NiB@NF catalyst was primarily attributed to the following reasons: (i) using a three-dimensional reticular structure of nickel foam as the substrate, enabling the in situ growth of uniform and dense spherical structures, effectively providing a larger specific surface area and abundant active sites; (ii) the synergistic effect between Ni and Pt effectively modulated the material's electronic structure, optimized the adsorption energy of intermediates, exposed more active sites, and possessed higher intrinsic catalytic activity; (iii) the surface reconstruction-induced Pt/Ni(OH)2 not only synergistically promoted catalytic activity with Pt–NiB, prevented further oxidation, and enhanced electrode stability; (iv) the boron-modified NF with abundant defects was utilized to disrupt the H–OH bonds, facilitating the HER by promoting the catalytic activity of Ni and Pt.
Expanded applications
The aim of this work is to achieve the industrial application of catalytic electrodes. By using the utilization efficiency of Pt as a measure, the economic benefits of the Pt-NiB@NF catalytic electrode were analyzed. Fig. 5a compares the Pt weight percentage (0.58 wt%) of Pt-NiB@NF with that of recent platinum-based electrodes and their HER performance (η10) in a neutral solution. The results showed that the Pt weight percentage of Pt-NiB@NF was significantly lower than that of most recently reported platinum-based electrodes, and its HER performance (η10 = 70 mV) exceeded that of most platinum-based electrodes (Tables S4 and S5†). The extremely high Pt utilization efficiency of Pt-NiB@NF implied significant economic benefits. With the rapid industrialization requirements, achieving the mild and rapid preparation of large-area electrode sheets with high activity and long-term stability had greater commercialization potential. This work achieved the preparation of large-area Pt-NiB@NF electrode sheets (5.0 cm × 5.0 cm) through mild electroless plating (298 K and 3 h) and rapid electroplating processes (298 K and 5 min). These electrodes could be assembled into a proton exchange membrane electrolyzer cell (PEMEC) for the HER via water electrolysis. Fig. 5b presents the device structure of the PEMEC, including: (I) the anode terminal plate connected to the inlet and outlet of water; (II) the cathode terminal plate connected to the hydrogen outlet; (III) the insulating pad to prevent the current from diffusing outward; (IV) the anode electrode plate connected to the positive pole of the power supply; (V) the cathode electrode plate connected to the negative pole of the power supply; (VI) the rubber gasket to prevent water vapor leakage; (VII) the anode catalyst layer (Pt-NiB@NF) catalyzing water electrolysis and transferring protons; (VIII) the cathode catalyst layer (Pt-NiB@NF) catalyzing water electrolysis and transferring electrons; (IX) the proton exchange membrane (PEM), conducting protons and separating hydrogen and oxygen. The specific device configuration and applications are shown in Fig. 5c and Video 1†. The hydrogen production rate of the PEMEC could reach 128 mL min−1 in a neutral environment (deionized water) (Video 2†). The hydrogen production rate of the PEMEC could reach 133 mL min−1 in 1.0 M KOH (Video 3†). The prepared PEMEC could achieve rapid and stable hydrogen production in a wide pH range system. Furthermore, the hydrogen production rate met the standard of hydrogen production devices on the market (100 mL min−1) (Video 4†). These results indicated that the high Pt utilization efficiency of Pt-NiB@NF achieved stable HER performance and its mild electroless plating and rapid electroplating processes highlighted industrial advantages, greatly expanding its potential prospects in hydrogen production through water electrolysis. It was expected to be applied in hydrogen therapy machines, new energy vehicles, spacecraft, hydrogen fuel cell machines, and other equipment (Fig. 5d).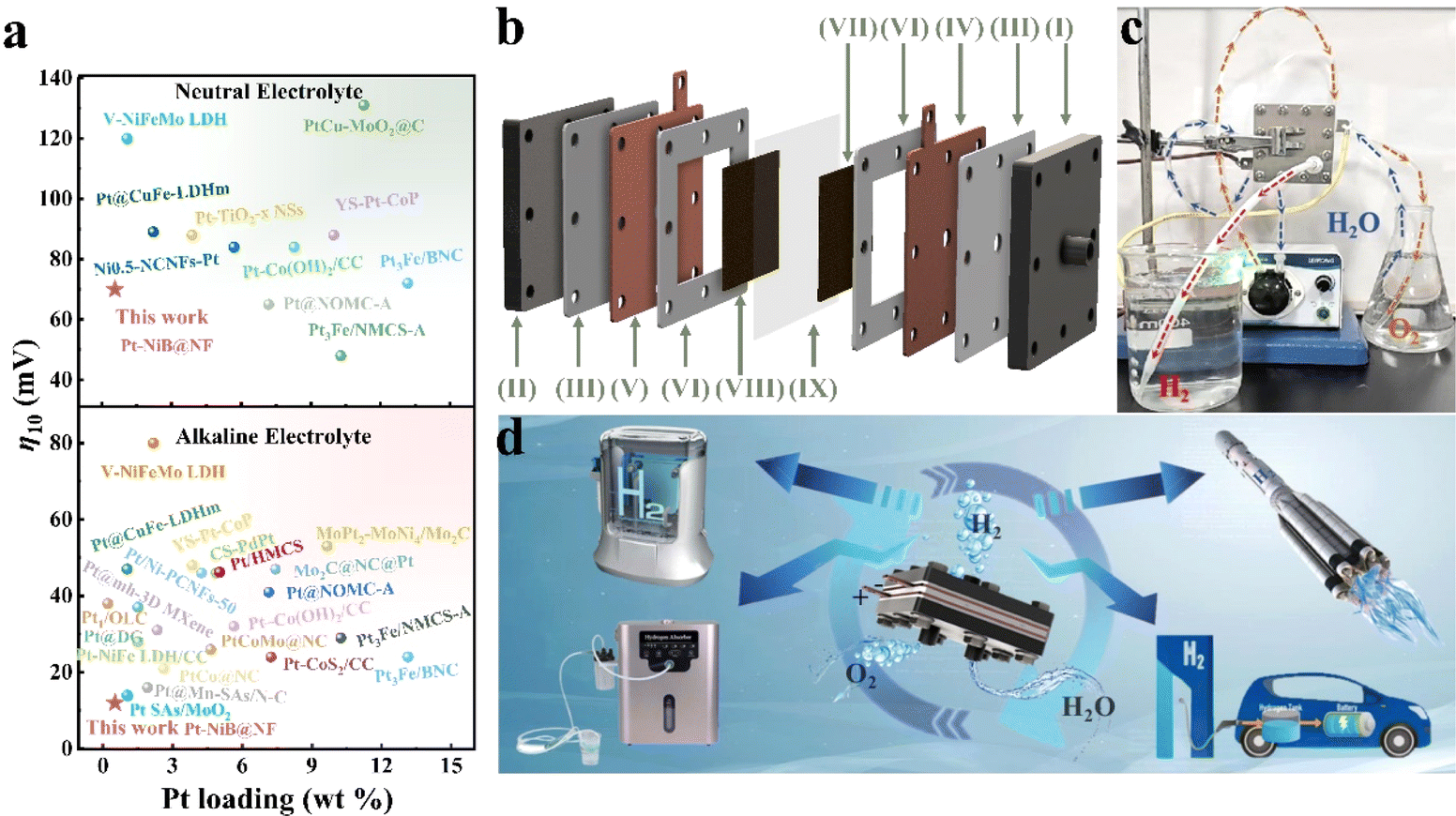 | ||
| Fig. 5 (a) Comparison of the HER performance and Pt loading of Pt-NiB@NF with other Pt-loading electrocatalysts. (b) Structure of the PEMEC: (I) anode end plate; (II) cathode end plate; (III) insulating mat; (IV) anode current collector; (V) cathode current collector; (VI) gasket; (VII) anode catalyst layer (Pt-NiB@NF); (VIII) cathode catalyst layer (Pt-NiB@NF); (IX) proton exchange membrane. (c) Photograph of the assembled PEMEC generating hydrogen. (d) Application fields of the PEMEC. | ||
Conclusions
This work successfully synthesized efficient and stable Pt-NiB@NF catalytic electrodes on nickel foam through mild electroless plating (298 K and 3 h) and rapid electroplating (298 K and 5 min). A trace amount of Pt (0.58 wt%) “decorated” the surface to achieve wide pH range applicability and high catalytic activity. In neutral high-salt environments (1.0 M NaCl) and alkaline media (1.0 M KOH), only 70 mV and 12 mV overpotentials are required to reach a current density of 10 mA cm−2, respectively. In addition, the catalyst operates stably for 120 hours at a high current density of 100 mA cm−2 in 1.0 M NaCl and for 1200 hours without attenuation at a high current density of 500 mA cm−2 in 1.0 M KOH. The mild electroless plating and rapid electroplating processes enabled the preparation of large-area (5.0 cm × 5.0 cm) electrodes, which could be used to assemble PEM electrolysis cells for hydrogen production through water electrolysis. This device achieved a commercial level of hydrogen release in a unit of time. This work provides an efficient and stable new solution for the design of industrial-scale water electrolysis hydrogen production catalytic electrodes.Experimental
Materials
Nickel sulfate hexahydrate (NiSO4·6H2O, 98.5%, AR), anhydrous sodium sulfate (Na2SO4, 99%, AR), disodium succinate (C4H4Na2O4, 99%, AR), dimethylamine borane (C2H10BN, 97%, Adamas), sulfamic acid (NH3SO3, 99%), ammonium chloride (NH4Cl, 99.5%, AR), potassium chloroplatinate (K2PtCl6, 98%), sodium chloride (NaCl, 99.9%, AR), potassium hydroxide (KOH, 90%, AR), ethanol (C2H5OH, 99.7%, AR), and hydrochloric acid (HCl, 36%, GR) were purchased from Shanghai Titan Technology Co. Ltd. Deionized water (18 MΩ cm) and nickel foam (NF, thickness: 0.3 mm, Shanghai Zhengyang Foam Co. Ltd, porosity 60%) were also used.Substrate activation
The nickel foam (NF) was used as the support substrate material and cut into 1.0 cm × 1.0 cm pieces. The NF was placed in an ethanol solution, sonicated for 10 minutes, and then removed and rinsed with deionized water to remove surface oils and impurities. After air drying at 298 K, the NF was immersed in a 2.0 M HCl solution for 30 minutes for activation. After rinsing with deionized water, the NF was air dried at 298 K.Preparation of the NiB@NF precursor
The experimental procedure involved dissolving 10.0 grams (g) of hexahydrate nickel sulfate, 1.0 g of anhydrous sodium sulfate, 1.0 g of disodium succinate, and 2.28 g of dimethylamine borane in 100 mL of deionized water, followed by thoroughly mixing the solution to obtain a nickel–boron (Ni–B) electroless plating solution. The activated NF was immersed in the Ni–B electroless plating bath at 298 K, reactions were conducted for 1, 3, 5, and 7 hours (h), and the resulting samples were named NiB@NF-1 h, NiB@NF-3 h, NiB@NF-5 h, and NiB@NF-7 h, respectively.Preparation of the Pt-NiB@NF electrode
0.2 g of potassium chloroplatinate, 1.0 g of aminosulfonic acid, 1.0 g of anhydrous sodium sulfate, and 0.25 g of ammonium chloride were dissolved in 100 mL of deionized water. 50 microliters of 2 M HCl solution were added and thoroughly mixed to prepare the Pt electroplating solution. NiB@NF served as the cathode and a carbon rod as the anode; both were immersed in the potassium chloroplatinate (K2PtCl6) electroplating solution. The current was set at 0.015 A (current density of 30 mA cm−2). After electroplating for 1, 5, and 10 min, the samples were removed, resulting in electrode samples named Pt-NiB@NF-1 min, Pt-NiB@NF-5 min, and Pt-NiB@NF-10 min.Data availability
The data supporting this article (Journal of Materials Chemistry A – TA-ART-08-2024-005770.R1) have been included as part of the ESI.†Author contributions
Xunwei Ma and Yifan Zhang contributed equally. Xunwei Ma and Yifan Zhang: methodology, investigation, formal analysis, data analysis, and writing – original draft. Liugang Wu, Zijun Huang, Jiyuan Yang, Chunguang Chen and Shengwei Deng: conceptualization, resources, methodology, review & editing, and revision. Lincai Wang and Jian Chen: conceptualization, resources, methodology, and review & editing. Weiju Hao: conceptualization, methodology, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the Natural Science Foundation of Shanghai (23ZR1443900) and the National Natural Science Foundation of China (Grant No. 22109098). The authors would like to thank Yuwei Zhou for the scanning electron microscope images and grazing-incident XRD analysis and Nannan Han for the XPS analysis from Shiyanjia Lab (https://www.shiyanjia.com/). The authors would like to thank Youxian Hu for the ICP-OES analysis from Ceshihui Lab (https://www.ceshihui.cn/).References
- Q. R. Liang, Y. N. Huang, Y. X. Guo, X. Zhang, X. M. Hu, H. Zeng, K. Liang, D. Y. Zhao, L. Jiang and B. Kong, Nat. Sustain., 2024, 7, 628–639 CrossRef
.
- Y. Y. Yan, J. Y. Du, C. Y. Li, J. Yang, Y. K. Xu, M. L. Wang, Y. P. Li, T. Wang, X. S. Li, X. M. Zhang, H. Zhou, X. Hong, Y. Wu and L. X. Kang, Energy Environ. Sci., 2024, 17, 6024–6033 RSC
- R. Fan, C. Liu, Z. Li, H. Huang, J. Feng, Z. Li and Z. Zou, Nat. Sustain., 2024, 7, 158–167 CrossRef
- D. Guan, B. Wang, J. Zhang, R. Shi, K. Jiao, L. Li, Y. Wang, B. Xie, Q. Zhang, J. Yu, Y. Zhu, Z. Shao and M. Ni, Energy Environ. Sci., 2023, 16, 4926–4943 RSC
- Y. Wang, H. Lv, L. Sun, F. Jia and B. Liu, Adv. Energy Mater., 2022, 12, 2201478 CrossRef CAS
- H. G. Han, J. W. Choi, M. Son and K. C. Kim, eScience, 2024, 4, 100204 CrossRef
- A. H. Shah, Z. Zhang, Z. Huang, S. Wang, G. Zhong, C. Wan, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Catal., 2022, 5, 923–933 CrossRef CAS
- Y. Li, W. Wang, M. Cheng, Y. Feng, X. Han, Q. Qian, Y. Zhu and G. Zhang, Adv. Mater., 2023, 35, e2206351 CrossRef
- K. Sun, X. Wu, Z. Zhuang, L. Liu, J. Fang, L. Zeng, J. Ma, S. Liu, J. Li, R. Dai, X. Tan, K. Yu, D. Liu, W. C. Cheong, A. Huang, Y. Liu, Y. Pan, H. Xiao and C. Chen, Nat. Commun., 2022, 13, 6260 CrossRef CAS PubMed
- P. Siva and K. Vasu, J. Mater. Chem. A, 2024, 12, 11101–11108 RSC
- Y. Xu, C. Wang, Y. Huang and J. Fu, Nano Energy, 2021, 80, 105545 CrossRef CAS
- Y. Ren, J. Wang, W. Wang, H. Wen, M. Chen, Y. Qiu, G. Li, Z. Yang and P. Wang, J. Mater. Chem. A, 2023, 11, 13282–13288 RSC
- L. Jiang, R. Wang, D. Zhang, G.-F. Wei and X. Wang, Chem. Eng. J., 2024, 485, 149920 CrossRef CAS
- N. S. Peighambardoust, E. Hatipoglu and U. Aydemir, ACS Sustain. Chem. Eng., 2022, 10, 15909–15925 CrossRef CAS
- C. Wang, J. Zhang, K. Miao, M. Long, S. Lai, S. Zhao and X. Kang, Adv. Mater., 2024, 36, e2400433 CrossRef
- L. Chen, L. Kang, S. Geng, L. Cheng, D. Cai, S. Song, H. Jia and Y. Wang, Adv. Funct. Mater., 2024, 34, 2403467 CrossRef CAS
- Y. Zeng, M. T. Zhao, H. L. Zeng, Q. Jiang, F. W. Ming, K. Xi, Z. C. Wang and H. F. Liang, eScience, 2023, 3, 100156 CrossRef
- L. Zhai, X. She, L. Zhuang, Y. Li, R. Ding, X. Guo, Y. Zhang, Y. Zhu, K. Xu, H. J. Fan and S. P. Lau, Angew. Chem., Int. Ed., 2022, 61, e202116057 CrossRef CAS PubMed
- T. Qiu, J. Cheng, Z. Liang, H. Tabassum, J. Shi, Y. Tang, W. Guo, L. Zheng, S. Gao, S. Xu and R. Zou, Appl. Catal., B, 2022, 316, 121626 CrossRef CAS
- T. Gao, X. Tang, X. Li, S. Wu, S. Yu, P. Li, D. Xiao and Z. Jin, ACS Catal., 2022, 13, 49–59 CrossRef
- Y. Li, S. Zhang, W. Xu, C. Jiang, L. Shao, S. Wang and J. Wang, J. Mater. Chem. A, 2023, 11, 4000–4006 RSC
- H. Zhang, W. Wang, Z. Dai, Y. Zhu, M. Cheng, B. Zhang, Y. Feng, Y. Zhang and G. Zhang, J. Mater. Chem. A, 2023, 11, 14674–14681 RSC
- J. Fan, X. Ma, J. Xia, L. Zhang, Q. Bi and W. Hao, J. Colloid Interface Sci., 2024, 657, 393–401 CrossRef CAS PubMed
- A. Salah, L. Zhang, H. Tan, F. Yu, Z. Lang, N. Al-Ansi and Y. Li, Adv. Energy Mater., 2022, 12, 2200332 CrossRef CAS
- H. You, D. Wu, D. Si, M. Cao, F. Sun, H. Zhang, H. Wang, T. F. Liu and R. Cao, J. Am. Chem. Soc., 2022, 144, 9254–9263 CrossRef CAS PubMed
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu, Z. Liu, Q. Wang, W. Ren, C. Sun, H. M. Cheng and B. Liu, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed
- X. Xu, Y. Lu, J. Shi, X. Hao, Z. Ma, K. Yang, T. Zhang, C. Li, D. Zhang, X. Huang and Y. He, Nat. Commun., 2023, 14, 7708 CrossRef CAS PubMed
- Z. Li, Y. Yao, S. Sun, J. Liang, S. Hong, H. Zhang, C. Yang, X. Zhang, Z. Cai, J. Li, Y. Ren, Y. Luo, D. Zheng, X. He, Q. Liu, Y. Wang, F. Gong, X. Sun and B. Tang, Angew. Chem., Int. Ed., 2024, 63, e202316522 CrossRef CAS
- H. X. Liu, X. Zhou, C. M. Ye, M. X. Ye and J. F. Shen, Appl. Catal., B, 2024, 343, 123560 CrossRef CAS
- F. Chengyu, W. Shuo, F. Jinli, Z. Yiran, G. Yanhui and H. Weiju, Chem. Eng. J., 2021, 430, 132881 Search PubMed
- J. Xia, L. Zhang, Y. Wang and W. Hao, Inorg. Chem. Front., 2024, 11, 1153–1166 RSC
- S. Ye, F. Luo, T. Xu, P. Zhang, H. Shi, S. Qin, J. Wu, C. He, X. Ouyang, Q. Zhang, J. Liu and X. Sun, Nano Energy, 2020, 68, 104301 CrossRef CAS
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C. W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 33, e2008599 CrossRef
- B. Lu, R. Dun, W. Wang, J. Huang, J. Wu, Z. Hua and J. Shi, Appl. Catal., B, 2023, 342, 123343 CrossRef
- T. Ma, H. Cao, S. Li, S. Cao, Z. Zhao, Z. Wu, R. Yan, C. Yang, Y. Wang, P. A. van Aken, L. Qiu, Y. G. Wang and C. Cheng, Adv. Mater., 2022, 34, e2206368 CrossRef PubMed
- H. Hu, Z. Zhang, Y. Zhang, T. Thomas, H. Du, K. Huang, J. P. Attfield and M. Yang, Energy Environ. Sci., 2023, 16, 4584–4592 RSC
- Y. Zhu, Y. Chen, Y. Feng, X. Meng, J. Xia and G. Zhang, Adv. Mater., 2024, 36, e2401694 CrossRef
- F. S. Chen, M. Sakthivel, Z. X. Jin, L. Y. Lin and K. C. Ho, J. Colloid Interface Sci., 2024, 678, 1022–1035 CrossRef PubMed
- D. C. Cha, T. I. Singh, A. Maibam, T. H. Kim, D. H. Nam, R. Babarao and S. Lee, Small, 2023, 19, e2301405 CrossRef PubMed
- W. J. Hao, D. X. Yao, Q. Y. Xu, R. R. Wang, C. L. Zhang, Y. H. Guo, R. M. Sun, M. X. Huang and Z. L. Chen, Appl. Catal., B, 2021, 292, 120188 CrossRef CAS
- Y. Zhuo, D. Liu, L. Qiao, S. Chen, J. Lu, W. F. Ip, H. Pan and Z. Wang, Adv. Energy Mater., 2023, 13, 2301921 CrossRef CAS
- R. Zhou, X. Han, Q. Chen, L. Peng, X. Qiu, P. Wang, C. Guo, J. Wang, Z. Wang and J. Hao, J. Mater. Chem. A, 2024, 12, 5719–5730 RSC
- Z. Zhu, Y. Lin, P. Fang, M. Wang, M. Zhu, X. Zhang, J. Liu, J. Hu and X. Xu, Adv. Mater., 2024, 36, e2307035 CrossRef PubMed
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef
- B. Zhang, Z. Dai, Y. Chen, M. Cheng, H. Zhang, P. Feng, B. Ke, Y. Zhang and G. Zhang, Nat. Commun., 2024, 15, 2816 CrossRef CAS PubMed
- T. S. Munonde and H. Zheng, Ultrason. Sonochem., 2021, 76, 105664 CrossRef CAS PubMed
- Y. Da, Z. Tian, R. Jiang, G. Chen, Y. Liu, Y. Xiao, J. Zhang, S. Xi, W. Chen, X. Han and W. Hu, ACS Nano, 2023, 17, 18539–18547 CrossRef CAS
- Z. Li, X. He, Q. Qian, Y. Zhu, Y. Feng, W. Wan and G. Zhang, Adv. Funct. Mater., 2023, 33, 2304079 CrossRef CAS
- K. Eiler, S. Suriñach, J. Sort and E. Pellicer, Appl. Catal., B, 2020, 265, 118597 CrossRef CAS
- D. Janisch, F. Igoa Saldana, E. De Rolland Dalon, C. V. M. Inocêncio, Y. Song, P. O. Autran, A. Miche, S. Casale and D. Portehault, J. Am. Chem. Soc., 2024, 146, 21824–21836 CrossRef PubMed
- L. Tan, H. Wang, C. Qi, X. Peng, X. Pan, X. Wu, Z. Wang, L. Ye, Q. Xiao, W. Luo, H. Gao, W. Hou, X. Li and T. Zhan, Appl. Catal., B, 2024, 342, 123352 CrossRef CAS
- Y. Zhang, W. Zhang, X. Zhang, X. Wang, J. Wang, Q. Zhao, Y. Sun, J. Li, G. Liu and X. Han, Energy Environ. Sci., 2024, 17, 3347–3357 RSC
- S. Zhou, H. He, J. Li, Z. Ye, Z. Liu, J. Shi, Y. Hu and W. Cai, Adv. Funct. Mater., 2023, 34, 2313770 CrossRef
- W. Liu, J. Yu, M. G. Sendeku, T. S. Li, W. Q. Gao, G. T. Yang, Y. Kuang and X. M. Sun, Angew. Chem., Int. Ed., 2023, 62, 202309882 CrossRef PubMed
- X. Shi, M. Xie, K. Yang, Y. Niu, H. Ma, Y. Zhu, J. Li, T. Pan, X. Zhou, Y. Cui, Z. Li, Y. Yu, X. Yu, J. Ma and H. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202406750 CrossRef CAS PubMed
- H. Shi, T. Wang, J. Liu, W. Chen, S. Li, J. Liang, S. Liu, X. Liu, Z. Cai, C. Wang, D. Su, Y. Huang, L. Elbaz and Q. Li, Nat. Commun., 2023, 14, 3934 CrossRef CAS
- Y. H. Wang, S. Zheng, W. M. Yang, R. Y. Zhou, Q. F. He, P. Radjenovic, J. C. Dong, S. Li, J. Zheng, Z. L. Yang, G. Attard, F. Pan, Z. Q. Tian and J. F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed
- M. Tang, W. Yuan, Y. Ou, G. Li, R. You, S. Li, H. Yang, Z. Zhang and Y. Wang, ACS Catal., 2020, 10, 14419–14450 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05770h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |