
Advances in photoinduced radical–polar crossover cyclization (RPCC) of bifunctional alkenes
Meng
Liu
a,
Xinke
Ouyang
a,
Chenglong
Xuan
a and
Chao
Shu
 *ab
*ab
aNational Key Laboratory of Green Pesticide, CCNU-uOttawa Joint Research Centre, Engineering Research Center of Photoenergy Utilization for Pollution Control and Carbon Reduction, College of Chemistry, Central China Normal University (CCNU), 152 Luoyu Road, Wuhan, Hubei 430079, China. E-mail: chaoshu@ccnu.edu.cn
bWuhan Institute of Photochemistry and Technology, 7 North Bingang Road, Wuhan, Hubei, 430083 China
First published on 20th December 2023
Abstract
The term “cyclic architectures” refers to a class of molecules that are highly valuable and play significant roles in organic compounds. Therefore, there is growing interest in researching the development of efficient and straightforward methods for constructing these complex structures. Photoinduced radical–polar crossover cyclization (RPCC) of bifunctional alkenes represents a class of reactions that are of great synthetic utility because they show high chemoselectivity, broad functional group tolerance and proceed under mild conditions to produce high-value cyclic products with precise alkene design. This mini-review summarizes the recent representative advances in the development of RPCC over the past two decades through different synthetic strategies in the reactions, highlighting their product diversity, selectivity and applicability, and the mechanistic rationale where possible. The intention is to provide readers with a comprehensive understanding of the current state-of-play in this field and contribute to future research efforts.
 Meng Liu | Meng Liu was born in 2001 in Shanxi province, China. She is presently a student in Prof. Chao Shu's research group at the College of Chemistry, Central China Normal University, China. Her current research interests mainly focus on the development and applications of novel synthetic methods based on radical–polar crossover cyclization reactions. |
 Xinke Ouyang | Xinke Ouyang graduated from Jiangxi Normal University in 2022 with his B.S. in Chemistry. He is currently pursuing his M.S. degree under the supervision of Professor Chao Shu at the Central China Normal University. His specific research interests are directed towards exploring new photoinduced radical–polar crossover cyclization reactions. |
 Chenglong Xuan | Chenglong Xuan was born in 2004 in Anhui province, China. He is presently a student in Prof. Chao Shu's research group at the College of Chemistry, Central China Normal University, China. His current research interests mainly focus on the development of radical sulfur chemistry. |
 Chao Shu | Chao Shu was born in Anhui province, China. He obtained his Ph.D. in 2017 from Xiamen University, under the supervision of Prof. Long-wu Ye in the field of transition-metal catalyzed alkyne tandem reactions. Then, he joined the lab of professor V. K. Aggarwal at the University of Bristol as a postdoctoral research associate, working on radical boron chemistry. In 2021, he began his independent career at the Central China Normal University as a Full Professor. His current research interests focus on the development of novel reactions, new reagents and strategies for organic synthesis. |
1. Introduction
The chemistry of functionalized cyclic architectures is one of the most extensively studied branches of organic chemistry. Such cyclic architectures are not only present in various biological macro-molecules that are essential for all forms of life (e.g., DNA and RNA), but they are also involved in many types of well-known organic compounds with industrial and pharmacological importance, such as compounds for optics, agrochemicals, electronics, and material sciences, as well as in the development of anticancer, antihypertensives, analgesics, anti-inflammatory agents, antivirals, antifungals, anti-depressants, antileishmanial agents, antibacterial agents, etc. Therefore, developing novel and facile strategies to efficiently and selectivity build such cyclic frameworks has become a subject of momentous significance.1Visible light photoredox catalysis has been widely applied as a useful platform for the development of synthetic techniques, as it exhibits a pioneering approach for molecular functionalization and has made a significant impact on organic synthesis since the end of the 2000s with the breakthrough works by the MacMillan, Yoon, Stephenson, Xiao group and many other groups from the viewpoints of cost, safety, and availability.2,3 The importance of using environment-friendly light to form chemical bonds under benign conditions has grown significantly and attracted huge attention in many fields, such as renewable energy and chemical feedstocks, new reaction development, natural product synthesis, materials and biological applications.
Radical–polar crossover reactions, first mentioned in 1993, are a class of powerful chemical reactions that involve a splicing of radical species and polar/ionic species in one pot. This type of reaction expands the synthetic toolbox and provides new opportunities for the installation of novel molecules, as it enables the conversion of simple starting materials into complex structures under mild conditions, making it an important strategy in the field of chemical synthesis.3g
Because of the elaborate design of alkene substrates, the RPCC of bifunctional alkenes is arguably one of most important aspects of homogeneous visible-light-mediated photoredox catalysis, which can enable the rapid incorporation of cyclic molecular complexity from simple and readily accessible starting materials.
In this review, a focus will be placed on representative transformations involving typical bifunctional alkenes as readily available building blocks through radical–polar crossover cyclizations to form the desired structurally diverse cyclic products through dislodging the tethered leaving group (LG), trapping by a nucleophile or electrophile, or other reaction types (Scheme 1). It is noteworthy that radical–polar crossover reactions in the field of photoredox catalysis are mentioned in only a few reviews,4 which emphasize transformations in which both radical and ionic intermediates were involved in the reaction pathways; however, no review has sought to highlight the interesting and key roles of bifunctional-alkene-mediated radical–polar crossover for the construction of cyclic compounds in a net-neutral manner.
 | ||
| Scheme 1 Photoinduced radical–polar crossover cyclization (RPCC) of bifunctional alkenes. | ||
2. Reductive RPCC reactions of bifunctional alkenes
The reductive quenching of RPCC reactions starts with an initial reduction of an excited-state photo-catalyst by a radical precursor, and then, an oxidatively generated radical is formed. Following radical-mediated reaction with bifunctional alkenes, such as leaving-group-tethered alkenes, the suitable radical intermediates are reduced to the corresponding anion, and then cyclization occurs to produce the desired cyclized products by intramolecular alkylation with the elimination of leaving groups including halides (F, Cl, Br, I), tosylates, etc. Although reductive terminations are operative in a number of photoredox-catalysed transformations, the anion generated after single-electron reduction of the radical intermediate is invariably protonated to give hydrofunctionalisation product; as a result, the desired RPCC may be affected by the reaction conditions, the nature of the bifunctional alkene substrates selected, and the radical precursors.2.1. Synthesis of cyclopropanes
The cyclopropyl group is one of the most important groups found in pharmaceutical compounds, because 10 of the top 200 drugs contain a cyclopropyl group, as do many natural products.5 In 2018, Molander and co-workers developed an annulation process for the construction of 1,1-disubstituted cyclopropanes though an RPCC process.6 The bifunctional alkene tethered with an extra electrophile/leaving group was applied in this transformation with various radical precursors catalysed by the organo-photocatalyst 4CzIPN under visible light irradiation (30 W blue LEDs), leading to a variety of densely functionalized cyclopropanes in moderate-to-excellent yields. In this reaction, trifluoroborates, dihydropyridines, and silicates all are suitable radical precursors. The RPCC method allows for the rapid and efficient production of 1,1-disubstituted cyclopropanes with various complex functional groups from readily available homoallylic tosylates as bifunctional substrates.As displayed in Scheme 2, the alkyl radical 4 generated through single-electron transfer (SET) between radical precursor 2 and the photo-excited 4CzIPN* undergoes Giese-type addition with the alkene 1 to give the new radical intermediate 5. After single-electron transfer reduction, the carbanion 6 is formed, and the ground-state photocatalyst is regenerated. Finally, the key in situ intramolecular nucleophile substitution occurs via the departure of the tosyl group, 1,1-disubstituted cyclopropane compound 3 (Scheme 2).
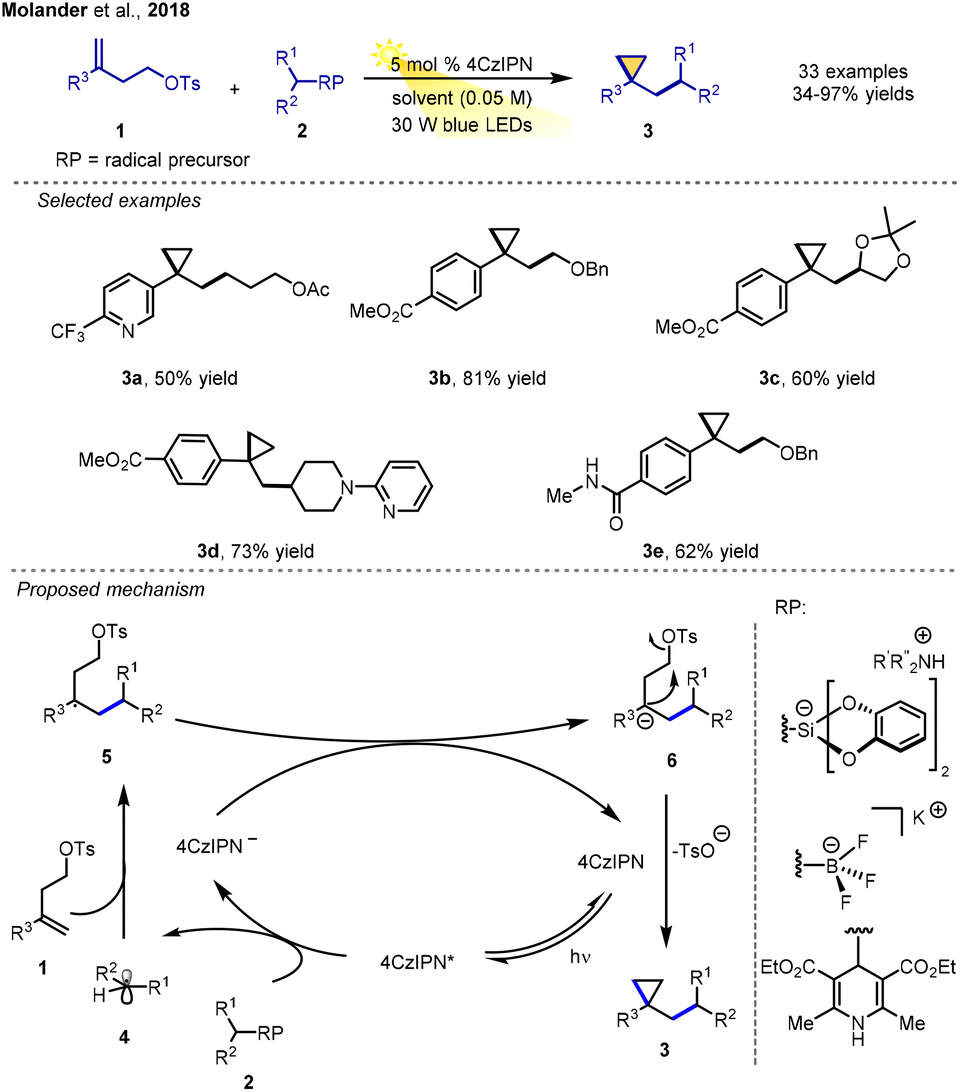 | ||
| Scheme 2 1,1-Disubstituted cyclopropane synthesis via RPCC. | ||
In 2019, Kelly, Molander and co-workers described a method for the cyclopropanation of benzo-fused alkenes embedded in bicyclic scaffolds via RPCC. Benzo-fused alkenes and radical precursors (RP) were reacted under visible light irradiation (30 W blue LEDs) in DMSO in the presence of 4CzIPN as the photocatalyst, resulting in a variety of polycyclic cyclopropanes in moderate-to-good yields. The current reaction was quite general and was tolerant to a wide range of bifunctional alkenes as well as radical precursors, leading to a mild, metal-free strategy for assembling an array of diverse bicyclic cyclopropane structures with versatile and structurally diverse functional groups.
The proposed mechanism involves a Giese-type addition of the radical intermediate 10 from the substrate 8 to the double bond of 7, followed by a radical–polar crossover step converting the resulting radical adduct 11 to anion intermediate 12, which undergoes an anionic 3-exo-tet ring closure to lead to the formation of the product. Tosylates, as the key leaving group, ensured that the cyclisation took place to yield strained polycyclic system 9 (Scheme 3).7
 | ||
| Scheme 3 Polycyclic cyclopropane synthesis via RPCC. | ||
In 2018, Aggarwal and co-workers developed a photoredox-catalyzed synthesis of functionalized cyclopropanes through a decarboxylative radical–polar crossover cyclization process. The reaction of readily available aliphatic carboxylic acids, distal halosubstituted olefins and cesium carbonate in the presence of 4CzIPN (1–5 mol%) as a photocatalyst in DMF at room temperature under blue LED light irradiation was conducted to form the respective functionalized cyclopropanes in moderate- to-excellent yields. The mild conditions, with catalysis by an inexpensive organic photocatalyst, were found to tolerate a wide range of functional groups on both the carboxylic acid and the homoallyl or allyl chloride substrates. In addition, a broad range of complex carboxylic acids directly were converted into structurally and functionally diverse cyclopropanes in excellent yields under the current conditions, such as the densely functionalized cholic acid; even the dipeptides Z-Gly-Phe-OH and Z-Phe-Leu-OH were suitable substrates.
The authors experimentally confirmed that the ring cyclisation occurred via an anionic pathway and postulated that reductive RPCC was involved. The reaction mechanism suggests that single-electron oxidation of carboxylic acid 16 by the excited state photocatalyst produces radical intermediate 17 and releases CO2. Then, the carbon-centered radical 17 adds to homoallyl chloride 14 to give the stabilized alkyl radical species 18. After single-electron reduction of 18 by the reduced photocatalyst, the resulting carbanion 19 subsequently undergoes polar 3-exo-tet cyclization to form the cyclopropane product 20. Again, the halide-tethered alkene plays a crucial role as a bifunctional alkene for the success of this RPCC reaction (Scheme 4).8a α-MIDA-boryl styrenes, 2-propenyl BMIDA, and ethenyl–BMIDA were demonstrated to be viable substrates, giving a variety of cyclopropyl MIDA boronates through photoinduced RPCC, by Jin, Li, Fang and co-workers in 2020.8b
 | ||
| Scheme 4 Decarboxylative cyclopropanation via RPCC. | ||
In 2020, a photoredox-catalysed cyclopropanation of 1,1-disubstituted alkenes via an RPCC process was presented by Fang, Li, Jin, and co-workers. 1,1-Disubstituted alkenes and bis-catecholato silicates were reacted in the presence of 2 mol% Ir[dF(CF3)ppy]2(dtbbpy)PF6 as a photocatalyst in DMSO under 9 W blue LEDs, leading to the corresponding cyclization products in moderate-to-excellent yields with broad functional group tolerance. A range of alkyl and acyl radicals from the corresponding radical precursor, bis-catecholato silicates and 1,4-dihydropyridines could participate this radical addition–anionic cyclization cascade.
Mechanistic studies confirmed that the reaction proceeds via a similar decarboxylative radical–polar crossover cyclization mechanism involving a radical addition to a double bond followed by single-electron reduction and cyclization of the resulting carbanion intermediate by cutting the leaving group (Cl, Br, OTs, OMs) (Scheme 5).9a Using alkyl silicates or 4-alkyl-1,4-dihydropyridines as the radical sources, silylcyclopropanes, (cyclopropylmethyl)trimethylsilanes and other cycles could be prepared via photoinduced RPCC reactions as well.9b–d
 | ||
| Scheme 5 Cyclopropane synthesis via RPCC from bis-catecholato silicates. | ||
The phosphorylation of olefins is important for the formation of valuable organophosphorus compounds. In 2021 Yu, Yu, Fu and co-workers reported a visible-light-driven redox-neutral phosphonoalkylation of alkenes, providing an expedient synthesis of diverse pharmaceutically interesting organophosphorus-containing cyclopropane derivatives from readily available starting materials.10 Alkene-bearing mesityl group and H–P(O) compounds were reacted under visible-light irradiation in the presence of 4CzIPN (2 mol%) resulting in various organophosphorus-containing three-membered carbocyclic scaffolds. Moreover, the advantages of this redox-neutral method are mild reaction conditions, good functional group tolerance, and short reaction time (Scheme 6).
 | ||
| Scheme 6 Phosphorus-containing cyclopropane synthesis via RPCC. | ||
A proposed mechanism is displayed in Scheme 6. The phosphonyl radical species 30, which is generated from H–P(O) compound 28 with the assistance of a base via a SET process with excited-state 4CzIPN*, adds to the alkene 27 regioselectively to give the tertiary carbon radical 31. Subsequently, the benzyl radical undergoes reduction through a SET process, leading to the formation of the benzyl carbanion 32, which eventually undergoes intramolecular nucleophilic substitution with the release of the leaving group (MsO–) to afford phosphorus-containing cyclopropane derivatives.
In 2023, Fang, Luo and co-workers discovered a protocol for the preparation of functionalized alkynylcyclopropane derivatives from readily available 1,3-enynes via an RPCC process.11 Using alkyl silicates as the radical precursors, a variety of primary and secondary radicals bearing different functional groups could undergo the desired cyclopropanation reactions to give the cyclization products in moderate-to-good yields.
The authors presented a possible reaction mechanism, in which the alkyl radical species 36, which is generated from silicate via a SET process with the excited-state photocatalyst, adds to the double bond of 1,3-enynes 33 regioselectively to give the tertiary carbon radical 37. Then, radical 37 is reduced via a SET process, resulting in carbanion intermediate 38. Intramolecular nucleophilic substitution of intermediate 38 with the release of the leaving group (Br–) gives the alkyne-substituted cyclopropane derivatives (Scheme 7).
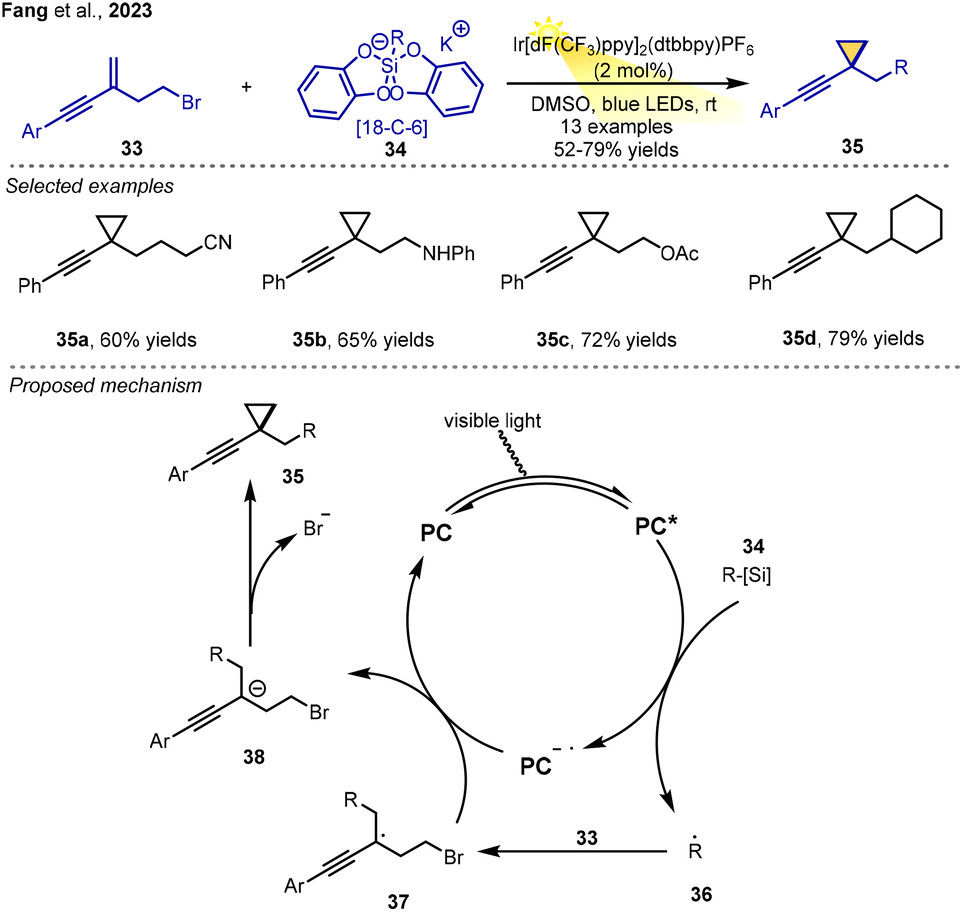 | ||
| Scheme 7 Functionalized alkynylcyclopropane synthesis via RPCC. | ||
2.2. Synthesis of cyclobutanes
In 2019, Aggarwal and co-workers described a synthesis of photoredox-catalyzed cyclobutanes by a deboronative radical addition–polar cyclization cascade. The key to the success of the methodology was that easily oxidizable arylboronate complexes, which are generated in situ by reaction of aryl pinacol boronic esters with a slight excess of phenyllithium at low temperature, proceed to generate alkyl radicals in situ through single-electron transfer under rigorously anhydrous conditions. The treatment of arylboronate complexes with iodide-tethered alkenes in the presence of 4CzIPN (5 mol%) as an organic photocatalyst in DMSO solvent at room temperature under blue LED irradiation allowed the efficient formation of functionalized cyclobutanes in generally good-to-excellent yields. The mild reaction conditions enabled densely fused cyclobutanes with a variety of structures to be built, demonstrating good functional group tolerance. Moreover, the broad variety of bifunctional alkenes renders the attractive method easily applicable for the formation of other ring systems such as 3-, 5-, 6-, and 7-membered rings.A plausible mechanism based on polar-radical crossover cyclization process has been postulated in Scheme 8. Single-electron oxidation of arylboronate complex 40 by the excited photoredox catalyst gives alkyl radical species 43. This radical intermediate 43 adds to the alkene 44 to generate the stabilized carbon radical species 46. After single-electron reduction of 46 by the reduced photocatalyst, a polar 4-exo-tet cyclization is involved to produce the cyclobutane product. Additionally, with SET between PC˙− and an iodine radical, an alternative pathway involving SH2 cyclization of radical 46 to form cyclobutane in DMSO solvent is also possible (Scheme 8).12
 | ||
| Scheme 8 Cyclobutane synthesis via RPCC. | ||
2.3. Synthesis of cyclohexenes
In 2013, the Zhou group reported an interesting procedure for the synthesis of mono-fluorocyclohexenes via two consecutive reactions based on two different radical–polar crossover reactions with the initial 1,3-benzodioxoles as radical precursors. In the presence of 4CzIPN, 1,3-benzodioxoles and α-CF3 alkenes were reacted under irradiation with a 5 W blue LED in MeCN to produce a variety of mono-γ,γ-difluoroallylation products 51 in 45–78% yields through a C–F bond cleavage process. Subsequently, reaction of mono-γ,γ-difluoroallylated 1,3-benzodioxoles, alkenes and excess 2,4,6-collidine in the presence of a more-oxidizing iridium photocatalyst in MeCN at room temperature under 5 W blue LED irradiation produced functionalized mono-fluorocyclohexenes in 38–88% yields via a second C–F bond cleavage process. This reaction is compatible with a wide range of different substrates under mild conditions, demonstrating the nice generality of this protocol.The authors gave a possible pathway for this reaction. 1,3-Benzodioxole 54 undergoes SET by the excited photocatalyst 4CzIPN* to give radical cation 55. Radical 56 generated from the deprotonation of 55 adds to the α-CF3 alkene, leading to the radical 57. Single-electron transfer reduction of 57 forms –CF3 carbanion 58. β-Fluoride elimination of 58 leads to mono-γ,γ-difluoroallylated 1,3-benzodioxole 59. In the second photoredox catalytic cycle, carbanion 62 is generated with the Ir complex as the photocatalyst, which is similar to the formation of 58. The γ,γ-difluoroalkene could trap the carbanion intramolecularly because of the high electron deficiency on γ,γ-difluoromethylene carbon, leading to the anion intermediate 63 (path A). Intramolecular radical addition to γ,γ-difluoroalkene 61 gives a new radical 64, which then undergoes single-electron reduction to form 63 (path B). Finally, β-fluoride elimination of intermediate 63 affords the product 65 (Scheme 9).13
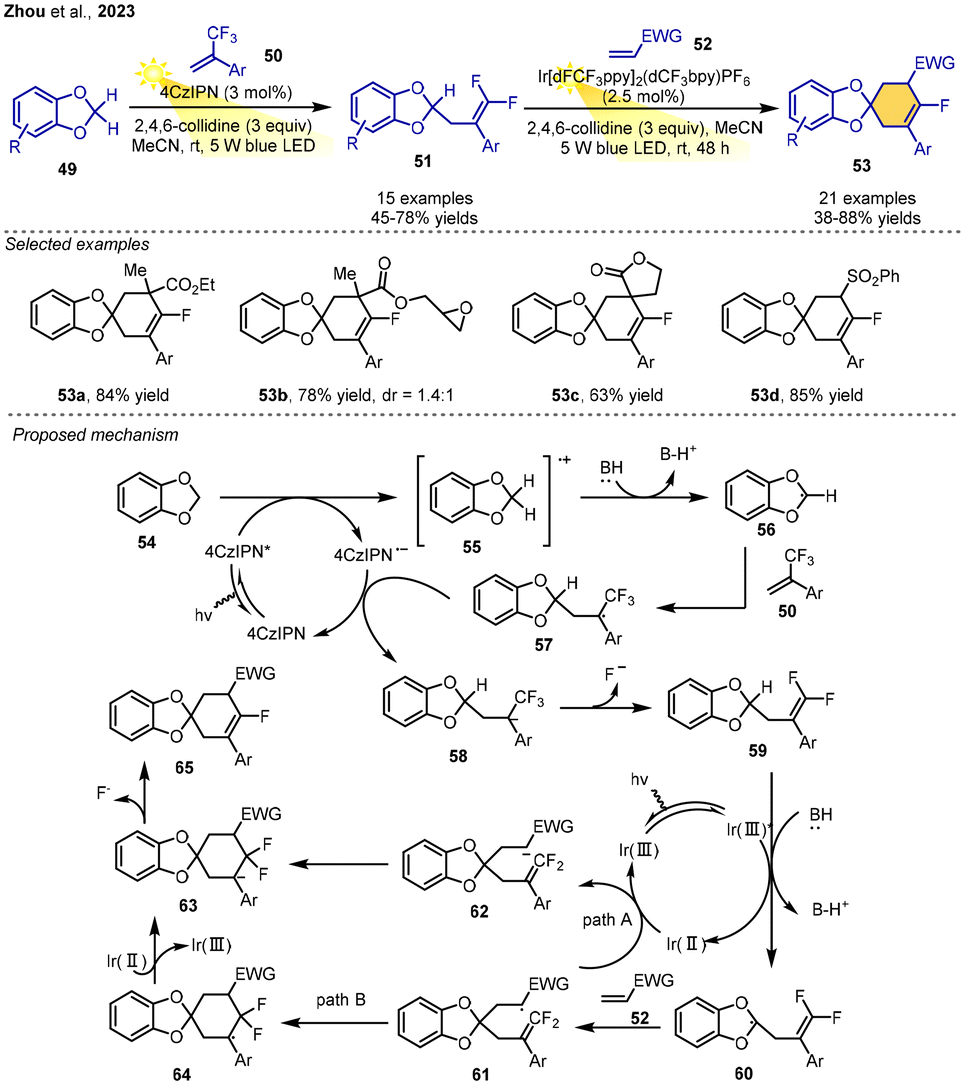 | ||
| Scheme 9 Monofluorocyclohexene synthesis via PRCC. | ||
2.4. Synthesis of heterocycles
In 2022, the Zhou group reported a one-pot, two-step protocol to synthesize mono-fluorocyclopentenes by photocatalytic RPCC. The three-component reaction of α-trifluoromethyl alkenes, electron-rich alkenes, and sodium sulfinates was catalysed by 4CzIPN under irradiation by a blue LED in DMSO to give gem-difluoroalkenes, which could be transformed into various monofluorinated cyclopentenes under base conditions (LiHMDS) at a lower temperature in one pot. A wide range of bicyclic monofluorinated cyclopentenes were constructed in satisfactory yields with a cis configuration. Of note, using t-BuOK as the base in DMF, monofluorinated hexahydropentalene could be produced in 57% yield (X = CH2).As shown in the proposed mechanism in Scheme 10, single-electron oxidation of the sodium sulfinate by the excited state photocatalyst produces sulfonyl radical 70. Then, radical 70 adds to alkene 66 to give β-sulfonyl alkyl radical 71. α-Trifluoromethyl alkene 67 reacts with 71 to produce α-CF3 carbon radical 72. Finally, SET reduction of radical 72 and β-fluoride elimination give gem-difluoroalkene 73. The intramolecular SNV reaction could give the final product monofluorocyclopentene in the presence of base at low temperature (Scheme 10).14
 | ||
| Scheme 10 Mono-fluorocyclopentene synthesis via RPCC. | ||
The same group reported a protocol to synthesize 5-fluoro-dihydroindolizines from pyrrole-2-acetic acids and trifluoromethyl alkenes in 2022. Pyrrole-2-acetic acids, trifluoromethyl alkenes and DABCO were reacted in the presence of 3 mol% 4DPAIPN as a photoredox catalyst under visible-light irradiation from 5 W blue LEDs in DMSO to synthesize gem-difluoroalkenes bearing pyrrole motifs in moderate-to-good yields, and then a base-mediated intramolecular SNV reaction cleaved the second C–F bond and formed a C–N bond to give the 5-fluoro-dihydroindolizines in a one-pot or two-step manner.
The reaction mechanism suggests single-electron oxidation of the carboxylate 81 by the excited state photocatalyst of 4DPAIPN* in the presence of DABCO produces radical intermediate 82. The radical then undergoes decarboxylation to afford pyrrole-2-methyl radical 83, followed by the addition of the radical to α-trifluoromethyl alkene 77 to give radical 84. Intermediate 84 undergoes SET reduction to give the α-CF3 carbanion 85, and then β-fluoride elimination to give gem-difluoroalkene 86. Finally, intramolecular nucleophilic substitution reaction of the double-bond carbon atom leads to 5-fluoro-dihydroindolizine 87 under base conditions (Scheme 11).15
 | ||
| Scheme 11 5-Fluoro-dihydroindolizine synthesis via RPCC. | ||
In 2018, Zhou and co-workers discovered a strategy to synthesize fluorinated dihydrobenzoxepines from o-hydroxyphenylacetic acids and trifluoromethyl alkenes by a photocatalytic RPCC. The reaction of o-hydroxyphenylacetic acids, α-CF3 alkenes and Cs2CO3 in the presence of 5 mol% 4DPAIPN as a photoredox catalyst under visible-light irradiation from 5 W blue LEDs could synthesize fluorinated dihydrobenzoxepines in moderate-to-good yields. Styryl systems and o-hydroxyphenylacetic acids containing a variety of functional groups can be successfully converted into the corresponding products via a decarboxylative/defluorinative [4 + 3] annulation process.
On the basis of the experimental results and literature reports, a proposed mechanism was developed, as displayed in Scheme 12. Single-electron oxidation of the anionic –COO– group of 92 by the excited-state photocatalyst 4DPAIPN* produces radical 93. Then radical undergoes the loss of CO2 to form a new carbon-centered radical 94, which adds to styrene 89 to give radical intermediate 95. Subsequent single-electron reduction leads to the α-CF3 carbanion 96. Finally, β-fluoride elimination of 96 and intramolecular SNV reaction yield fluorinated dihydrobenzoxepine 98 (Scheme 12).16
 | ||
| Scheme 12 Fluorinated dihydrobenzoxepine synthesis via RPCC. | ||
Cascade-type reactions have been established as a general strategy for the synthesis of highly functionalized compounds. Sultines play a very important role in materials, medicine, synthesis and other areas. In 2023, the Shu group developed a unique strategy to synthesize multifluoromethylated γ-sultines via photoinduced RPCC. This process includes a radical multifluoromethylation/SO2 incorporation/polar cyclization cascade. The reaction of homoallylic tosylates and sodium multifluoro-alkanesulfinates (RfSO2Na) in the presence of 5 mol% 4CzIPN as a photoredox catalyst under visible-light irradiation from 420 nm blue LEDs in MeCN could produce structurally diverse multifluoromethylated γ-sultines in moderate-to-good yields and moderate-to-good trans/cis diastereoselectivities.
In the proposed mechanism, the multifluoroalkanesulfinate anion undergoes single-electron transfer with the excited state photocatalyst 4CzIPN*, leading to multifluoroalkyl radical 103 and releasing SO2. The radical 103 adds to alkene 99 to generate the stabilized radical 104. Then, the pivotal incorporation of SO2 with 104 affords the sulfonyl radical 105, followed by SET between 105 and the reduced state of the photocatalyst to give sulfonyl anion 106. In a polar aprotic solvent, 106 quickly translates into 107, which bears a negative charge on the O-atom. Finally, 107 undergoes a polar 5-exo-tet cyclization to yield sultine 101. In alternative pathway, radical 104 could be reduced to give carbanion intermediate 108, followed by SO2 insertion and cyclization to yield product (Scheme 13).17
 | ||
| Scheme 13 Sultine synthesis via RPCC. | ||
Very recently, the Shu group also reported a general strategy to build challenging-to-access polycyclic γ-sultines by a RPCC procedure. The reaction of the inexpensive Langlois reagent and alkene derivatives in the presence of 3 mol% 4CzIPN as the photoredox catalyst under visible-light irradiation from 30 W blue LEDs under an N2 atmosphere in THF could form the corresponding highly substituted polycyclic γ-sultines in good-to-excellent yields with excellent diastereoselectivities. The mild reaction conditions give the reaction good substrate scope and functional group tolerance. Sodium perfluoroalkanesulfinates with long-chain perfluoroalkyl groups as substrates also give the corresponding products. In addition, the transformation could be extended to the preparation of difluoroalkylated polycyclic sultines.
The proposed reaction mechanism suggests that multifluoroalkanesulfinate anion 113 undergoes single-electron transfer with the excited state photocatalyst 4CzIPN*, leading to multifluoroalkyl radical 114 and releasing SO2. The addition of radical intermediate 114 to alkene 115 generates the stabilized radical 116. Then, the pivotal incorporation of SO2 with 116 affords the sulfonyl radical 117, and then SET between 117 and the reduced state of the photocatalyst gives sulfonyl anion 118, which could resonate with sulfinyl anion 119 in polar solvent. Finally, 119 undergoes a polar 5-exo-tet cyclization to furnish polycyclic sultine 120 (Scheme 14).18
 | ||
| Scheme 14 Polycyclic sultine synthesis via RPCC. | ||
In 2023, Mattia Silvi and co-workers developed a nice one-pot, two-step approach for the synthesis of oxetanes through radical–polar crossover cyclization with vinyl sulfonium compounds as difunctional alkenes. The reaction of alcohol substrates, vinyl sulfonium triflates, quinuclidine (10 mol%) and tetrabutyl ammonium dihydrogen phosphate (25 mol%) in the presence of 4CzIPN (5 mol%) as the photoredox catalyst under visible-light irradiation in CH3CN followed by the addition of KOtBu could afford a variety of corresponding oxetanes in moderate-to-excellent yields. The mild reaction conditions make this method suitable for simple or complex substrates with a wide range of functional groups. Spirocyclooxides are obtained from different bicyclic and polycyclic substrates. For example, norborneol, adamantanol, and aza-byciclooctane heterocyclic alcohol substrates afford the desired products in good yields. However, this method has limitations; benzylic, allylic, and propargylic alcohols could not give the corresponding products.
A possible mechanism is shown in Scheme 15. Activated H-bonded alcohol 124 undergoes HAT to give nucleophilic radical 125. Then, the addition of radical 125 to 126 leads to radical cation 127. Subsequent single-electron reduction and protonation give compound 128. Finally, intramolecular nucleophilic substitution cyclization yields oxetane 129 in the presence of KOtBu (Scheme 15).19
 | ||
| Scheme 15 Oxetane synthesis via PRCC. | ||
In 2023, the Wang group described a method for synthesizing polysubstituted fluoropyrazole derivatives via a photoinduced radical–polar crossover cyclization of enamines. The reaction of CF2Br2, enamines and aryl sulfonyl hydrazines in the presence of 2 mol% fac-Ir(ppy)3 as the photoredox catalyst under visible-light irradiation from blue LEDs under an N2 atmosphere in DMA could afford the desired fluoropyrazole products in moderate-to-good yields. Expansion of the substrate scope proved that this method has good functional group compatibility and widespread practicality. Complex compounds such as (+)-fenchol and dehydroepiandrosterone derivatives were also suitable substrates for this strategy, giving the corresponding products in moderate yields.
A plausible reaction mechanism for these multicomponent reactions was discussed. Firstly, single-electron reduction of CF2Br2 by the excited-state photocatalyst IrIII* generates radical species ˙CF2Br. Then, the radical adds to enamine 134 to form a new radical 135, which undergoes single-electron oxidation to deliver 136. An HBr elimination step from 136 affords the key intermediate 137. The intermolecular nucleophilic addition between 137 and TsNHNH2 leads to 139, which undergoes elimination to give 140. Finally, intramolecular cyclization and elimination give the target product 142 (Scheme 16).20
 | ||
| Scheme 16 Polysubstituted fluoropyrazole derivative synthesis via PRCC. | ||
3. Oxidative RPCC reactions of bifunctional alkenes
In contrast to the reductive RPCC reactions of bifunctional alkenes, the oxidative process for RPCC of bifunctional alkenes with a nucleophile has been more widely explored. The radical precursor is reduced to form a radical-based intermediate, which could induce subsequent diversification reactions. The open-shell radical intermediate undergoes single-electron transfer and is oxidized to generate the corresponding carbocation intermediate. Subsequently, the carbocation intermediate reacts with the nucleophile tethered to the alkene to produce the cyclic product. These oxidative RPCC reactions have led to the development of numerous efficient methods for ring products from designed alkene substrates, the majority of which proceed via oxidative termination of the radical cascade and trapping of the resulting carbocation with a heteroatom-based nucleophile.3.1. Synthesis of cyclopentanes
In 2019, the Jiang group and co-workers reported a photocatalytic annulation–alkynyl migration strategy to generate cyclopentane carboxylates. This is an interesting radical–polar crossover cyclization in which both 1,4-enynes and α-allyl-α-bromomalonate play a role as bifunctional alkenes. The reaction of 1,4-enynes, α-allyl-α-bromomalonate substrates and K2HPO4 in the presence of 1 mol% Ir(dFppy)3 as the photoredox catalyst under visible-light irradiation from 10 W blue LEDs in HFIP could give highly substituted cyclopentane carboxylate derivatives in moderate-to-good yields. Under the mild conditions, the system demonstrates good functional group tolerance and moderate-to-good diastereoselectivities. Complex naturally derived tertiary alcohols, such as estrone and isopulegol, can be transformed into the corresponding products in satisfactory yields.A plausible reaction mechanism is proposed in Scheme 17. Single-electron reduction of α-allyl-bromomalonate 146 by the excited-state photocatalyst IrIII* complex produces radical intermediate 147. Then, the radical adds to 1,4-enyne 148 to form a new radical intermediate 149. 5-exo-trig/5-exo-dig bicyclization of 149 delivers 151. Ring-opening of the cyclopentane intermediate 151 affords hydroxyalkyl radical 152, which is oxidized by the IrIV species to yield carbocation 153. Finally, intermediate 153 loses a proton to generate the product 154 (Scheme 17).21
 | ||
| Scheme 17 Cyclopentane carboxylate synthesis via RPCC. | ||
3.2. Synthesis of lactones
The Xiao group in 2014 developed an oxidative RPCC reaction for the synthesis of γ,γ-disubstituted butyrolactones under visible-light irradiation with olefinic carboxylic acids as bifunctional alkene substrates. Alkenoic acids were reacted with the aryldiazonium salts and excess H2O in CH3CN in the presence of [Ru(bpy)3](PF6)2 under visible-light irradiation from 3 W white LEDs at very low temperature, leading to a variety of γ,γ-disubstituted butyrolactones in 31–83% yields. This method can be adapted to a variety of readily available alkenoic acids bearing either electron-donating or electron-withdrawing groups on the aryl substituent, and could synthesize various sterically hindered lactones rapidly. This process can also be applied to the synthesis of six-membered lactone compounds, such as δ-valerolactone, which was formed in 54% yield.The proposed mechanism of this reaction suggested that SET reduction of aryldiazonium salts 156 by the excited photo-catalyst gives an aryl radical species 158 and PC+. This aryl radical regioselectively adds to the double-bond part of substrate 155 to generate tertiary carbon radical intermediate 159. This is followed by an oxidative radical–polar crossover reaction to form reactive carbocation intermediate 160. The carbocation is then trapped by the neighbouring carboxyl group, providing the desired γ,γ-disubstituted lactone 157 (Scheme 18).22
 | ||
| Scheme 18 γ,γ-Disubstituted butyrolactone synthesis via RPCC. | ||
Acyl radicals play an important role in synthesizing useful natural products and medicinal molecules in organic synthesis. A visible-light-driven RPCC reaction of alkenoic acids with acyl chlorides in toluene was discovered by Cao, Rao and co-workers in 2023. The reaction of acyl chloride, alkenoic acid and Na2CO3 in toluene using Ir(ppy)3 as a photoredox catalyst under blue LED visible-light irradiation at room temperature could afford the products in satisfactory yields. Mild reaction conditions enabled the synthesis of a wide variety of acyl lactones from a series of aroyl and heteroaroyl chlorides and diverse unsaturated carboxylic acids, reflecting the good functional group tolerance and substrate scope. Also, the simple protocol allows the efficient construction of the biologically important phthalide framework.
In terms of the reaction mechanism, as depicted in Scheme 19, benzoyl radical intermediate 165 is first produced from benzoyl chloride 164via SET reduction with reductive photoexcited IrIII*. The radical then adds to the double bond of alkene 166, leading to stable benzyl-type radical species 167. SET between radical 167 and the oxidative IrIV species furnishes corresponding carbocation intermediate 168. Finally, the lactone 169 is delivered through an intramolecular nucleophilic substitution under base conditions (Scheme 19).23
 | ||
| Scheme 19 Benzo-fused lactone synthesis via RPCC. | ||
In 2014, Koike, Akita, and co-workers discovered a strategy for the highly regio- and diastereoselective synthesis of CF3-substituted lactones from both terminal and internal alkenoic acids by a photoredox catalysis RPCC process. The reaction of alkenoic acids, CF3 reagents and K2CO3 in MeCN using 5 mol% of [Ru(bpy)3](PF6)2 as a photoredox catalyst under visible-light irradiation from 425 nm blue LEDs at room temperature could deliver the highly regio- and diastereoselective CF3-substituted lactones bearing various functional groups smoothly in moderate-to-excellent yields. Remarkably, this is the first example of a highly endo- and diastereoselective synthesis of CF3-substituted five-, six-, and seven-membered ring lactones from a series of internal alkenoic acids.
Mechanistic investigations suggested that a radical–polar cyclization process should be involved in the transformation. The trifluoromethyl radical generated from CF3 reagents (Umemoto's reagent and Togni's reagents) under the visible light catalysis of the Ru photocatalyst adds to the terminal or internal double bond of alkenoic acids, giving the stabilized radical intermediates. Then, SET with the highly oxidized state of the photocatalyst produces a β-CF3-substituted carbocation intermediate in a highly regioselective manner. Finally, nucleophilic attack of the dangling carboxylic acids produces the CF3-substituted lactones (Scheme 20).24
 | ||
| Scheme 20 CF3-substituted lactone synthesis via PRCC. | ||
3.2. Synthesis of spirooxazolines
In 2015, Munetaka Akita, Koike and co-workers proposed a useful strategy to give CF3-containing spirooxazolines and spirooxazines in anti-fashion through regiospecific trifluoromethylative spirocyclization by photoredox catalysis. Cyclic alkenes bearing an amide pendant were reacted with Umemoto's reagent and 2,6-lutidine in DCM in the presence of 0.5 mol% of [Ru(bpy)3](PF6)2 under 425 nm LED irradiation, leading to CF3-containing spirooxazolines and spirooxazines in moderate-to-good yields and diastereoselectivities. This modular photocatalytic system could synthesize a diverse range of CF3-containing heterocycles from benzo-fused alkenes with various amide pendants. The substituents of the amide groups and the structures of the cyclic alkenes influence the diastereoselectivity of the products. Amides with longer chains could also afford spirooxazolines with a six-membered ring in good yields.The reaction appears to proceed through a mechanism similar to the previous lactone synthesis. Initially, the trifluoromethyl radical is generated via SET between Umemoto's reagent and the excited photocatalyst. Subsequently, SET oxidation generates a α-CF3-substituted carbocationic intermediate regiospecifically after a Giese-type addition. Finally, intramolecular nucleophilic attack of the dangling amide moiety in an anti-fashion results in regiospecific product 181 due to the steric factor of the CF3 substituent. This is an interesting example of an oxygen atom on an amide moiety as a nucleophile in the oxidative RPCC of bifunctional alkenes (Scheme 21).25
 | ||
| Scheme 21 Spirooxazoline and spirooxazine synthesis via RPCC. | ||
3.3. Synthesis of 2-oxazolidinones
In 2018, the Yu group described a procedure for the utilization of CO2 and BrCF2CO2Et in the difluoroalkylation of allylamines and subsequent carboxylative cyclization to build highly substituted 2-oxazolidinones. The reactions of allylamines, difluoroalkyl reagents, DABCO and CO2 in DMF using Ru(bpy)3Cl2·6H2O as a photoredox catalyst under visible-light irradiation from 30 W blue LEDs furnish a series of 2-oxazolidinones with functionalized difluoroalkyl groups in 20–86% yields. This mild system exhibits good functional group tolerance; however, allylamines without aromatic groups are not suitable substrates, which might be because the intermediate is unstable. Under the same conditions, the reaction could give six-membered cyclization products in moderate yields as well. Furthermore, the successful production of the phosphonyldifluoromethyl group, which is useful in medicinal chemistry, shows a potential application of this approach.A possible mechanism was proposed, as shown in Scheme 22. Firstly, the excited ruthenium catalyst is reduced to a RuI complex by DABCO. Then, single-electron reduction of difluoroalkyl reagent BrCF2COOEt 183 with the RuI complex produces radical intermediate 187. The radical 187 adds to the double bond of carbamate 186 generated from 185 and CO2, leading to the stabilized radical 188. Finally, oxidation of radical 188 by photoexcited RuII and intramolecular cyclization give cyclic product 190 (Scheme 22).26
 | ||
| Scheme 22 2-Oxazolidinone synthesis via RPCC. | ||
The same group further developed a novel oxy-alkylation of allylamines with CO2 and unactivated alkyl bromides through visible-light-driven palladium catalysis in 2018. The reactions of allylamines, unactivated alkyl bromides, and TBD under 1 atm of CO2 in DMSO in the presence of Pd(PPh3)4 as a photocatalyst under visible-light irradiation from 10 W blue LEDs provide a series of 2-oxazolidinones in good-to-high yields and selectivity. Allylamines bearing different functional groups and a variety of tertiary, secondary, and primary alkyl bromides were applied as reactants, showing the good compatibility of this reaction. However, allylamines without an aryl substituent were found to show no reactivity, which may be due to the low reactivity of electron-rich alkenes towards tertiary alkyl radicals and the challenges in obtaining a less-stable secondary radical.
A plausible mechanism was proposed as well based on mechanistic study. Single-electron reduction of alkyl bromide 192 under the catalysis of the photoexcited Pd(0) complex produces alkyl radical species 196. The addition of radical 196 to the double bond of carbamate 195 generated from 194 and CO2 gives the stabilized benzylic radical 197, which is subsequently oxidized to carbocation 198 by Pd(I) and regenerates the Pd(0) species. Finally, the intramolecular cyclization of 198 affords desired 2-oxazolidinone 199 (Scheme 23).27
 | ||
| Scheme 23 2-Oxazolidinone synthesis via RPCC with CO2. | ||
3.4. Synthesis of cyclic ethers
In 2012, Xia, Yang and co-workers introduced an approach for the synthesis of tetrahydrofurans and tetrahydropyrans by the visible-light-induced radical–polar crossover cyclization of electron-enriched styrenes. The reaction of styrenes, diethyl-2-bromomalonates and K2HPO4 in DMF using fac-Ir(ppy)3 as a photoredox catalyst under visible-light irradiation from 1 W blue LEDs gave the corresponding products in moderate-to-excellent yields. In addition to the 5-endo cycloetherification, alkenols with a longer chain could undergo 6-endo cycloetherification to furnish the corresponding disubstituted tetrahydropyrans. This RPCC method for forming various and complex substituted tetrahydrofurans and tetrahydropyrans features operational simplicity, high efficiency and regioselectivity under mild and environmentally friendly conditions.The proposed reaction mechanism suggests that single-electron reduction of the diethyl 2-bromomalonate 201 by the excited-state photocatalyst under visible-light irradiation produces the radical ˙CH(CO2Et)2 (203) and an Ir4+ complex. Then, radical 203 adds to alkene 204 to form intermediate 205, which could undergo oxidation by Ir4+ to afford benzylic carbocation 206. Rapid trapping by a tethered hydroxyl achieves the formation of the desired product. Another possible pathway is the formation of an oxygen radical though hydrogen atom transfer. Next, the oxygen radical could be rapidly trapped by the alkene to form the tetrahydrofuran ring radical 209. After SET oxidation, the corresponding carbocation 210 would react with the diethyl malonate carbon anion or Br− to provide the product 207 (Scheme 24).28
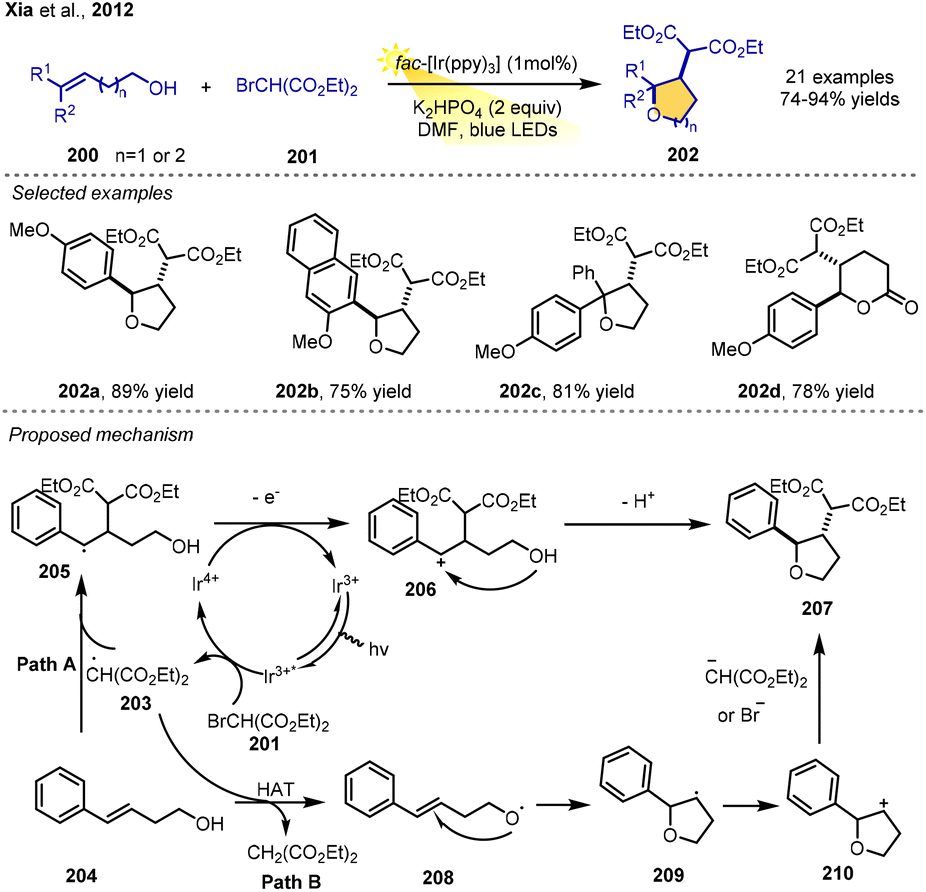 | ||
| Scheme 24 Tetrahydrofuran and tetrahydropyran synthesis via PRCC. | ||
In 2016, Koike, Akita and co-workers realized a nice method for the highly diastereoselective synthesis of CF3- and CF2H-substituted spiroethers from aryl-fused cycloalkenylal–kanol derivatives via photoredox catalysed RPCC reaction. Aryl-fused cycloalkenylalkanols reacted with Umemoto's reagent and 2,6-lutidine in DCM in the presence of 5 mol% of [Ru(bpy)3](PF6)2 under visible-light irradiation from 470 nm LEDs, leading to a versatile synthesis of CF3-containing spiroethers. Single-crystal X-ray crystallography confirmed that this reaction has high diastereoselectivity. The thiacyclic derivative and seven-membered cycloalkene could also give the corresponding products, but the yields and/or diastereoselectivities declined. By changing the fluoromethylating reagent to N-tosyl-S-difluoromethyl-S-phenylsulfoximine and photocatalysts, this photocatalytic system could also form the anti-CF2H-spiroethers from aryl-fused cycloalkenylalkanols under similar reaction conditions.
A possible RPCC mechanism is presented in Scheme 25. Firstly, SET between the photoexcited Ru or Ir photocatalyst and the fluoromethylating reagents gives the fluoromethyl radical (˙CF2X) and oxidized photocatalyst species. The generated fluoromethyl radical then adds to cycloalkenylalkanol 215 in a regioselective manner, giving the stabilized radical 216. SET with the highly oxidized state of the photocatalyst gives the α-fluoromethyl substituted carbocation intermediate 217. The key intermediate 217 undergoes intramolecular nucleophilic attack of the hydroxyl group in an anti-fashion, resulting in the formation of the anti-fluoromethylated spiroether 218 (Scheme 25).29
 | ||
| Scheme 25 Spiroether synthesis via PRCC. | ||
4. Elaborate radical RPCC reactions of bifunctional alkenes
As an important complement to the previously described reductive and oxidative radical–polar crossover cyclization reactions with bifunctional alkenes tethered to an electrophile or nucleophile, the scope of such RPCC reactions can be much expanded by employing a more in-depth reaction design, in which the open-shell intermediate undergoes single-electron transfer to generate a carbocation or carbanion that is subsequently trapped using different reaction patterns, unlike that straightforward approach using an intramolecular electrophile or nucleophile. Selected recent representative examples are reviewed here.4.1. Synthesis of five-membered ring compounds
In 2015, Frank Glorius and co-workers developed a visible-light photoredox-catalyzed semipinacol-type rearrangement procedure, which proceeds via 1,2-alkyl migration, leading to novel cycloalkanones with all-carbon quaternary centers. The reaction proceeded through trifluoromethylation and ring expansion by a radical–polar mechanism. 1-(1-Arylvinyl)cyclobutanols 219 reacted with the electrophilic trifluoromethylating reagents and a slight excess of TMSOTf in DMF in the presence of [Ru(bpy)3](PF6)2 (1 mol%) under visible-light irradiation, yielding a variety of semipinacol-type rearrangement cycloalkanone products in 27–90% yields. The method provides an efficient way to synthesize densely functionalized CF3-containing cycloalkanones under mild photochemistry conditions.A plausible mechanism was proposed based on mechanistic study. Firstly, SET reduction of Umemoto's reagent 220 by the excited photocatalyst gives an electrophilic ˙CF3 radical and [Ru(bpy)3]3+. The former adds to the double bond of the silyl-protected intermediate 223 formed from 222 and TMSOTf to generate the stabilized radical intermediate 224. Subsequently, SET with the oxidized state of the photocatalyst gives the tertiary cation 225 and regenerates the ground-state photocatalyst by a radical–polar crossover process. This step could proceed via a radical chain reaction between the radical and 220 to give 225, which was supported by the reaction quantum yield value. Finally, ring expansion with THE formation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond would afford the product 227 upon elimination of the silyl group (Scheme 26).30
O bond would afford the product 227 upon elimination of the silyl group (Scheme 26).30
 | ||
| Scheme 26 Cycloalkanone synthesis via RPCC. | ||
In 2016, the group of Kim proposed an approach based on the visible-light-mediated photocatalytic arylation/ring expansion of alkenylcyclobutanols to form functionalized cyclic ketones. This is another radical–polar crossover cyclization trapped by a key ring-expansion step. The reaction of alkenylcyclobutanols and aryldiazonium salts in DMSO using 3 mol% Ru(bpy)3(PF6)2 as a photoredox catalyst under visible-light irradiation from blue LEDs furnished the corresponding cyclic ketones with moderate-to-good yields. Various aryldiazonium tetrafluoroborates and 1-(1-arylvinyl)cyclobutanols with electron-withdrawing or electron-donating aryl groups are all suitable substrates. The reaction provides a modular route to densely functionalized cyclopentanones under mild conditions and uses visible light from readily available sources.
A possible mechanism was proposed, as shown in Scheme 27. SET from excited-state Ru2+(bpy)3(PF6)2* species to the aryldiazonium tetrafluoroborate 229 would generate [Ru(bpy)3]3+ species and an electrophilic aryl radical 231. Addition to the double bond of the 1-(1-arylvinyl)-cyclobutanol 228 leads to stabilized radical intermediate 232. Then, the key radical–polar crossover step can occur with SET from 232 to the oxidized state of the photocatalyst, thereby regenerating the ground-state photocatalyst and yielding the carbocation intermediate 233. A radical-chain transfer process is also possible in this step. Finally, the cation undergoes 1,2-carbon migration via a ring expansion to yield the product 230 (Scheme 27).31
 | ||
| Scheme 27 Cyclic ketone synthesis via RPCC. | ||
Later, in 2019, an effective photocatalytic methodology for the synthesis of β-sulfonated cyclopentanones bearing a quaternary carbon center was explored through an RPCC procedure by the same group. The reaction is carried out with a vast scope of vinyl cyclobutanols and sulfonyl chlorides in acetonitrile in the presence of 3 mol% fac-Ir(ppy)3 as a photocatalyst under visible-light irradiation to obtain the desired cyclopentanones in 45–84% yields. The cyclohexanone product was obtained as well, albeit in reduced yield. The gram-scale reaction was established to show the practicability of this convenient approach.
The similar reaction mechanism is shown in Scheme 28. Single-electron reduction of the sulfonyl chloride 235 by the photoexcited photocatalyst produces sulfonyl radical 237. The radical reacts with the vinyl cyclobutanol 238, leading to a new radical intermediate 239. SET between radical intermediate 239 and the oxidized state of the photocatalyst forms reactive carbocation 240, which then undergoes ring expansion via a semipinacol-type rearrangement to give the β-sulfonated cyclopentanone 241 (Scheme 28).32
 | ||
| Scheme 28 β-Sulfonated cyclopentanone synthesis via RPCC. | ||
In 2013, the Zhu group developed an efficient visible-light-induced tandem trifluoromethylation/arylation of electron-withdrawing alkenes to form CF3-containing oxindoles bearing a quaternary carbon center. The reaction of N-aryl acrylamides and Togni's reagents in the presence of 1 mol% [Ru(phen)3Cl2] as a photoredox catalyst under visible-light irradiation from 5 W blue LEDs in DCM could synthesize a variety of CF3-containing oxindoles effectively in moderate-to-excellent yields. The preparative-scale experiment was performed under the standard conditions with a moderate yield, demonstrating the scalability of this method.
A possible mechanism for this reaction is presented in Scheme 29. The initial reduction of Togni's reagent by [Ru(phen)3]2+* via a single-electron transfer process should generate radical anion 244. Then, the CF3 radical generated from the collapse of 244 adds to the double-bond of 246 to yield radical intermediate 247, which undergoes a radical C–H functionalization to give radical intermediate 248. Finally, radical species 248 is oxidized via SET with the oxidized photocatalyst to generate carbocation 249, and subsequent deprotonation affords product 250 (Scheme 29).33
 | ||
| Scheme 29 CF3-containing oxindole synthesis via RPCC. | ||
In 2015, Xia and co-workers developed a protocol to synthesize CF3-containing oxindoles and isoquinolinediones by visible-light-induced RPCC cascade reactions with alkenyl amides as bifunctional alkenes. The reaction of alkenyl amides, CF3SO2Cl and K2HPO4 in the presence of 5 mol% Ru(bpy)3Cl2 as a photoredox catalyst under visible-light irradiation from 5 W blue LEDs in anhydrous MeCN could synthesize trifluoromethyl oxindoles and isoquinolinediones in moderate-to-good yields. A range of substrates containing various substituents on the N atom, such as butyl, isopropyl and methyl, are tolerated in these trifluoromethylarylation/1,4-aryl shift/desulfonylation cascade reactions to give the corresponding cyclic products.
A reasonable mechanism is depicted in Scheme 30. Initially, RuII is transformed into the excited state RuII* species under visible light irradiation, which then reduces CF3SO2Cl by single-electron transfer to give a CF3 radical after the release of a Cl− and SO2. The CF3 radical then undergoes addition to the double bond of the substrate to form a new radical, 256. When X is SO2, the radical undergoes an aryl migration/desulfonylation cascade reaction to give the intermediate 257. If the substituent on the N atom is alkyl, cyclization of the amidyl radical to the aryl ring of intermediate 257 leads to the final product. For X = CO, the intramolecular radical cyclization of 256 gives the intermediate 255, and the subsequent oxidation and deprotonation yield isoquinolinedione. Direct hydrogen abstraction from intermediate 257 generates trifluoromethylated amide, which might be due to the stability of the nitrogen radical when R is an aryl group (Scheme 30).34
 | ||
| Scheme 30 Oxindole and isoquinolinedione synthesis via RPCC. | ||
In 2013, the Zou group reported a general visible-light-mediated radical–polar cross-over cyclization reaction of N-arylacrylamides and aryl diazonium salts to form 3,3-disubstituted oxindoles. The reaction of N-arylacrylamides and aryl diazonium salts in the presence of 5 mol% Ru(bpy)3(PF6)2 as a photoredox catalyst under visible-light irradiation for 12 h in MeOH could afford the desired oxoindoles in moderate-to-good yields. Various N-protected substrates, such as isopropyl, butyl, or benzyl could be used as an effective substituent group in this reaction; however, acetyl or N-free N-arylacrylamides failed to give products. The characteristics of this method are product diversity, simplicity of the procedure, and mild reaction conditions.
A possible reaction mechanism is shown in Scheme 31. An aryl radical 262 is generated from diazonium salt 261 under the visible light catalysis via SET with the excited photocatalyst. This aryl radical then adds to the CC bond of N-arylacrylamides 263 to give the alkyl carbon radical 264. Intramolecular radical cyclization forms intermediate radical 265. At this point, the key radical–polar crossover step can happen with single-electron transfer from the radical 265 to the [Ru(bpy)]3+ species, leading to intermediate cation 266 and the ground-state photocatalyst. Finally, intermediate cation 266 undergoes dehydrogenation to yield the 3,3-disubstituted oxoindole 267 (Scheme 31).35
 | ||
| Scheme 31 Oxindole synthesis via RPCC. | ||
In 2016, Dolbier and co-workers described a nice photoredox-catalyzed radical–polar crossover cyclization of N-benzylacrylamides with HCF2SO2Cl as the HCF2 radical precursor to access CF2H-containing compounds. The reaction involves a tandem cyclization/dearomatization process. The reaction was performed with substituted N-benzylacrylamides, fluoroalkyl radical sources, sulfonyl chlorides, K2HPO4 and H2O in the presence of Ir(ppy)3 as a photoredox catalyst under visible-light irradiation in CH3CN and delivered the corresponding CF2H-containing 2-azaspiro[4.5]deca-6,9-diene-3,8-diones in moderate-to-good yields. The mild and efficient approach accommodates various substituted N-benzylacrylamides and a series of sulfonyl chlorides such as commercially available BrCF2COOEt. The steric influence of the N-substituent has a significant effect on the cyclization of the reaction. Interestingly, when CH2FSO2Cl and CF3CH2SO2Cl were used as radical sources, the reaction could afford sulfonyl products that retained the SO2 group.
The proposed mechanism is shown in Scheme 32. Initially, a difluoromethyl radical is generated from sulfonyl chloride under visible-light catalysis via SET with the excited Ir photocatalyst. Then, the radical adds to the CC bond of 271 to form intermediate radical 272. Dearomative cyclization then yields intermediate radical 273. Single-electron oxidation of this radical by the oxidized Ir catalyst affords intermediate cation 274. Finally, under the basic conditions, water reacts with 274 to form the product 275 upon loss of the benzyl group (Scheme 32).36
 | ||
| Scheme 32 CF2H-containing heterocycle synthesis via RPCC. | ||
A visible-light-induced RPCC of N-benzylacrylamides using the low-cost perfluorinated reagents RfI or RfBr as fluorine sources to afford perfluorinated azaspirocyclic cyclohexadienones was demonstrated by Sheng, Tang and co-workers in 2017. The reaction of N-benzylacrylamides, perfluoroalkyl iodides or bromides, K2CO3 and H2O in the presence of 2 mol% Ir(ppy)3 as a photoredox catalyst under visible-light irradiation from 5 W blue LEDs in DMF could afford a variety of corresponding dearomative cyclization products containing perfluorinated groups in 59–84% yields. The mild reaction conditions could tolerate a wide range of different N-benzylacrylamides and a series of perfluoroalkyl iodides or bromides, such as CF3I, C3F7I, C6F13I, C8F17I, CH2CF2I, BrCF2CO2Et and so on.
The putative mechanism is displayed in Scheme 33 with C4F9I as an example. Firstly, the C4F9I is reduced by the excited Ir photocatalyst, yielding the C4F9 radical 281. The radical then adds to the CC bond of N-benzylacrylamide 279 to generate the radical 282, which would quickly undergo cyclization to give intermediate radical 283. Single-electron oxidation of 283 by Ir(ppy)3+ affords intermediate cation 284. Finally, under basic conditions, water reacts with the 284 to generate the final product 285 by elimination of the methyl group (Scheme 33).37
 | ||
| Scheme 33 Perfluorinated azaspirocyclic cyclohexadienone synthesis via RPCC. | ||
In 2017, the Zhu group reported a novel and mild tandem radical–polar crossover cyclization of alkenyl aldehydes to construct cyclic ketones including indanones, cyclopentenones, 3,4-dihydronaphthalen-1(2H)-ones, and chroman-4-ones. Utilizing alkenyl aldehydes and activated bromides as substrates, the cyclization reactions were carried out smoothly in the presence of 2 mol% Ru(bpy)3Cl2 as the photoredox catalyst and additives (pyridine and LiBF4) under visible-light irradiation from 15 W blue LEDs in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of DMF/H2O at room temperature to afford a variety of cyclic ketones in moderate-to-high yields. Under the standard conditions, the reaction demonstrates good tolerance of functional groups, such as F, Br, Cl, OMe, CF3, CO2Me, CN, and Ac.
:4 mixture of DMF/H2O at room temperature to afford a variety of cyclic ketones in moderate-to-high yields. Under the standard conditions, the reaction demonstrates good tolerance of functional groups, such as F, Br, Cl, OMe, CF3, CO2Me, CN, and Ac.
The proposed reaction mechanism starts with single-electron transfer between activated bromide 289 and the excited-state photocatalyst Ru(II)*. This transfer results in the formation of an electrophilic carbon radical 290 and Ru(III) species. The radical 290 then adds to the electron-rich CC bond of 291 to form intermediate radical 292, followed by an intramolecular addition to aldehyde, yielding alkoxyl radical species 293. The alkoxyl radical is transformed into the carbon-centered radical 294via a 1,2-hydrogen atom transfer (1,2-HAT). Finally, the SET oxidation of 294 provides the cyclic product 295 in the presence of pyridine and regenerates the Ru(II) catalyst (Scheme 34).38
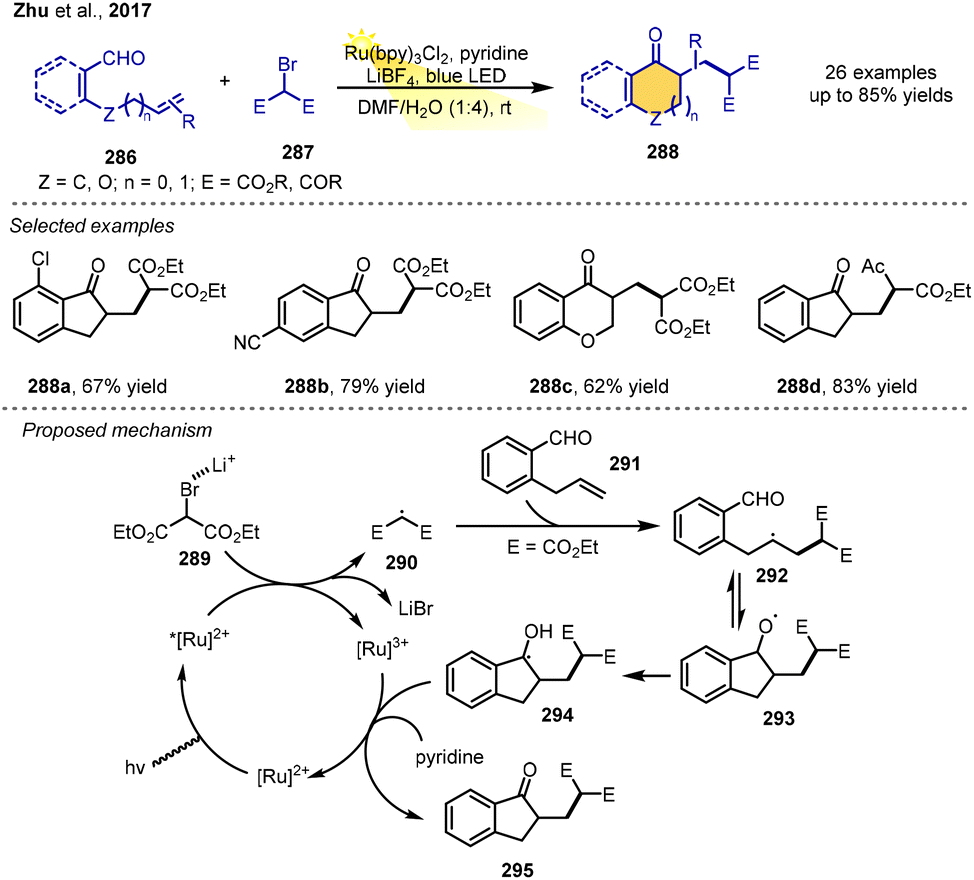 | ||
| Scheme 34 α-Bromocarbonyl compound synthesis via RPCC. | ||
4.2. Synthesis of six-membered ring compounds
The Sheng group in 2015 reported a good protocol to synthesize perfluorinated isoquinolinediones via visible-light-induced RPCC of bifunctional alkenes. The reaction of methacryloyl benzamides, perfluoroalkyl iodides and K3PO4 in the presence of 1 mol% fac-Ir(ppy)3 as a photoredox catalyst under visible-light irradiation from 5 W blue LEDs in DMF could synthesize the desired isoquinoline-1,3-diones in moderate-to-good yields. The facile and mild protocol accommodates a wide range of perfluoroalkyl halides and introduces different perfluorinated groups such as CF3, C3F7, C4F9, C6F13, C8F17, C10F21, and CF2CO2Et, leading to the useful perfluorinated isoquino-line-1,3-diones efficiently. The N-substituents of methacryloyl benzamides have an important effect on cyclization reactions, as the reaction did not work well with aryl or hydrogen on the N atom.Mechanistically, perfluororoalkylation of the methacryloyl benzamide derivative 299 leads to the carbon radical intermediate 300, followed by the cyclization on the aromatic ring of intermediate 300, which is then oxidized to carbocation intermediate 302 through a typical oxidative radical–polar crossover mechanism. Finally, this carbocation is quenched by base to yield the desired isoquinoline-1,3-dione 303 (Scheme 35).39
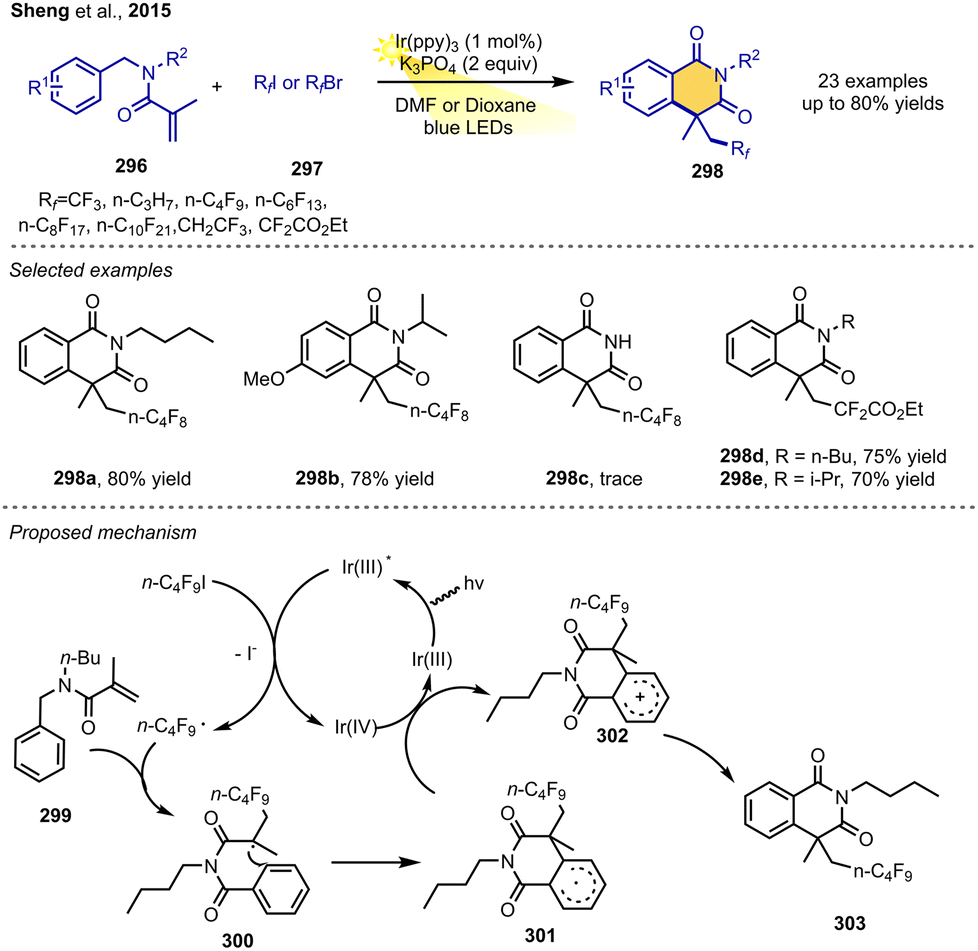 | ||
| Scheme 35 Isoquinoline-1,3-dione synthesis via RPCC. | ||
4.3. Synthesis of seven-membered ring compounds
Carboxylic acids are a class of abundant and inexpensive chemical feedstocks. In 2018, Xiao and co-workers reported an elegant approach to synthesize benzazepine derivatives via visible-light photocatalytic decarboxylative alkyl RPCC protocol. The reaction of acrylamide-tethered styrenes and alkyl radical precursors such as NHP ester, pyridine, and H2O in the presence of 1 mol% Ir(ppy)2(dtbbpy)PF6 as a photoredox catalyst under visible-light irradiation from blue LEDs in MeCN could produce a range of diverse benzazepine derivatives in moderate to good yields. The reaction is operationally simple, proceeds under relatively mild conditions, and demonstrates good functional group compatibility. The acryla-tetethered styrene could be used as a substrate for this reaction to obtain the product in a reduced 31% yield. Using the Ac-protected ursolic acid as starting material could afford “two privileged scaffolds” between the ursolic group and benzazepine, leading to the desired product in a satisfactory 58% isolated yield.A plausible mechanistic framework for this RPCC is suggested in Scheme 36. Initially, the complex 308 bound by hydrogen bonding undergoes single-electron transfer with the photoexcited Ir photocatalyst to give radical intermediate 309 and oxidation of the photocatalyst. Extrusion of CO2 and the phthalimide anion provides the carbon-centred radical, which subsequent adds to the double bond of 310, followed by the chemoselective radical addition cyclization process to form benzyl radical intermediate 312. A second single-electron transfer between radical 312 and the oxidized state of the photocatalyst (IrIV) completes the photoredox catalytic cycle and results in oxidative termination of the radical process. Finally, the resulting stabilised carbanion 313 undergoes deprotonation under base conditions to produce the product 314 (Scheme 36).40
 | ||
| Scheme 36 Benzazepine derivative synthesis via RPCC. | ||
5. Conclusions
As demonstrated in this review, the photoinduced radical–polar crossover cyclization (RPCC) of bifunctional alkenes protocol has been emerging continuously as a highly attractive strategy for the synthesis of valuable cyclic natural or non-natural products due to (1) the initiation of radicals in a gentle, green and controllable way under visible light photoredox conditions; (2) the scope of functional groups that can be incorporated into such reactions has been widely expanded by employing an RPCC process, in which the open-shell radical intermediate undergoes single-electron transfer to generate a carbocation or carbanion that subsequently is quenched with a nucleophile or electrophile or other reaction types to yield a cyclic product. A wide range of cyclization reactions were discovered with good functional group tolerance and broad substrate scope under mild and operationally simple conditions, which greatly benefit from the elaborate design of the key bifunctional alkene precursors, such as alkenes tethered to an electrophile, nucleophile, strained ring and so on. Previous review articles about photoinduced radical–polar crossover reactions mainly dealt with outlining radical–polar crossover, and no reviews before this one have aimed to highlight the significance of logical bifunctional alkene design in the area of radical polar crossover for cyclic product construction.4As a rapidly growing field in photoredox catalysis, there is still much more that remains to be explored in future research, such as (1) how to expand the scope of compatible nucleophiles and electrophiles tethered to the alkene; (2) how to design new bifunctional alkene substrates that could mediate RPCC in new forms; (3) how to control the enantioselectivity in the RPCC reaction of bifunctional alkenes using novel chiral catalysts or catalysis systems; (4) how to develop more atom-economic and environmentally friendly reactions, as this cyclization still exhibits low atom economy. We anticipate that studies of the RPCC of bifunctional alkenes will attract more interest from chemists to contribute to the development of this research field on the larger stage.
Abbreviations
| RPCC | Radical–polar crossover cyclization |
| 4CzIPN | 2,3,5,6-Tetrakis(carbazol-9-yl)-1,4-dicyanobenzene |
| TBD | 2,3,4,6,7,8-Hexahydro-1H-pyrimido[1,2-a]pyrimidine |
| SET | Single-electron transfer |
| DMSO | Dimethyl sulfoxide |
| DCM | 1,2-Dichloromethane |
| DMF | N,N-Dimethylformamide |
| TMSOTf | Trimethylsilyl trifluoromethanesulfonate |
| HFIP | Hexafluoroisopropanol |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| Dtbbpy | 4,4′-Di-tert-butyl-2,2′-bipyridine |
| HAT | Hydrogen atom transfer |
| DME | 1,2-Dimethoxyethane |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Key R&D Program (2023YFD1700500), National Natural Science Foundation of China (22301093), the Fundamental Research Funds for the Central Universities, the Central China Normal University (CCNU) and Key Laboratory of Functional Molecular Solids, Ministry of Education.References
-
(a) J. Xuan, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Visible-Light-Driven Photoredox Catalysis in the Construction of Carbocyclic and Heterocyclic Ring Systems, Eur. J. Org. Chem., 2013, 6755–6770 CrossRef CAS
; (b) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Exploration of Visible-Light Photocatalysis in Heterocycle Synthesis and Functionalization: Reaction Design and Beyond, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed
-
(a) J. M. R. Narayanam and C. R. J. Stephenson, Visible Light Photoredox Catalysis: Applications in Organic Synthesis, Chem. Soc. Rev., 2011, 40, 102–113 RSC
-
(a) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Visible Light-Driven Radical-Mediated C-C Bond Cleavage/Functionalization in Organic Synthesis, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed
-
(a) K. Donabauer and B. König, Strategies for the Photocatalytic Generation of Carbanion Equivalents for Reductant-Free C-C Bond Formations, Acc. Chem. Res., 2021, 54, 242 CrossRef CAS PubMed
- T. T. Talelea, The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules, J. Med. Chem., 2016, 59, 8712–8756 CrossRef PubMed
- J. A. Milligan, J. P. Phelan, V. C. Polites, C. B. Kelly and G. A. Molander, Radical/Polar Annulation Reactions (RPARs) Enable the Modular Construction of Cyclopropanes, Org. Lett., 2018, 20, 6840–6844 CrossRef CAS PubMed
- J. A. Milligan, K. L. Burns, A. V. Le, V. C. Polites, Z.-J. Wang, G. A. Molander and C. B. Kelly, Radical-Polar Crossover Annulation: A Platform for Accessing Polycyclic Cyclopropanes, Adv. Synth. Catal., 2020, 362, 242–247 CrossRef CAS PubMed
-
(a) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Synthesis of Functionalized Cyclopropanes from Carboxylic Acids by a Radical Addition-Polar Cyclization Cascade, Angew. Chem., Int. Ed., 2018, 57, 15430 CrossRef CAS PubMed
-
(a) W. Luo, Y. Yang, Y. Fang, X. Zhang, X. Jin, G. Zhao, L. Zhang, Y. Li, W. Zhou, T. Xia and B. Chen, Photoredox-Catalyzed Cyclopropanation of 1,1-Disubstituted Alkenes via Radical-Polar Crossover Process, Adv. Synth. Catal., 2019, 361, 4215–4221 CrossRef CAS
- Y.-M. Jiang, J. Liu, Q. Fu, Y.-M. Yu and D.-G. Yu, Visible-Light-Driven Phosphonoalkylation of Alkenes, Synlett, 2021, 378–382 CrossRef CAS
- B. Zhang, J. Luo and Y. Fang, Access to functionalized alkynylcyclopropanes via reductive radical-polar crossover-based reactions of 1,3-enynes with alkyl radicals, Org. Biomol. Chem., 2023, 21, 732–737 RSC
- C. Shu, A. Noble and V. K. Aggarwal, Photoredox-Catalyzed Cyclobutane Synthesis by a Deboronative Radical Addition-Polar Cyclization Cascade, Angew. Chem., Int. Ed., 2019, 58, 3870–3874 CrossRef CAS PubMed
- J. Tian and L. Zhou, Photoredox radical/polar crossover enables C-H gem-difunctionalization of 1,3-benzodioxoles for the synthesis of monofluorocyclohexenes, Chem. Sci., 2023, 14, 6045–6051 RSC
- W. Li, X. Chen and L. Zhou, Photocatalytic Defluorinative Three-Component Reaction of α-TrifluoromethyI Alkenes, Alkenes, and Sodium Sulfinates: Synthesis of Monofluorocyclopentenes, Org. Lett., 2022, 24, 5946–5950 CrossRef CAS PubMed
- Z. Sun and L. Zhou, Synthesis of 5-Fluoro-dihydroindolizines from Pyrrole-2-acetic Acids and TrifluoromethyI Alkenes via Dual C-F Bond Cleavage in a CF3 Group, J. Org. Chem., 2022, 87(7), 4801–4812 CrossRef CAS PubMed
- H. Chen, Y. He and L. Zhou, A photocatalytic decarboxylativeldefluorinative [4 + 3] annulation of o-hydroxyphenylacetic acids and trifluoromethyI alkenes: synthesis of fluorinated dihydrobenzoxepines, Org. Chem. Front., 2018, 5, 3240–3244 RSC
- H. Li, Y. Zhang, X. Yang, Z. Deng, Z. Zhu, X. Ouyang, Y. Yuan, X. Chen, L. Yang, M. Liu and C. Shu, Synthesis of Multifluoromethylated γ-Sultines by a Photoinduced Radical Addition-Polar Cyclization, Angew. Chem., Int. Ed., 2023, 62, e202300159 CrossRef CAS PubMed
- Y. Zhang, P. Zhou, X. Yang, H. Li, Z. Deng, M. Ren and C. Shu, Photoinduced Synthesis of Polycyclic γ-Sultines by a Radical-Polar Crossover Cyclization, Adv. Synth. Catal., 2023, 365, 3478–3483 CrossRef CAS
- S. Paul, D. Filippini, F. Flicarra, H. Melnychenko, C. Janot and M. Silvi, Oxetane Synthesis via Alcohol C-H Functionalization, J. Am. Chem. Soc., 2023, 145, 15688–15694 CrossRef CAS PubMed
- W. Zuo, L. Zuo, X. Geng, Z. Li and L. Wang, Radical-Polar Crossover Enabled Triple Cleavage of CF2Br2: A Multicomponent Tandem Cyclization to 3-Fluoropyrazoles, Org. Lett., 2023, 25, 6062–6066 CrossRef CAS PubMed
- Q. Zhao, W.-J. Hao, H.-N. Shi, T. Xu, S.-J. Tu and B. Jiang, Photocatalytic Annulation-Alkynyl Migration Strategy for Multiple Functionalization of Dual Unactivated Alkenes, Org. Lett., 2019, 21, 9784–9789 CrossRef CAS PubMed
- W. Guo, H.-G. Cheng, L.-Y. Chen, J. Xuan, Z.-J. Feng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao,
De Novo Synthesis of γ,γ-Disubstituted Butyrolactones through a Visible Light Photocatalytic Arylation-Lactonization Sequence, Adv. Synth. Catal., 2014, 356, 2787–2793 CrossRef CAS
- W.-H. Rao, Q. Li, Y.-G. Li, L.-L. Jiang, M.-X. Yue, G.-D. Zou and X. Cao, Visible-Light Photoredox-Catalyzed Acyl Lactonization of Alkenes with AcyI Chlorides, Eur. J. Org. Chem., 2023, e202300191 CrossRef CAS
- Y. Yasu, Y. Arai, R. Tomita, T. Koike and M. Akita, Highly Regio- and Diastereoselective Synthesis of CF3-Substituted Lactones via Photoredox-Catalyzed Carbolactonization of Alkenoic Acids, Org. Lett., 2014, 16, 780–783 CrossRef CAS PubMed
- N. Noto, K. Miyazawa, T. Koike and M. Akita, Anti-Diastereoselective Synthesis of CF3-Containing Spirooxazolines and Spirooxazines via Regiospecific Trifluoromethylative Spirocyclization by Photoredox Catalysis, Org. Lett., 2015, 17, 3710–3713 CrossRef CAS PubMed
- Z.-B. Yin, J.-H. Ye, W.-J. Zhou, Y.-H. Zhang, L. Ding, Y.-Y. Gui, S.-S. Yan, J. Li and D.-G. Yu, Oxy-Difluoroalkylation of Allylamines with CO2 via Visible-Light Photoredox Catalysis, Org. Lett., 2018, 20, 190–193 CrossRef CAS PubMed
- L. Sun, J.-H. Ye, W.-J. Zhou, X. Zeng and D.-G. Yu, Oxy-Alkylation of Allylamines with Unactivated Alkyl Bromides and CO2 via Visible-Light-Driven Palladium Catalysis, Org. Lett., 2018, 20, 3049–3052 CrossRef CAS PubMed
- R. Lin, H. Sun, C. Yang, W. Shen and W. Xia, Visible light-induced difunctionalization of electron-enriched styrenes: synthesis of tetrahydrofurans and tetrahydropyrans, Chem. Commun., 2015, 51, 399–401 RSC
- N. Noto, T. Koike and M. Akita, Diastereoselective Synthesis of CF3- and CF2H-Substituted Spiroethers from Aryl-Fused Cycloalkenylalkanols by Photoredox Catalysis, J. Org. Chem., 2016, 81, 7064–7071 CrossRef CAS PubMed
- B. Sahoo, J.-L. Li and F. Glorius, Visible-Light Photoredox-Catalyzed Semipinacol-Type Rearrangement: Trifluoromethylation/Ring Expansion by a Radical-Polar Mechanism, Angew. Chem., Int. Ed., 2015, 54, 11577–11580 CrossRef CAS PubMed
- S. J. Kwon and D. Y. Kim, Visible Light Photoredox-Catalyzed Arylative Ring Expansion of 1-(1-Arylvinyl)cyclobutanol Derivatives, Org. Lett., 2016, 18, 4562–4565 CrossRef CAS PubMed
- J. H. Kim and D. Y. Kim, Photocatalytic reductive sulfonylation and 1,2-alkyl migration cascades of vinyl cyclobutanols: A synthesis of β-sulfonated cyclopentanones, Synth. Commun., 2019, 50, 207 CrossRef
- P. Xu, J. Xie, Q. Xue, C. Pan, Y. Cheng and C. Zhu, Visible-Light-Induced Trifluoromethylation of N-Aryl Acrylamides: A Convenient and Effective Method To Synthesize CF3-Containing Oxindoles Bearing a Quaternary Carbon Center, Chem. – Eur. J., 2013, 19, 14039–14042 CrossRef CAS PubMed
- L. Zheng, C. Yang, Z. Xu, F. Gao and W. Xia, Difunctionalization of Alkenes via the Visible-Light-Induced Trifluoromethylarylation/1,4-Aryl Shift/Desulfonylation Cascade Reactions, J. Org. Chem., 2015, 80, 5730–5736 CrossRef CAS PubMed
- W. Fu, F. Xu, Y. Fu, M. Zhu, J. Yu, C. Xu and D. Zou, Synthesis of 3,3-Disubstituted Oxindoles by Visible-Light-Mediated Radical Reactions of Aryl Diazonium Salts with N-Arylacrylamides, J. Org. Chem., 2013, 78, 12202–12206 CrossRef CAS PubMed
- Z. Zhang, X.-J. Tang and W. R. Dolbier, Photoredox-Catalyzed Intramolecular Difluoromethylation of N-Benzylacrylamides Coupled with a Dearomatizing Spirocyclization: Access to CF2H-Containing 2-Azaspiro[4.5]deca-6,9-diene-3,8-diones, Org. Lett., 2016, 18, 1048–1051 CrossRef CAS PubMed
- S. Tang, L. Yuan, Z.-Z. Li, Z.-Y. Peng, Y.-L. Deng, L.-N. Wang, G.-X. Huang and R.-L. Sheng, Visible-light-induced dearomative spirocyclization of N-benzylacrylamides toward perfluorinated azaspirocyclic cyclohexadienones, Tetrahedron Lett., 2017, 58, 2127–2130 CrossRef CAS
- D. Lu, Y. Wan, L. Kong and G. Zhu, Visible-Light-Induced Tandem Radical Addition-Cyclization of AlkenyI Aldehydes Leading to Indanones and Related Compounds, Org. Lett., 2017, 19, 2929–2932 CrossRef CAS PubMed
- S. Tang, Y.-L. Deng, J. Li, W. X. Wang, G.-L. Ding, M.-W. Wang, G.-L. Ding, M.-W. Wang, Z.-P. Xiao, Y.-C. Wang and R.-L. Sheng, Synthesis of Perfluorinated Isoquinolinediones through Visible-Light-Induced Cyclization of Alkenes, J. Org. Chem., 2015, 80, 12599–12605 CrossRef CAS PubMed
- Y. Zhao, J.-R. Chen and W.-J. Xiao, Visible-Light Photocatalytic Decarboxylative Alkyl Radical Addition Cascade for Synthesis of Benzazepine Derivatives, Org. Lett., 2018, 20, 224–227 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2024 |