
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
TEMPO-mediated late stage photochemical hydroxylation of biaryl sulfonium salts†
Yue
Zhao
,
Congjun
Yu
,
Wenjing
Liang
,
Iuliana L.
Atodiresei
and
Frederic W.
Patureau
 *
*
Institute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, Aachen 52074, Germany. E-mail: frederic.patureau@rwth-aachen.de; Web: https://www.patureau-oc-rwth-aachen.de
First published on 7th February 2022
Abstract
The late stage photochemical hydroxylation of biaryl sulfonium salts was enabled with a TEMPO derivative as a simple oxygen source, in metal free conditions. The scope and mechanism of this exceptionally simple synthetic method, which constructs important arylated phenols from aromatic C–H bonds, are herein discussed.
Hydroxylated arenes are frequently found in important pharmaceuticals, agrochemicals, polymers, and biologically active natural compounds. Thus, owing to the ubiquity of the phenol motif, the hydroxylation of simple arenes is an important chemical transformation in various metabolic pathways and organic synthesis.1–4 However, traditional hydroxylation often requires either onerous metal catalysts and sophisticated ligands and/or multiple synthetic steps.5–12 Moreover, these do not always provide the desired regioselectivity. Therefore, the development of simple, affordable, and highly regio-selective synthetic methods that would allow the direct hydroxylation of aromatic C–H bonds constitutes a strategic priority. For example, Siegel and co-authors utilized in 2013 an activated cyclic peroxide in hexafluoroisopropanol (HFIP) to furnish the corresponding phenols upon hydrolysis (Scheme 1, eqn (1)).13 Ritter and co-authors utilized a similar strategy in 2018 with a non-cyclic activated peroxide, also in HFIP (eqn (2)).14 Most recently, Yu and co-workers even managed the benzoic acid directed Pd-catalyzed ortho hydroxylation of aromatic C–H bonds (eqn (3)).15 Moreover, they impressively utilized molecular oxygen as both the terminal oxidant and the hydroxyl source. Of course, this strategy requires a specific directing group. In terms of selectivity, the 2019 aryl sulfonium salts approach of Ritter stands out because of the highly regio-selective character of the prior bulky thianthrenium installation at the target C–H bond (eqn (4)).16 However, and while they impressively utilized simple water as the hydroxyl source, the method still requires an onerous Iridium photocatalyst as well as a Copper additive. Consequently, we decided to investigate the feasibility of a simpler metal-free approach for the photochemical hydroxylation of aryl sulfonium salts (eqn (5)).
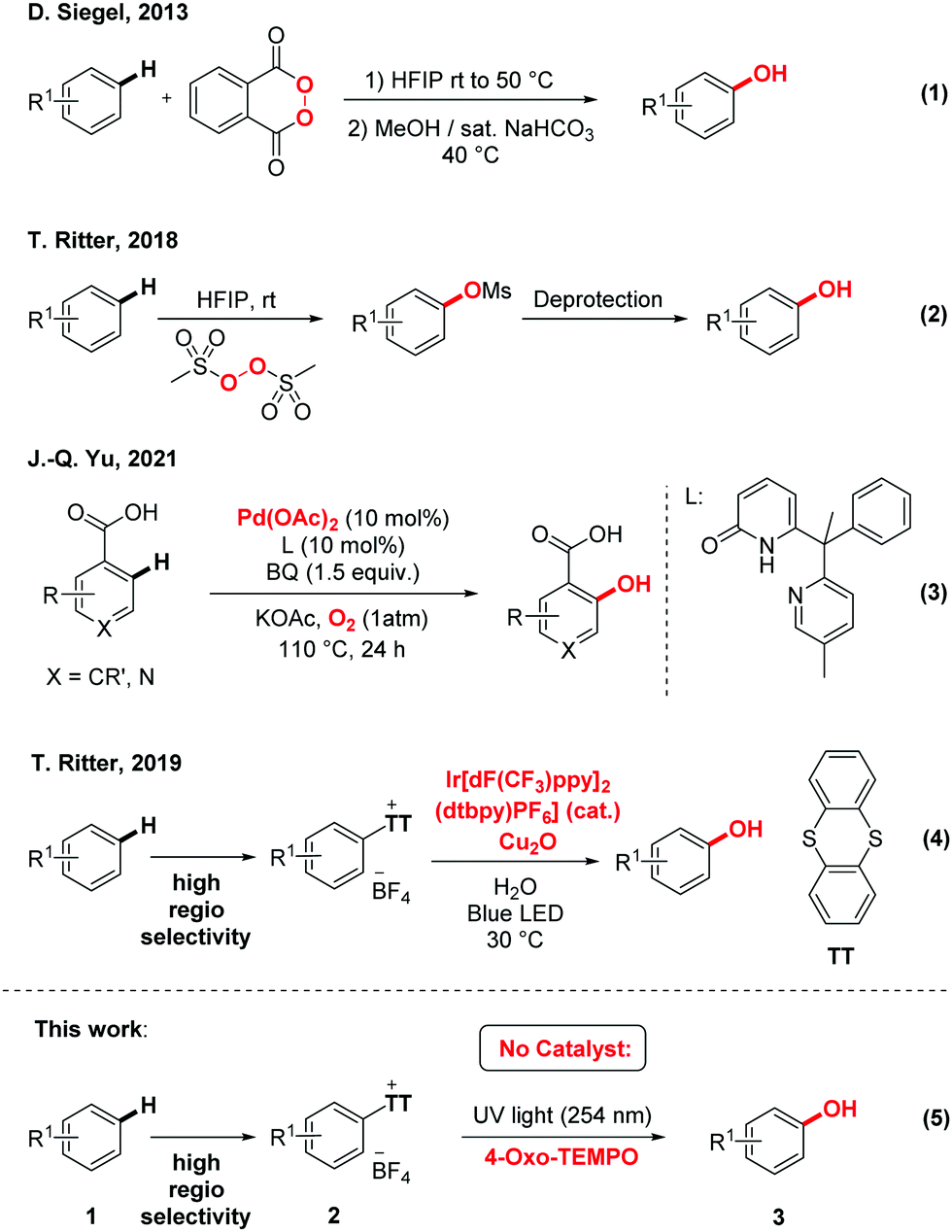 | ||
| Scheme 1 Recent aromatic C–H hydroxylations. | ||
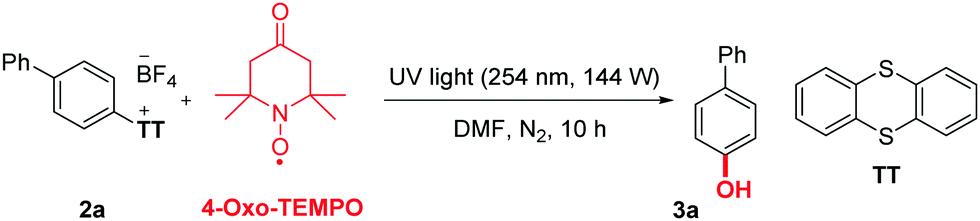
We commenced our study with a photo-reactor developed by us (254 nm, 144 W, see ESI† for technical details and pictures), which recently allowed the metal-free photochemical (hetero)-arylation of aryl sulfonium salts.17 In this prior study, we noticed in a control experiment that the addition of TEMPO, a well-known radical scavenger as well as oxidant, did not deliver the expected heteroarene coupled product. Instead, it delivered the corresponding phenol, arising from the hydroxylation of the thiantrenium substrate. A very interesting feature of this new reaction is that it does not simply deliver the expected O–N TEMPO-trapped aryl radical intermediate, but rather directly the corresponding O–H free phenol. Thus, the O–N bond is readily reduced in situ. This direct hydroxylation of an aromatic position without the need for a hydrolytic work up or deprotection is rare for metal free conditions (Scheme 1, eqn (1) and (2)). This initial result17 therefore encouraged us to further investigate this reaction.
At the initial stage of the investigation, biphenyl was chosen as a substrate to form aryl thianthrenium18–24 salt 2a, which was utilized to explore the optimal conditions for a site-selective hydroxylation of simple arenes (Table 1). The type of light, power, additive, reaction time, solvent and hydroxyl sources were notably evaluated. Typically, a 50 mL flat-bottom cylindrical quartz vial equipped with a magnetic stir bar is charged with aryl thianthrenium salts 2a (0.4 mmol, 1.0 equiv.), 4-oxo-TEMPO (3.2 mmol, 8 equiv.), and DMF (3 mL) under inert N2 atmosphere. The tube is then sealed, and the mixture is stirred at room temperature under UV-light (254 nm, 144 W) for 10 h. Phenol 3a was thus obtained in 82% isolated yield (Table 1, entry 1). In the absence of light or when 390 nm UV-light was used instead of 254 nm UV light, 3a was not detected (Table 1, entries 2 and 3). When the power of the light was reduced by half (72 W), the yield of 3a dropped to 56% (Table 1, entry 4). None of the additives tested improved the yield (Table 1, entries 5 and 6). Interestingly, when the reaction was carried out under air, the yield dropped to 74% (Table 1, entry 7). Among all other tested solvents, none afforded more than 46% of the desired product (Table 1, entry 8). There is moreover no obvious benefit to extend or shorten the reaction time (Table 1, entries 11 and 12). Finally, no other hydroxylation reagents could deliver the desired product, with the exception of TEMPO (60% yield, Table 1, entries 9 and 10).
|
|
||
|---|---|---|
| Entry | Variations from the optimal conditionsa | Yieldb (%) of 3a |
| a Reaction conditions: 2a (0.4 mmol, 1 equiv.), 4-oxo-TEMPO (3.2 mmol, 8 equiv.), DMF (3 mL), UV-light (254 nm, 144 W), quartz vial, r.t., N2, 10 h. b Yield of isolated product. c Other solvent: 1,4-dioxane, DMSO, CH3CN, DCM, DCE, THF, HFIP. d Other hydroxyl sources: H2O2, pyridine-N-oxide, 4-methylmorpholine N-oxide. | ||
| 1 | None | 82 |
| 2 | No light | 0 |
| 3 | 390 nm instead of 254 nm | 0 |
| 4 | 72 W instead of 144 W | 56 |
| 5 | Al2O3 additive (0.2 mmol) | 50 |
| 6 | SiO2 additive (0.2 mmol) | 44 |
| 7 | Air instead of N2 | 74 |
| 8 | Other solvents instead of DMFc | <46 |
| 9 | TEMPO instead of 4-oxo-TEMPO | 60 |
| 10 | Other hydroxyl sourcesd | Traces |
| 11 | 8 h reaction time | 71 |
| 12 | 16 h reaction time | 78 |
With the optimized reaction conditions in hand, we set out to explore the scope of compatible arenes (Scheme 2). Initially, a wide range of biphenyl derivatives was studied (3a–l), which afforded promising yields. Various functional groups were well tolerated under standard conditions, such as methyl, isopropyl, methoxy, ester, carbonyl, phenoxy, imidazole, cyano and various halogens. Even an iodo derivative was obtained (product 3f), albeit in a reduced 24% yield, which is presumably due to limited compatibility with the UV light. Interestingly, two important drugs were successfully engaged as substrates in this reaction. Bifonazole 1k, an antifungal drug, as well as Flurbiprofen 1l, a nonsteroidal anti-inflammatory drug, delivered C–H hydroxylated products 3k and 3l in respectively 62% and 56% yield on the second step. These results demonstrate the utility of the method. A series of important heterocycles were otherwise well tolerated, such as dibenzofuran (3m), dibenzothiophene (3n), and several carbazoles (3o–r). Interestingly, when engaging the structurally similar but considerably more electron-withdrawing fluorenone (1t), not only does the thianthrenium group move from the 3- to the 2-position (2t), but the oxo-TEMPO-adduct is isolated instead of the corresponding free phenol (product 3t). While it is unclear why the electronic situation in 3t would prevent in situ O–N reduction, it does at least prove the TEMPO adduct to be a plausible intermediate in this reaction. The structure of 3t was confirmed by X-ray crystallography (Fig. 1).
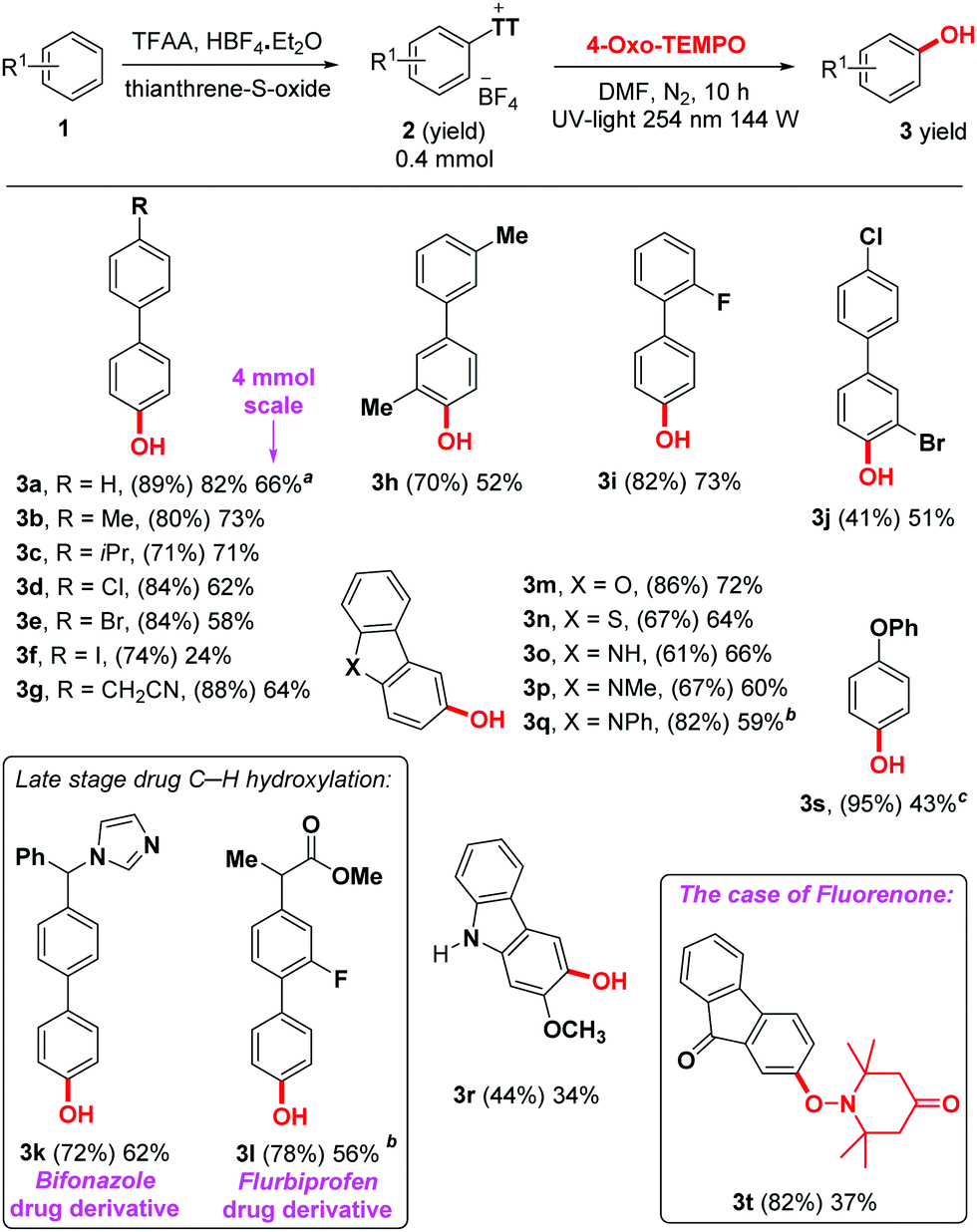 | ||
| Scheme 2 Reaction scope, isolated yields. Reaction conditions for the first step: see ESI;† reaction conditions for the second step: 2 (0.4 mmol), 4-oxo-TEMPO (3.2 mmol), DMF (3 mL), UV light (254 nm, 144 W), quartz vial, r.t., N2, 10 h. Yield of isolated product; yield in parentheses for the first step. a 66% Yield of isolated product was obtained by increasing the reaction scale to 4 mmol, reaction time: 24 h, DMF (10 mL), 4-oxo-TEMPO (32 mmol). b Hydroxyl source = TEMPO. c Hydroxyl source = 20 equiv. TEMPO. | ||
 | ||
| Fig. 1 X-Ray structure of product 3t, ORTEP25 view, 30% probability level. | ||
Furthermore, interestingly, aromatic substrates that do not possess a biaryl unit perform often poorly in this reaction. While this constitutes a clear limitation of the synthetic method, it may also indicate a certain electronic contribution of the extended π-system to the reaction mechanism. In order to gain additional mechanistic insights, we performed some control experiments in the presence of either 1,4-dinitrobenzene, 1,1-diphenylethylene, or BHT (Scheme 3). This reduced the yield of 3a from 82% to 23%, 41% and 31%, respectively. In the case of 1,1-diphenylethylene, a trace of the trapped adduct 4a could be detected by GCMS analysis of the crude reaction mixture (m/z = 332, see ESI†). These results further highlight the probable radical character of the reaction. Finally, compound 2a on the one hand, as well as a combination of 2a with 4-oxo-TEMPO (8 equiv.) on the other hand, have almost no absorption under a 254 nm wavelength (Fig. 2). Thus, autocatalysis can be reasonably ruled out.
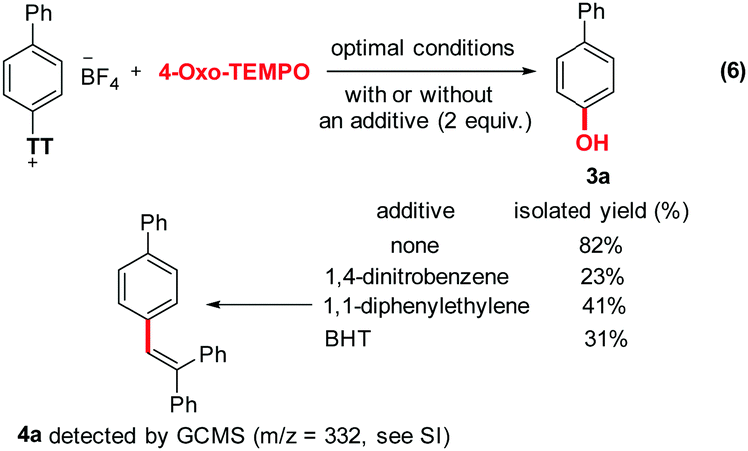 | ||
| Scheme 3 Mechanistic experriments. | ||
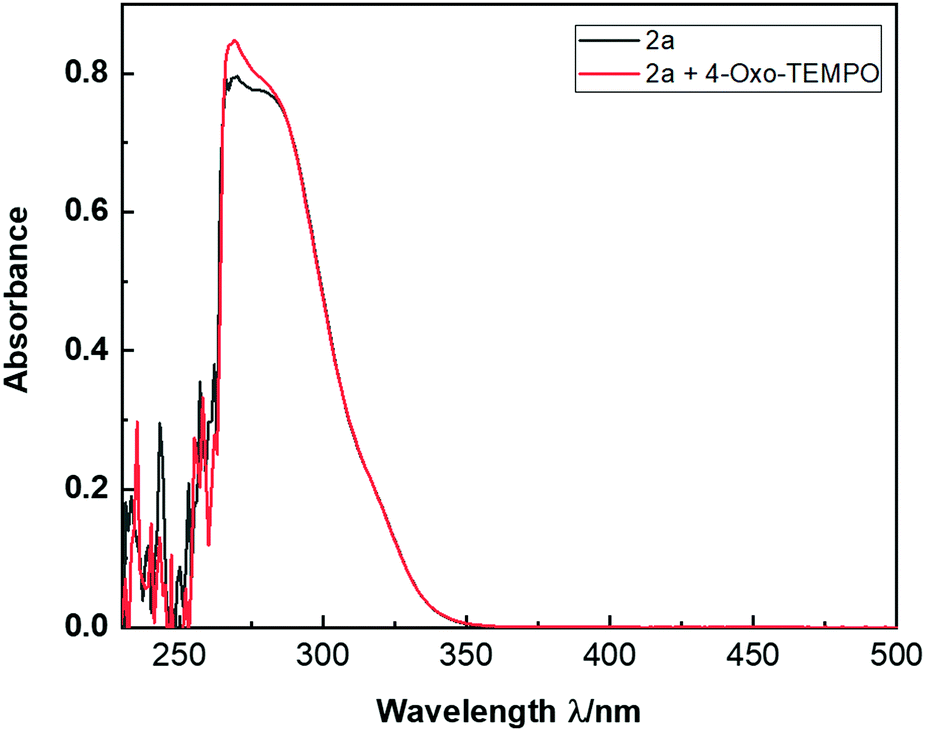 | ||
| Fig. 2 UV absorption experiments with 2a and a mixture of 2a combined to 4-oxo-TEMPO (8 equiv. in DMF). | ||
Combining the above mechanistic insights as well as previous reports,17,26 we conjectured a possible mechanism (Scheme 4). The UV-light would first disrupt the aryl thianthrenium C–S bond to form the thianthrene radical cation A and aryl radical B. The latter species would then be captured by 4-oxo-TEMPO to form C–O–N intermediate C, a structure which was isolated in the case of product 3t. The UV-light would then disrupt the O–N bond to generate semi-persistent phenol radical D and N-centered radical E. The recombination of phenol radical D with the thianthrenium radical A to form sulfonium–phenol intermediate F (or alternatively F′) is a possibility that we envisioned in order to explain the isolation of phenol product 3 rather than O–N intermediate C. However, our attempts to experimentally detect it were unsuccessful. Nevertheless, similar sulfonium–phenol intermediates were previously proposed in the recent literature.27
 | ||
| Scheme 4 Proposed mechanism. | ||
The limitation of the method to arene substrates featuring extended π-systems might arise in part from the need for a sufficiently stabilized radical intermediate (B, D). The workup of the reaction through simple silica column chromatography under air would then directly deliver phenol product 3 as well as byproduct G, which we could detect through APCI-HRMS (Scheme 3).
In conclusion, we reported here the first TEMPO mediated catalyst-free and metal-free photochemical hydroxylation of aryl sulfonium salts. TEMPO is already known as an oxidant28–34 as well as a radical scavenger.35–39 Nevertheless, its use as a hydroxyl source for the synthesis of phenols is to our knowledge unprecedented. The key to this approach is the UV-light, which can disrupt the C–S bond to form thianthrene radical cations and aryl radicals. The latter are then captured by TEMPO, further generating phenol radicals under UV light irradiation. The method has moreover great potential for the late-stage hydroxylation of medicines, as we have demonstrated with antifungal drug Bifonazole40 or nonsteroidal anti-inflammatory drug Flurbiprofen41,42 (respectively products 3k and 3l).
ERC project 716136:2O2ACTIVATION is acknowledged for generous financial support. We are also thankful to the Chinese Scholarship Council (CSC) for financial support to Yue Zhao (No. 201908320377) and Wenjing Liang (No. 201906740012).
Conflicts of interest
There are no conflicts to declare.Notes and references
-
Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, Weinheim, 2003 Search PubMed
.
-
J. H. P. Tyman, Synthetic and Natural Phenols, Studies in Organic Chemistry 52, Elsevier, Amsterdam, 1996 vol. 5 Search PubMed
-
C. A. Fyfe, in The Chemistry of the Hydroxyl Group, ed. S. Patai, Wiley-Interscience, New York, 1971, vol. 1, pp. 83–127 Search PubMed
-
J. F. Hartwig, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-I. Negishi, Wiley-Interscience, New York, 2002, vol. 1, p. 1097 Search PubMed
- S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912 RSC
- D. A. Alonso, C. Nájera, I. M. Pastor and M. Yus, Chem. – Eur. J., 2010, 16, 5274 CrossRef CAS PubMed
- Z. He and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 3353 CrossRef CAS PubMed
- S. Taktak, M. Flook, B. M. Foxman, L. Que Jr and E. V. Rybak-Akimova, Chem. Commun., 2005, 5301 RSC
- T. Jintoku, K. Nishimura, K. Takaki and Y. Fujiwara, Chem. Lett., 1990, 1687 CrossRef CAS
- H. Gao, Z. Zhou, D. H. Kwon, J. Coombs, S. Jones, N. E. Behnke, D. H. Ess and L. Kürti, Nat. Chem., 2017, 9, 681 CrossRef CAS PubMed
- R. J. Schmidt, Appl. Catal., A, 2005, 280, 89 CrossRef CAS
- L. Cheng, H. Wang, H. Cai, J. Zhang, X. Gong and W. Han, Science, 2021, 374, 77 CrossRef CAS PubMed
- C. Yuan, Y. Liang, T. Hernandez, A. Berriochoa, K. N. Houk and D. Siegel, Nature, 2013, 499, 192 CrossRef CAS PubMed
- J. Börgel, L. Tanwar, F. Berger and T. Ritter, J. Am. Chem. Soc., 2018, 140, 16026 CrossRef PubMed
- Z. Li, Z. Wang, N. Chekshin, S. Qian, J. X. Qiao, P. T. Cheng, K. S. Yeung, W. R. Ewing and J. Q. Yu, Science, 2021, 372, 1452 CrossRef CAS PubMed
- R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161 CrossRef CAS PubMed
- Y. Zhao, C. Yu, W. Liang and F. W. Patureau, Org. Lett., 2021, 23, 6232 CrossRef CAS PubMed
- Y.-L. Zhang, G.-H. Wang, Y. Wu, C.-Y. Zhu and P. Wang, Org. Lett., 2021, 23, 8522 CrossRef CAS PubMed
- X. Li, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2021, 60, 6943 CrossRef CAS PubMed
- Y. Ye, J. Zhu and Y. Huang, Org. Lett., 2021, 23, 2386 CrossRef CAS PubMed
- X.-X. Nie, Y.-H. Huang and P. Wang, Org. Lett., 2020, 22, 7716 CrossRef CAS PubMed
- L. Liang, H.-Y. Niu, R.-L. Li, Y.-F. Wang, J.-K. Yan, C.-G. Li and H.-M. Guo, Org. Lett., 2020, 22, 6842 CrossRef CAS PubMed
- Y. Wu, Y.-H. Huang, X.-Y. Chen and P. Wang, Org. Lett., 2020, 22, 6657 CrossRef CAS PubMed
- X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang, Chem. Commun., 2020, 56, 5058 RSC
- L. J. Farrugia, WinGX and ORTEP for Windows: an update, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS PubMed
- See for example: K. Higuchi, T. Tago, Y. Kokubo, M. Ito, M. Tayu, S. Sugiyama and T. Kawasaki, Org. Chem. Front., 2018, 5, 3219 RSC
- A. E. De Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1153 CrossRef CAS
- C. Bolm, A. S. Magnus and J. P. Hildebrand, Org. Lett., 2000, 2, 1173 CrossRef CAS PubMed
- A. L. Chauvin, S. A. Nepogodiev and R. A. Field, J. Org. Chem., 2005, 70, 960 CrossRef CAS PubMed
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730 CrossRef CAS PubMed
- C. Perusquía-Hernández, G. R. Lara-Issasi, B. A. Frontana-Uribe and E. Cuevas-Yañez, Tetrahedron Lett., 2013, 54, 3302 CrossRef
- X. Zhu, Y. F. Wang, W. Ren, F. L. Zhang and S. Chiba, Org. Lett., 2013, 15, 3214 CrossRef CAS PubMed
- Z. He, B. Wibbeling and A. Studer, Adv. Synth. Catal., 2013, 355, 3639 CrossRef CAS
- Y. Zhao, J. H. Lin, X. C. Hang and J. C. Xiao, J. Org. Chem., 2018, 83, 14120 CrossRef CAS PubMed
- J. Li, J. Chen, R. Sang, W.-S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56 CrossRef CAS PubMed
- M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163 CrossRef CAS
- S. Heindl, M. Riomet, J. Matyasovsky, M. Lemmerer, N. Malzer and N. Maulide, Angew. Chem., Int. Ed., 2021, 60, 19123 CrossRef CAS PubMed
- M. Scherübl, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 711 CrossRef PubMed
- T. E. Lackner and S. P. Clissold, Bifonazole. A review of its antimicrobial activity and therapeutic use in superficial mycoses, Drugs, 1989, 38, 204 CrossRef CAS PubMed
- J. L. Eriksen, S. A. Sagi, T. E. Smith, S. Weggen, P. Das, D. C. McLendon, V. V. Ozols, K. W. Jessing, K. H. Zavitz, E. H. Koo and T. E. Golde, J. Clin. Invest., 2003, 112, 440 CrossRef CAS PubMed
- M. R. Wester, J. K. Yano, G. A. Schoch, C. Yang, K. J. Griffin, C. D. Stout and E. F. Johnson, J. Biol. Chem., 2004, 279, 35630 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Optimization, methods, characterization and spectra. CCDC 2118729. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc07057f |
| This journal is © The Royal Society of Chemistry 2022 |