
A novel hydrophobic tag leads to the efficient degradation of programmed death-ligand 1†
Jieke
Gao‡
a,
Yongli
Xie‡
b,
Jiantao
Zhang‡
a,
Huirong
Chen
a,
Yan
Zou
a,
Shan
Cen
b and
Jinming
Zhou
 *a
*a
aKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, Department of Chemistry, Zhejiang Normal University, 688 Yingbin Road, Jinhua 321004, P. R. China. E-mail: zhoujinming@zjnu.edu.cn
bInstitute of Medicinal Biotechnology, Chinese Academy of Medical Science, Beijing, China
First published on 4th July 2024
Abstract
The interaction of PD-L1 and PD-1 transmits the inhibitory signal to reduce the proliferation of antigen-specific T-cells in lymph nodes. The expression of PD-L1 confers a potential escaping mechanism of tumors from the host immune system. Blocking the interaction of PD-1 and PD-L1 enables tumor-reactive T cells to overcome regulatory mechanisms and induce an effective antitumor response. The hydrophobic tag tethering degrader (HyTTD) contains a hydrophobic moiety, binding to the protein of interest (POI) to mimic the misfolding state of the POI, thereby inducing the degradation of POI. In this work, using the HyTTD strategy, we selected the diphenylmethyl derivatives as the PD-L1 binding motif for PD-L1 to develop the degraders for PD-L1, and multiple hydrophobic tags were attached. As a result, two HyTTDs Z2d and Z3d efficiently decreased the protein level of PD-L1 in both NCI-H460 and HT-1080 cells with low cytotoxicity. Meanwhile, the reduction of PD-L1 protein levels by Z2d/Z3d was counteracted by MG132, which indicated that Z2d/Z3d degraded PD-L1 through the proteasome pathway. Moreover, the molecular modeling results indicated that the HyT group of Z2d or Z3d extended the surface of the protein to mimic the misfold. Importantly, our work also identified a novel HyT, which could be applied to develop the HyTTD for other target proteins.
Introduction
Overexpression of programmed cell death protein 1 (PD-1) leads to the inactivation of T cell effectors and causes the immune escape of cancer cells.1,2 Programmed death-ligand 1 (PD-L1) is the principal ligand of PD-1 (Fig. 1A), which binds to PD-L1 and transmits the inhibitory signal to reduce the proliferation of antigen-specific T-cells in lymph nodes.3,4 Therefore, the expression of PD-L1 confers a potential escaping mechanism from the host immune system.5,6 Blocking the interaction of PD-1 and PD-L1 removes the inhibitory signals of T-cell activation, which enables tumor-reactive T cells to induce an effective antitumor response.4,6 The anti-PD-L1 antibodies like avelumab, atezolizumab, or durvalumab exhibit significant clinical efficacy by blocking the binding of PD-L1 with PD-1.7,8 However, antibody-based drugs have multiple drawbacks, including low oral bioavailability, high price, and inducing immune-related adverse effects, thereby limiting the clinical application of PD-L1/PD-1 monoclonal antibodies. Therefore, the development of small-molecular PD-1/PD-L1 inhibitors has attracted huge attention in oncoimmunotherapy.9,10 | ||
| Fig. 1 The strategies to develop PD-L1 degraders. A) The complex of PD-1 and PD-L1 (PDB-ID:4ZQK); B) the PD-L1 degraders linked with the cereblon ligand; C) the proposed mechanism for HyTTD to degrade the targeted protein. | ||
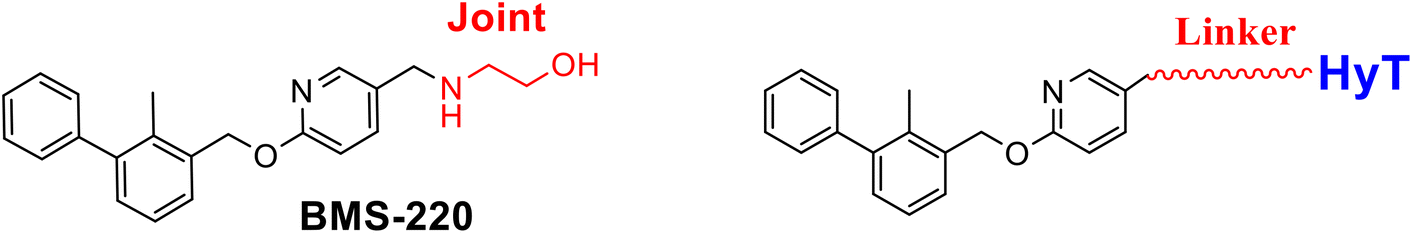
Various small-molecular PD-1/PD-L1 blockers have been reported, especially the compounds with the biphenyl core structure, which bind to PD-L1 and induce the formation of the PD-L1 dimmer to block the interaction between PD-1 and PD-L1.10–15 Targeted protein degradation (TPD) is an efficient strategy to develop degraders, which not only inhibit the protein's function via the traditional occupying mode but disable the target through catalytically degrading the target.16–19 Previously, based on the PROTACs technology, two types of PD-L1 degraders linked with the cereblon ligand were developed and indicated moderate degradation potency (Fig. 1).20,21 Hydrophobic tag tethering degrader (HyTTD) contains a moiety specifically binding with the protein of interest (POI), a hydrophobic moiety (such as HyT13, HyT16, HyT21, or HyT22, shown in Fig. 1C), and a linker between two moieties, which binds to POI to mimic the protein misfolding, thereby inducing the degradation of POI (Fig. 1).17,22–24 Herein, using the HyTTD strategy, we developed the degraders for PD-L1 protein. Importantly, a novel hydrophobic tag (HyT) was developed, which led to the efficient degradation of PD-L1.
Design and chemical synthesis of PD-L1 HyTTDs
Various PD-L1 binders have been reported. Among the binders, the biphenyl derivatives exhibit high affinities to PD-L1 by inducing the formation of PD-L1 dimers, thereby blocking the interaction between PD-L1 and PD-1. Therefore, similar to P22 or 21a (Fig. 1B), we selected the biphenyl derivatives BMS-220 and R3 as the PD-L1 binding motif for PD-L1 considering the high binding affinities with PD-L1 and ease of synthesis (structures shown in Tables 1 and 3).15,25 It has been identified that the biphenyl group of these compounds is buried in the interface site of the PD-L1 dimer,26 and the other terminal of the compounds extends to the outside of the site, which is suitable for inducing the linker and HyTs to mimic the unnatural state of PD-L1.|
|
|||
|---|---|---|---|
| Compounds | Structure | Compounds | Structure |
| L2a |

|
L3c |
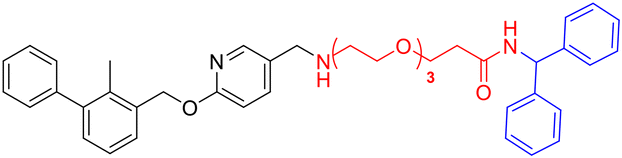
|
| L3a |

|
L4c |

|
| L4a |
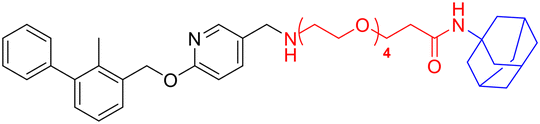
|
L5c |

|
| L5a |

|
LC1a |
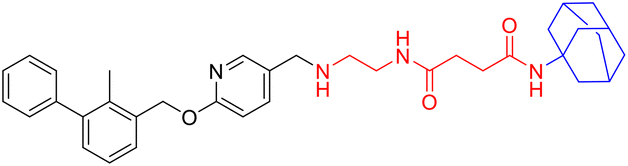
|
| L2b |

|
LC2a |

|
| L3b |
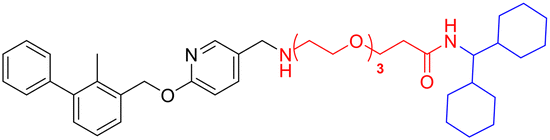
|
L2h |

|
| L4b |

|
L3h |

|
| L5b |
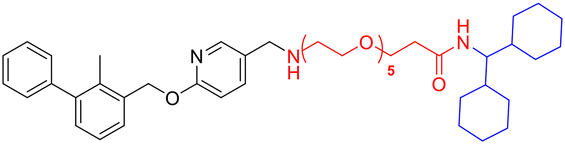
|
L4h |

|
| L2c |
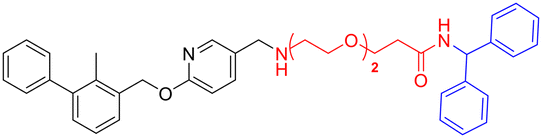
|
L5h |
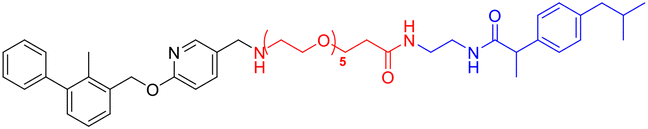
|

Firstly, we synthesized the PD-L1 binding motifs derived from BMS-220 and R3 according to Schemes S1 and S2 (ESI† data), and the corresponding aldehydes (compound 7 and compound 11) were prepared for further attaching. The linkers (NLn) were synthesized according to Scheme S3 (ESI† data) through Gabriel's reaction. While linkers CLn were obtained by cyclic dianhydrides reacting with mono-Boc-protection diamines (Scheme S3†). The PD-L1 binding motifs were attached to the linker through indirect reductive amination, and the HyT motifs were joined to the other end of the linkers through the amide condensation reaction to get the final HyTDDs (Scheme S4, ESI† data).
The introducing of the diphenylmethyl HyT leads to the decrease of PD-L1 protein level
Firstly, we try to develop the PROTACs from the PD-L1 ligand BMS-220 linked with the von Hippel–Lindau (VHL) ligand, such as L1V–L5V (Table S1†). However, such molecules exhibited no degradation activity (Fig. S1†). As the hydrophobic tag (HyT) strategy is more robust and less dependent on the unique E3 ligase than PROTAC technology, we then tried to develop HyTTD which targets the PD-L1 protein. According to the reported HyTs, such as HyT12, HyT13, HyT16, HyT21, and HyT22,24 we designed the HyTTD compounds in Table 1. We chose the PD-L1 binder BMS-220 as the motif to bind with PD-L1, and poly (ethylene glycol) (PEG) as the linker to attach the different HyTs (Table 1) to generate compounds L2a–L5a, L2b–L5b, L2c–L5c, and L2h–L5h. Furthermore, we designed two other HyTTDs, LC1a and LC2a, of which the linkers are alkyl chains. From the structures in Table 1, we would like to identify the suitable HyT that induces the degradation activity.To determine the degradation efficacy of compounds in Table 1, we further checked PD-L1 protein levels in NCI-H460 cells under 10 μM compounds treated through western blot. CDK5 inhibitor would lead to the reduction of PD-L1.27 Therefore, we selected CDK5 inhibitor dinaciclib as the positive control. As demonstrated in Fig. 2A, compounds L2c, L3c, L4c, and L5a reduced PD-L1 protein levels after the treatment for 24 h, compared to DMSO treated group. Further dose-dependent assay indicated that L2c and L3c showed mild degradation potency of PD-L1. From the results, we found that the inducing of HyTs HyT13, HyT16, HyT21, and HyT22 to the designed compounds would not lead to the reducing of PD-L1 protein level. The introducing of HyT12, the diphenylmethyl group, to compounds L2c and L3c exhibited a decrease in the PD-L1 protein levels. The results indicated that different HyTs may show diverse effects on the degradation of the protein, which was consistent with our previous summary that HyTTDs with different HyTs would lead to distinct degradation effects.17 Moreover, such results provided a clue in the optimization of the HyTTDs of PD-L1s by the modification of the diphenylmethyl HyTs.
 | ||
| Fig. 2 The effects of designed HyTTDs on the PD-L1 protein level. A) The protein level of PD-L1 treated by various HyTTDs; B) the effects of L2c, L3c, L4c, or L5a on the PD-L1 protein level with different concentrations. | ||
The substituted diphenylmethyl HyT make better degradation activity for PD-L1 HyTTDs
The fluorine substitution is often used in drug development to improve the potency and metabolic stability of lead compounds. Considering that the introducing of HyT12 (diphenylmethyl group) to the HyTTDs will lead to the decrease of PD-L1 level, we next designed and synthesized the HyTTDs with the fluorine or trifluoromethyl substituted diphenylmethyl HyTs such as L2d–L4d, L2e–L4e, and L2f–L4f (Table 2). Moreover, it was reported that the binding affinity to the POI would play a key role in the degradation potency of HyTTDs.17,28 Therefore, we designed and synthesized the HyTTDs with the binding motif to PD-L1 replaced BMS-220 by R3, which includes Z2c–Z4c, Z2d–Z3d, Z2e–Z4e, and Z2f–Z4f (Table 3).Next, we detected PD-L1 protein levels in NCI-H460 cells treated with 10 μM compounds listed in Tables 2 and 3 through western blot. As a result, compared to the DMSO or BMS-220 treated group, compound Z3c, Z4c, Z2d, Z3d, or L2d treated group showed a decrease in PD-L1 protein level (Fig. 3A). The Z2d, Z3d, or L2d treated group exhibited a comparable potency to that of dinaciclib as the result of the quantification by ImageJ (Fig. 3B). These three compounds Z2d, Z3d, and L2d all bear 4,4′-bifluorobenzhydrylpiperazinyl HyT, which belong to a novel HyT. Interestingly, through the query from the Drugbank (https://go.drugbank.com),29 we found that two approved drugs flunarizine and almitrine bearing 4,4′-bifluorobenzhydrylpiperazinyl motifs, which indicated that such a HyT group could be applied in the drug development.
|
|
|||
|---|---|---|---|
| Compounds | Structure | Compounds | Structure |
| L2d |
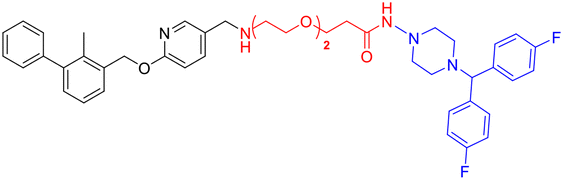
|
L4e |

|
| L3d |

|
L2f |
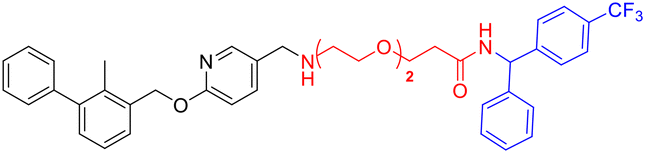
|
| L4d |
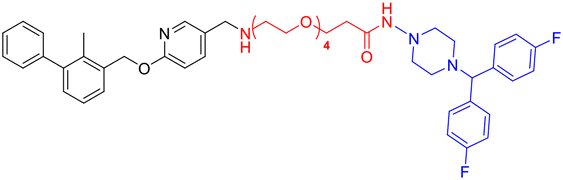
|
L3f |

|
| L2e |

|
L4f |

|
| L3e |
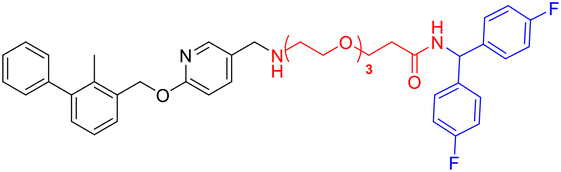
|
||
|
|
|||
|---|---|---|---|
| Compounds | Structure | Compounds | Structure |
| Z2c |

|
Z2e |

|
| Z3c |

|
Z3e |

|
| Z4c |
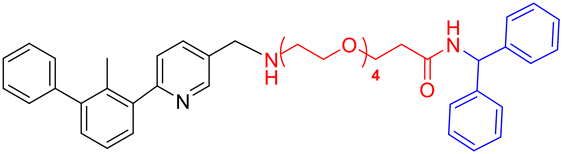
|
Z4e |

|
| Z2d |

|
Z2f |
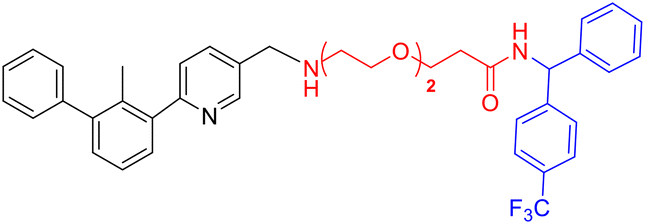
|
| Z3d |

|
Z3f |

|
| Z4d |

|
Z4f |

|

 | ||
| Fig. 3 The effects of designed HyTTDs with substituted diphenylmethyl HyT on the PD-L1 protein level. A) The protein level of PD-L1 treated by various HyTTDs; B) The protein levels were semi-quantified through the ImageJ software. | ||
The confirmation assay for HyTTDs activities
Further western blot assay indicated that compounds Z2d, Z3d, and L2d could reduce the protein levels of PD-L1 in a dose-dependent mode (Fig. 4A), which confirmed previous results. Moreover, Z2d showed a significant decrease in PD-L1 level at the concentration of 1 μM. To verify if the decrease in PD-L1 was caused by the cytotoxicity, the MTT assay was performed for the NCI-H460 cells treated by Z2d, Z3d, or L2d. As a result, the compound L2d showed mild cytotoxicity at 10 μM, while Z2d and Z3d indicated almost no cytotoxicity at the concentration range from 0.5 μM to 10 μM (Fig. 4B), which indicated that the decrease in the PD-L1 level was not caused by the toxicity of Z2d or Z3d. In our previous work, we identified a cell line HT-1080 expressing a significantly higher level of PD-L1 than other cell lines including NCI-H460.30 Therefore, we tested if Z2d and Z3d would downregulate the PD-L1 protein level in HT-1080. As a result, Z2d or Z3d treated groups showed a decrease in PD-L1 level in a dose-dependent mode (from 1 to 10 μM, Fig. S2†), which indicated that Z2d or Z3d could reduce the protein level of PD-L1 in diverse cell lines. | ||
| Fig. 4 The confirmation assay for HyTTDs activities. A) The effects of Z2d, Z3d, or L2d on the PD-L1 protein level with different concentrations; B) the cytotoxicity of Z2d, Z3d, or L2d by the MTT assay for the NCI-H460 cells. | ||
Next, the immunofluorescence assay was further performed to evaluate the effects of the compounds Z2d and Z3d on the level of PD-L1. NCI-H460 cells were treated with Z2d or Z3d in different concentrations. The cells were incubated with primary antibody to PD-L1 and followed by Alexa Fluor-conjugated secondary antibody, and nuclei were visualized by DAPI staining. As results, the immunofluorescence staining experiments showed that both Z2d and Z3d could inhibit the PD-L1 protein levels of NCI-H460 cells in a dose-dependent manner (Fig. 5). Z2d and Z3d could down-regulate the protein levels of PD-L1 at the concentration of 10 μM, which was comparable to the positive control dinaciclib. Therefore, both western blot and immunofluorescence assay indicated that Z2d and Z3d could down-regulate the PD-L1 protein levels.
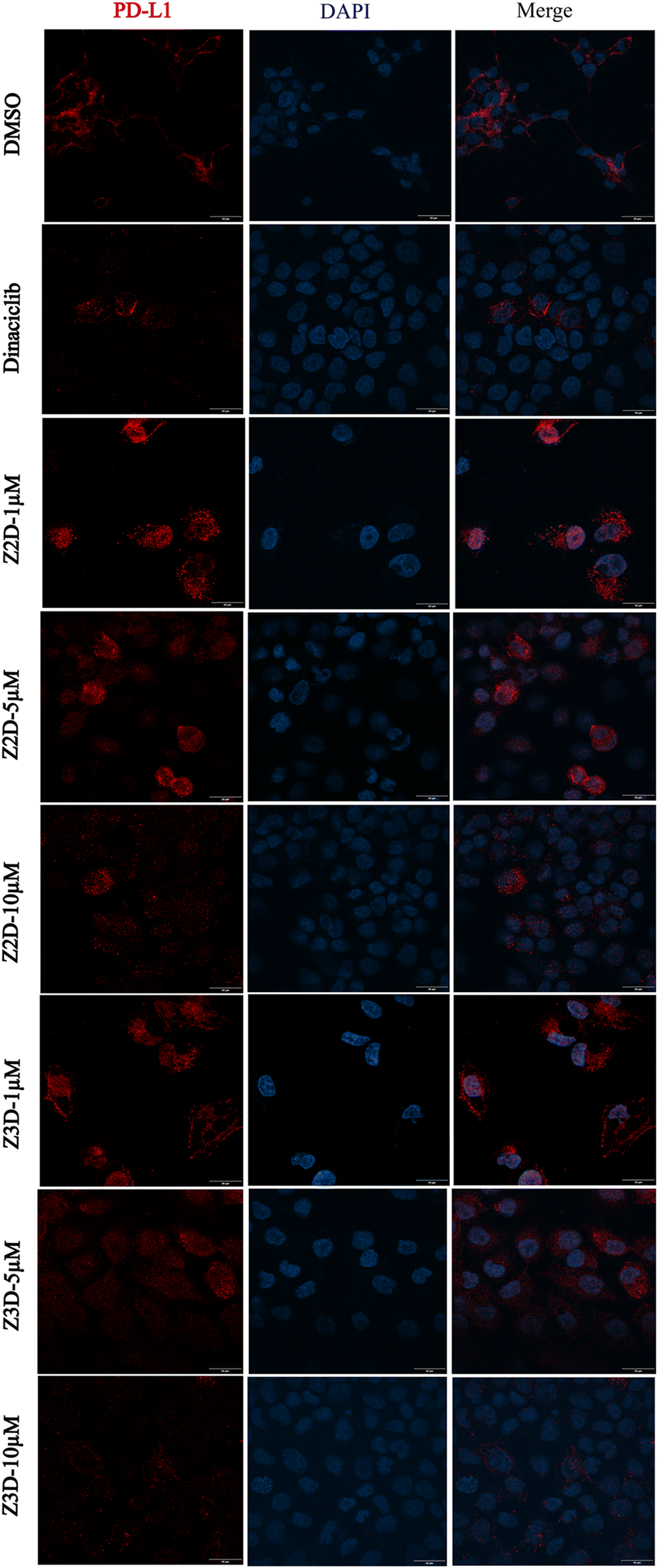 | ||
| Fig. 5 Immunofluorescence staining of PD-L1 (red) in NCI-H460 cells treated with Z2d and Z3d in different concentrations. Nuclei were visualized by DAPI staining (blue). | ||
Z2d/Z3d promoted PD-L1 degradation through proteasome pathway
The proteasome pathway is the main participant that regulates intracellular protein degradation. It was reported that HyTTD-induced protein degradation was dependent on the proteasome pathway.28 To explore whether Z2d/Z3d promoted PD-L1 degradation through the proteasome pathway, NCI-H460 cells were treated with Z2d/Z3d in the presence or absence of proteasome inhibitor MG132. As a result (Fig. 6A), the reduction of PD-L1 protein levels by Z2d/Z3d was counteracted by MG132, which indicated that Z2d/Z3d degraded PD-L1 through the proteasome pathway. Meanwhile, the time-course effects of Z2d/Z3d for PD-L1 down-regulation were evaluated at the concentration of 10 μM. Unfortunately, either Z2d or Z3d exhibited reduced degradation activity for PD-L1 in NCI-H460 cells over time, and there was almost no degradation activity for 72 h, which indicated that the Z2d or Z3d might be unstable in cell culture conditions. | ||
| Fig. 6 A) Western blot analysis of PD-L1 protein levels 10 μM Z2d/Z3d or/and 5 μM MG132; B) the time-course effects of Z2d/Z3d for PD-L1 down-regulation. | ||
The predicted binding mode between Z2d or Z3d with PD-L1
We further investigated the binding mode of Z2d or Z3d with PD-L1, and the binding modes of Z2d or Z3d in the PD-L1 site were predicted through molecular docking using the DOCK module in MOE. Using the crystal structure of the PD-L1 and compound HOU (PDB code: 7DY7)31 as the receptor structure for docking, both Z2d and Z3d were docked into the binding site occupied by compound HOU, and the results were shown in Fig. 7. The two compounds adopted similar orientations in the binding site composed by PD-L1 dimer with the biphenyl group at the subsite formed by hydrophobic residues including Ile 54 (A chain), Ala 121 (B chain), Met 115 (A and B chains), and Ile 116 (A and B chains), also similar to that of compound HOU. The PEG linkers of both compounds were extended to the upper part of the binding site and formed a hydrogen bond with Lys 124 (B chain). Particularly, the HyTs were stretched into the solvent for both compounds, which was supposed to mimic unnatural state proteins to induce the degradation (Fig. 7). Our molecular modeling results indicated that the attachment of two or three glycol-linked chains and 4,4′-bifluorobenzhydrylpiperazinyl HyT would not affect the binding mode of the PD-L1 binding motif (compared to the compound HOU), and such a long linker (two or three glycol linked chains) also allowed the HyT group extending the surface of the protein to mimic the misfold. | ||
| Fig. 7 The predicted binding mode between Z2d or Z3d with PD-L1. A) The binding mode and detailed interactions between the Z2d and PD-L1. The PD-L1 is in surface and the Z2d is in stick; B) the binding mode and detailed interactions between the Z3d and PD-L1. The PD-L1 is in the surface and the Z3d is in stick. The ligand–protein interaction illustrations were performed with UCSF ChimeraX. The hydrogen bond is indicated by a blue dash. | ||
Discussion
Herein we designed and synthesized a series of PD-L1 HyTTDs with different linkers, different HyTs, and the different PD-L1 binder motifs derived from BMS-220 and R3. Among the compounds, Z2d and Z3d showed good degradation activity for PD-L1 in multiple cell lines including NCI-H460 and H-1080. Moreover, our work indicated that the linker type or HyT type has a great effect on the degradation activity of the PD-L1 HyTTDs. We found that the inducing of the diphenylmethyl HyTs had the degradation potency for PD-L1, and further modification of the diphenylmethyl HyTs led to the HyTTD with the better degradation potency. Importantly, we developed several HyTs through the modification of the diphenylmethyl group and identified the 4,4′-bifluorobenzhydrylpiperazinyl group as the novel HyT for the development of PD-L1 HyTTD. Such a HyT could be applied to develop the HyTTD for other target proteins.TPD strategy has been applied to develop the inhibitor of PD-1 and PD-L1 interaction, such as recently reported compounds 21a, P22, and F4.20,21,32 The HyTTD technology is a valuable complement to PROTAC technology with more robust and less dependent on the unique E3 ligase than PROTAC technology. According to the current degrading mechanism, the HyTTD could degrade the intracellular PD-L1 rather than the cell-surface PD-L1. Interestingly, the degradation of intracellular PD-L1 also led to a decrease of cell surface PD-L1.20 The intracellular PD-L1 is a reservoir for the replenishment of antibody-degraded membrane PD-L1, the degradation of intracellular PD-L1 could lead to more effective cancer immunotherapy. Meanwhile, it is reported that the cytoplasmic domain of PD-L1 can regulate PD-L1 stability and function through multiple pathways. Therefore, the intracellular domain of PD-L1 and its regulatory pathways could be promising targets for cancer therapy, expanding available strategies for combined immunotherapy.33 Moreover, the intracellular PD-L1 increases mRNA expression of factors regulating the DNA damage response (DDR), which in turn contributes to tumor cell resistance to radiotherapy and chemotherapy.34 Combined targeting of the intracellular and extracellular functions of PD-L1 may represent a promising approach that enhances the antitumor immune activity.
However, the degradation potency of HyTTDs is not significant compared to PROTAC.17,28 In the case of PD-L1 as POI, our developed HyTTDs exhibited promising activity in degrading the protein of PD-L1, which indicated that HyTTD technology was a potential strategy to develop the PD-L1 degraders, and the further optimization and in vivo evaluation should be performed to obtain the HyTTD PD-L1 degraders with better potency.17 It was reported that PROTAC 21a downregulated PD-L1 protein in a proteasome-dependent manner.21 Surprisingly, although designed as the PROTAC, P22 reduces the protein levels of PD-L1 in a lysosome-dependent manner.20 As for our HyTTDs, current data support that both Z2d and Z3d downregulate PD-L1 levels in a proteasome-dependent manner, however, the precise mechanisms of HyTTD-induced PD-L1 degradation remain unclear.
Methods
General procedure for the synthesis of HyTTDs
The synthetic routes for PD-L1 binder motifs and linkers were referred to Schemes S1–S3 (ESI† data). The detailed synthesis procedures of the HyTTDs (Scheme S4†) and the detailed spectral data were listed in the ESI.† Herein, we listed the procedure of the synthesis of L2a–L5i as examples.Compound 7 (PD-L1 binder motif derived from BMS-220) was dissolved in methanol under a nitrogen atmosphere, then the linkers NL2–NL5 were added dropwise to the solution and stirred overnight at 40 °C. NaBH4 (3.0 eq.) was added and the solvent was evaporated under reduced pressure, and the crude product was separated by column chromatography to give compound 17.
Compound 17 (compound 18) was dissolved in DCM, then TFA was added dropwise to form TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM (1:1), stirred at room temperature for 2 h. The solvent was evaporated under reduced pressure to obtain the crude product compound 19, and the crude product was used directly in the next step.
:DCM (1:1), stirred at room temperature for 2 h. The solvent was evaporated under reduced pressure to obtain the crude product compound 19, and the crude product was used directly in the next step.
Compound 19, HATU (1.5 eq.), and DIPEA (5.0 eq.) were dissolved in DCM, and R3–NH2 (R3 = a–i, 1.0 eq.) dissolved in DCM was added dropwise and stirred at room temperature for 3 hours. The organic phase extract was dried over anhydrous sodium sulfate, the solvent was evaporated under reduced pressure, and the crude product was separated by column chromatography to give hydrophobic labeled compounds L2a–L5i.
Cell culture
NCI-H460 and HT-1080 cell lines were purchased from American Type Culture Collection (ATCC). HT-1080 cells were cultured in minimum essential medium (MEM; Thermo Fisher Scientific) with 10% fetal bovine serum (FBS; Gibco) at 37 °C in a 5% CO2 incubator, and H-460 cells were cultured in Roswell Park Memorial Institute – 1640 (RPMI-1640; Thermo Fisher Scientific) with 10% fetal bovine serum (FBS; Gibco) at 37 °C in a 5% CO2 incubator.Western blot
HT-1080 cells were seeded at a density of 1 × 106 cells per well in a 6-well plate. After overnight incubation, 2 μL DMSO (Sigma-Aldrich) or 2 μL chemical compounds with different concentrations were added to each well at the designated concentration. After another 24 h incubation, the cells were lysed with RIPA. Then the protein lysis was denatured at 100 °C in SDS-PAGE loading buffer for 15 min was treated and separated by 10% SDS-PAGE. The membranes were probed with primary antibodies against PD-L1 overnight at 4 °C. All antibodies were purchased from Cell Signaling Technology and diluted at a ratio of 1:1000. Mouse anti-β-actin (1:5000, Abcam) was used as a loading control. Proteins were visualized using anti-mouse or anti-rabbit HRP-conjugated secondary antibodies (1:5000, Zhongshan Jinqiao Biotechnology Company) and ECL-Plus (Millipore).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (in 1× PBS) for 15 min at room temperature, and then washed with 1× PBS three times. After blocking with 0.5% BSA for 1 h, the cells were incubated with primary antibody to PD-L1 (ab205921, 1:200, Abcam, USA) overnight at 4 °C with gentle shaking. After washing with 1× PBS three times, Alexa Fluor-conjugated secondary antibodies (Alexa Fluor 555 – donkey anti-rabbit) (1:1000, Life Technologies, Shanghai China) were added for a further incubation of 30 min in dark. Nucleus were stained with DAPI (4′,6-diamidino-2-phenylindole). Images were recorded with a PerkinElmer Ultra View VoX confocal imaging system.
MTT assay
NCI-H460 cells were seeded at a density of 4 × 103 cells per well in 96-well plates in the complete RPMI-1640 growth medium, after 24 h incubation, 1 μL DMSO (Sigma-Aldrich) or 1 μL chemical compounds at different concentration were added to each well. After 24 h, 48 h, and 72 h incubation, 20 μL MTT solution (5 mg mL−1 in PBS) was added to each well and incubated for another 4 h. The MTT formazan formed by metabolically viable cells was dissolved in 100 μL isopropanol. The absorbance was measured at 570 nm wavelength on a plate reader (EnSpire 2300, PerkinElmer). Experiments were performed in triplicate. The value of the DMSO group was defined as 100%.Molecular modeling
The binding mode for Z2d or Z3d in the binding site of PD-L1 (PDB code: 7DY7) was generated through molecular docking using MOE (Molecular Operating Environment) version 2009.10. In general, the docking was performed through the ‘DOCK’ module in MOE using the alpha-triangle placement method. 2000 poses were generated during molecular docking. Refinement of the docked poses was carried out using the force field refinement scheme and scored using both the affinity dG and London dG scoring system. The pose with the highest docking score was returned for further analysis. The ligand–protein interaction illustrations were performed with UCSF ChimeraX.35Statistical analysis
Data are presented as mean standard deviation (SD) from at least three independent experiments. Statistical analyses were performed using a two-tailed Student's t-test, or one-way ANOVA with Dunnet's post hoc test for multiple comparisons by using SPSS software (version 25.0.IBM Corp., Armonk, NY, USA) as appropriate. P values ≦ of 0.05 were considered significant. * denotes p ≦ 0.05, ** indicates p ≦ 0.01.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was in part supported by the Nature Science Foundation of China (22077115, 81672559, 81311120299). Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.References
- R. H. Thompson, H. Dong, C. M. Lohse, B. C. Leibovich, M. L. Blute, J. C. Cheville and E. D. Kwon, Clin. Cancer Res., 2007, 13, 1757–1761 CrossRef CAS PubMed.
- Y. Zhang, S. Huang, D. Gong, Y. Qin and Q. Shen, Cell. Mol. Immunol., 2010, 7, 389–395 CrossRef PubMed.
- N. Boisgerault and P. Bertrand, Eur. J. Med. Chem., 2023, 256, 115465 CrossRef CAS PubMed.
- P. Sharma, S. Goswami, D. Raychaudhuri, B. A. Siddiqui, P. Singh, A. Nagarajan, J. Liu, S. K. Subudhi, C. Poon, K. L. Gant, S. M. Herbrich, S. Anandhan, S. Islam, M. Amit, G. Anandappa and J. P. Allison, Cell, 2023, 186, 1652–1669 CrossRef CAS PubMed.
- C. Sun, R. Mezzadra and T. N. Schumacher, Immunity, 2018, 48, 434–452 CrossRef CAS PubMed.
- F. K. Dermani, P. Samadi, G. Rahmani, A. K. Kohlan and R. Najafi, J. Cell. Physiol., 2019, 234, 1313–1325 CrossRef CAS PubMed.
- G. I. Vladimer, B. Snijder, N. Krall, J. W. Bigenzahn, K. V. M. Huber, C. H. Lardeau, K. Sanjiv, A. Ringler, U. W. Berglund, M. Sabler, O. L. de la Fuente, P. Knöbl, S. Kubicek, T. Helleday, U. Jäger and G. Superti-Furga, Nat. Chem. Biol., 2017, 13, 681–690 CrossRef CAS PubMed.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- H. F. Zhu and Y. Li, Nat. Prod. Bioprospect., 2018, 8, 297–301 CrossRef CAS PubMed.
- B. Cheng, Y. Xiao, M. Xue, H. Cao and J. Chen, J. Med. Chem., 2020, 63, 15389–15398 CrossRef CAS PubMed.
- P. Zhu, J. Zhang, Y. Yang, L. Wang, J. Zhou and H. Zhang, Mol. Diversity, 2022, 26, 245–264 CrossRef CAS PubMed.
- J. Guo, L. Luo, Z. Wang, N. Hu, W. Wang, F. Xie, E. Liang, X. Yan, J. Xiao and S. Li, J. Med. Chem., 2020, 63, 13825–13850 CrossRef CAS PubMed.
- S. Basu, J. Yang, B. Xu, K. Magiera-Mularz, L. Skalniak, B. Musielak, V. Kholodovych, T. A. Holak and L. Hu, J. Med. Chem., 2019, 62, 7250–7263 CrossRef CAS PubMed.
- K. Guzik, K. M. Zak, P. Grudnik, K. Magiera, B. Musielak, R. Torner, L. Skalniak, A. Domling, G. Dubin and T. A. Holak, J. Med. Chem., 2017, 60, 5857–5867 CrossRef CAS PubMed.
- T. Wang, S. Cai, M. Wang, W. Zhang, K. Zhang, D. Chen, Z. Li and S. Jiang, J. Med. Chem., 2021, 64, 7390–7403 CrossRef CAS PubMed.
- R. R. Paudel, D. Lu, S. Roy Chowdhury, E. Y. Monroy and J. Wang, Biochemistry, 2023, 62, 564–579 CrossRef CAS PubMed.
- J. Gao, J. Zhang, X. Han and J. Zhou, Curr. Med. Chem., 2023, 30, 3137–3155 CrossRef CAS PubMed.
- L. Zhao, J. Zhao, K. Zhong, A. Tong and D. Jia, Signal Transduction Targeted Ther., 2022, 7, 113 CrossRef CAS PubMed.
- P. P. Chamberlain and L. G. Hamann, Nat. Chem. Biol., 2019, 15, 937–944 CrossRef CAS PubMed.
- B. Cheng, Y. Ren, H. Cao and J. Chen, Eur. J. Med. Chem., 2020, 199, 112377 CrossRef CAS PubMed.
- Y. Wang, Y. Zhou, S. Cao, Y. Sun, Z. Dong, C. Li, H. Wang, Y. Yao, H. Yu, X. Song, M. Li, J. Wang, M. Wei, G. Yang and C. Yang, Bioorg. Chem., 2021, 111, 104833 CrossRef CAS PubMed.
- S. Xie, F. Zhan, J. Zhu, Y. Sun, H. Zhu, J. Liu, J. Chen, Z. Zhu, D. H. Yang, Z. S. Chen, H. Yao, J. Xu and S. Xu, Angew. Chem., Int. Ed. Engl., 2023, 62, e202217246 CrossRef CAS PubMed.
- S. Xie, J. Zhu, J. Li, F. Zhan, H. Yao, J. Xu and S. Xu, J. Med. Chem., 2023, 66, 10917–10933 CrossRef CAS PubMed.
- T. K. Neklesa, H. S. Tae, A. R. Schneekloth, M. J. Stulberg, T. W. Corson, T. B. Sundberg, K. Raina, S. A. Holley and C. M. Crews, Nat. Chem. Biol., 2011, 7, 538–543 CrossRef CAS PubMed.
- Bristol-Myers Squibb Company, USA Pat., WO2015034820A1, 2015 Search PubMed.
- K. Guzik, K. M. Zak, P. Grudnik, K. Magiera, B. Musielak, R. Törner, L. Skalniak, A. Dömling, G. Dubin and T. A. Holak, J. Med. Chem., 2017, 60, 5857–5867 CrossRef CAS PubMed.
- R. D. Dorand, J. Nthale, J. T. Myers, D. S. Barkauskas, S. Avril, S. M. Chirieleison, T. K. Pareek, D. W. Abbott, D. S. Stearns, J. J. Letterio, A. Y. Huang and A. Petrosiute, Science, 2016, 353, 399–403 CrossRef CAS PubMed.
- Q. He, X. Zhao, D. Wu, S. Jia, C. Liu, Z. Cheng, F. Huang, Y. Chen, T. Lu and S. Lu, Eur. J. Med. Chem., 2023, 260, 115741 CrossRef CAS PubMed.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–D1082 CrossRef CAS PubMed.
- Y. Xie, J. Ding, X. Cui, M. Wu, C. Huang, R. Zhang, J. Wang, X. Li, S. Cen and J. Zhou, Eur. J. Pharm. Sci., 2020, 142, 105088 CrossRef CAS PubMed.
- T. Wang, S. Cai, Y. Cheng, W. Zhang, M. Wang, H. Sun, B. Guo, Z. Li, Y. Xiao and S. Jiang, J. Med. Chem., 2022, 65, 3879–3893 CrossRef CAS PubMed.
- C. Pan, M. Luo, Y. Lu, X. Pan, X. Chen, L. Ding, J. Che, Q. He and X. Dong, Bioorg. Chem., 2022, 125, 105820 CrossRef CAS PubMed.
- F. Chai, P. Li, X. Liu, Z. Zhou and H. Ren, J. Mol. Cell Biol., 2023, 15(11), mjad070 CrossRef PubMed.
- X. Tu, B. Qin, Y. Zhang, C. Zhang, M. Kahila, S. Nowsheen, P. Yin, J. Yuan, H. Pei, H. Li, J. Yu, Z. Song, Q. Zhou, F. Zhao, J. Liu, C. Zhang, H. Dong, R. W. Mutter and Z. Lou, Mol. Cell, 2019, 74, 1215–1226.e1214 CrossRef CAS PubMed.
- E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, Protein Sci., 2023, 32, e4792 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00320a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |