
Engineering sulfated polysaccharides and silk fibroin based injectable IPN hydrogels with stiffening and growth factor presentation abilities for cartilage tissue engineering†
Akansha
Dixit‡
ab,
Aman
Mahajan‡
ab,
Rakshita
Saxena§
ab,
Saptomee
Chakraborty§
 ab and
Dhirendra S.
Katti
*ab
ab and
Dhirendra S.
Katti
*ab
aDepartment of Biological Sciences and Bioengineering, Indian Institute of Technology-Kanpur, Kanpur-208016, Uttar Pradesh, India. E-mail: dsk@iitk.ac.in
bThe Mehta Family Centre for Engineering in Medicine, Indian Institute of Technology-Kanpur, Kanpur-208016, Uttar Pradesh, India
First published on 28th February 2024
Abstract
The extracellular matrix (ECM) presents a framework for various biological cues and regulates homeostasis during both developing and mature stages of tissues. During development of cartilage, the ECM plays a critical role in endowing both biophysical and biochemical cues to the progenitor cells. Hence, designing microenvironments that recapitulate these biological cues as provided by the ECM during development may facilitate the engineering of cartilage tissue. In the present study, we fabricated an injectable interpenetrating hydrogel (IPN) system which serves as an artificial ECM and provides chondro-inductive niches for the differentiation of stem cells to chondrocytes. The hydrogel was designed to replicate the gradual stiffening (as a biophysical cue) and the presentation of growth factors (as a biochemical cue) as provided by the natural ECM of the tissue, thus exemplifying a biomimetic approach. This dynamic stiffening was achieved by incorporating silk fibroin, while the growth factor presentation was accomplished using sulfated-carboxymethyl cellulose. Silk fibroin and sulfated-carboxymethyl cellulose (s-CMC) were combined with tyraminated-carboxymethyl cellulose (t-CMC) and crosslinked using HRP/H2O2 to fabricate s-CMC/t-CMC/silk IPN hydrogels. Initially, the fabricated hydrogel imparted a soft microenvironment to promote chondrogenic differentiation, and with time it gradually stiffened to offer mechanical support to the joint. Additionally, the presence of s-CMC conferred the hydrogel with the property of sequestering cationic growth factors such as TGF-β and allowing their prolonged presentation to the cells. More importantly, TGF-β loaded in the developed hydrogel system remained active and induced chondrogenic differentiation of stem cells, resulting in the deposition of cartilage ECM components which was comparable to the hydrogels that were treated with TGF-β provided through media. Overall, the developed hydrogel system acts as a reservoir of the necessary biological cues for cartilage regeneration and simultaneously provides mechanical support for load-bearing tissues such as cartilage.
1. Introduction
Articular cartilage (AC) is a connective tissue present at the end of bones which helps in the smooth functioning of the joint and facilitates load transfer. Due to its load-bearing function, articular cartilage is highly prone to chronic and acute injuries and its associated defects which can result in the impairment of joint function and at times even disability. Owing to a sparse population of resident chondrocytes and progenitor cells, a lack of vasculature and dense extracellular matrix (ECM), AC has a limited ability to self-repair.1,2 Current treatment methods used in the clinics for cartilage repair are microfracture, mosaicplasty, autografts or allografts and autologous chondrocyte implantation. While these approaches have demonstrated some clinical success, they are invasive and have limitations like extended rehabilitation time and low patient compliance. Additionally, the tissue regenerated by these methods often results in cartilage with inferior functional properties.3,4 Hence, engineering of AC using tissue engineering approaches has emerged as an alternative for the regeneration of functionally mature articular cartilage.Amongst the available tissue engineering strategies, cell-based therapies have shown promise in the regeneration and repair of cartilage tissue.5,6 Mesenchymal stem cells (MSCs) owing to their multilineage potential and high proliferative capacity are the desirable cells for cartilage tissue engineering. However, they require specific signals either chemical or physical, to effectively differentiate into chondrocytes.7 In the natural tissue environment, these chemical and physical cues are presented by the ECM of the tissue during both the developing and mature stages, facilitating tissue formation, remodeling and ultimately maintaining its homeostasis.8 Therefore, it is important to create a microenvironment that imparts the essential biophysical and biochemical signals required for the differentiation of MSCs to chondrocytes, thereby facilitating cartilage regeneration.
During cartilage development and maturation, a gradual increase in biomechanical properties has been observed due to an increase in collagen density.9,10 This transition of the biomechanical properties of the ECM from the embryonic stage to adulthood signifies the importance of physical factors in cartilage development. Importantly, the mechanical cues generated during this process play a pivotal role in chondrocyte geometry and arrangement, thereby guiding proper tissue morphogenesis and skeletal development.11,12 Hence, creating a microenvironment that offers dynamic mechanical properties could be an important fundamental aspect that can be considered while designing scaffolds aimed at stimulating the differentiation of progenitors to chondrocytes. Recent studies have shown that mimicking the natural processes during cartilage development such as cellular condensation enhances cartilage regeneration.13,14 In addition, other studies have also developed hydrogels capable of increasing stiffness over time. However, in these cases, the temporal stiffening was achieved through external stimuli at pre-determined time points and as a result, these failed to recapitulate the gradual increase in stiffness observed during natural tissue development.15
Another important attribute that should be considered while designing these microenvironments is how growth factors or biochemical cues are presented to the cells. In native cartilage, growth factors remain electrostatically bound to heparan sulfate which is a sulfated glycosaminoglycan (sGAG) present in the ECM of the tissue.8 However, in most studies involving growth factor-based approaches for cartilage tissue engineering, growth factors are normally provided by the supplementation of cell culture media, which does not mimic the natural presentation of the factors by the ECM. In addition, under 3D culture conditions, the diffusion of these soluble factors becomes challenging, leading to their reduced presentation to the cells and consequently diminished regenerative potential.16 Although, in many studies, covalent binding of growth factors and incorporation of heparan sulfate to the matrix were shown to be effective in sequestering growth factors, both the strategies are associated with limitations. Covalent binding is known to alter the conformation of the bound growth factor and thereby its biological activity, whereas, exogenously provided heparan sulfate may induce inflammatory responses and blood-thinning effects in the body.17,18 Therefore, it may be desirable to develop materials that provide growth factor binding abilities similar to the natural tissues.
Hydrogels serve as a mimic of extracellular matrices that not only provide support to the encapsulated cells but can also bestow a microenvironment with cues that regulate the differentiation of progenitor cells to desired lineages.19,20 Moreover, injectable hydrogels due to their minimal invasiveness and ability to fill irregular defects have better translational potential compared to pre-formed scaffolds.21 Therefore, in a biomimetic approach, it would be desirable to engineer a next generation hydrogel system which possesses two distinct attributes simultaneously: (i) dynamically achieving stiffness and (ii) providing binding moieties to confer it with growth factor sequestration and long-term presentation ability, as seen in tissue development and homeostasis in situ. Thus, we hypothesized that designing a hydrogel with microenvironments that mimic the biophysical (temporal ECM stiffening) and biochemical (growth factor presentation) cues provided by the ECM during development would facilitate engineering of cartilage tissue. Additionally, the introduction of temporal stiffening within these hydrogels would address a critical need for enhancing their mechanical strength to facilitate joint stability. Moreover, making these hydrogels injectable in nature would enhance their translational potential and overcome the challenge of invasiveness associated with current treatment modalities.
In the present study, we fabricated an injectable interpenetrating hydrogel (IPN) system using silk fibroin, sulfated carboxymethyl cellulose (s-CMC) and tyraminated carboxymethyl cellulose (t-CMC). Silk has a unique property in that its polymeric chains transform spontaneously from random coils to β-sheets.22 This spontaneous transformation under physiological conditions is associated with a gradual increase in the mechanical properties of silk. Therefore, incorporating silk fibroin in the hydrogels would enable the formation of the desired dynamically stiffening microenvironment. Similar to heparan sulfate, s-CMC effectively binds and presents cationic growth factors such as transforming growth factor-β (TGF-β).21,23,24 TGF-β is a commonly used growth factor for chondrogenic differentiation of MSCs. The long-term exposure of TGF-β to MSCs is critical for various steps involved in chondrogenesis.25,26 Therefore, in order to mimic the natural presentation of growth factors, incorporating s-CMC into a hydrogel would be desirable to electrostatically bind to TGF-β, ensuring its prolonged presentation, thereby promoting chondrogenic differentiation of the encapsulated stem cells. To facilitate crosslinking of IPN hydrogels, t-CMC was used as the third polymeric system along with silk fibroin and s-CMC. Overall, we have utilized a biomimetic approach to develop an injectable IPN hydrogel system using silk fibroin, s-CMC and t-CMC synthesized by enzymatic crosslinking using horseradish peroxidase (HRP) and hydrogen peroxide (H2O2). The developed hydrogel system demonstrated the desired properties i.e. a dynamic increase in mechanical strength and the ability to bind to cationic molecules/growth factors, while having the advantage of injectability. Together, these properties are expected to provide the desired biophysical and biochemical cues to stem cells and facilitate the regeneration of cartilage when injected in situ.
2. Materials and methods
2.1 Materials
Carboxymethyl cellulose (CMC) (700 kDa), tyramine, N-(3-dimethylaminopropyl)-N-ethylcarbodimide (EDC), hydroxysuccinamide (NHS), hydroxybenzotriazole (HoBt), horseradish peroxidase, lysozyme, hyaluronidase, hydrogen peroxide, and 4′,6-diamidino-2-phenylindole (DAPI) were procured from Sigma-Aldrich, USA. Sodium azide and MES buffer were procured from Merck Millipore, USA. Human recombinant TGF-β1 growth factor was obtained from Sino Biological Inc., China. Lithium bromide was procured from Avra Chemicals, India. Anti-collagen 2 and anti-chondroitin sulfate antibodies were procured from the Developmental Studies Hybridoma Bank, University of Iowa, USA, anti-TGF-β antibody was purchased from Invitrogen, and IgG secondary antibodies were procured from Jackson Immuno Research Laboratories. Sodium pyruvate, non-essential amino acids, N-2-hydroxyethylpiperazine-N′-ethanesulfonic acid (HEPES) buffer, proline, ascorbate-2-phosphate, bovine serum albumin (BSA) and penicillin–streptomycin, and bovine serum albumin (BSA) and pepsin were procured from Himedia, India. Trizol reagent was procured from Sigma-Aldrich, USA. cDNA reverse transcriptase kit and SYBR green were purchased from Genetix Biotech Asia Pvt. Ltd. Additionally, diaminobenzidine (DAB)-enhanced liquid substrate system used was obtained from Sigma-Aldrich, USA. Collagenase type II was procured from Gibco, USA.2.2 Modification of CMC with tyramine and sulfate groups
CMC was modified with tyramine (phenolic) groups for enzymatic crosslinking based on a previously reported method.27 Briefly, CMC (∼700 kDa) and tyramine hydrochloride were dissolved in 50 nM MES (morpholino ethane sulfonic acid) buffer (pH 6.0) at a concentration of 1% (w/v). Subsequently, NHS (N-hydroxy succinimide), EDC (ethyl 3-dimethylaminopropyl carbodiimide), and hydroxybenzotrizole (HoBt) were added to this solution at a final concentration of 7 mg mL−1, 0.47 mg mL−1, 3.94 mg mL−1 and 3.15 mg mL−1, respectively. The reaction mixture was stirred continuously for 24 h at room temperature. The polymer solution thus obtained was then dialyzed (cutoff ∼13 kDa) against deionized water for 72 h and finally lyophilised.CMC was chemically modified with sulfate groups to impart it with growth factor binding ability, as reported previously.28 Briefly, 1 g of CMC was dissolved in N,N-dimethylformamide (DMF) and then mixed with 1 mL of anhydrous p-toluenesulfonic acid in DMF. The mixture was then stirred for 30 min at 65 °C. Next, a solution containing 15 mL of sulfation reagent, prepared by mixing chlorosulfonic acid with DMF, was gradually added to the polymer solution and was kept for 3 h at 25 °C. The modified polymer was precipitated with chilled absolute ethanol and collected using centrifugation at 8000 rpm for 15 min (4 °C). The modified polymer was resuspended in distilled water, neutralised using NaOH, freeze-dried and stored at 25 °C until used further. The modification of polymers (t-CMC and s-CMC) was confirmed using FTIR spectroscopy (PerkinElmer, USA), 1H Nuclear magnetic resonance spectroscopy (500 MHz, ECX-500, JEOL Delta, USA), energy dispersive X-ray spectroscopy (Carl Zeiss, Germany) and UV-visible spectroscopy (200–400 nm, Thermofisher, USA) and differential scanning calorimetry (5.0 °C min−1, TA Instruments, USA) as reported previously.29,30
2.3 Differential scanning calorimetry (DSC)
The polymer samples were analyzed using a temperature-modulated double-scan method in conventional DSC (TMDSC TA2920, TA Instruments, US). The samples were heated across the temperature range of 0–400 °C at the rate of 10.0 °C min−1 in the presence of nitrogen. The heat flow rate versus temperature graph was plotted to obtain the DSC curve for the samples. The determination of the glass transition temperature (Tg) was then performed using the tangent line method as described previously.29,312.4 Isolation of silk fibroin
Silk fibroin was isolated from the cocoons of Bombyx mori silkworms according to a previously reported protocol.32 Briefly, 5 g of cocoons was cut into small pieces and boiled in 0.02 M Na2CO3 for 30 min to separate silk fibroin from sericin protein. The degummed silk fibres were thoroughly washed with distilled water to remove any remaining sericin and Na2CO3. The fibres were then dried overnight at 37 °C. The dried silk fibres were solubilized in 9.3 M lithium bromide (LiBr) solution at 60 °C for approximately 3 h. The resulting solution of silk fibroin was dialyzed to remove the residual LiBr for 48 h at 4 °C. The solution was further centrifuged and stored for the fabrication of hydrogels.2.5 Fabrication of CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels
CMC/t-CMC/silk hydrogels were fabricated by mixing CMC, t-CMC, and silk in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 to obtain a final concentration of 4% (w/v). To fabricate s-CMC/t-CMC/Silk hydrogels, CMC was replaced with s-CMC. An enzyme horseradish peroxidase (HRP) at a concentration of 0.25 U mL−1 was added to the polymer solution. The resulting polymer solution was poured into the mold having dimensions of 8 mm × 4 mm (diameter × height). The polymer solution was crosslinked by the addition of hydrogen peroxide (H2O2) at a final concentration of 2 mM. The fabricated hydrogels were analyzed by FTIR spectroscopy (PerkinElmer, USA) for determining the changes in the molecular structure of silk as a function of time. The detailed methodology of FTIR is as provided in section 1.3 of the ESI.†
:1:2 to obtain a final concentration of 4% (w/v). To fabricate s-CMC/t-CMC/Silk hydrogels, CMC was replaced with s-CMC. An enzyme horseradish peroxidase (HRP) at a concentration of 0.25 U mL−1 was added to the polymer solution. The resulting polymer solution was poured into the mold having dimensions of 8 mm × 4 mm (diameter × height). The polymer solution was crosslinked by the addition of hydrogen peroxide (H2O2) at a final concentration of 2 mM. The fabricated hydrogels were analyzed by FTIR spectroscopy (PerkinElmer, USA) for determining the changes in the molecular structure of silk as a function of time. The detailed methodology of FTIR is as provided in section 1.3 of the ESI.†
2.6 Gelation kinetics
The gelling time of CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels in both the compositions was assessed using a magnetic bead rotation test as reported previously.33 The mixture of polymer precursors and HRP (0.25 U mL−1) was stirred within a glass vial utilizing a magnetic bead rotated at a speed of 200 rpm. The point at which the bead stopped rotating due to the formation of a solid mass of the polymer solution upon the addition of H2O2, was considered as the gelling time.2.7 Swelling ratio
The swelling ratio of the fabricated hydrogels was determined after 24 h of incubation in PBS. The weight of CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels, just after fabrication, was measured and recorded as the initial weight. Subsequently, the hydrogels were immersed in phosphate buffered saline (PBS) and were allowed to incubate for 24 h at 37 °C. After 24 h, the wet weight of the hydrogels was measured and recorded. The weight swelling ratio was then calculated using the following formula:where Ws is the wet weight after swelling of the hydrogel and W0 is the initial weight of the hydrogel. Similarly, the volumetric swelling ratio was determined by measuring the dimensions and calculating the volume of the fabricated hydrogels before and after incubation in PBS.
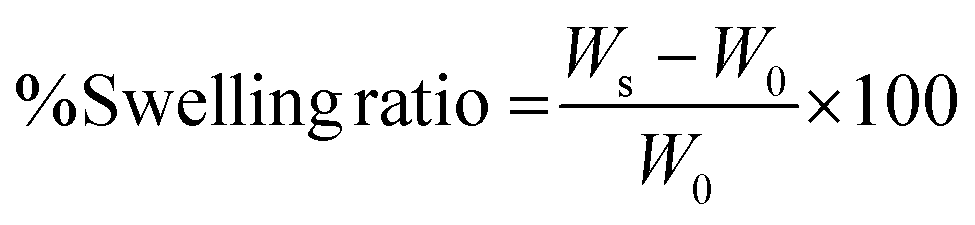
2.8 Mechanical testing
CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels were fabricated and incubated in phosphate buffered saline at 37 °C for 24 h. The mechanical properties of the swollen hydrogels were assessed by measuring their compressive strength using a Bose Electro Force 3200 instrument equipped with a 20N load cell. The hydrogels were subjected to uniaxial compression between flat parallel plates at a constant rate of 0.05 mm s−1 up to 50% strain. The counterforce applied by the hydrogel during compression was recorded. The compressive modulus was then calculated by analyzing the stress–strain curve between 5% and 15% strain using Origin software (Origin Lab Corporation). In addition, the fabricated hydrogels were characterized for rheological properties, degradation and chemical groups (FTIR analysis) (ESI 1.1-1.3†).2.9 Turbidimetric analysis for studying the interactions of polymers
The interaction between the polymers CMC or s-CMC with the two model proteins BSA (anionic) and lysozyme (cationic) was studied to understand the binding efficiency of cationic molecules with GAG mimics. For this, varying amounts of protein (0–1800 μg) were added to 250 μg mL−1 CMC or s-CMC solution and mixed in a 96 microwell plate to achieve a final volume of 200 μL. The increase in the turbidity of the solution due to the interaction between protein and polymer was measured at 420 nm using a multi-mode reader (Synergy H4, Biotek instruments Inc. USA).2.10 Toluidine blue staining
Toluidine blue dye (metachromatic dye) was prepared in acetate buffer (0.4% w/v). The prepared dye was mixed and filtered before using. The fabricated CMC/t-CMC/Silk and s-CMC/t-CMC/Silk IPN hydrogels were incubated in the prepared toluidine blue dye for 4 h at room temperature. Finally, the hydrogels were washed with distilled water and the gross images were captured to evaluate the change in the color of the dye due to the presence of negatively charged sulfate groups in hydrogels.2.11 Cell isolation and culture
Infrapatellar fat pad-derived mesenchymal stem cells (IFP-MSCs) were isolated as per a previously reported protocol.34,35 Briefly, IFP-MSCs were isolated from the infrapatellar fat pad of goat tibia-femoral joints collected from a local abattoir. The isolated fat pad was minced and enzymatically digested overnight using collagenase (1.5 mg mL−1) in DMEM low glucose medium supplemented with 2% FBS. The cell suspension obtained was filtered through a 70 μm cell strainer and then centrifuged, washed, and resuspended in DMEM complete culture media supplemented with 5 ng mL−1 bFGF. IFP-MSCs from Passage 2 to Passage 4 were used in the experiments.The isolated IFP-MSCs were encapsulated in CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels. The cell encapsulated hydrogels were then cultured in complete cell culture media in vitro. Live/dead assay was performed using fluorscein diacetate (FDA) and propidium iodide (PI) which stain for live and dead cells respectively. After day-1 and day-7, the hydrogels were incubated in FDA/PI solution and incubated at room temperature for 2–5 min and finally imaged under a confocal microscope (Carl Zeiss, Germany). Chondrogenic differentiation of the isolated IFP-MSCs was performed to study the activity of the growth factor (TGF-β1) either bound to the fabricated hydrogel or supplemented in the media. IFP-MSCs (2 × 106 cells per hydrogel) were resuspended in HRP (0.25 U mL−1) containing precursor polymer solutions prepared to fabricate CMC/t-CMC/silk or s-CMC/t-CMC/silk hydrogels. 50 μL of this solution was placed on a polystyrene plate followed by the addition of 40 ng (for 14 days culture) or 80 ng (for 28 day culture) of TGF-β1. The gelation was initiated by the addition of H2O2 to achieve the final concentration of 2 mM in hydrogels. The fabricated hydrogels were incubated for ∼30 min at 37 °C and then transferred to chondrogenic media.34 The chondrogenic medium was changed every three days for 14 and 28 days. For TGF media hydrogels, the chondrogenic medium supplemented with 10 ng mL−1 TGF-β1 was used, whereas, for TGF loaded hydrogels, the chondrogenic medium without TGF-β1 was used. After 14 and 28 days of culture, the hydrogels were harvested for gene expression, histology and immunohistochemistry.
2.12 Histology and immunohistochemistry
The samples harvested after 14 and 28 days of chondrogenic differentiation were fixed and embedded in optimum cutting temperature (OCT) solution. The samples were cryo-sectioned at a thickness of 10 μm (Leica Biosystems, Germany). For histological analysis, these sections were then stained with safranin O and Alcian blue to detect sulfated glycosaminoglycan (sGAG) deposition, and picrosirius red to detect total collagen deposition as previously reported.36 For the immunohistochemistry analysis, the sections were fixed and treated with pepsin (2 mg mL−1) and hyaluronidase (2 mg mL−1) for antigen retrieval. The sections were then incubated overnight with primary antibodies against hyaline cartilage markers namely, collagen 2 and chondroitin sulfate. Subsequently, the samples were incubated with anti-mouse HRP conjugated IgG secondary antibodies (dilution 1:250) for 2 h. The sections were finally developed using a DAB (diaminobenzidine) enhanced liquid substrate system. The sections were mounted using a resin-based DPX medium and imaged using a brightfield microscope (Carl Zeiss, Germany). For TGF-β immunofluorescence staining, sections were incubated overnight at 4 °C with anti TGF-β antibodies. Following this the sections were stained with anti-mouse IgG secondary antibodies labelled with Alexa Fluor 488 (Jackson Immuno Research Laboratories Inc., USA). The stained sections were mounted and then imaged using a confocal microscope (Carl Zeiss, Germany).
2.13 Gene expression analysis
The expression of cartilage and hypertrophic markers was studied after 14 and 28 days of chondrogenic differentiation of IFP-MSCs encapsulated within the fabricated IPN hydrogels. The harvested samples were digested in TRIZOL using a motorised pellet pestle followed by the addition of chloroform, gentle vortexing and centrifugation at 13000 rpm (4 °C) for 15 min. The aqueous phase was carefully collected into fresh tubes and an equal volume of isopropanol was added and incubated at room temperature for 10 min. The samples were then centrifuged at 13000 rpm (4 °C) for 15 min to pellet down the total RNA. The pellet was washed twice with 75% chilled ethanol and resuspended in RNAse free water. cDNA was then synthesized from total RNA (500 ng) using a First Strand cDNA synthesis kit (Genetix Biotech Asia Pvt. Ltd) using the manufacturer's protocol. Real time RT-PCR (StepOne Plus, Applied Biosystems) was performed using SYBR Green Master Mix (Genetix Biotech Asia Pvt. Ltd) and the relative expression of the target genes with respect to the control group was calculated by the ΔΔCt method using HPRT as a house keeping gene. The primer sequences are listed in Table S1.†
2.14 Statistical analysis
All statistical analyses were performed using GraphPad Prism software. The results were presented as mean ± standard deviation. Statistical significance was determined using either one-way or two-way analysis of variance (ANOVA), followed by a multiple comparison test. A significance level of P < 0.05 was considered statistically significant.3. Results and discussion
3.1 Synthesis and characterization of modified polymers
The main aim of this study was to design an injectable interpenetrating hydrogel system that has the ability to dynamically stiffen while providing the advantage of long-term presentation of TGF-β to the encapsulated cells. We opted for a biomimetic approach for the choice of materials to fabricate the IPN hydrogel that possesses these properties. The IPN hydrogel was fabricated majorly using two polymers: carboxymethyl cellulose (CMC) and silk fibroin.CMC is a plant derived polysaccharide having the structure and properties very similar to hyaluronic acid, which is an abundantly present GAG in the ECM of articular cartilage. CMC has widely been used in biomedical applications such as lubricant in eye formulations and as a bone autograft substitute.37,38 Hence employing CMC for cartilage regeneration would provide increased hydration and subsequent lubrication similar to GAGs. In addition, introducing CMC to the hydrogel would provide resilience and shock absorbing property essential for load-bearing tissues such as cartilage.21 CMC was modified with tyramine moieties to form tyraminated-CMC (t-CMC) which imparts the ability of enzymatic crosslinking for the formation of hydrogel networks (Fig. 1A). Even though many different approaches have been investigated for the crosslinking of hydrogels, enzymatic cross-linking offers several advantages. Due to the substrate specificity of the enzyme, this type of crosslinking does not cause undesirable reactions. Moreover, enzymatic crosslinking is highly tunable and cyto-compatible and therefore, has been used widely for many tissue engineering applications.39,40
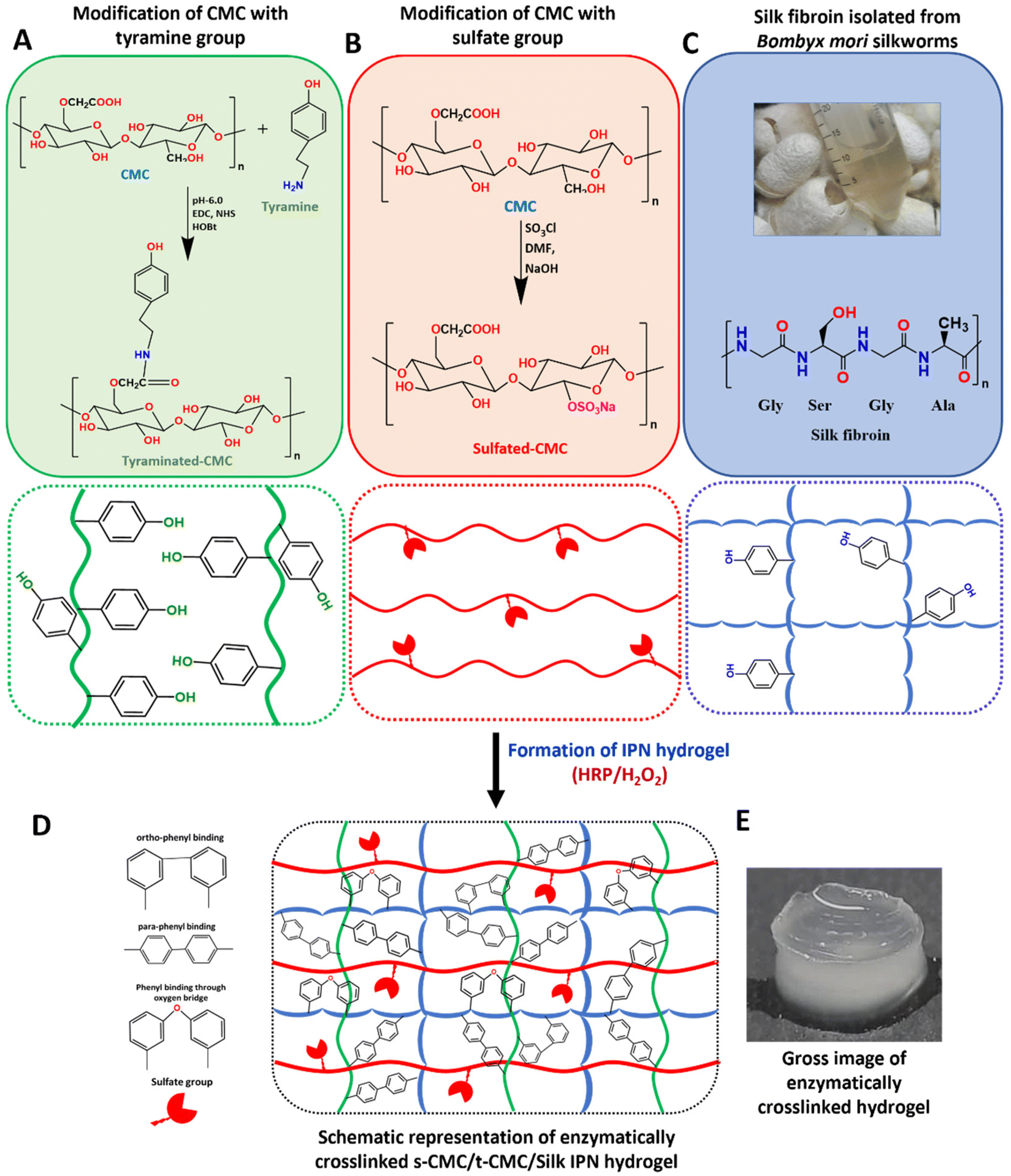 | ||
| Fig. 1 Schematic representation of IPN hydrogel formation using, tyraminated-CMC (t-CMC), sulfated-CMC (s-CMC) and Silk fibroin via enzymatic crosslinking (HRP/H2O2). (A) Chemical modification of CMC to t-CMC (upper box) and schematic representation of the polymeric chains of t-CMC (lower box). (B) Chemical modification of CMC to s-CMC (upper box) and schematic representation of the polymeric chains of s-CMC (lower box). (C) Isolated silk fibroin from Bombyx mori silkworms (upper box) and the schematic representation depicting the tyrosine residues present on silk fibroin (lower box). (D) Enzymatically crosslinked polymeric network of a s-CMC/t-CMC/silk IPN hydrogel. (E) Gross image of the enzymatically crosslinked s-CMC/t-CMC/silk IPN hydrogel. | ||
Although the presence of CMC in scaffolds is known to provide resilience, it lacks the ability to bind to growth factors. Therefore, to enable growth factor binding ability and its long-term presentation to cells, CMC was subjected to chemical sulfation to obtain sulfated-CMC (s-CMC) (Fig. 1B). s-CMC is a sulfated GAG mimic and has a structure similar to that of heparan sulfate present in tissues. In the cartilage tissue, growth factors are usually available in the bound state to the ECM via electrostatic interactions with heparan sulfate. Thus, modifying CMC with sulfate groups presents a biomimetic strategy for effective cartilage regeneration. In addition, the incorporation of sulfate groups on polysaccharides has also shown anti-inflammatory and pro-chondrogenic effects, which make them a suitable candidate for hydrogel-mediated growth factor delivery.41,42
Silk fibroin, a natural polymer, is recognized for its ability to enhance the mechanical properties of scaffolds.43 Many studies have demonstrated that even in its unmodified state, silk fibroin significantly improves the mechanical strength of scaffolds. In the present study, silk fibroin was isolated from Bombyx mori silkworms and was used without modification (Fig. 1C). The enzymatic crosslinking of intrinsically present tyrosine in silk protein is known to generate highly elastomeric and cyto-compatible hydrogels.44 Silk fibroin has the tendency to self-assemble into large β-sheet aggregates which is driven by entropy induced recoiling of the silk polymeric chains.43 Previous studies have demonstrated that by controlling the extent of crosslinking of the silk polymeric chains, self-assembly of β-sheets can be regulated which in turn can be used to impart a dynamic microenvironment to the hydrogel system.13,45 Therefore, we hypothesized that incorporating t-CMC, s-CMC and silk fibroin in a single system via enzymatic crosslinking (Fig. 1D and E) would enable formation of a resilient IPN hydrogel that not only will possess the capabilities of injectability and growth factor binding but will also have the ability to stiffen over time leading to the regeneration of functionally matured cartilage tissue.
After modification of CMC, FTIR spectroscopy was performed to validate the presence of tyramine and sulfate groups in t-CMC and s-CMC respectively. The FTIR spectra showed two new characteristic peaks at 1536 cm−1 and 1066 cm−1 in t-CMC, which represent the N–H bending and C–N stretching peaks due to the presence of tyramine groups (Fig. 2A and C). However, these peaks were absent in unmodified CMC. Additionally, the FTIR spectra of s-CMC showed two new characteristic peaks at 1253 cm−1 which correspond to the asymmetric stretching of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and at 843 cm−1 corresponding to the symmetrical vibration of the S–O–C bond due to the presence of sulfate groups (Fig. 2B and C). Furthermore, UV-visible spectroscopy was performed to confirm the conjugation of tyramine groups to CMC. The UV-visible spectra of t-CMC showed an absorbance peak at ∼275 nm by virtue of the presence of a phenol ring in the tyramine group which was absent in unmodified CMC as well as s-CMC, indicating the successful conjugation of tyramine groups to CMC (Fig. 2D).
O bond and at 843 cm−1 corresponding to the symmetrical vibration of the S–O–C bond due to the presence of sulfate groups (Fig. 2B and C). Furthermore, UV-visible spectroscopy was performed to confirm the conjugation of tyramine groups to CMC. The UV-visible spectra of t-CMC showed an absorbance peak at ∼275 nm by virtue of the presence of a phenol ring in the tyramine group which was absent in unmodified CMC as well as s-CMC, indicating the successful conjugation of tyramine groups to CMC (Fig. 2D).
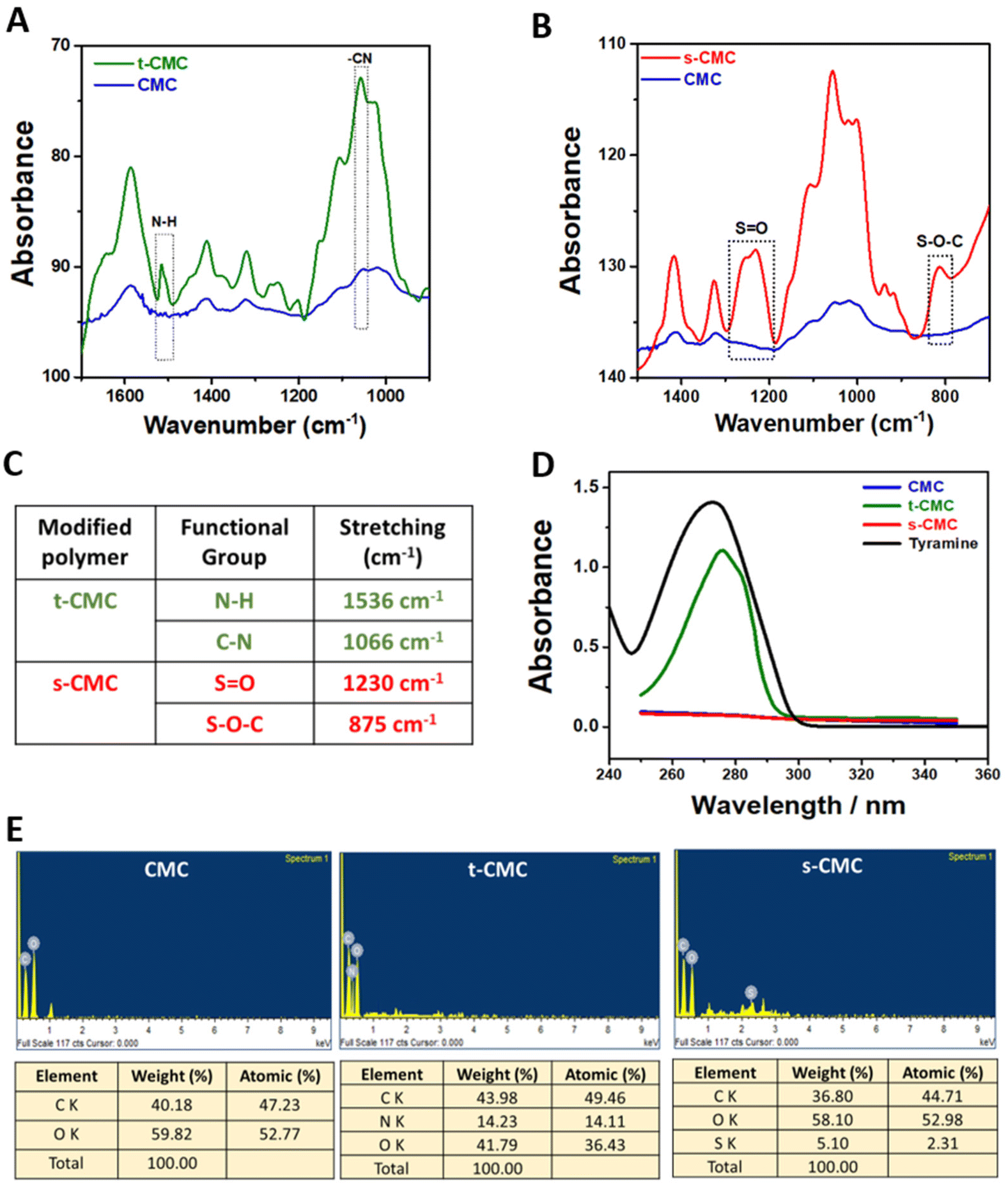 | ||
| Fig. 2 Chemical characterization of modified polymers. FTIR spectra of (A) CMC and tyraminated-CMC (t-CMC), (B) CMC and sulfated-CMC (s-CMC). (C) Tabular representation of characteristic peaks of t-CMC and s-CMC. (D) UV-visible spectra of CMC, t-CMC, s-CMC and tyramine. (E) EDX spectra for elemental analysis of CMC, t-CMC and s-CMC polymers. | ||
The elemental composition of CMC, t-CMC and s-CMC was characterized by EDX spectroscopy. The EDX spectra of CMC showed two element signals corresponding to carbon and oxygen. The weight percentages of these elements were 40.1% and 59.8%, while the atomic percentages were 47.2% and 52.7%, respectively (Fig. 2E). However, in the case of t-CMC, along with the peaks corresponding to carbon and oxygen, a peak corresponding to nitrogen due to the amine group of tyramine was also observed. The weight percentages for these elements were 43.9%, 14.2%, and 41.6%, with corresponding atomic percentages being 49.4%, 14.1%, and 36.4%, respectively (Fig. 2E). The s-CMC EDX spectra also showed elemental peaks for carbon and oxygen and an additional peak corresponding to sulfur due to the modification of CMC with sulfate groups. The weight percentages for these elements were 36.8%, 58.1%, and 5.1%, with corresponding atomic percentages being 44.7%, 52.9%, and 2.3%, respectively (Fig. 2E). The presence of nitrogen in t-CMC and sulfur in the s-CMC spectrum when compared to CMC served as a definitive confirmation of the successful modification of CMC.
Furthermore, we performed 1H NMR spectroscopy to confirm the modification of CMC. The 1H NMR spectrum of t-CMC along with unmodified CMC within the range of 2.5–4.5 ppm, shows the presence of seven proton peaks corresponding to the glucose monomer present in CMC (Fig. 3A). Additionally, the 1H NMR spectra of t-CMC also showed two characteristic peaks at 7.06 ppm and 6.75 ppm of two specific protons Hc and Hd present in the phenol ring of tyramine. The alkyl protons of tyramine, Ha and Hb, yield two more identifiable NMR signals, observed at 2.73 ppm and 3.00 ppm, respectively (Fig. 3A). These peaks are consistent with the results reported previously.13,46 Remarkably, it was observed that these four new proton peaks were absent in the unmodified polymer thereby confirming the modification of CMC to t-CMC. The 1H NMR spectra of s-CMC exhibited overlapped resonance signals corresponding to seven protons from the glucose monomer in CMC except for the α-anomeric proton (5.09 ppm) present on the C-2 atom of the CMC polymer (Fig. 3B). Due to the addition of the (SO3)− group at the C-2 position, a downfield shift of the peak from 5.09–5.12 ppm to 5.11–5.14 ppm was observed, thereby confirming the modification of CMC to s-CMC. (Fig. 3B, dotted circle region).47,48 The merged spectra of CMC, t-CMC and s-CMC show the shifting of the α-anomeric proton peaks for both t-CMC and s-CMC. Multiple previous studies have shown that the modification at various positions of carbon atoms in CMC causes shifts in the electron density and as a consequence result in the shielding or deshielding of the proton at the α-anomeric position.47–49 Due to the presence of a highly electronegative group in s-CMC, the α-anomeric proton at the C-2 position was more shielded and hence there was a downfield shift in the peak (Fig. 3C, violet and cyan). On the other hand, the addition of the tyramine group on the C-5 position of CMC causes the deshielding of the α-anomeric proton which resulted in an upfield shift of the peak towards 5.06–5.09 ppm (Fig. 3C, violet and green).
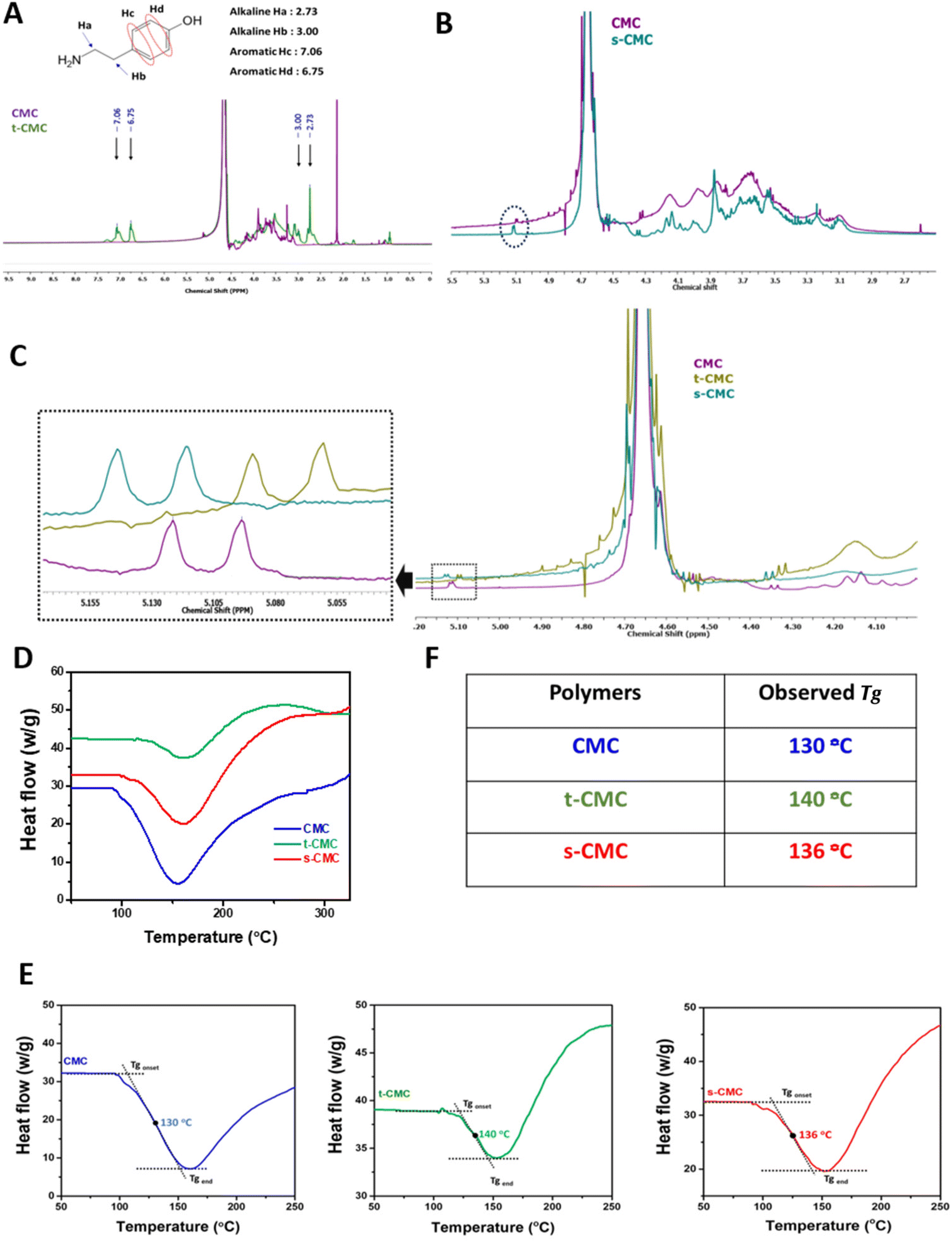 | ||
| Fig. 3 Chemical characterization of modified polymers. 1H NMR spectra of (A) CMC and tyraminated-CMC (t-CMC), (B) CMC and sulfated-CMC (s-CMC). (C) Merged 1H NMR spectra of CMC, t-CMC, and s-CMC for peak shift identification after modification of CMC to t-CMC and s-CMC. (D) Differential scanning calorimetry (DSC) spectra of CMC, t-CMC and s-CMC. (E) Determination of glass transition temperature of CMC, t-CMC and s-CMC from their respective DSC spectrum using tanget line method. (F) Table representing glass transition temperature (Tg) of modified polymers. | ||
We further characterized the modified polymers using differential scanning calorimetry (Fig. 3D). The glass transition temperature (Tg) was calculated from the curves generated by differential scanning calorimetry (DSC). In corroboration with previous literature,50 the glass transition temperature of unmodified CMC was found to be 130.95 °C (Fig. 3E and F). An increase in the glass transition temperature to 140.39 °C and 136.40 °C was observed for t-CMC and s-CMC respectively (Fig. 3E and F). The significant alteration in Tg observed in t-CMC when compared to CMC can be attributed to the incorporation of tyramine groups which resulted in diminished flexibility due to inherent molecular rigidity, restricted molecular motion and polymer packing associated with phenyl rings.51,52 On the other hand, the increase in Tg of s-CMC was attributed to the introduction of a polar (SO3)− group having multiple lone pairs of electrons resulting in altered interactions within the polymeric chains. In addition, modification of CMC with sulfate groups introduces SO (double bond), which also contributes to the increase in Tg of s-CMC.53 Overall, the modification of CMC led to a restricted and less flexible molecular environment which significantly contributed to the elevation of Tg of both t-CMC and s-CMC, thereby also verifying that the polymer was successfully modified.
3.2 Fabrication and characterization of s-CMC/t-CMC/silk IPN hydrogels
For the synthesis of IPN hydrogels that could dynamically stiffen and at the same time endow growth factor binding abilities, s-CMC, t-CMC and silk polymers were mixed in a 1:1:2 weight ratio, respectively to form an enzymatically crosslinked s-CMC/t-CMC/silk hydrogel network with a final polymer concentration of 4% (w/v). The CMC/t-CMC/silk IPN hydrogel was fabricated as a control hydrogel system, wherein, s-CMC was replaced with the unmodified CMC. Horseradish peroxidase (HRP) and hydrogen peroxide (H2O2) were used for enzymatic crosslinking. The process of employing horseradish peroxidase and hydrogen peroxide-mediated enzymatic crosslinking has previously been utilized to introduce orthogonal crosslinking in polymers containing phenol groups to develop hydrogel systems for various tissue engineering applications.40,54 Since phenol groups are present in silk fibroin and in t-CMC, the radicals generated due to the action of an enzyme are responsible for the emergence of tyrosine–tyrosine, tyramine–tyramine and tyrosine–tyramine crosslinks in between the polymeric chains giving rise to a crosslinked hydrogel network.55
Firstly, FTIR spectroscopy was employed to verify the presence of functional groups of the modified polymers in the fabricated CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels. In both the IPN hydrogels, common peaks corresponding to N–H bending at 1526 cm−1 and C–N stretching at 1066 cm−1 were observed, indicative of amine groups of tyramine moieties. On the other hand, a discernible peak associated with S–O–C bending at 827 cm−1, and a peak for SO stretching at 1235 cm−1 were identified only in s-CMC/t-CMC/silk and were absent in CMC/t-CMC/silk IPN hydrogels (Fig. 4A), signifying the incorporation of sulfate groups in hydrogels. Furthermore, the morphology of the fabricated CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels were analyzed by scanning electron microscopy. The scanning electron micrograph of both hydrogel systems showed a well crosslinked porous network with no evidence of phase separation between the protein and polysaccharide present in the hydrogels (ESI Fig. 1†).
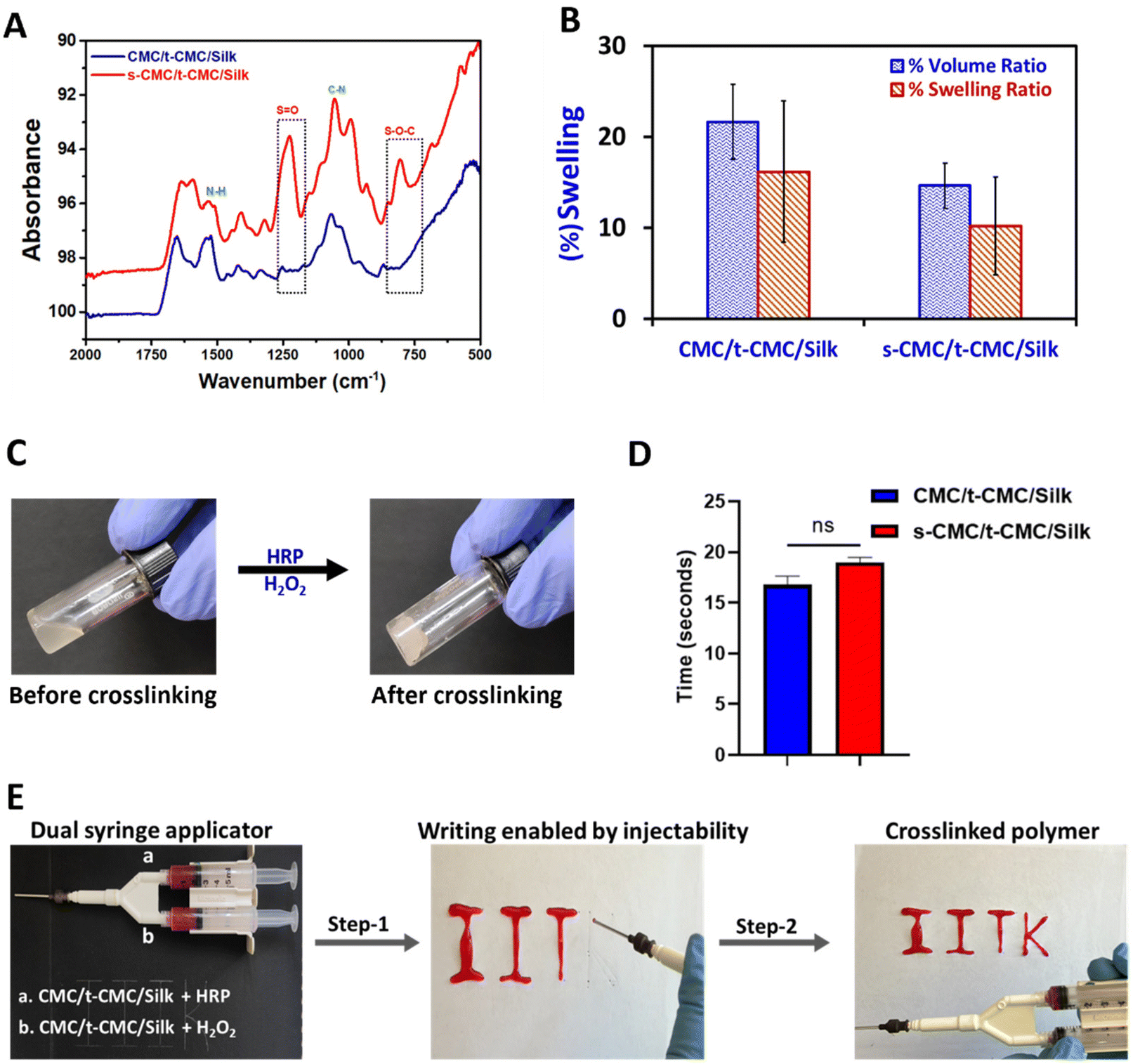 | ||
| Fig. 4 Physicochemical characterization of fabricated IPN hydrogels. (A) FTIR spectra depicting the presence of tyramine group in CMC/t-CMC/Silk and s-CMC/t-CMC/silk IPN hydrogels and the presence of sulfate group in s-CMC/t-CMC/silk IPN hydrogels. (B) Weight swelling and volumetric swelling of CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels after incubation in PBS for 24 h. (C) Gross image of the s-CMC/t-CMC/silk IPN hydrogel before and after enzymatic crosslinking using HRP/H2O2. (D) Gelation time of CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels determined using the magnetic bead rotation test. Error bar depicts the standard error of mean (n = 3). ‘ns’ indicates no statistical significance between groups. (E) Gross images depicting injectability and gelation of s-CMC/t-CMC/silk IPN hydrogels using dual syringe applicator. | ||
The weight and volumetric swelling ratios of the IPN hydrogels were studied after incubation in PBS for 24 h. The volumetric and weight swelling for CMC/t-CMC/silk was found to be 21.6% and 16.2% while that of s-CMC/t-CMC/silk was found to be 14.6% and 10.2%, respectively (Fig. 4B). Although s-CMC/t-CMC/silk hydrogels showed reduced swelling behavior, the difference was not significant. The gelling time of hydrogels was calculated by the magnetic bead rotation test. The time taken by the polymer solution to gelate fully and form a solid lump after the addition of H2O2 was measured (Fig. 4C). We observed that the CMC/t-CMC/silk polymer solution gelated in ∼17 seconds, whereas s-CMC/t-CMC/silk gelated in ∼19 seconds (Fig. 4D) with the difference being non-significant. The results demonstrated that the addition of s-CMC did not significantly alter the gelation kinetics of the hydrogels. This is attributed to the equal amounts of crosslinking groups imparted by t-CMC (tyramine) and silk (tyrosine) in both the hydrogels. We also studied the degradation of hydrogel systems in the presence of collagenase in vitro which mimicked the enzymatic concentration present in a healthy and injured/diseased knee.56 It was observed that all the hydrogels showed slightly higher degradation in the presence of collagenase; however, these differences were non-significant (ESI Fig. 2A–D†). Hence, the decreasing trend in the dry weight for both CMC/t-CMC/silk as well as s-CMC/t-CMC/silk hydrogels suggests that the fabricated hydrogels were degradable.
To assess the injectability of the developed hydrogel systems, a dual syringe applicator (18 G needle) was used to inject the precursor polymer solutions. The gross images show the steps which depict the injectability of the s-CMC/t-CMC/silk hydrogel and its immediate crosslinking to form a solid hydrogel network in the form of a set of letters (Fig. 4E and ESI video 1†). The results indicate the potential of IPN hydrogels to fill the irregular defect sites and crosslink immediately, thereby making them suitable as an in situ gelling and minimally invasive injectable hydrogel system for cartilage repair. These properties along with tunability and cytocompatibility of enzymatic crosslinking would make the developed hydrogel system more clinically translatable.39
3.3 Mechanical characterization of the s-CMC/t-CMC/silk IPN hydrogels
The unconfined compression test was performed to study the compressive modulus of CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels. The IPN hydrogels were compressed up to 50% of their original length. The gross images depict IPN hydrogels in the uncompressed state and after 10%, 30% and 50% compression (Fig. 5A). To study the dynamic increase in the stiffness, mechanical properties of the fabricated IPN hydrogels were studied after day-1, day-14 and day-28 of incubation in PBS. The stress–strain curve generated at day-1, day-14 and day-28 presented no significant difference in the slope of CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels; however, a significant increase in the peak stress of both the hydrogels from ∼6 kPa to ∼60 kPa was observed over the duration of 28 days (Fig. 5B and Table S2†). Compressive modulus was calculated on day-1, day-14 and day-28 from the slope of the stress–strain curve. On day-1, the compressive modulus of the s-CMC/t-CMC/silk hydrogels at 10% strain was ∼1.9 kPa. The compressive modulus increased to ∼3.7 kPa and ∼5.1 kPa at 20% and 30% strain respectively, due to the increase in resistance to compression at higher strains. No significant difference was observed between the compressive modulus of CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels measured at different strains at day-1, indicating that sulfation does not adversely affect the mechanical properties of IPN hydrogel. (Fig. 5C and Table S2†). Interestingly, over the duration of 28 days, the modulus of s-CMC/t-CMC/silk hydrogels at 30% strain increased from ∼35 kPa at day-14 to ∼95 kPa at day-28. A similar increase in the compressive modulus of CMC/t-CMC/silk hydrogels was also observed. Furthermore, we performed rheological analysis to study mechanical properties of hydrogels. It was observed that the storage modulus of CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels increased as a function of time over a 28 day period, demonstrating an increase in the mechanical strength of the hydrogels. The storage modulus for both the hydrogels at day 1 and day 28 was higher than the loss modulus, leading to tan δ < 1, thereby suggesting that the hydrogels showed solid-like behavior from 0.1 to 50 rad per s (ESI Fig. 3†). Taken together, this suggests that the fabricated s-CMC/t-CMC/silk and CMC/t-CMC/silk IPN hydrogels undergo an increase in the stiffness as a function of time.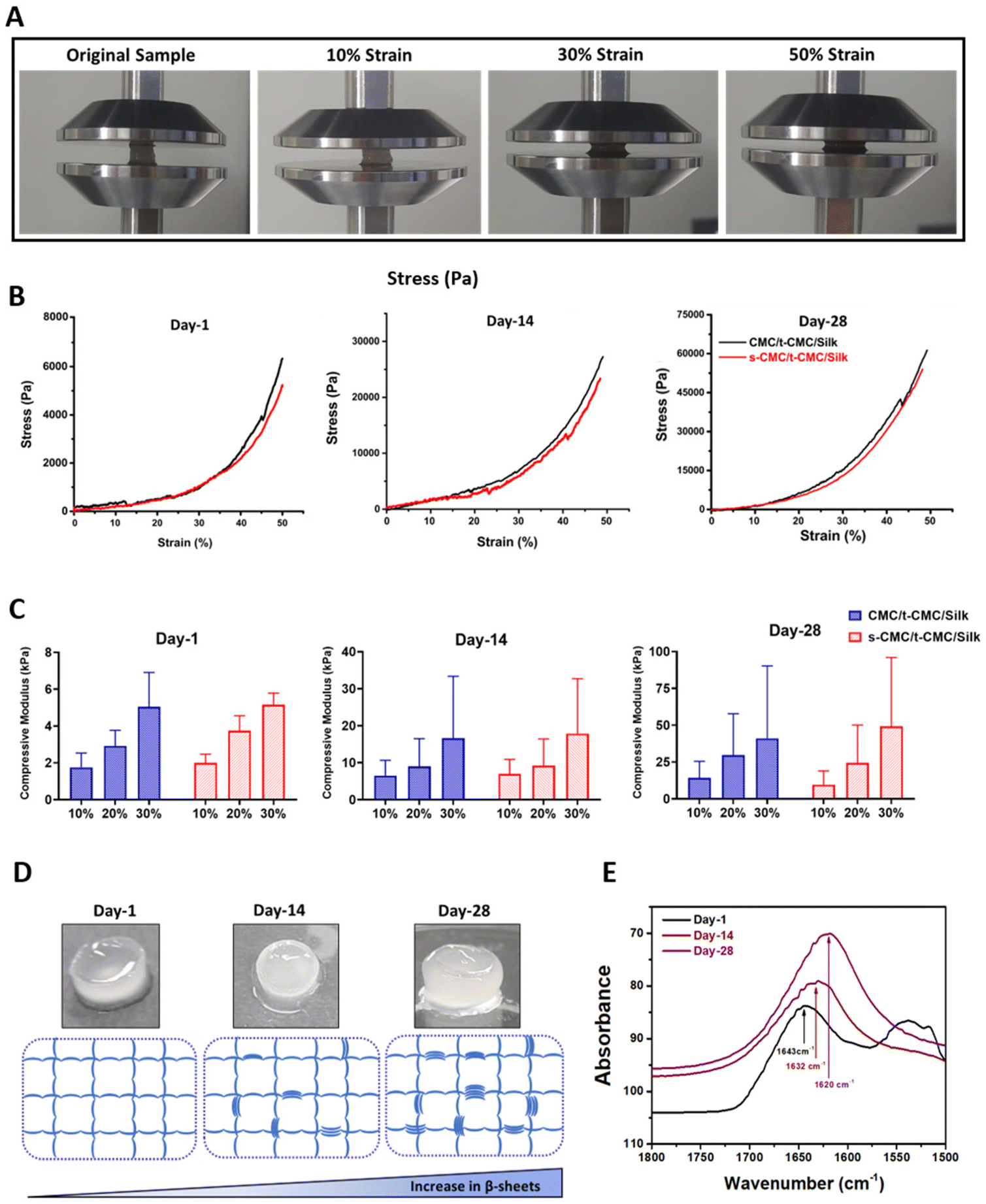 | ||
| Fig. 5 Mechanical characterization of CMC/t-CMC/Silk and s-CMC/t-CMC/silk IPN hydrogels. (A) Representative images of hydrogel constructs before and after compression to 10%, 30% and 50% strain. (B) Stress–strain curve of fabricated IPN hydrogels generated during compression after day-1, day-14 and day-28 of incubation in PBS. (C) Compressive modulus of fabricated IPN hydrogels calculated at 10%, 20% and 30% strain after day-1, day-14 and day-28 of incubation in PBS. (D) Gross image of s-CMC/t-CMC/silk hydrogels depicting loss in transparency from day-1 to day-28. (E) FTIR spectra of s-CMC/t-CMC/silk hydrogels on day-1, day-14 and day-28 showing a peak shift from 1643 cm−1 to 1620 cm−1. | ||
Notably, when the gross images of the fabricated IPN hydrogels were captured, an increase in the opacity of these hydrogels with time was observed (Fig. 5D). The increase in the opacity indicates formation of microstructures which has been associated with the secondary structural changes in the silk polymeric chains.22 Therefore, to confirm the secondary structural changes in silk polymeric chains, we performed FTIR spectroscopy. On day 1, the silk present in s-CMC/t-CMC/silk hydrogels displayed a peak at 1643 cm−1 which corresponds to the random coil within the carbonyl stretching of the amide-I region. On day 14, a chemical shift in the peak towards 1632 cm−1 was observed, which suggests the formation of β-sheets. The chemical shift was more pronounced at day 28 with a peak at 1620 cm−1, indicating a further increase in β-sheet formation, thereby confirming secondary structural changes in the silk polymeric chains present in IPN hydrogels as a function of time (Fig. 5E). Additionally, the degree of β-sheet formation in the s-CMC/t-CMC/silk hydrogels was investigated over a 28-day period (ESI Fig. 4A†). The reduction in the peak at 1660–1640 cm−1 (corresponding to random coil), coupled with an increase in the peak at 1620 cm−1 (corresponding to beta sheets), serves as an indicative measure of the evolving molecular structure. It was observed that the ratio of peak area and peak width progressively increased over the period of 28 days (ESI Fig. 4B and C†). This transition of random coil structure to β sheets confers the hydrogels with the ability to stiffen as a function of time. The exceptional property of silk fibroin to self-assemble into β-sheet arises from the uniformly arranged poly(Gly-Ala) and poly(Ala) domains in its structure and has been exploited previously to fabricate hydrogels with improved mechanical strength or achieving dynamic stiffening microenvironment in hydrogels.57 Our results demonstrated that β-sheet formation contributes significantly to the enhancement of mechanical properties of the s-CMC/t-CMC/silk IPN hydrogels. Previously, hydrogels with time-dependent stiffening have been studied mostly to understand the behavior of cells on dynamic matrices. The time-dependent stiffening is achieved majorly by the use of external stimuli such as UV or blue light in situ, which limits the stiffening only at predetermined time points and can have adverse effect on cell viability.15,58,59 The s-CMC/t-CMC/silk IPN hydrogel developed in the study mimics the dynamic changes seen during cartilage development, hence, making it a more suitable hydrogel candidate for cartilage tissue regeneration.9,60 Such a hydrogel system would provide a soft microenvironment to the encapsulated cells initially which is known to enhance chondrogenesis and gradually stiffen with time to mechanically support the joint. Overall, the developed hydrogel system would provide a conducive matrix for effective chondrogenic differentiation of stem cells.
3.4 Interaction of sCMC with cationic molecules and growth factors
To investigate the ability of s-CMC/t-CMC/silk hydrogels to interact with cationic growth factors, we first studied the binding efficiency of s-CMC and CMC to model molecules. Lysozyme has been used as a model protein for growth factors to study their interactions with polyelectrolytes.61 Lysozyme (pI ∼ 11.35) attains positive charge at physiological pH and electrostatically binds to the negatively charged sulfate groups on s-CMC. These interactions lead to the formation of complexes resulting in increased turbidity of the solution. Hence, the increase in absorbance due to turbidity serves as an indirect measure of the extent of polymer-protein interactions. Therefore, we studied the interaction of different concentrations of lysozyme with GAG mimics, CMC and s-CMC. A significant increase in the absorbance of s-CMC solution was observed with the increase in lysozyme concentrations. However, no such increase in absorbance was observed when different concentrations of lysozyme were added to CMC (Fig. 6A and ESI Fig. 5†). Bovine serum albumin (pI ∼ 5.1) attains a negative charge at physiological pH and was used as an anionic control. As expected, there was no increase in the turbidity of CMC and s-CMC solutions with increasing concentrations of BSA (Fig. 6A and ESI Fig. 5†). These observations showed that the sulfation of CMC conferred it with the ability to preferentially interact with cationic proteins like lysozyme.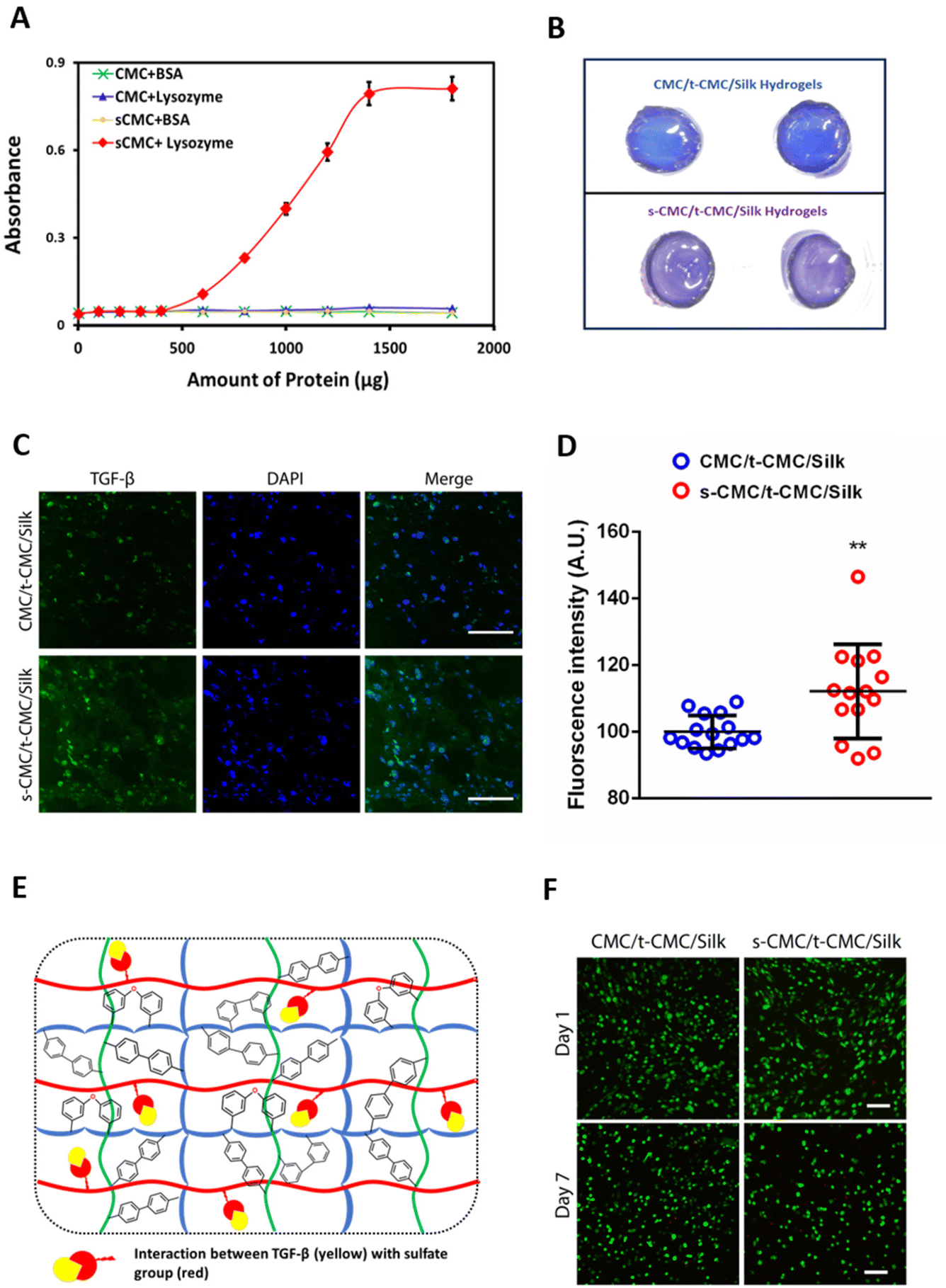 | ||
| Fig. 6 Interaction of sCMC with cationic proteins. (A) Turbidimetric titration curve of CMC and s-CMC with lysozyme (pI ∼ 11.5) or BSA (pI ∼ 5.2) (n = 6). (B) Gross images of two technical repeats of toluidine blue stained CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels. (C) Immunofluorescence micrographs of anti-TGF-β stained sections of TGF-β1 loaded CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels after 28 days of culture (Background green staining depicts loaded TGF-β) (Scale bar: 100 μm). (D) Quantification of fluorescence intensity of TGF-β stained sections. Each dot represents the intensity of a single image, number of images quantified >10 for each group. Error bar depicts standard deviation (n = 3), ** (P < 0.01) indicates statistically significant difference with respect to a TGF-β loaded CMC/t-CMC/silk IPN hydrogel. (E) Schematic representation depicting the interaction of TGF-β (cationic growth factor) with the sulfate groups of s-CMC present in the fabricated s-CMC/t-CMC/silk IPN hydrogel. (F) Live/dead staining of IFP-MSCs encapsulated within CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels after day-1 and day-7 of culture (Scale bar: 100 μm). | ||
Additionally, the ability of s-CMC containing hydrogels (s-CMC/t-CMC/silk) to effectively sequester growth factors was investigated by studying the interaction of fabricated hydrogels with toluidine blue dye, which is another model molecule. Toluidine blue is a metachromatic dye that undergoes a color change when it binds to negatively charged molecules, and therefore can be used to indirectly correlate the interaction between growth factors and s-CMC in a hydrogel.62 After incubation of s-CMC/t-CMC/silk and CMC/t-CMC/silk hydrogels in toluidine blue, the macroscopic images of the hydrogels were captured. It was observed that the color of dye bound to the s-CMC/t-CMC/silk hydrogel turned purple due to the presence of negatively charged sulfate groups in s-CMC. The absence of s-CMC in CMC/t-CMC/silk hydrogel had no effect on the color of the bound dye (no change from blue to purple) (Fig. 6B). The change in color of toluidine blue dye in the s-CMC/t-CMC/silk hydrogel further demonstrated its ability to bind to cationic molecules due to electrostatic interactions. Thereafter, we loaded TGF-β (pI ∼ 9.0) and encapsulated IFP-MSCs into s-CMC/t-CMC/silk and CMC/t-CMC/silk IPN hydrogels. The hydrogels were cultured for 14 and 28 days in cell culture media and thereafter, antibody staining for TGF-β was performed. Immunofluorescence micrographs demonstrated no difference in the intensity of TGF-β staining in s-CMC/t-CMC/silk and CMC/t-CMC/silk IPN hydrogels after 14 days (ESI Fig. 6†). However, after 28 days of culture an increased intensity of TGF-β staining in s-CMC/t-CMC/silk IPN hydrogels compared to CMC/t-CMC/silk IPN hydrogels was observed, indicating higher retention of TGF-β in s-CMC containing hydrogels due to electrostatic interactions between the negatively charged sulfate groups and cationic growth factor (Fig. 6C and E). Interestingly, we observed higher TGF-β expression inside the cells in s-CMC/t-CMC/silk hydrogels. This observation could be speculated to the increased retention of growth factor in these hydrogels subsequently causing higher expression of TGF-β inside the cells. The quantification of fluorescence intensity of TGF-β also verified the observations (Fig. 6D). The incorporation of s-CMC in hydrogels is anticipated to not only sequester the exogenously loaded growth factors but would also efficiently bind to the cationic factors released endogenously by the encapsulated cells, subsequently facilitating their long-term presentation. Previously, polysaccharides like alginate modified with sulfate groups have been demonstrated to have high affinity to cationic growth factors such as TGF-β and FGFs.63–65 Apart from being a growth factor sequestering agent, sulfated polysaccharides like pentosan polysulfate have shown to upregulate cartilage matrix markers in mesenchymal stem cells which can be an added benefit in cartilage tissue engineering applications.41 The sulfated polysaccharides including s-alginate and s-CMC have been utilized to fabricate a variety of scaffolds including, injectable hydrogels, fibres, pre-formed injectable scaffolds and 3-D printed scaffolds for cartilage tissue engineering applications.21,23,24,63 However, in the present study injectable interpenetrating hydrogels having the ability to stiffen with time along with ability to bind to cationic growth factors were fabricated and are hitherto not explored for cartilage tissue engineering.
3.5 Chondrogenic differentiation of IFP-MSCs in s-CMC/t-CMC/silk IPN hydrogels
After demonstrating that the s-CMC/t-CMC/silk IPN hydrogel can dynamically attain mechanical strength and bind to cationic growth factors, we next sought to investigate their potential as a delivery vehicle for TGF-β to induce chondrogenic differentiation of stem cells. Firstly, IFP-MSCs were encapsulated in CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels and cultured in vitro. After day-1 and day-7 of culture, live/dead assay was performed to study the viability of the encapsulated IFP-MSCs. It was observed that most of the encapsulated cells remained viable after both day-1 and day-7, as can be appreciated from fluorescence micrographs of FDA/PI-stained hydrogels (Fig. 6E). The results demonstrated that the composition and the process of crosslinking used to fabricate IPN hydrogels are both cyto-compatible and have negligible toxic effects.Further, IFP-MSCs encapsulated CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels were cultured either with loaded TGF-β (TGF loaded) or TGF-β in media (TGF media) to induce chondrogenic differentiation. After 14 days of chondrogenic differentiation, histological and immunohistochemical staining was performed to study the deposition of major cartilage extracellular matrix components. It was observed that all the constructs, irrespective of their composition and mode of availability of TGF-β, showed intense and similar levels of deposition of sGAG as depicted from Alcian blue and safranin O staining (Fig. 7A). Similarly, immunohistochemical staining for collagen 2 and chondroitin sulfate showed no difference in the accumulation of matrix components in all the constructs (Fig. 7B). The observations were also validated by quantifying the intensities of brightfield images of stained sections (Fig. 7C). Importantly, it was observed that both CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels where TGF-β was provided through media and replenished with every media change showed similar levels of deposition of sGAG and collagen 2 compared to the s-CMC/t-CMC/silk hydrogel where an equivalent amount of TGF-β was loaded only once. The gene expression of various cartilage-specific makers (COL2A1 and ACAN) and hypertrophic markers (COL10, RUNX2 and COL1) confirmed the observation that the TGF-β loaded in the s-CMC/t-CMC/silk hydrogel was active and had similar levels compared to the s-CMC/t-CMC/silk hydrogel with TGF-β supplemented in the media (ESI Fig. 7A†).
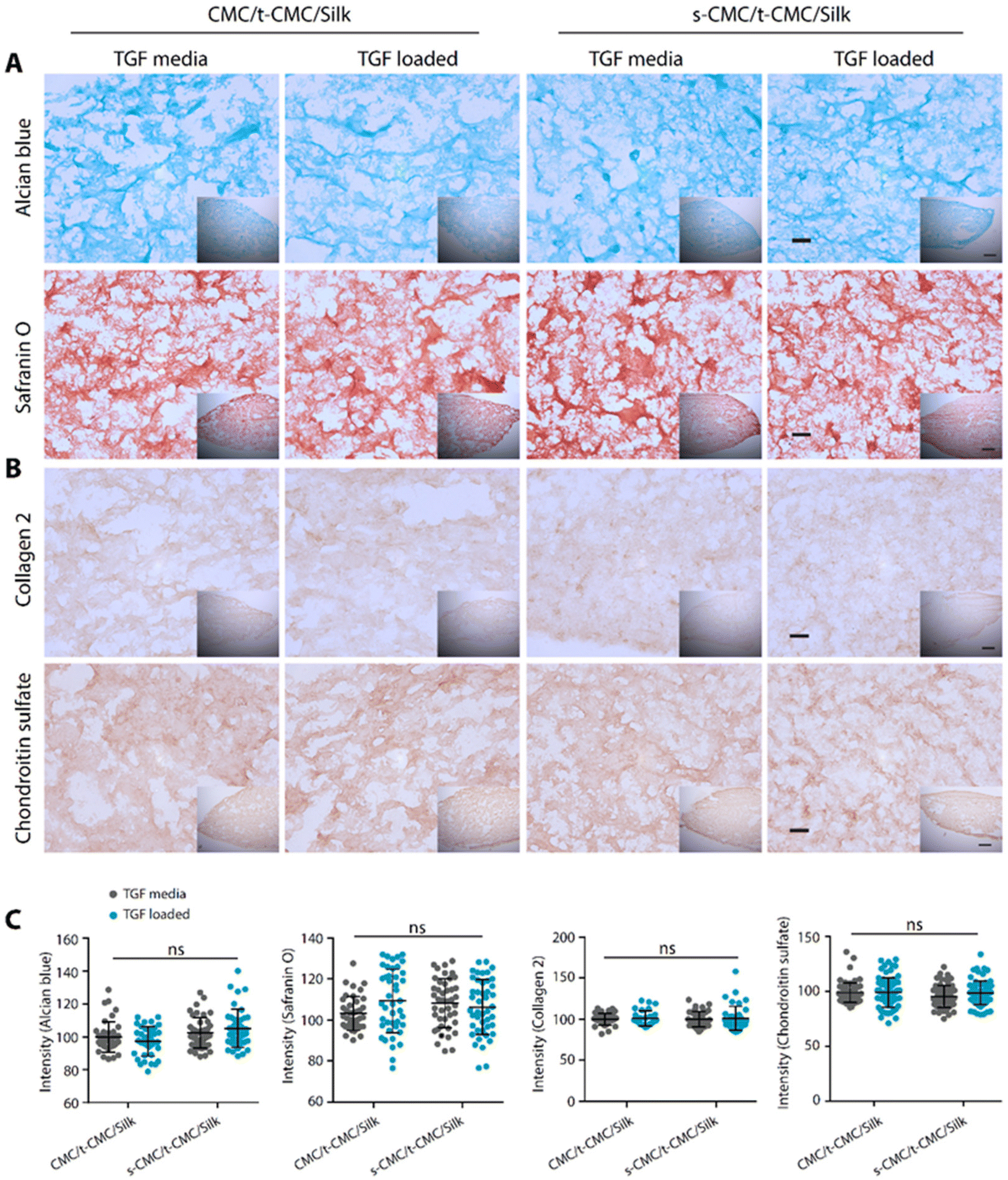 | ||
| Fig. 7 Histological and immunohistochemical staining of IFP-MSCs seeded in CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels after 14 days of chondrogenic differentiation with TGF-β present either in media or loaded in IPN hydrogels. (A) Histological analysis of hydrogel sections stained with Alcian blue and safranin O (stains sGAG), (insets – low magnification images). (B) Immunohistochemical staining of sections stained for collagen 2 and chondroitin sulfate (cartilage matrix components), high magnification scale bar: 100 μm and low magnification (inset) scale bar: 200 μm. (C) Normalised brightfield intensity measured using Image J software. Each dot represents the intensity of a single image, number of images quantified >20 for each group. Error bar depicts standard deviation (n = 3), ‘ns’ indicates no statistically significant difference between groups. | ||
We then studied the deposition of cartilage ECM components after 28 days of chondrogenic differentiation of IFP-MSCs by histological and immunohistochemical staining. In contrast to the results obtained at earlier time points, TGF-β loaded CMC/t-CMC/silk hydrogel showed reduced intensity of Alcian blue and safranin O staining compared to all the other constructs (Fig. 8A). Additionally, when immunostaining for collagen 2 and chondroitin sulfate was performed, a similar reduction in the deposition of both the ECM components was observed in TGF-β loaded CMC/t-CMC/silk hydrogel (Fig. 8B). TGF-β is a critical and thus far indispensable factor for chondrogenic differentiation of stem cells.26 The absence of sulfate groups in the CMC/t-CMC/silk hydrogel led to the decreased retention of TGF-β in hydrogels and hence, incomplete chondrogenic differentiation of IFP-MSCs. On the other hand, s-CMC/t-CMC/silk IPN hydrogels due to their ability to bind to cationic growth factors, enabled greater retention of TGF-β and hence, more pronounced chondrogenesis compared to CMC/t-CMC/silk IPN hydrogels. The results were corroborated with the quantification of brightfield intensities of histologically and immunohistochemically stained sections (Fig. 8C). Interestingly, it was observed that both CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels where TGF-β was replenished with every medium change showed similar levels of deposition of sGAG and collagen 2, especially, when compared to TGF-β loaded s-CMC/t-CMC/silk hydrogels. The gene expression of various cartilage-specific makers and hypertrophic markers validated the observation that the TGF-β loaded in s-CMC/t-CMC/silk hydrogel was active and had similar levels compared to the s-CMC/t-CMC/silk hydrogel with TGF-β supplemented in the media (ESI Fig. 7B†). Notably, no significant difference in the extent of chondrogenesis was observed between the CMC/t-CMC/silk and s-CMC/t-CMC/silk hydrogels when TGF-β was loaded or continuously supplemented into the media, suggesting a negligible role of s-CMC in directing the process of chondrogenesis. Overall, the results after day-14 and day-28 of chondrogenic differentiation, demonstrated that the TGF-β loaded in s-CMC/t-CMC/silk hydrogels showed higher retention and induces chondrogenesis compared to TGF-β loaded in CMC/t-CMC/silk hydrogels, hence signifying the importance of incorporating sulfated polysaccharides in the hydrogel as a growth factor delivery system for cartilage tissue engineering.
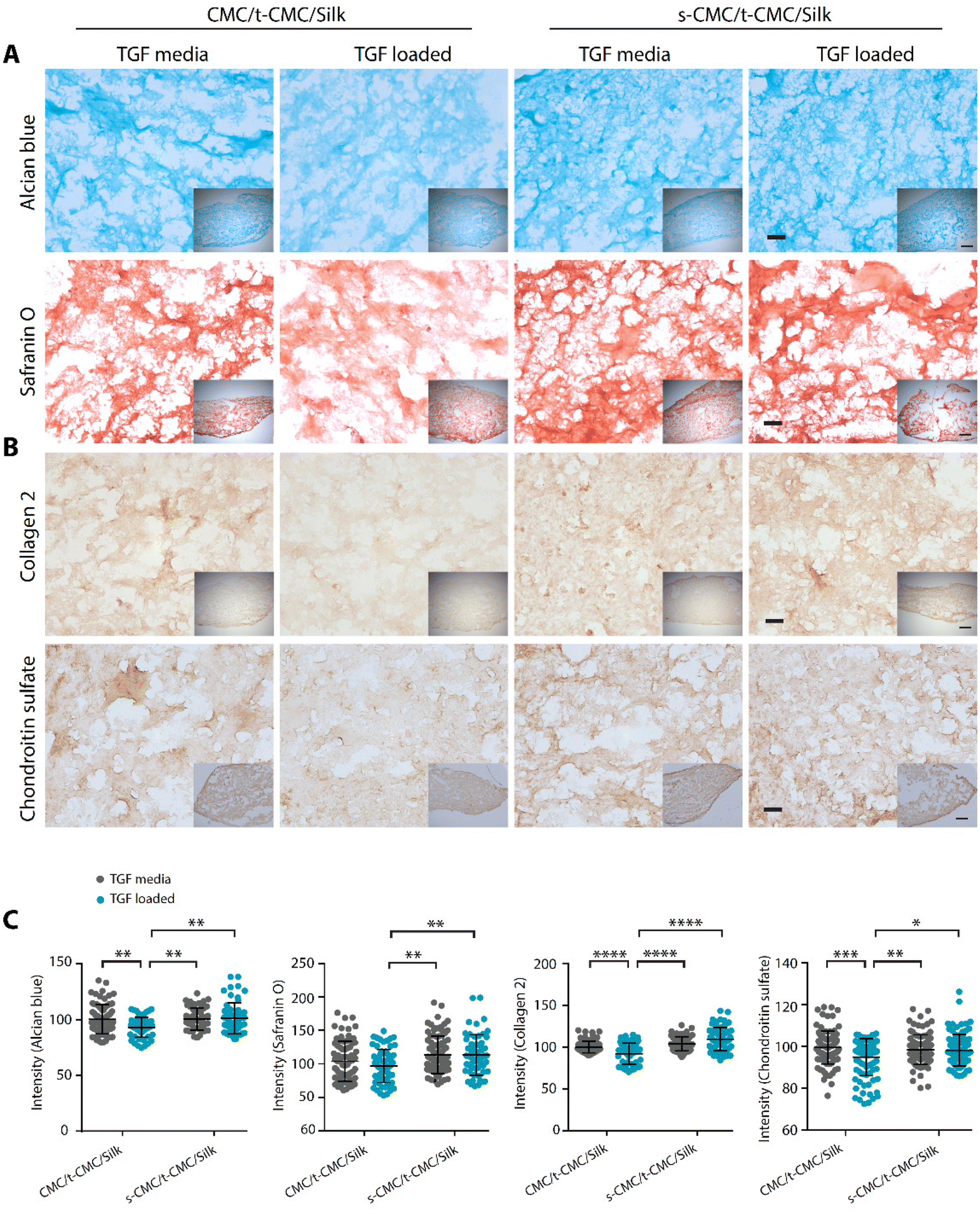 | ||
| Fig. 8 Histological and immunohistochemical staining of IFP-MSCs seeded in CMC/t-CMC/silk and s-CMC/t-CMC/silk IPN hydrogels after 28 days of chondrogenic differentiation with TGF-β present either in media or loaded in hydrogels. (A) Histological analysis of hydrogel sections stained with Alcian blue and safranin O (stains sGAG), (insets – low magnification images). (B) Immunohistochemical staining of sections stained for collagen 2 and chondroitin sulfate (cartilage matrix components), high magnification scale bar: 100 μm and low magnification (inset) scale bar: 200 μm. (C) Normalised brightfield intensity measured using Image J software. Each dot represents the intensity of a single image; number of images quantified >20 for each group. Error bar depicts standard deviation (n = 3), *(P < 0.05), **(P < 0.01), ***(P < 0.001) and ****(P < 0.0001) indicate statistically significant difference with respect to TGF-β loaded CMC/t-CMC/silk hydrogel. | ||
4. Conclusions
In the current study, we have utilized a biomimetic strategy to design and develop injectable hydrogels for the purpose of cartilage tissue engineering. The approach involves two key components. First, we have engineered a dynamic hydrogel system that gradually stiffens over time and mimics the natural process of matrix stiffening during cartilage development. Second, we have introduced a sulfated polysaccharide in the hydrogel which replicates the growth factor binding ability of heparan sulfate present in the native extracellular matrix of tissues. These remarkable properties in hydrogels were achieved through the use of silk fibroin and sulfated CMC respectively. Such a hydrogel system would provide a growth factor binding ability and negate the requirement of exogenous administration of growth factors. At the same time, the hydrogels would initially provide a soft microenvironment to cells that promotes chondrogenesis and finally stiffen with time to mechanically support the load-bearing joint. Overall, this biomimetic approach to develop an injectable hydrogel system that endows chondro-inductive niches is more translatable, induces chondrogenic differentiation and represents a substantial advancement in the field of injectable hydrogel systems for cartilage regeneration.Conflicts of interest
All the authors declare no conflict of interest.Acknowledgements
The authors thank the following individuals from IIT Kanpur: G. Karmakar for FTIR, S. Singh for EDX and R. Babu for NMR experiments. Furthermore, the authors acknowledge the live-cell imaging facility at ACMS, IIT-Kanpur, and Prof. N. Verma for his generous support with DSC experiments. Funding: the work was funded by a Science and Engineering Research Board (SERB), India (PDF/2020/000211) grant to AD and DSK. AM and SC thank the Indian Institute of Technology-Kanpur (IIT-Kanpur) for the institute fellowship and RS thanks Prime Minister's Research Fellowship (PMRF) India for thefellowship. All the authors thank The Mehta Family Centre for Engineering in Medicine at IIT-Kanpur for their generous support. DSK gratefully acknowledges the Gireesh Jankinath Chair Professorship (2018–2021), the Rajeeva and Sangeeta Lahri Chair Professorship (2022–2025) and the Tata Innovation Fellowship from the Department of Biotechnology, Government of India (2023–2028).References
- L. Zhang, J. Hu and K. A. Athanasiou, The role of tissue engineering in articular cartilage repair and regeneration, Crit. Rev. Biomed. Eng., 2009, 37(1–2), 1–57 CrossRef PubMed.
- A. J. S. Fox, A. Bedi and S. A. Rodeo, The basic science of articular cartilage: structure, composition, and function, Sports Health, 2009, 1(6), 461–468 CrossRef PubMed.
- H. Kwon, W. E. Brown, C. A. Lee, D. Wang, N. Paschos, J. C. Hu and K. A. Athanasiou, Surgical and tissue engineering strategies for articular cartilage and meniscus repair, Nat. Rev. Rheumatol., 2019, 15(9), 550–570 CrossRef PubMed.
- E. A. Makris, A. H. Gomoll, K. N. Malizos, J. C. Hu and K. A. Athanaiou, Repair and tissue engineering techniques for articular cartilage, Nat. Rev. Rheumatol., 2015, 11(1), 21–34 CrossRef CAS PubMed.
- A. Arora, A. Bhattacharjee, A. Mahaja and D. S. Katti, Cartilage tissue engineering: scaffold, cell, and growth factor-based strategies, Regener. Med., 2017, 233–257 Search PubMed.
- A. Jayakumar, V. K. Jose and J. M. Lee, Hydrogels for medical and environmental applications, Small Methods, 2020, 4(3), 1900735–1900731 CrossRef CAS.
- F. H. Chen, K. T. Rousche and R. S. Tuan, Technology Insight: adult stem cells in cartilage regeneration and tissue engineering, Nat. Clin. Pract. Rheumatol., 2006, 2(7), 373–382 CrossRef CAS PubMed.
- B. Yue, Biology of the extracellular matrix: an overview, J. Glaucoma, 2014, S20–S23 CrossRef PubMed.
- C. Prein, N. Warmbold, Z. Farkas, M. Schieker, A. Aszodi and H. Clausen-Schaumann, Structural and mechanical properties of the proliferative zone of the developing murine growth plate cartilage assessed by atomic force microscopy, Matrix Biol., 2016, 50, 1–15 CrossRef CAS PubMed.
- A. K. Williamson, A. C. Chen and R. L. Sah, Compressive properties and function—composition relationships of developing bovine articular cartilage, J. Orthop. Res., 2001, 19(6), 1113–1121 CrossRef CAS PubMed.
- J. L. Allen, M. E. Cooke and T. Alliston, ECM stiffness primes the TGFβ pathway to promote chondrocyte differentiation, Mol. Biol. Cell, 2012, 23(18), 3731–3742 CrossRef CAS PubMed.
- J. Henderson and D. R. Carter, Mechanical induction in limb morphogenesis: the role of growth-generated strains and pressures, Bone, 2002, 31(6), 645–653 CrossRef CAS PubMed.
- A. Mahajan, A. Singh, D. Datta and D. S. Katti, Bioinspired injectable hydrogels dynamically stiffen and contract to promote mechanosensing-mediated chondrogenic commitment of stem cells, ACS Appl. Mater. Interfaces, 2022, 14(6), 7531–7550 CrossRef CAS PubMed.
- Y. Yang, Y. Liu, Z. Lin, H. Shen, C. Lucas, B. Kuang, R. S. Tuan and H. Lin, Condensation–Driven Chondrogenesis of Human Mesenchymal Stem Cells within Their Own Extracellular Matrix: Formation of Cartilage with Low Hypertrophy and Physiologically Relevant Mechanical Properties, Adv. Biosyst., 2019, 3(12), 1900229 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Stiffening hydrogels to probe short-and long-term cellular responses to dynamic mechanics, Nat. Commun., 2012, 3(1), 792 CrossRef PubMed.
- S. Ramanujan, A. Pluen, T. D. McKee, E. B. Brown, Y. Boucher and R. K. Jain, Diffusion and convection in collagen gels: implications for transport in the tumor interstitium, Biophys. J., 2002, 83(3), 1650–1660 CrossRef CAS PubMed.
- B. L. Farrugia, M. S. Lord, J. Melrose and J. M. Whitelock, The role of heparan sulfate in inflammation, and the development of biomimetics as anti-inflammatory strategies, J. Histochem. Cytochem., 2018, 66(4), 321–336 CrossRef CAS PubMed.
- M. B. Karam, J. El Khoury, C. Chakar, S. Changotade, D. Lutomski, N. Naaman, G. Godeau, A. Elm'selmi, R. Younes and K. Senni, Heparan-mimetics: potential agents of tissue regeneration for bone and periodontal therapies, Med. Nov. Technol. Devices., 2021, 11, 100066 CrossRef.
- B. K. Mann, R. H. Schmedlen and J. L. West, Tethered-TGF-β increases extracellular matrix production of vascular smooth muscle cells, Biomaterials, 2001, 22(5), 439–444 CrossRef CAS PubMed.
- J. Nicolas, S. Magli, L. Rabbachin, S. Sampaolesi, F. Nicotra and L. Russo, 3D extracellular matrix mimics: fundamental concepts and role of materials chemistry to influence stem cell fate, Biomacromolecules, 2020, 21(6), 1968–1994 CrossRef CAS PubMed.
- A. Arora, A. Mahajan and D. S. Katti, TGF-β1 presenting enzymatically cross-linked injectable hydrogels for improved chondrogenesis, Colloids Surf., B, 2017, 159, 838–848 CrossRef CAS PubMed.
- A. Matsumoto, J. Chen, A. L. Collette, U.-J. Kim, G. H. Altman, P. Cebe and D. L. Kaplan, Mechanisms of silk fibroin sol− gel transitions, J. Phys. Chem. B, 2006, 110(43), 21630–21638 CrossRef CAS PubMed.
- N. A. Waghmare, A. Arora, A. Bhattacharjee and D. S. Katti, Sulfated polysaccharide mediated TGF-β1 presentation in pre-formed injectable scaffolds for cartilage tissue engineering, Carbohydr. Polym., 2018, 193, 62–72 CrossRef CAS PubMed.
- S. S. Bhutada, M. Sriram and D. S. Katti, Sulfated carboxymethylcellulose conjugated electrospun fibers as a growth factor presenting system for tissue engineering, Carbohydr. Polym., 2021, 268, 118256 CrossRef CAS PubMed.
- X. Du, L. Cai, J. Xie and X. Zhou, The role of TGF-beta3 in cartilage development and osteoarthritis, Bone Res., 2023, 11(1), 2 CrossRef CAS PubMed.
- A. Tekari, R. Luginbuehl, W. Hofstetter and R. J. Egli, Transforming growth factor beta signaling is essential for the autonomous formation of cartilage-like tissue by expanded chondrocytes, PLoS One, 2015, 10(3), e0120857 CrossRef PubMed.
- Y. Ogushi, S. Sakai and K. Kawakami, Synthesis of enzymatically-gellable carboxymethylcellulose for biomedical applications, J. Biosci. Bioeng., 2007, 104(1), 30–33 CrossRef CAS PubMed.
- Z. M. Wang, L. Li, B. S. Zheng, N. Normakhamatov and S. Y. Guo, Preparation and anticoagulation activity of sodium cellulose sulfate, Int. J. Biol. Macromol., 2007, 41(4), 376–382 CrossRef CAS PubMed.
- T. Li, X. Song, C. Weng, X. Wang, L. Gu, X. Gong, Q. Wei, X. Duan, L. Yang and C. Chen, Silk fibroin/carboxymethyl chitosan hydrogel with tunable biomechanical properties has application potential as cartilage scaffold, Int. J. Biol. Macromol., 2019, 137, 382–391 CrossRef CAS PubMed.
- S. Banerjee and K. K. Kar, Impact of degree of sulfonation on microstructure, thermal, thermomechanical and physicochemical properties of sulfonated poly ether ether ketone, Polymer, 2017, 109, 176–186 CrossRef CAS.
- C. Leyva-Porras, P. Cruz-Alcantar, V. Espinosa-Solís, E. Martínez-Guerra, C. I. Piñón-Balderrama, I. C. Martínez and M. Z. Saavedra-Leos, Application of differential scanning calorimetry (DSC) and modulated differential scanning calorimetry (MDSC) in food and drug industries, Polymers, 2019, 12(1), 5 CrossRef PubMed.
- D. N. Rockwood, R. C. Preda, T. Yücel, X. Wang, M. L. Lovett and D. L. Kaplan, Materials fabrication from Bombyx mori silk fibroin, Nat. Protoc., 2011, 6(10), 1612–1631 CrossRef CAS PubMed.
- S. Sakai, K. Hirose, K. Taguchi, Y. Ogushi and K. Kawakami, An injectable, in situ enzymatically gellable, gelatin derivative for drug delivery and tissue engineering, Biomaterials, 2009, 30(20), 3371–3377 CrossRef CAS PubMed.
- A. Arora, M. Sriram, A. Kothari and D. S. Katti, Co-culture of infrapatellar fat pad–derived mesenchymal stromal cells and articular chondrocytes in plasma clot for cartilage tissue engineering, Cytotherapy, 2017, 19(7), 881–894 CrossRef CAS PubMed.
- A. Mahajan, S. Hazra, A. Arora and D. S. Katti, Isolation, Expansion, and Differentiation of Mesenchymal Stem Cells from the Infrapatellar Fat Pad of the Goat Stifle Joint, J. Visualized Exp., 2022,(186), e63617 CAS.
- N. Schmitz, S. Laverty, V. Kraus and T. Aigner, Basic methods in histopathology of joint tissues, Osteoarthritis Cartilage, 2010, 18, S113–S116 CrossRef PubMed.
- J. H. Lee, H. S. Ahn and E. K. Kim, Efficacy of sodium hyaluronate and carboxymethylcellulose in treating mild to moderate dry eye disease, Cornea, 2011, 30(2), 175–179 CrossRef PubMed.
- G. Pluhar, A. Turner, A. Pierce, C. Toth and D. Wheeler, A comparison of two biomaterial carriers for osteogenic protein-1 (BMP-7) in an ovine critical defect model, J. Bone Jt. Surg., 2006, 88(7), 960–966 CrossRef CAS PubMed.
- Y. Kim, E. E. Gill and J. C. Liu, Enzymatic cross-linking of resilin-based proteins for vascular tissue engineering applications, Biomacromolecules, 2016, 17(8), 2530–2539 CrossRef CAS PubMed.
- M. E. Carnes, C. R. Gonyea, R. G. Mooney, J. W. Njihia, J. M. Coburn and G. D. Pins, Horseradish peroxidase-catalyzed crosslinking of fibrin microthread scaffolds, Tissue Eng., Part C, 2020, 26(6), 317–331 CrossRef CAS PubMed.
- J. E. Frith, A. R. Cameron, D. J. Menzies, P. Ghosh, D. L. Whitehead, S. Gronthos, A. C. Zannettino and J. J. Cooper-White, An injectable hydrogel incorporating mesenchymal precursor cells and pentosan polysulphate for intervertebral disc regeneration, Biomaterials, 2013, 34(37), 9430–9440 CrossRef CAS PubMed.
- S. Hauck, P. Zager, N. Halfter, E. Wandel, M. Torregrossa, A. Kakpenova, S. Rother, M. Ordieres, S. Räthel and A. Berg, Collagen/hyaluronan based hydrogels releasing sulfated hyaluronan improve dermal wound healing in diabetic mice via reducing inflammatory macrophage activity, Bioact. Mater., 2021, 6(12), 4342–4359 CAS.
- K. Luo, Y. Yang and Z. Shao, Physically crosslinked biocompatible silk–fibroin–based hydrogels with high mechanical performance, Adv. Funct. Mater., 2016, 26(6), 872–880 CrossRef CAS.
- B. P. Partlow, C. W. Hanna, J. Rnjak-Kovacina, J. E. Moreau, M. B. Applegate, K. A. Burke, B. Marelli, A. N. Mitropoulos, F. G. Omenetto and D. L. Kaplan, Highly tunable elastomeric silk biomaterials, Adv. Funct. Mater., 2014, 24(29), 4615–4624 CrossRef CAS PubMed.
- D. Su, M. Yao, J. Liu, Y. Zhong, X. Chen and Z. Shao, Enhancing mechanical properties of silk fibroin hydrogel through restricting the growth of β-sheet domains, ACS Appl. Mater. Interfaces, 2017, 9(20), 17489–17498 CrossRef CAS PubMed.
- S. Xu, Q. Li, H. Pan, Q. Dai, Q. Feng, C. Yu, X. Zhang, Z. Liang, H. Dong and X. Cao, Tubular silk fibroin/gelatin-tyramine hydrogel with controllable layer structure and its potential application for tissue engineering, ACS Biomater. Sci. Eng., 2020, 6(12), 6896–6905 CrossRef CAS PubMed.
- N. D. N. Marques, R. D. C. Balaban, S. Halila and R. Borsali, Synthesis and characterization of carboxymethylcellulose grafted with thermoresponsive side chains of high LCST: The high temperature and high salinity self-assembly dependence, Carbohydr. Polym., 2018, 184, 108–117 CrossRef CAS PubMed.
- G. Joshi, S. Bisht, K. Pandey, S. Naithani, S. Monga, B. Thapliyal and M. Bist, New route for carboxymethylation of cellulose: synthesis, structural analysis and properties, Cellul. Chem. Technol., 2017, 7–8 Search PubMed.
- Y. Huang, G. Sun, L. Lyu, Y. Li, D. Li, Q. Fan, J. Yao and J. Shao, Dityrosine-inspired photocrosslinking technique for 3D printing of silk fibroin-based composite hydrogel scaffolds, Soft Matter, 2022, 18(19), 3705–3712 RSC.
- G. Priya, U. Narendrakumar and I. Manjubala, Thermal behavior of carboxymethyl cellulose in the presence of polycarboxylic acid crosslinkers, J. Therm. Anal. Calorim., 2019, 138, 89–95 CrossRef CAS.
- C. Mengdi, L. Wei, Y. Changping, X. Suli, J. Su and S. Liping, The influence of molecular structure on the thermal properties and processability of nitrile-based resin molecules, Journal of Physics: Conference Series, IOP Publishing, 2021, p. 012002.
- D. M. Murali and G. Shanmugam, The aromaticity of the phenyl ring imparts thermal stability to a supramolecular hydrogel obtained from low molecular mass compound, New J. Chem., 2019, 43(31), 12396–12409 RSC.
- J. O. Valderrama, R. A. Campusano and R. E. Rojas, Glass transition temperature of ionic liquids using molecular descriptors and artificial neural networks, C. R. Chim., 2017, 20(5), 573–584 CrossRef CAS.
- M. R. Arkenberg, H. D. Nguyen and C.-C. Lin, Recent advances in bio-orthogonal and dynamic crosslinking of biomimetic hydrogels, J. Mater. Chem. B, 2020, 8(35), 7835–7855 RSC.
- F. F. Karageorgos and C. Kiparissides, Prediction of viscoelastic properties of enzymatically crosslinkable tyramine–modified hyaluronic acid solutions using a dynamic monte carlo kinetic approach, Int. J. Mol. Sci., 2021, 22(14), 7317 CrossRef CAS PubMed.
- P. Gysen, M. Malaise, S. Caspar and P. J. C. R. Franchimont, Measurement of proteoglycans, elastase, collagenase and protein in synovial fluid in inflammatory and degenerative arthropathies, Clin. Rheumatol., 1985, 4, 39–50 CrossRef CAS PubMed.
- S. Keten, Z. Xu, B. Ihle and M. J. Buehler, Nanoconfinement controls stiffness, strength and mechanical toughness of β-sheet crystals in silk, Nat. Mater., 2010, 9(4), 359–367 CrossRef CAS PubMed.
- J. L. Young and A. J. Engler, Hydrogels with time-dependent material properties enhance cardiomyocyte differentiation in vitro, Biomaterials, 2011, 32(4), 1002–1009 CrossRef CAS PubMed.
- I.-N. Lee, O. Dobre, D. Richards, C. Ballestrem, J. M. Curran, J. A. Hunt, S. M. Richardson, J. Swift and L. S. Wong, Photoresponsive hydrogels with photoswitchable mechanical properties allow time-resolved analysis of cellular responses to matrix stiffening, ACS Appl. Mater. Interfaces, 2018, 10(9), 7765–7776 CrossRef CAS PubMed.
- B. K. Hall and T. Miyake, All for one and one for all: condensations and the initiation of skeletal development, Bioessays, 2000, 22(2), 138–147 CrossRef CAS PubMed.
- Y.-I. Chung, G. Tae and S. H. Yuk, A facile method to prepare heparin-functionalized nanoparticles for controlled release of growth factors, Biomaterials, 2006, 27(12), 2621–2626 CrossRef CAS PubMed.
- G. Sridharan and A. A. Shankar, Toluidine blue: A review of its chemistry and clinical utility, Int. J. Oral Maxillofac. Pathol., 2012, 16(2), 251 CrossRef PubMed.
- B. Wang, P. J. Diaz-Payno, D. C. Browe, F. E. Freeman, J. Nulty, R. Burdis and D. J. Kelly, Affinity-bound growth factor within sulfated interpenetrating network bioinks for bioprinting cartilaginous tissues, Acta Biomater., 2021, 128, 130–142 CrossRef CAS PubMed.
- M. Kunou, M. Koizumi, K. Shimizu, M. Kawase and K. Hatanaka, Synthesis of sulfated colominic acids and their interaction with fibroblast growth factors, Biomacromolecules, 2000, 1(3), 451–458 CrossRef CAS PubMed.
- I. Freeman, A. Kedem and S. Cohen, The effect of sulfation of alginate hydrogels on the specific binding and controlled release of heparin-binding proteins, Biomaterials, 2008, 29(22), 3260–3268 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01466e |
| ‡ The first group of equally contributing authors. |
| § The second group of equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2024 |