
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExchange of equatorial ligands in protein-bound paddlewheel Ru25+ complexes: new insights from X-ray crystallography and quantum chemistry†
Aarón Terán a,
Francesca Fasuloa,
Giarita Ferrarob,
Ana Edilia Sánchez-Peláezc,
Santiago Herrerocd,
Michele Pavoneb,
Ana Belén Muñoz-García*a and
Antonello Merlino*b
a,
Francesca Fasuloa,
Giarita Ferrarob,
Ana Edilia Sánchez-Peláezc,
Santiago Herrerocd,
Michele Pavoneb,
Ana Belén Muñoz-García*a and
Antonello Merlino*b
aDepartment of Physics “Ettore Pancini”, University of Naples Federico II, Complesso Universitario di Monte Sant'Angelo, Via Cinthia, 21, Naples 80126, Italy. E-mail: anabelen.munozgarcia@unina.it
bDepartment of Chemical Science, University of Naples Federico II, Complesso Universitario di Monte Sant'Angelo, Via Cinthia, 21, Naples 80126, Italy. E-mail: antonello.merlino@unina.it
cMatMoPol Research Group, Department of Inorganic Chemistry, Faculty of Chemical Sciences, Complutense University of Madrid, Avda. Complutense s/n, 28040 Madrid, Spain
dKnowledge Technology Institute, Complutense University of Madrid, Campus de Somosaguas, 28223 Pozuelo de Alarcón, Madrid, Spain
First published on 6th September 2024
Abstract
Here we report the binding of the diruthenium complex [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n (D-p-CNPhF− = N,N′-bis(4-cyanophenyl)formamidinate) to the model protein bovine pancreatic ribonuclease (RNase A), investigated for the first time by means of X-ray crystallography and Quantum Chemistry. The crystal structure reveals that the compound binds a histidine side chain with the diruthenium core anchored to RNase A at the axial site, without significantly altering the overall protein structure. The protein binding to the diruthenium core is associated with the replacement of an equatorial acetate ligand by two water molecules. This species is expected to be highly reactive in the absence of the protein. Thus, the Ru2/RNase A structure here reported can be associated with the entatic state of the artificial metalloenzyme produced upon reaction of RNase A with [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n. Quantum chemical investigations unveil the possible reaction mechanisms and help in dissecting the role of the imidazole group axial ligands on the convenient replacement of equatorial acetate ligands by water molecules.
Introduction
Protein metalation concerns the interaction between metal ions or metal compounds and proteins, resulting in the formation of metal/protein adducts where metal centers are generally coordinated to specific residue side chains. This process has a major role in biology,1 since it is involved in the correct folding and function of metalloproteins, which are estimated to be a large fraction of proteins within the cells.2 Artificial protein metalation is an important tool for protein chemical modification, creating therapeutic conjugates,3,4 structural models,5 and artificial metalloenzymes.6,7 Furthermore, it plays an essential role in the design and development of protein-based metallodrug delivery systems and significantly influences the absorption, transportation, storage and mechanism of action of these metal-based therapeutics.8,9Over the past decades, different methodologies have emerged to introduce exogenous metal complexes into proteins via non-covalent binding,10 direct metal-coordination,11,12 direct ligand-coordination,13,14 metal substitution,15 supramolecular assembly,16,17 or metal-mediated ligand activation.18 The metal binding process is a multifactorial event that depends on the nature of the metal ion, the physico-chemical properties of the protein and the reaction conditions (i.e. protein![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal ratio, pH, ionic strength, buffer, etc.). As a result, the metalation mechanism of many compounds is not clearly understood, being essential to regulate their activity and function.9,19
:metal ratio, pH, ionic strength, buffer, etc.). As a result, the metalation mechanism of many compounds is not clearly understood, being essential to regulate their activity and function.9,19
Here we focus on artificial diruthenium metalloproteins as structural models to understand the biological response described for diverse diruthenium compounds (Ru25+), e.g. cytotoxic activity,20–25 molecular carriers,26–28 RNA probing compounds,29 or anti-amyloid-β (Aβ) aggregation activity.30,31 We have recently solved the X-ray structures of several Ru2/protein adducts using Hen Egg White Lysozyme (HEWL) as a model protein. The aim of these studies has been to discern how the charge and the steric hindrance affect the biological response of diruthenium derivatives.32–35 The charge of diruthenium complexes affects their capacity to interact with a protein: cationic compounds have a strong preference for acidic residue side chains, while anionic species tend to bind proteins non-covalently, remaining on the surface.32,33 The diverse protein binding ability of differently-charged diruthenium compounds explains why only cationic species exhibit anti-Aβ aggregation activity.30,31 For species with different steric hindrance and equal charge, a similar metalation capacity has been observed.30,33 However, higher steric hindrance in those complexes implies a higher number of bulky groups, and, consequently, an increase in the lipophilicity of the compounds and a decrease of the capacity as Lewis acid of the axial positions which can influence absorption, distribution, metabolism, and elimination processes, thus affecting their cytotoxic activity and bioaccumulation.33
The coordination versatility of the diruthenium core and the stability of the metal–metal bond motivated us to explore the formation of a novel diruthenium-based metalloprotein, i.e. that formed upon reaction of the diruthenium compound [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n (D-p-CNPhF− = N,N′-bis(4-cyanophenyl)formamidinate), depicted in Fig. 1 and already characterized in a previous study,36 with the model protein bovine pancreatic ribonuclease (RNase A). In this work, we have carried out single-crystal X-ray diffraction experiments to obtain detailed information regarding the geometric parameters of the bimetallic compound and its coordination environment in the presence of the protein. In addition, we unveil new insights into the stability of these species and the mechanism of Ru2/RNase A recognition process with state-of-the-art Density Functional Theory (DFT) calculations.
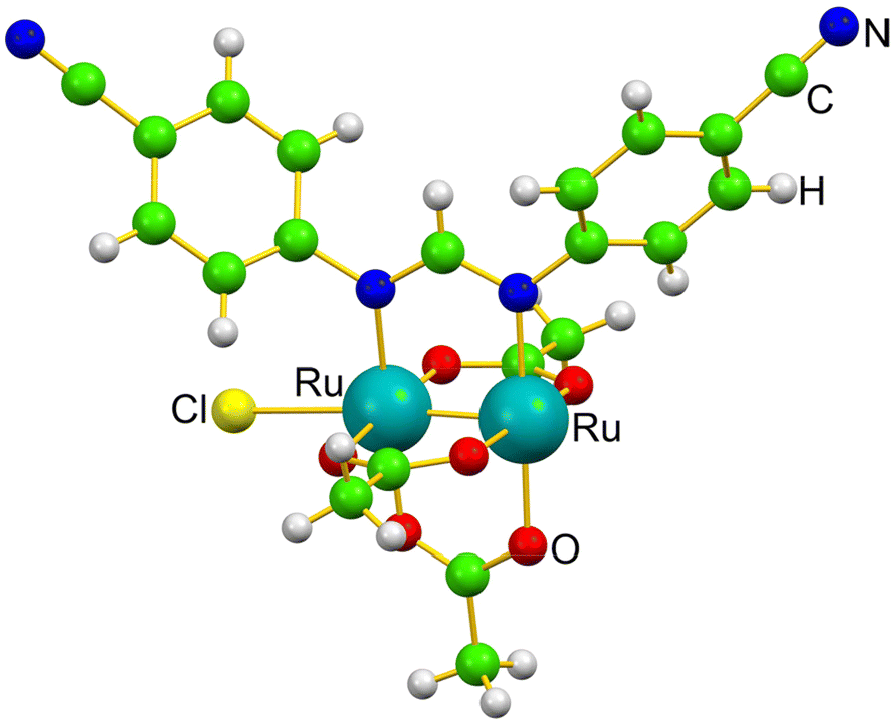 | ||
| Fig. 1 Monomer structure of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n in the solid state.36 | ||
Results and discussion
The three-dimensional (3D) structure of the adduct formed upon reaction of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n with RNase A was solved at 1.40 Å resolution. The Ru2/RNase A adduct was formed by using the soaking procedure,37 i.e. exposing metal-free monoclinic RNase A crystals, containing two protein molecules in the asymmetric unit (a.u.), hereafter denoted as chains A and B, to a stabilizing solution containing the diruthenium complex (see Methods section for further details). The overall content of the a.u. is reported in Fig. 2a.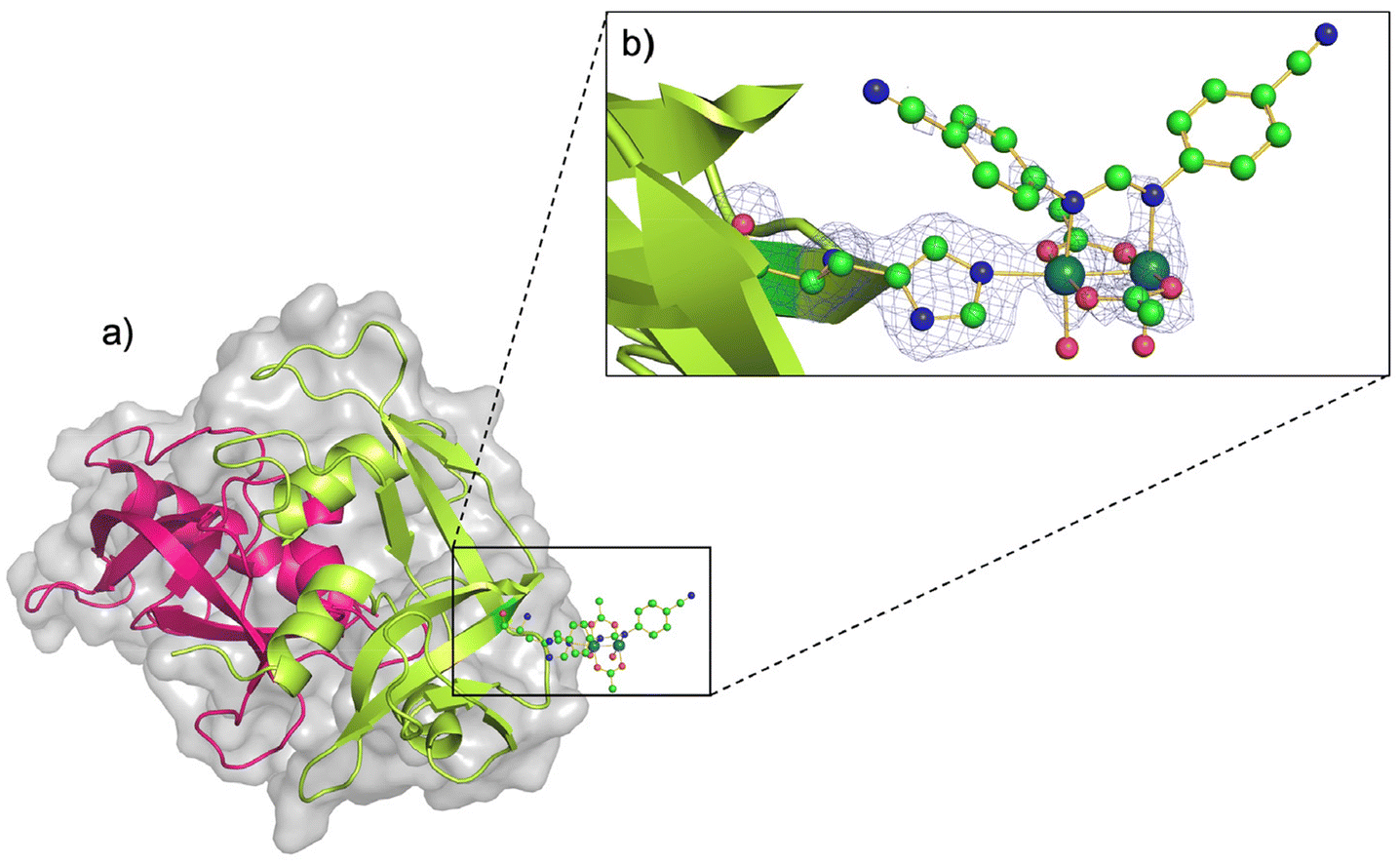 | ||
| Fig. 2 (a) Asymmetric unit content of the crystal of the artificial diruthenium metalloprotein formed upon reaction of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n with RNase A (chain A in pink and chain B in green). (b) Diruthenium binding site close to the side chain of residue His105 in chain B. 2Fo − Fc electron density map at 1.0σ level is reported in brown. C atoms are in green, N in blue, O in red and Ru atoms are in turquoise (PDB code: 9FYW). | ||
The inspection of the electron density maps reveals that only one of the two protein molecules in the a.u. is metalated (occupancy = 0.35). The Ru2 species was found close to the side chain of residue His105 of chain B (Fig. 2b). Thus, we have a metal-free RNase A (chain A) and a metalated protein (chain B) within the same crystal. This represents an ideal system to study structural variations induced by metal compound binding, since structural alterations that can be due to differences in the experimental conditions are reduced.
The overall Cα root mean square deviation (RMSD) between chains A and B shows that the metal compound binding does not significantly influence the overall structure of the protein. Indeed, the RMSD between chain A and B in the present structure is 0.46 Å, to be compared to the value obtained comparing A and B chains of metal-free RNase A from isomorphous crystals (PDB code 1JVT, RMSD = 0.29 Å).
In the metal complex binding site, Nε atom of His105 imidazole is coordinated to one of the axial positions of the bimetallic complex. The coordination sphere of the diruthenium core is completed at the equatorial sites by the D-p-CNPhF− and two remaining acetate ligands, and also by two water molecules trans to the D-p-CNPhF− ligand. The other axial position is not occupied.
The partial hydrolysis of the diruthenium compound, i.e. the replacement of an acetate equatorial ligand by two water molecules, has been observed in the crystal structure of other artificial diruthenium metalloproteins (PDB code: 4OOO,35 8BPH, 8BPU, 8BPJ, 8BQM, 8PFT, and 8PFX).32 This species with two water ligands at the equatorial positions have been proposed as an intermediate state in the substitution of equatorial acetate ligands in aqueous solution.38
Diruthenium compounds with six equatorial positions occupied by three bidentate bridging ligands and two equatorial positions occupied by two monodentate terminal ligands have been previously described as open-paddlewheel diruthenium compounds.39 Usually, these species are very reactive and tend to rebuild the paddlewheel structure by introducing a new bridging ligand,26–28 or even fabricate the fourth paddle.40 Otherwise, they decompose in solution over the time,39 especially in non-coordinating solvents. Nevertheless, for Ru2/HEWL adducts, all measurements performed by UV-vis spectroscopy showed no time-dependent changes (up to seven days) in the characteristic bands associated with the electronic transitions of the diruthenium compounds, suggesting high stability in aqueous solution even for those with water ligands at the equatorial positions.32–34 The stabilization of energized diruthenium species due to their interaction with protein atoms can be related to the entatic state of the metalloenzymes.41 This suggests the use of the artificial metalloenzyme produced upon reaction of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n with RNase A as a promising biocatalyst.
In any case, it is evident that the interaction of the diruthenium species with HEWL or RNase A enhances the reactivity of their equatorial positions. In other words, the interaction with the protein facilitates the replacement of the equatorial ligands at room temperature.33 In the absence of proteins, the substitution of acetate ligands by other bridging ligands in paddlewheel diruthenium compounds requires activation energies based on the use of heat, ultrasounds or microwaves radiation.36,42–44 However, it is important to note that the presence of a formamidinate ligand already increases the reactivity of these species compared to complexes with only carboxylate ligands at the equatorial positions. This is caused by the increase of the electron density in the bimetallic core which have a major effect on the ligand substitution rate.27,33,45
To better understand the binding mechanism of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n to RNase A and the origin of the formation of hydrolyzed species and to clarify the effect of the presence of the protein environment on the reactivity of the diruthenium core, a thorough quantum chemical investigation based on state-of-the-art DFT calculations was carried out. The crystal structure of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n (Fig. S1†)36 was used to evaluate the compound geometry and the energy of the ligand substitution reactions as depicted in Scheme 1. The imidazole (Im) molecule was used as a simplified model for the histidine residue. All the optimized structures are reported in the ESI (Fig. S2–S8†). Structurally speaking, the introduction of the imidazole ligand at the axial position of the diruthenium core produces slight variations with respect to the experimental parameters of compound [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n (Fig. S1†), mainly related to the Ru–Ru distance (Fig. S4–S7†). For derivatives 4–6, the Ru–Ru distance increase can be ascribed to the greater σ-donation from the axial ligands (imidazole and water) while the reduced Ru–Ru distance in compound 3 can be explained by the labile nature of the axial chloride ligand. Nevertheless, the obtained values for the Ru–Ru distance fall within the expected ranges for Ru25+ compounds in all cases.43
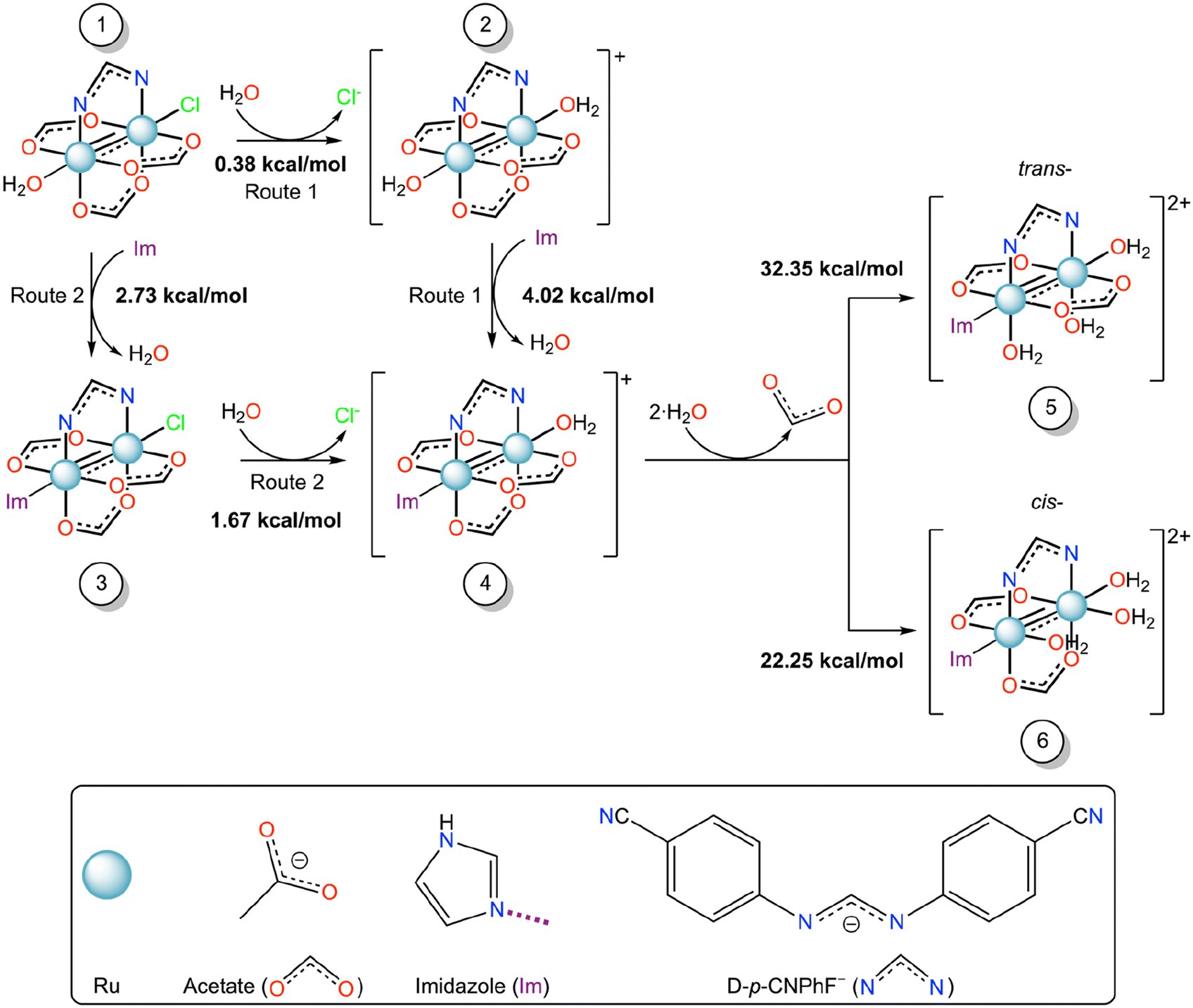 | ||
| Scheme 1 Gibbs free energy profile (ΔG, kcal mol−1) of the equatorial/axial substitution mechanism of diruthenium compounds from 1 to 6 in water solution. | ||
Various studies have tried to understand how the interaction between the diruthenium core and biological species takes place.46–48 Computational research focused on the Gibbs free energies for ligand exchange between [Ru2(O2CCH3)4(OH2)2]+, [Ru2Cl(O2CCH3)4(OH2)], and [Ru2Cl(O2CCH3)4(OH)]− and simplified models of protein residue side chains46,47 proposed that the axial ligands are promptly substituted by suitable binding sites with high exergonicity. These studies also demonstrated that the substitution of the axial water molecule is favored over the chloride ligand. Kinetic experimental studies indicated that the axial chloride ligand in [Ru2Cl(O2CCH3)4] is quite labile in aqueous media and that, in the presence of amino acids (aa), such as glycine, cysteine, histidine, and tryptophan, axial-(H2O)(aa)Ru2 species is the predominant form.49,50 Our crystal structures of Ru2/HEWL adducts support this idea, since after the axial or equatorial coordination of the protein to the Ru2 core, the presence of chloride ligands at the axial or equatorial sites has never been observed.32–35
Here, we used the DFT calculations to discern the most favorable substitution route to form the adduct observed in the crystallographic experiment, starting from [Ru2Cl(D-p-CNPhF)(O2CCH3)3(OH2)] (1), which is the first species formed when the polymer [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n is dissolved in water. To obtain this information we have used the reaction scheme reported in Scheme 1.
Computational data indicate that both routes, Route 1 and Route 2, to form compound 4 starting from compound 1 are endergonic processes. Nevertheless, the Gibbs free energy in the first substitution step suggests that Route 1 could be the most favorable pathway. These results suggest that the protein environment favors the diruthenium binding, i.e. the protein axial coordination, by decreasing the activation energy of the process.
After the protein axial coordination, the ligand substitution process continues. In the crystal structure of the Ru2/RNase A adduct here reported, the formation of the hydrolyzed compound [Ru2(D-p-CNPhF)(O2CCH3)2(OH2)2,eq(Im)ax] was observed (Fig. 2a). In principle, two isomers with trans- (5) or cis- (6) configurations with respect to the formamidinate ligand can be obtained from compound 4. In the synthesis of diruthenium compounds, usually the second equatorial substitution step in diruthenium core results in the formation of the cis-isomer.51,52 However, this is not the case when sterically hindered ligands are employed.52,53 In the adduct of other diruthenium compounds with HEWL, we have observed that the equatorial coordination could occur either cis- or trans- to the formamidinate ligand.32–34 At Asp119 binding site, cis-coordination has always been observed. However, close to the side chain of Asp101, both types of configuration have been found.32,33 The Asp101 position in HEWL is most sterically hindered than Asp119 due to crystalline packing. The steric hindrance is a decisive factor in the second step of equatorial substitution, which could have resulted in a hydrolyzed species with trans- configuration.
According to DFT calculations the formation of compound 6 should be favored with respect to that of compound 5. Thus, water molecules should replace acetate ligands in cis configuration with respect to D-p-CNPhF−. However, compound 5 mimics the structure experimentally observed. This discrepancy can be explained considering the crystal packing. Indeed, equatorial acetates are in contact with symmetry mate atoms and thus less accessible that the acetate that is in trans with respect to the formamidinate ligand (Fig. S10 in the ESI†).
To understand the effect of axial ligand on the substitution of equatorial ligands, we have set up the scenario reported in Scheme 2, i.e., we have studied the energetics of the formation of compounds 7 or 8 starting from compound 2, where the imidazole ligand is replaced by a water molecule.
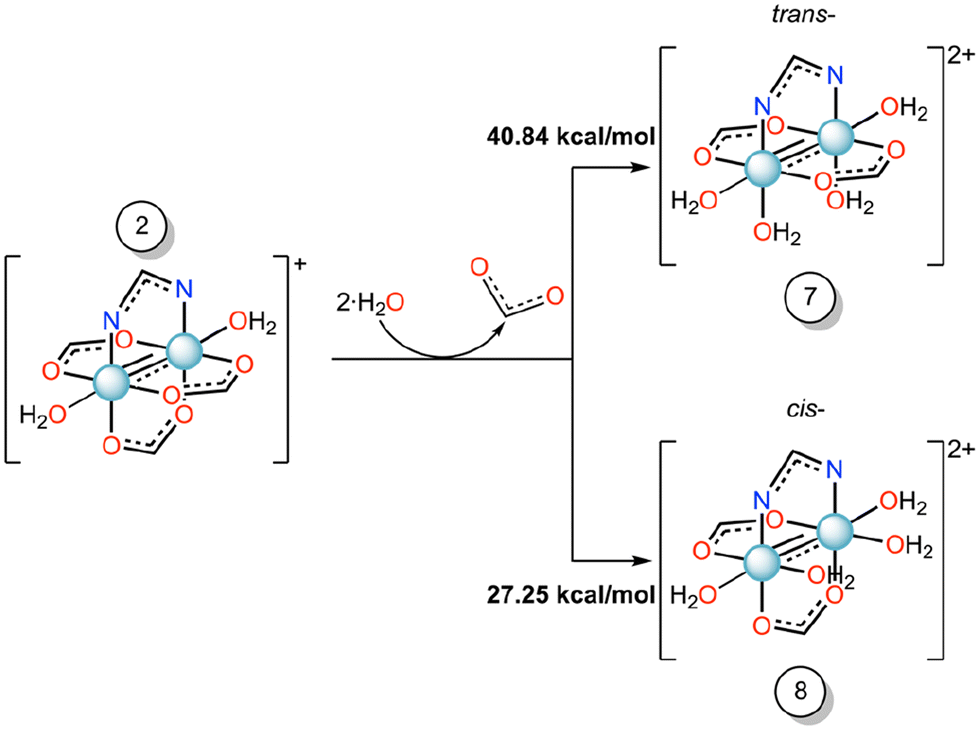 | ||
| Scheme 2 Gibbs free energy profile (ΔG, kcal mol−1) of the equatorial substitution mechanism of diruthenium compound 2 to form 7 and 8 in water solution. | ||
For these reactions, the substitution energy for both isomers is much higher than that formed when the imidazole is bound at the axial site. This suggests that the coordination of the imidazole at the axial site facilitates the equatorial ligand substitution around the coordination sphere of ruthenium atoms. Previously, in some Ru2-HEWL structures (PDB codes: 8PH7 and 8PH8), where only an axial coordination occurred with the protein, we observed the substitution of equatorial acetate ligands by other bidentate ligands found in the crystallization medium (e.g. formate or succinate ions).33
These results could help in the design of new synthetic strategies for diruthenium compounds. Our data support the idea that the protein environment could play a fundamental role in the reactivity of the bimetallic center and that the axial coordination ligand replacement could be a way to enhance the reactivity of equatorial diruthenium ligands.48
Conclusions
We have engineered the model protein bovine pancreatic ribonuclease A by incorporating the diruthenium core of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n through a direct covalent bond with the side chain of His105.Previously, theoretical calculations for other diruthenium compounds showed a high binding affinity between the inorganic core and the imidazole group of histidine.46,47 In addition, NMR results suggested that the binding of the Ru2 core to the Aβ peptide occurred through the side chain of His residues.30 However, this is the first time that the binding of a diruthenium compound to the side chain of a histidine protein residue has been observed at the atomic level. Data reveal that when the imidazole of the His is coordinated at the axial diruthenium site of [Ru2(D-p-CNPhF)(O2CCH3)3(OH2)]+, one acetate equatorial ligand is replaced by two water molecules. This substitution leads to the formation of species that would be unstable in the absence of the protein. Thus, the structure of the metalloprotein here reported seems to be an entatic state. This suggests its use as an artificial metalloenzyme.
The crystallographic results have been explained on the basis of first principles calculations, which reveal the important role played by protein environment in the binding of the diruthenium compound to the protein. Computational results also highlight the important role of the imidazole axial coordination in the reactivity of the diruthenium core. In addition, the results indicate that axial ligand substitution significantly affects the equatorial ligand exchange process. These findings suggest that proteins can be used as activators of diruthenium compounds carrying pharmacologically active ligand at the equatorial sites.25 On the other hand, after the release of the bioactive molecules, the Ru2 core remains attached to the biomolecule, giving rise to other cellular mechanisms that could affect to the cell survival, development or regulation.
Methods
Materials
The synthesis of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n was performed as previously described.36 The formamidine HD-p-CNPhF ligand precursor was prepared according the procedure reported in literature, using 4-cyanoaniline and triethyl orthoformate as reagents.54 RNase A was purchased from Sigma-Aldrich (type XIIA) and used without further purification.Crystallization, X-ray diffraction data collection and structure refinement
Metal-free RNase A crystals in the C2 space group were grown by the hanging drop vapor diffusion method mixing equal volumes of the protein (22 mg mL−1) and reservoir solutions (22% PEG4K and 10 mM sodium citrate at pH 5.1) at 20 °C. Then, the metal-free protein crystals were exposed to solid aliquots of the diruthenium complex [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n to obtain the Ru2/RNase A adduct. These crystals were captured with a nylon loop and cryoprotected using a solution containing the reservoir and 25% glycerol, after 7 days of soaking. Cryoprotected crystals were then flash-frozen in liquid nitrogen and shipped to Elettra Synchrotron in Trieste (Italy).X-ray diffraction data were collected at 100 K using the XRD2 beamline. All data were indexed, integrated, and scaled using the Global Phasing AutoPROC pipeline.55 The data collection statistics are reported in Table S1 (ESI†). The structure of the adduct was solved by molecular replacement with Phaser implemented in the CCP4 suite,56 using the structure of metal-free RNase A deposited in the PDB under the accession code 1JVT (chain A)57 as starting model. The structures were refined using Refmac.58 Manual interventions on the models based on observation of the electron density map were carried out using Coot.59 The electron density map of the protein chain is rather well defined, with the exclusion of residues 17–21 and 18–20 of chains A and B, respectively. The final structure of the adduct was validated using the PDB validation server60 and deposited in the PDB under the accession code 9FYW.
Quantum calculations
All the DFT calculations were performed with Gaussian 16.61 The structural optimization of the diruthenium complexes was computed at the level of theory B3LYP. Geometries were fully optimized without symmetry constraints. The Def2TZVP basis set was used for C, H, N, O, and Cl atoms while the SDD effective core potential and basis set was used for Ru atoms. The water solvent is considered with the Solvent Model Density (SMD) implicit solvation model. Such approach has been proven to be very effective for studying the energetics of interaction transition metal complexes and protein moieties.62 Since our solvation Free energies are computed within the continuum solvation model, no gas-to-solution corrections have been applied to the energetics of the hydrolysis processes, as recommended by recent specialized literature in the field.63 The crystal structure of [Ru2Cl(D-p-CNPhF)(O2CCH3)3]n compound (deposited in the Cambridge Structural Database64 with number 2098740) revealed a polymeric structure consisting of a linear chain of diruthenium units linked by the axial chloride ligand. However, the physicochemical characterization of this compound suggests that compound [Ru2Cl(D-p-CNPhF)(O2CCH3)3(OH2)] (1) is found as the unique form and that the polymeric compound is merely obtained under the anhydrous conditions used to crystallize this species.36 For this reason, we have taken as a starting point the structure of compound 1 for the reaction Scheme 1.Data availability
X-ray diffraction data have been deposited in the Protein Data Bank under the accession code 9FYW.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Elettra staff for technical assistance. This work was supported by the Human Frontier Science Program (Grant Number RGY0056/2022). A. M. thanks MIUR PRIN 2022- Cod. 2022JMFC3X, “Protein Metalation by Anticancer Metal-based Drugs” for financial support. Comunidad de Madrid (Project S2017/BMD-3770-CM) and Complutense University of Madrid (GRFN32/23 and Program PR3/23) are gratefully acknowledged for financial support. A. T. also acknowledges the Complutense University for a Predoctoral Grant (CT63/19-CT64/19) and Research Stay Grant (EB25/22) and the Spanish Ministry of Science and Innovation for a Postgraduate Fellowship at Residencia de Estudiantes (2021–2022).References
- D. Osman and N. J. Robinson, Protein Metalation in a Nutshell, FEBS Lett., 2023, 597(1), 141–150, DOI:10.1002/1873-3468.14500.
- A. W. Foster, D. Osman and N. J. Robinson, Metal Preferences and Metallation, J. Biol. Chem., 2014, 289(41), 28095–28103, DOI:10.1074/jbc.R114.588145.
- K. Tanaka and K. Vong, Unlocking the Therapeutic Potential of Artificial Metalloenzymes, Proc. Jpn. Acad., Ser. B, 2020, 96(3), 79–94, DOI:10.2183/pjab.96.007.
- S. Eda, I. Nasibullin, K. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. Tanaka, Biocompatibility and Therapeutic Potential of Glycosylated Albumin Artificial Metalloenzymes, Nat. Catal., 2019, 2(9), 780–792, DOI:10.1038/s41929-019-0317-4.
- A. Merlino, Recent Advances in Protein Metalation: Structural Studies, Chem. Commun., 2021, 57(11), 1295–1307, 10.1039/D0CC08053E.
- M. Wittwer, U. Markel, J. Schiffels, J. Okuda, D. F. Sauer and U. Schwaneberg, Engineering and Emerging Applications of Artificial Metalloenzymes with Whole Cells, Nat. Catal., 2021, 4(10), 814–827, DOI:10.1038/s41929-021-00673-3.
- H. J. Davis and T. R. Ward, Artificial Metalloenzymes: Challenges and Opportunities, ACS Cent. Sci., 2019, 5(7), 1120–1136, DOI:10.1021/acscentsci.9b00397.
- D. M. Monti, D. Loreto, I. Iacobucci, G. Ferraro, A. Pratesi, L. D'Elia, M. Monti and A. Merlino, Protein-Based Delivery Systems for Anticancer Metallodrugs: Structure and Biological Activity of the Oxaliplatin/β-Lactoglobulin Adduct, Pharmaceuticals, 2022, 15(4), 425, DOI:10.3390/ph15040425.
- A. Merlino, T. Marzo and L. Messori, Protein Metalation by Anticancer Metallodrugs: A Joint ESI MS and XRD Investigative Strategy, Chem. – Eur. J., 2017, 23(29), 6942–6947, DOI:10.1002/chem.201605801.
- A. Bauzá and A. Frontera, Noncovalent Interactions Involving Group 6 in Biological Systems: The Case of Molybdopterin and Tungstopterin Cofactors, Chem. – Eur. J., 2022, 28(50), e202201660, DOI:10.1002/chem.202201660.
- G. S. Biggs, O. J. Klein, S. L. Maslen, J. M. Skehel, T. J. Rutherford, S. M. V. Freund, F. Hollfelder, S. R. Boss and P. D. Barker, Controlled Ligand Exchange Between Ruthenium Organometallic Cofactor Precursors and a Naïve Protein Scaffold Generates Artificial Metalloenzymes Catalysing Transfer Hydrogenation, Angew. Chem., Int. Ed., 2021, 60(19), 10919–10927, DOI:10.1002/anie.202015834.
- J. Podtetenieff, A. Taglieber, E. Bill, E. J. Reijerse and M. T. Reetz, An Artificial Metalloenzyme: Creation of a Designed Copper Binding Site in a Thermostable Protein, Angew. Chem., 2010, 122(30), 5277–5281, DOI:10.1002/ange.201002106.
- S. A. Kerns, A. Biswas, N. M. Minnetian and A. S. Borovik, Artificial Metalloproteins: At the Interface between Biology and Chemistry, JACS Au, 2022, 2(6), 1252–1265, DOI:10.1021/jacsau.2c00102.
- E. Klemencic, R. C. Brewster, H. S. Ali, J. M. Richardson and A. G. Jarvis, Using BpyAla to Generate Copper Artificial Metalloenzymes: A Catalytic and Structural Study, Catal. Sci. Technol., 2024, 14(6), 1622–1632, 10.1039/D3CY01648J.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron, Nature, 2016, 534(7608), 534–537, DOI:10.1038/nature17968.
- T. Heinisch and T. R. Ward, Artificial Metalloenzymes Based on the Biotin–Streptavidin Technology: Challenges and Opportunities, Acc. Chem. Res., 2016, 49(9), 1711–1721, DOI:10.1021/acs.accounts.6b00235.
- J. Bos, W. R. Browne, A. J. M. Driessen and G. Roelfes, Supramolecular Assembly of Artificial Metalloenzymes Based on the Dimeric Protein LmrR as Promiscuous Scaffold, J. Am. Chem. Soc., 2015, 137(31), 9796–9799, DOI:10.1021/jacs.5b05790.
- L. Skos, Y. Borutzki, C. Gerner and S. M. Meier-Menches, Methods to Identify Protein Targets of Metal-Based Drugs, Curr. Opin. Chem. Biol., 2023, 73, 102257, DOI:10.1016/j.cbpa.2022.102257.
- L. Messori and A. Merlino, Protein Metalation by Metal-Based Drugs: X-Ray Crystallography and Mass Spectrometry Studies, Chem. Commun., 2017, 53(85), 11622–11633, 10.1039/C7CC06442J.
- D. de Oliveira Silva, Perspectives for Novel Mixed Diruthenium-Organic Drugs as Metallopharmaceuticals in Cancer Therapy, Curr. Med. Chem. Anticancer Agents, 2010, 10(4), 312–323, DOI:10.2174/187152010791162333.
- E. Barresi, I. Tolbatov, T. Marzo, E. Zappelli, A. Marrone, N. Re, A. Pratesi, C. Martini, S. Taliani, F. Da Settimo and D. La Mendola, Two Mixed Valence Diruthenium(II,III) Isomeric Complexes Show Different Anticancer Properties, Dalton Trans., 2021, 50(27), 9643–9647, 10.1039/D1DT01492G.
- E. Barresi, I. Tolbatov, A. Pratesi, V. Notarstefano, E. Baglini, S. Daniele, S. Taliani, N. Re, E. Giorgini, C. Martini, F. Da Settimo, T. Marzo and D. A. La Mendola, Mixed-Valence Diruthenium(II,III) Complex Endowed with High Stability: From Experimental Evidence to Theoretical Interpretation, Dalton Trans., 2020, 49(41), 14520–14527, 10.1039/D0DT02527E.
- S. R. Alves, A. Z. Abbasi, G. Ribeiro, T. Ahmed, X. Y. Wu and D. de Oliveira Silva, Diruthenium(II,III) Metallodrugs of Ibuprofen and Naproxen Encapsulated in Intravenously Injectable Polymer–Lipid Nanoparticles Exhibit Enhanced Activity against Breast and Prostate Cancer Cells, Nanoscale, 2017, 9(30), 10701–10714, 10.1039/C7NR01582H.
- S. R. Alves, A. Colquhoun, X. Y. Wu and D. de Oliveira Silva, Synthesis of Terpolymer-Lipid Encapsulated Diruthenium(II,III)-Anti-Inflammatory Metallodrug Nanoparticles to Enhance Activity against Glioblastoma Cancer Cells, J. Inorg. Biochem., 2020, 205, 110984, DOI:10.1016/j.jinorgbio.2019.110984.
- I. Tolbatov, E. Barresi, S. Taliani, D. La Mendola, T. Marzo and A. Marrone, Diruthenium(II,III) Paddlewheel Complexes: Effects of Bridging and Axial Ligands on Anticancer Properties, Inorg. Chem. Front., 2023, 10(8), 2226–2238, 10.1039/D3QI00157A.
- I. Coloma, M. Cortijo, I. Fernández-Sánchez, J. Perles, J. L. Priego, C. Gutiérrez, R. Jiménez-Aparicio, B. Desvoyes and S. Herrero, pH- and Time-Dependent Release of Phytohormones from Diruthenium Complexes, Inorg. Chem., 2020, 59(11), 7779–7788, DOI:10.1021/acs.inorgchem.0c00844.
- I. Coloma, M. Cortijo, M. J. Mancheño, M. E. León-González, C. Gutierrez, B. Desvoyes and S. Herrero, Diruthenium Complexes as pH-Responsive Delivery Systems: A Quantitative Assessment, Inorg. Chem. Front., 2023, 10, 4402–4413, 10.1039/D3QI00399J.
- I. Coloma, J. Parrón-Ballesteros, M. Cortijo, C. Cuerva, J. Turnay and S. Herrero, Overcoming Resistance of Caco-2 Cells to 5-Fluorouracil through Diruthenium Complex Encapsulation in PMMA Nanoparticles, Inorg. Chem., 2024, 63(28), 12870–12879, DOI:10.1021/acs.inorgchem.4c01323.
- G. Lozano, R. Jimenez-Aparicio, S. Herrero and E. Martinez-Salas, Fingerprinting the Junctions of RNA Structure by an Open-Paddlewheel Diruthenium Compound, RNA, 2016, 22(3), 330–338, DOI:10.1261/rna.054353.115.
- S. La Manna, C. Di Natale, V. Panzetta, M. Leone, F. A. Mercurio, I. Cipollone, M. Monti, P. A. Netti, G. Ferraro, A. Terán, A. E. Sánchez-Peláez, S. Herrero, A. Merlino and D. Marasco, A Diruthenium Metallodrug as a Potent Inhibitor of Amyloid-β Aggregation: Synergism of Mechanisms of Action, Inorg. Chem., 2024, 63(1), 564–575, DOI:10.1021/acs.inorgchem.3c03441.
- S. La Manna, V. Panzetta, C. Di Natale, I. Cipollone, M. Monti, P. A. Netti, A. Terán, A. E. Sánchez-Peláez, S. Herrero, A. Merlino and D. Marasco, Comparative Analysis of the Inhibitory Mechanism of Aβ1–42 Aggregation by Diruthenium Complexes, Inorg. Chem., 2024, 63(21), 10001–10010, DOI:10.1021/acs.inorgchem.4c01218.
- A. Terán, G. Ferraro, A. E. Sánchez-Peláez, S. Herrero and A. Merlino, Charge Effect in Protein Metalation Reactions by Diruthenium Complexes, Inorg. Chem. Front., 2023, 10, 5016–5025, 10.1039/D3QI01192E.
- A. Terán, G. Ferraro, P. Imbimbo, A. E. Sánchez-Peláez, D. M. Monti, S. Herrero and A. Merlino, Steric Hindrance and Charge Influence on the Cytotoxic Activity and Protein Binding Properties of Diruthenium Complexes, Int. J. Biol. Macromol., 2023, 253, 126666, DOI:10.1016/j.ijbiomac.2023.126666.
- A. Terán, G. Ferraro, A. E. Sánchez-Peláez, S. Herrero and A. Merlino, Effect of Equatorial Ligand Substitution on the Reactivity with Proteins of Paddlewheel Diruthenium Complexes: Structural Studies, Inorg. Chem., 2023, 62(2), 670–674, DOI:10.1021/acs.inorgchem.2c04103.
- L. Messori, T. Marzo, R. N. F. Sanches, H. U. Rehman, D. de Oliveira Silva and A. Merlino, Unusual Structural Features in the Lysozyme Derivative of the Tetrakis(Acetato)Chloridodiruthenium(II,III) Complex, Angew. Chem., Int. Ed., 2014, 53(24), 6172–6175, DOI:10.1002/anie.201403337.
- A. Terán, M. Cortijo, Á. Gutiérrez, A. E. Sánchez-Peláez, S. Herrero and R. Jiménez-Aparicio, Ultrasound-Assisted Synthesis of Water-Soluble Monosubstituted Diruthenium Compounds, Ultrason. Sonochem., 2021, 80, 105828, DOI:10.1016/j.ultsonch.2021.105828.
- I. Russo Krauss, G. Ferraro, A. Pica, J. A. Márquez, J. R. Helliwell and A. Merlino, Principles and Methods Used to Grow and Optimize Crystals of Protein–Metallodrug Adducts, to Determine Metal Binding Sites and to Assign Metal Ligands, Metallomics, 2017, 9(11), 1534–1547, 10.1039/C7MT00219J.
- I. Tolbatov, P. Umari and A. Marrone, Diruthenium Paddlewheel Complexes Attacking Proteins: Axial versus Equatorial Coordination, Biomolecules, 2024, 14(5), 530, DOI:10.3390/biom14050530.
- M. C. Barral, T. Gallo, S. Herrero, R. Jiménez-Aparicio, M. R. Torres and F. A. Urbanos, The First Open-Paddlewheel Structures in Diruthenium Chemistry: Examples of Intermediate Magnetic Behaviour between Low and High Spin in Ru25+ Species, Chem. – Eur. J., 2007, 13(36), 10088–10095, DOI:10.1002/chem.200700494.
- M. C. Barral, S. Herrero, R. Jiménez-Aparicio, J. L. Priego, M. R. Torres and F. A. Urbanos, Activation of Isocyanate Ligands in Complexes, J. Mol. Struct., 2008, 890(1–3), 221–226, DOI:10.1016/j.molstruc.2008.04.018.
- J. Stanek, A. Hoffmann and S. Herres-Pawlis, Renaissance of the Entatic State Principle, Coord. Chem. Rev., 2018, 365, 103–121, DOI:10.1016/j.ccr.2018.03.009.
- M. Cortijo, P. Delgado-Martínez, R. González-Prieto, S. Herrero, R. Jiménez-Aparicio, J. Perles, J. L. Priego and M. R. Torres, Microwave and Solvothermal Methods for the Synthesis of Nickel and Ruthenium Complexes with 9-Anthracene Carboxylate Ligand, Inorg. Chim. Acta, 2015, 424, 176–185, DOI:10.1016/j.ica.2014.07.063.
- M. Cortijo, R. González-Prieto, S. Herrero, J. L. Priego and R. Jiménez-Aparicio, The Use of Amidinate Ligands in Paddlewheel Diruthenium Chemistry, Coord. Chem. Rev., 2019, 400, 213040, DOI:10.1016/j.ccr.2019.213040.
- M. A. S. Aquino, Recent Developments in the Synthesis and Properties of Diruthenium Tetracarboxylates, Coord. Chem. Rev., 2004, 248(11), 1025–1045, DOI:10.1016/j.ccr.2004.06.016.
- S. Herrero, R. Jiménez-Aparicio, J. Perles, J. L. Priego, S. Saguar and F. A. Urbanos, Microwave Methods for the Synthesis of Paddlewheel Diruthenium Compounds with N,N-Donor Ligands, Green Chem., 2011, 13(7), 1885–1890, 10.1039/C0GC00800A.
- I. Tolbatov and A. Marrone, Kinetics of Reactions of Dirhodium and Diruthenium Paddlewheel Tetraacetate Complexes with Nucleophilic Protein Sites: Computational Insights, Inorg. Chem., 2022, 61(41), 16421–16429, DOI:10.1021/acs.inorgchem.2c02516.
- I. Tolbatov and A. Marrone, Reaction of Dirhodium and Diruthenium Paddlewheel Tetraacetate Complexes with Nucleophilic Protein Sites: A Computational Study, Inorg. Chim. Acta, 2022, 530, 120684, DOI:10.1016/j.ica.2021.120684.
- I. Tolbatov, P. Umari and A. Marrone, The Binding of Diruthenium (II,III) and Dirhodium (II,II) Paddlewheel Complexes at DNA/RNA Nucleobases: Computational Evidences of an Appreciable Selectivity toward the AU Base Pairs, J. Mol. Graphics, 2024, 131, 108806, DOI:10.1016/j.jmgm.2024.108806.
- R. L. S. R. Santos, R. van Eldik and D. de Oliveira Silva, Thermodynamics of Axial Substitution and Kinetics of Reactions with Amino Acids for the Paddlewheel Complex Tetrakis(Acetato)Chloridodiruthenium(II,III), Inorg. Chem., 2012, 51(12), 6615–6625, DOI:10.1021/ic300168t.
- R. L. S. R. Santos, R. van Eldik and D. de Oliveira Silva, Kinetic and Mechanistic Studies on Reactions of Diruthenium(II,III) with Biologically Relevant Reducing Agents, Dalton Trans., 2013, 42(48), 16796–16805, 10.1039/C3DT51763B.
- P. Angaridis, F. A. Cotton, C. A. Murillo, D. Villagrán and X. Wang, Paramagnetic Precursors for Supramolecular Assemblies: Selective Syntheses, Crystal Structures, and Electrochemical and Magnetic Properties of Ru2(O2CMe)4(-n(Formamidinate)nCl Complexes, n = 1–4, Inorg. Chem., 2004, 43(26), 8290–8300, DOI:10.1021/ic049108w.
- T. Ikeue, Y. Kimura, K. Karino, M. Iida, T. Yamaji, I. Hiromitsu, T. Sugimori, D. Yoshioka, M. Mikuriya and M. Handa, Structural, Magnetic, and 1H NMR Spectral Study on Lantern-Type Cis- and Trans-Diruthenium(II,III) Complexes with Two Formamidinato and Two Acetato Bridges, Inorg. Chem. Commun., 2013, 33, 133–137, DOI:10.1016/j.inoche.2013.04.001.
- P. A. Angaridis, F. A. Cotton, C. A. Murillo, A. S. Filato and M. A. Petrukhina, Diruthenium Formamidinato Complexes, in Inorganic Syntheses, John Wiley & Sons, Ltd, 2014, vol. 36, pp. 114–121. DOI:10.1002/9781118744994.ch21.
- W. Bradley and I. Wright, 129. Metal Derivatives of NN′-Diarylamidines, J. Chem. Soc., 1956,(0), 640–648, 10.1039/JR9560000640.
- C. Vonrhein, C. Flensburg, P. Keller, A. Sharff, O. Smart, W. Paciorek, T. Womack and G. Bricogne, Data Processing and Analysis with the It autoPROC Toolbox, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 293–302, DOI:10.1107/S0907444911007773.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Overview of the CCP 4 Suite and Current Developments, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 235–242, DOI:10.1107/S0907444910045749.
- L. Vitagliano, A. Merlino, A. Zagari and L. Mazzarella, Reversible Substrate–induced Domain Motions in Ribonuclease A., Proteins, 2002, 46(1), 97–104, DOI:10.1002/prot.10033.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, REFMAC 5 for the Refinement of Macromolecular Crystal Structures, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67(4), 355–367, DOI:10.1107/S0907444911001314.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and Development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(4), 486–501, DOI:10.1107/S0907444910007493.
- H. Berman, K. Henrick and H. Nakamura, Announcing the Worldwide Protein Data Bank, Nat. Struct. Mol. Biol., 2003, 10(12), 980–980, DOI:10.1038/nsb1203-980.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- D. Loreto, F. Fasulo, A. B. Muñoz-García, M. Pavone and A. Merlino, Unexpected Imidazole Coordination to the Dirhodium Center in a Protein Environment: Insights from X-Ray Crystallography and Quantum Chemistry, Inorg. Chem., 2022, 61(22), 8402–8405, DOI:10.1021/acs.inorgchem.2c01370.
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis, ACS Catal., 2019, 9(8), 6803–6813, DOI:10.1021/acscatal.9b01537.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(2), 171–179, DOI:10.1107/S2052520616003954.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi01846j |
| This journal is © the Partner Organisations 2024 |