
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular mixing of benzene-1,3,5-tricarboxamide supramolecular hydrogelators allows tunable biomimetic hydrogels for control of cell aggregation in 3D†
Shahzad
Hafeez
 a,
Fiona R.
Passanha
b,
Antonio J.
Feliciano
a,
Floor A. A.
Ruiter
ab,
Afonso
Malheiro
a,
René P. M.
Lafleur
c,
Nicholas M.
Matsumoto
c,
Clemens
van Blitterswijk
a,
Lorenzo
Moroni
a,
Paul
Wieringa
a,
Vanessa L. S.
LaPointe
b and
Matthew B.
Baker
*a
a,
Fiona R.
Passanha
b,
Antonio J.
Feliciano
a,
Floor A. A.
Ruiter
ab,
Afonso
Malheiro
a,
René P. M.
Lafleur
c,
Nicholas M.
Matsumoto
c,
Clemens
van Blitterswijk
a,
Lorenzo
Moroni
a,
Paul
Wieringa
a,
Vanessa L. S.
LaPointe
b and
Matthew B.
Baker
*a
aDepartment of Complex Tissue Regeneration, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht University, P.O. Box 616, 6200 MD Maastricht, The Netherlands. E-mail: m.baker@maastrichtuniversity.nl
bDepartment of Cell Biology-Inspired Tissue Engineering, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht University, P.O. Box 616, 6200 MD Maastricht, The Netherlands
cInstitute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 21st July 2022
Abstract
Few synthetic hydrogels can mimic both the viscoelasticity and supramolecular fibrous structure found in the naturally occurring extracellular matrix (ECM). Furthermore, the ability to control the viscoelasticity of fibrous supramolecular hydrogel networks to influence cell culture remains a challenge. Here, we show that modular mixing of supramolecular architectures with slow and fast exchange dynamics can provide a suitable environment for multiple cell types and influence cellular aggregation. We employed modular mixing of two synthetic benzene-1,3,5-tricarboxamide (BTA) architectures: a small molecule water-soluble BTA with slow exchange dynamics and a telechelic polymeric BTA-PEG-BTA with fast exchange dynamics. Copolymerisation of these two supramolecular architectures was observed, and all tested formulations formed stable hydrogels in water and cell culture media. We found that rational tuning of mechanical and viscoelastic properties is possible by mixing BTA with BTA-PEG-BTA. These hydrogels showed high viability for both chondrocyte (ATDC5) and human dermal fibroblast (HDF) encapsulation (>80%) and supported neuronal outgrowth (PC12 and dorsal root ganglion, DRG). Furthermore, ATDC5s and human mesenchymal stem cells (hMSCs) were able to form spheroids within these viscoelastic hydrogels, with control over cell aggregation modulated by the dynamic properties of the material. Overall, this study shows that modular mixing of supramolecular architectures enables tunable fibrous hydrogels, creating a biomimetic environment for cell encapsulation. These materials are suitable for the formation and culture of spheroids in 3D, critical for upscaling tissue engineering approaches towards cell densities relevant for physiological tissues.
1. Introduction
Unlike traditional synthetic elastic hydrogels employed for tissue regeneration, the extracellular matrix (ECM) is fibrous and possesses a range of time-dependent viscoelastic properties.1,2 Stiffness can be tuned in synthetic elastic hydrogels to mimic soft tissues and control cell behaviour;3–6 however, static and uniform cross-links with mesh-like structures do not mimic the dynamicity and fibrous morphology of the ECM. Fibrous proteins in the ECM such as collagen, elastin, fibronectin, and laminin self-assemble via physical interactions to form highly organised superstructures.7,8 Furthermore, the ECM structure is highly dynamic, interacting with cells to control several functions such as growth, migration, proliferation, and differentiation,9 while undergoing continuous remodelling.10 These physical interactions between proteins and this remodelability are believed to be responsible for the dynamicity, viscoelasticity, and stress relaxation behaviour of the native ECM.2 Engineering both viscoelasticity and fibrous assembly in synthetic hydrogels remains a challenge.Stress relaxation is a measure of viscoelasticity, and indicates the ability of the ECM to dissipate cell stresses by either reorganisation of physical cross-links or remodelling by degradation. Stress relaxation has been shown to directly influence stem cell lineage and cell behaviour.1,2,11–18 Faster stress relaxing hydrogels generally enhance cell spreading,11 proliferation,18 and support the formation of actin stress fibres and focal adhesions.16 By tuning the stress relaxation/viscoelasticity of a hydrogel, the differentiation of mesenchymal stem cells (MSCs) can be controlled towards either the osteogenic or adipogenic lineages.18,19 Furthermore, therapeutic cells like primary chondrocytes have been shown to produce more cartilage-like ECM in fast relaxing hydrogels.20 Myoblasts (C2C12) have also shown sensitivity to stress relaxation by increased spreading and the formation of robust actin filaments in fast relaxing hydrogels.14 Such stark differences in cellular phenotype are observed largely due to the viscous component (viscoelasticity) of these hydrogels, which is responsible for dissipating stresses in the materials when a cell pulls or pushes against the matrix.1,21–23 While the effect of network dynamics on cell adhesion have been well documented,24 less is known about the effect of these network dynamics on cellular aggregation.
On the molecular scale, the rate of stress relaxation in a hydrogel is inherently dictated by the dynamics of the network cross-links.1,25 An early example of tunable stress relaxation involved grafting polyethylene glycol (PEG) on alginate to preturb the binding of the Ca2+ cross-linker.18 More recent approaches use knowledge from small molecules physical organic studies including dynamic covalent and supramolecular chemistry.1,26–29 These strategies offer a more rational way to tune the hydrogel properties by using the equilibrium constant Keq, the ratio of the rate constants of the on and off rates of binding. Exploiting dynamic covalent chemistry, the stress relaxation in hydrogels has been tuned by utilizing differences in k1 and k−1 of aliphatic (fast) versus aromatic (slow) aldehyde to form a hydrazone bond.14 Other strategies include leveraging the differences in equilibria and kinetics of esterification of boronate esters,30 mixing two different phenylboronic acid derivatives with unique diol complexation rates,31 and employing either distinctly different imine-type dynamic cross-links (oxime versus hydrazone)32 or modular mixing of dynamic cross-links (oxime and hydrazone).33 Host–guest chemistry has also been applied to engineer stress relaxation by using guest molecules with a range of affinity to host in both cucubiturils34 and cyclodextrins.35 Despite these advances, the control of viscoelasticity in fibrous hydrogels based on self-assembly remains elusive and is limited to a few examples.24,36
Among hydrogels, one-dimensional (1D) synthetic supramolecular biomaterials stand out, thanks to their fibrous structure, dynamicity, and ability to create a biomimetic ECM.37,38 Similar to the self-assembly of ECM proteins via physical interactions, the building blocks in a supramolecular fibre self-assemble via non-covalent interactions such as hydrogen bonding, hydrophobic interactions, and π–π stacking. The monomers in supramolecular fibres can be dynamic and can undergo exchange between fibres, which mimic the association and disassociation of physical cross-links between fibres in ECM proteins.37,39 Supramolecular polymerisation enables the creation of fibril structures with similar length scales to native ECM proteins, the tuning of dynamics on the molecular scale, and the building of structure–property relationships between molecular dynamics and bulk properties.37,39,40 Molecular engineering of peptide amphiphiles,41 short peptides,42 ureido-pyrimidinone (UPy)43 and BTAs36,44,45 has been employed to achieve fibrous structures and control the macroscale mechanical properties of hydrogels. Several recent reports have shown the ability for nanofibrillar hydrogels to support and facilitate cell aggregation;46,47 however, the role of viscoelastic timescales (or network exchange) on the speed or amount of cell aggregation has not been investigated.
Owing to the high specificity and directionality of supramolecular interactions, modular mixing via a mix-and-match approach with supramolecular monomers is a unique opportunity to create new materials on the bench-top without having to synthesise new molecules or polymers. This modular approach opens up possibilities to build generalizable platforms for the copolymerisation of supramolecular monomers for the tuning of material composition, mechanical properties, and bioactivity. Modular mixing has been used to create elastomeric scaffolds for tuning the bioactivity in UPy polymer platforms,24,48–50 and to carry different biological signals in peptide amphiphiles.51 Recent progress has shown that modular mixing also offers the potential to tune the exchange dynamics in UPy52 and BTA36,45 based supramolecular materials, which can be utilized to control hydrogel structure, dynamicity, and viscoelasticity. While both the UPy and peptide amphiphile architectures have a long history of use in hydrogel biomaterials, BTA systems have only been sparingly investigated.53
Here, we explore a simple and versatile supramolecular tuning strategy for cell culture applications. We build on the recently published work where BTA and BTA-PEG-BTA (a telechelic poly(ethylene glycol) (PEG) functionalized with BTA on both ends) were mixed in different ratios to create 1D fibrous structures with competitive interactions.36 By tuning the formulations at higher concentrations, we can achieve hydrogels with mechanical and viscoelastic properties in the physiological range for soft tissues. We investigated these BTA fibrous hydrogels for 3D cell culture with several different cell types. Since these BTA hydrogels are composed of non-covalent interactions, which are dynamic and can facilitate cell–cell contact and cell aggregation, we were able to employ hydrogels with different viscoelastic timescales to investigate the aggregation behaviour of chondrocytes (ATDC5) and primary hMSCs. To our best knowledge, this is the first report which studies the effect of dynamic timescales on cell aggregation and spheroid formation in viscoelastic hydrogels. BTA viscoelastic hydrogels offer alternatives to typical two-dimensional (2D) plastics and microwells for studying cell aggregation and spheroid formation, as they provide an ECM mimicking 3D fibril structure with physiological stiffness ranges and controlled viscoelasticity.
2. Results and discussion
2.1. Hydrogel formation and vial inversion
In this study, we used two BTA architectures (Fig. 1A and B): a small molecule BTA (slow exchange dynamics) and a telechelically functionalized PEG BTA-PEG-BTA (fast exchange dynamics). The BTA-PEG-BTA architecture was formed by connecting two BTA molecules with a bi-functional PEG20K using copper-catalysed azide–alkyne click chemistry.36 To start with, we investigated the ability of BTA and BTA-PEG-BTA to form hydrogels via a simple heat–cool approach. Both BTA and BTA-PEG-BTA formed hydrogels successfully at 10 wt% (Fig. 1C and D). The BTA formed an optically opaque gel, while the BTA-PEG-BTA formed an optically clear gel. During these experiments, we also observed that BTA-PEG-BTA hydrogels can also be made by mixing BTA-PEG-BTA powder with Milli-Q water, Dulbecco's phosphate-buffered saline (DPBS), and media and then placing overnight at 37 °C. When subjected to a vial inversion test, we noticed that the hydrogelators had markedly different behaviour (Fig. S1†). After 24 hours, the BTA hydrogel stayed at the top of the vial while the BTA-PEG-BTA hydrogel flowed to the bottom of the glass vial. This flow behaviour of BTA-PEG-BTA suggested greater viscoelasticity compared to BTA.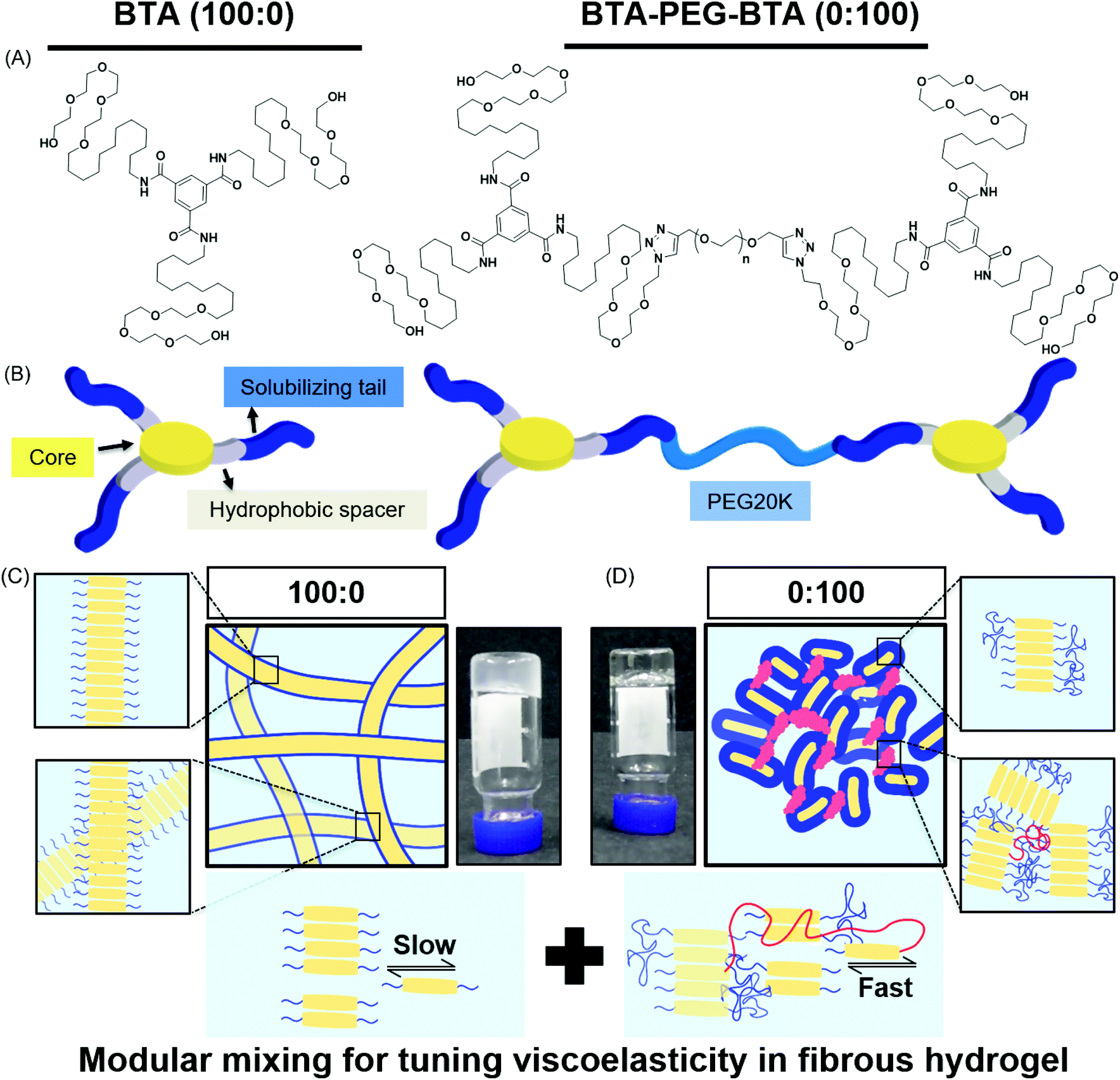 | ||
Fig. 1 Chemical structures and schematic representations of BTA and BTA-PEG-BTA: (A) The BTA chemical structure has a benzene core with dodecyl hydrophobic spacers and a tail of tetra-ethylene glycol units for solubilizing in water. The BTA-PEG-BTA chemical structure is formed by two BTAs connected to poly(ethylene glycol) 20 kg mol−1 (PEG20K) using copper-azide click chemistry. (B) Schematic representations of the supramolecular networks of BTA (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) and BTA-PEG-BTA (0:100) hydrogelators; the benzene core and hydrophobics are shown in yellow and gray, and blue represents the tetra-ethylene glycol units. PEG20K is shown in light blue. (C) A schematic representation of BTA units stacking over each other via 3-fold hydrogen bonding interactions, resulting in micron-length long fibres. The inset images are zoomed in areas, the top shows BTA stacks in a single fibre and the bottom demonstrates the BTA fibres physical cross-links/entanglements between fibres. The image with the black arrows shows the slow rate of molecular exchange dynamics. (D) A schematic representation of BTA-PEG-BTA forming micelles due to the steric hindrance of poly(ethylene glycol). The BTA units are shown in yellow and the PEG polymer in blue. Red represents the bridges between micelles. A faster rate of molecular exchange dynamics is shown in the image with black arrows for BTA-PEG-BTA. :0) and BTA-PEG-BTA (0:100) hydrogelators; the benzene core and hydrophobics are shown in yellow and gray, and blue represents the tetra-ethylene glycol units. PEG20K is shown in light blue. (C) A schematic representation of BTA units stacking over each other via 3-fold hydrogen bonding interactions, resulting in micron-length long fibres. The inset images are zoomed in areas, the top shows BTA stacks in a single fibre and the bottom demonstrates the BTA fibres physical cross-links/entanglements between fibres. The image with the black arrows shows the slow rate of molecular exchange dynamics. (D) A schematic representation of BTA-PEG-BTA forming micelles due to the steric hindrance of poly(ethylene glycol). The BTA units are shown in yellow and the PEG polymer in blue. Red represents the bridges between micelles. A faster rate of molecular exchange dynamics is shown in the image with black arrows for BTA-PEG-BTA. | ||
We then mixed the two hydrogelators in different ratios with a final concentration of 10 wt% (Fig. S1†). For this study, the different formulations of copolymerised hydrogels explored were 90:10, 75:25, 50:50, 25:75, and 10:90; this nomenclature indicates the % of BTA:BTA-PEG-BTA. For example, 200 μL of a 100:0 hydrogel contains 20 mg of BTA and zero mg of BTA-PEG-BTA, while a 75:25 gel contains 15 mg of BTA and 5 mg of BTA-PEG-BTA. All of the tested formulations formed stable hydrogels in water and DMEM using a simple heating–cooling procedure. We did observe that the transparency of the hydrogel increased with an increasing % of BTA-PEG-BTA (Fig. SI†). During a cursory vial inversion test, only the 10:90 hydrogel flowed to the bottom of the vial after 24 hours and all other tested copolymerised hydrogels stayed at the top of the vial.
2.2. Storage moduli
To investigate the mechanical properties, we turned to oscillatory rheology. First, we investigated BTA and BTA-PEG-BTA with a frequency sweep at 1% strain and with a final concentration of 10 wt%. The BTA hydrogel (100:0, grey colour Fig. 2A) showed an equilibrium storage modulus around 300 Pa, which stayed constant over almost five decades of angular frequency. This could be attributed to the very long (several micrometres in length) and entangled fibres observed under cryogenic transmission electron microscopy (cryo-TEM),44 which would result in a persistent network formation. The BTA-PEG-BTA hydrogel (0:100, black colour in Fig. 2A) showed a plateau storage modulus of 9000 Pa. This 0:100 formulation displayed a classic viscoelastic response; we observed a decreasing storage modulus with decreasing angular frequency. The elastic plateau at a higher frequency is believed to originate from the formation of elongated objects owing to the bridging of micelles by a BTA-PEG-BTA molecules.36
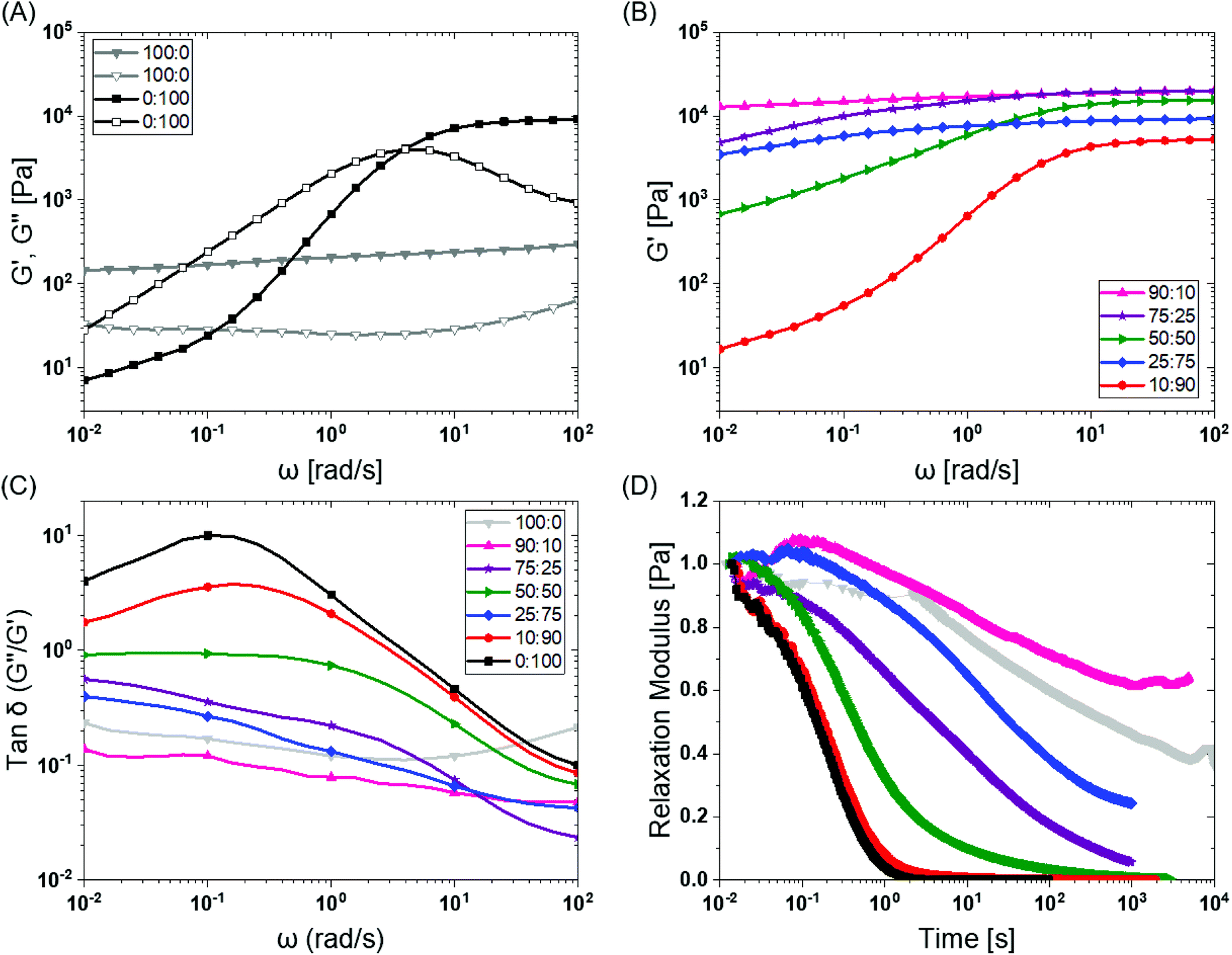 | ||
| Fig. 2 BTA modular mixing allows for tuning of the viscoelastic properties of hydrogels: (A) BTA (100:0, in grey) and BTA-PEG-BTA (0:100 in black) storage moduli (filled symbol) and loss moduli (empty symbol) as a function of frequency. (B) The tuning of the storage moduli by copolymerisation of BTA and BTA-PEG-BTA. (C) The tanδ (G′′/G′) as a function of frequency of the copolymerised hydrogels. (D) The stress relaxation profiles of the hydrogels when subjected to constant 1% strain. Duplicates were performed for all the hydrogel formulations in A, B, and C. Similar trends in viscoelasticity were found, and the tanδ (G′′/G′) values are reported for all runs in the ESI Fig S3E.† Duplicates were performed for 25:75 (BTA:BTA-PEG-BTA) in D in order to confirm the outlier behaviour and are reported in Fig. S4C.† | ||
To explore if BTA copolymerisation would result in a library of hydrogels with different mechanical properties, we mixed BTA and BTA-PEG-BTA in different proportions and recorded a frequency sweep. Starting with the pure BTA formulation, we observed a storage modulus of 300 Pa. When 10% BTA-PEG-BTA (90:10) was added, the storage modulus increased to ∼20 kPa, indicating a surprising increase in stiffness with a low amount of the telechelic architecture. The storage moduli stayed constant upon increasing the BTA-PEG-BTA to 25% (75:25) and then decreased to 15 kPa, 9 kPa, and 6 kPa (Fig. 2B) with increasing BTA-PEG-BTA (50:50, 25:75, and 10:90, respectively). These modularly mixed hydrogels have storage moduli in the range of soft tissues (shear moduli), such as cardiac muscle (5–50 kPa), kidney (4–8 kPa), spleen (15–20 kPa) and thyroid (1.3–1.9 kPa).4 Conveniently, these different stiffnesses can easily be tailored simply by mixing different proportions of the two components.
Non-linear dependence of the storage moduli has also been observed by Vereroudakis et al.36 of the BTA and BTA-PEG-BTA copolymerised binary hydrogels at low concentration (5 wt%). Here, they observed that the storage moduli gradually increased upon addition of BTA-PEG-BTA to BTA and reached a maximum of 10 kPa for the 50:50 formulation. A decrease in the supramolecular fibre length with an increasing BTA-PEG-BTA % and the resulting changes in the connectivity of the network have been attributed to be responsible for this observed non-monotonic dependence of the storage moduli.36
2.3. Viscoelasticity
Previous work has shown that BTA forms largely elastic hydrogels, while BTA-PEG-BTA is largely viscoelastic.44 We explored if this trend still holds true at the higher concentrations used for cell culture applications. The pure BTA largely showed frequency-independent storage and loss moduli, which is typical for a Hookean solid (see in Fig. 2A, grey colour). BTA-PEG-BTA showed a complex viscoelastic behaviour, with G′′ crossing G′ at around 5 rad s−1 (see in Fig. 2A, black colour), which is a typical transition (rubbery plateau to terminal region, going from high to low frequency) for viscoelastic materials (Fig. S2†). These results were in agreement with previously reported studies44 and with our visual observations during the vial inversion test.All modularly mixed hydrogel formulations showed characteristic viscoelastic behaviour as shown in Fig. S3A.† In general, we observed a shift towards a more viscoelastic character with higher weight fractions of BTA-PEG-BTA. The formulation 90:10 showed that G′, and G′′, remained largely independent of frequency (Fig. S3A).† The formulation 75:25 also did not show a crossover point between G′ and G′′ within the experimental window; however, the decreasing G′ as we moved to a lower frequency forecasts a convergence (shown in Fig. S3A†). This effect was more pronounced when we increased BTA-PEG-BTA to 50% (50:50) which showed the convergence point of G′′ and G′ around ω = 0.1 rad s−1, which is roughly 2 decades lower than the 0:100 crossover point. Interestingly, the 25:75 gel did not show any convergence or crossover point even though it has 25% more BTA-PEG-BTA compared to the 50:50 formulation. Upon increasing BTA-PEG-BTA to 90% (10:90), the crossover point reached 3 rad s−1 which is close to the 0:100 formulation (5 rad s−1). The shifting of crossover points to a higher frequency by adding BTA-PEG-BTA indicated that modular mixing of supramolecular BTA units allowed the tuning of viscoelasticity across a relatively broad range.
Tanδ, the ratio of G′′/G′, also is able to provide information on the viscous behaviour or stress dissipation characteristic of a material. We investigated tanδ of the different formulations as shown in Fig. 2C. Tanδ is highest for 0:100 (BTA-PEG-BTA) across all investigated frequencies, which indicated that it had the highest viscous component in all the formulations. In general, adding BTA in BTA-PEG-BTA led to a decrease in the tanδ indicating that the BTA added more elastic properties to the mixture. For example, at 0.1 rad s−1, 0:100 had a tanδ of 10, which decreased to 1 and 0.1 for 50:50 (50% BTA) and 90:10 (90% BTA). Interestingly, 90:10 had a lower tanδ than 100:0 (at all angular frequencies), indicating that 90:10 has more viscoelastic solid characteristics and less stress dissipation capacity compared to 100:0. Another exception to the general trend of adding BTA and decreasing tanδ was again 25:75 (75% BTA-PEG-BTA) which showed a tanδ value lower than 50:50. Important to note, the differences in the tanδ value increased as we gradually moved to lower frequencies (from 102 to 10−2 rad s−1); this observation indicated that the hydrogels behave very different at low frequencies, which points to different exchange dynamics of monomers in the BTA formulations.
Next, we investigated the stress relaxation behaviour of the hydrogels for in-depth analysis of the stress relaxation modes. BTA-PEG-BTA (0:100) relaxed fully in under just one second, while BTA (100:0) barely relaxed to 60% after 10000 seconds (Fig. 2D). As predicted by the viscoelastic frequency sweep, we found that the stress relaxation time scales can be fine-tuned by mixing BTA with BTA-PEG-BTA in different ratios. While the average stress relaxation times are often calculated using the Maxwell model (eqn (1)), this model only contains a single characteristic relaxation mode in which a Hookean spring and a Newtonian dashpot are connected in a series:
| G(t) = G0e−t/τ0 | (1) |
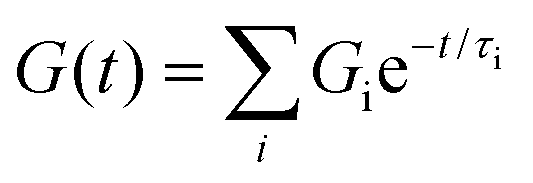 | (2) |
Using this approach, the average stress relaxation time can be obtained by summation of stress in each Maxwell-dashpot element over the time interval of the stress relaxation test.
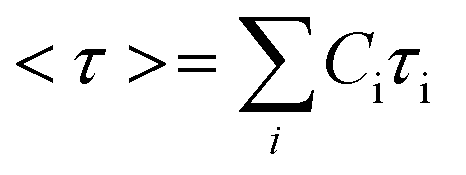 | (3) |
Using eqn (3) for two Maxwell elements in parallel, the average stress relaxation time <τ> was calculated as shown in Table 1. Increasing the BTA-PEG-BTA percentage led to a decrease in stress relaxation time and via the modular mixing of BTA and BTA-PEG-BTA the stress relaxation times could be tuned from under a few seconds to several thousand seconds. The average relaxation times ranged from several thousand to a few seconds and half relaxation times are between several hundred to a few seconds, shown in Table 1. Many of the BTA formulations have stress relaxation half times between 0.1–100 seconds, which are in the range of soft tissues such as brain, breast tumour, coagulated marrow, fracture hematoma, liver, and muscle.2 In addition, the stress relaxation half times (below 100 seconds) of our hydrogels are in the range shown to promote osteogenic differentiation18 and cartilage-like matrix formation.20
A general trend was observed, where increasing BTA-PEG-BTA resulted in faster-relaxing hydrogels (Fig. 2D). This is likely due to the presence of the telechelic PEG20K in BTA-PEG-BTA, which could result in less stable BTA aggregates and faster exchange dynamics. In previous studies, hydrogen/deuterium exchange (HDX) mass spectrometry (MS) experiments showed that mixing BTA-PEG-BTA with BTA weakens the association strength between BTA molecules within the assembly leading to less stable aggregates and faster exchange dynamics.36 In a similar study with a UPy supramolecular polymer, a bivalent UPy (UPy molecules connected with telechelic PEG20K) accelerated the exchange dynamics when mixed with a small-molecule UPy.52 These studies support that the faster exchange dynamics in the presence of telechelic PEG architectures are mainly responsible for the fast relaxation behaviour of BTA-PEG-BTA and that the different exchange dynamics, in combination with the differences in hydrogel morphology,36 are a likely source of the different relaxation behaviour in these multi-component supramolecular systems.
2.4. Self-healing
Initially, self-healing was visualized in the lab by placing two pieces of hydrogel, coloured with red and green food colouring dye, side by side and compressing them. The hydrogels with ≥75% of BTA-PEG-BTA self-healed after 24 hours, yet the 90:10 and 100:0 formulations did not (Fig. S5†). Next, the self-healing behaviour was quantitatively investigated on rheometer using a cone-plate configuration. Initially, a strain amplitude sweep was conducted to find the critical strain needed to break the hydrogel structure. BTA (100:0) showed strain at break around 100%, while strain at break sharply decreased to ∼2% for 90:10 (10% BTA-PEG-BTA), increased to ∼20% for 75:25 (25% BTA-PEG-BTA), and then stayed almost constant for 50:50 and 25:75. When the % of BTA-PEG-BTA increased to 90%, strain at break declined to 10% for 10:90 and ∼2% for pure BTA-PEG-BTA (0:100). The data showed that modular mixing of BTA architectures allowed tuning the strain at break; however, distinct trends are less clear.
We then investigated self-healing via a step strain shear rupture cycles (1% to 400% strain) at a constant frequency (ω = 10 rad s−1) for investigating the self-healing behaviour of the hydrogels (Fig. S5†). Hydrogel rupture was observed upon applying a high strain (G′ > G′′) and the moduli quickly recovered to original values during the low strain cycle. Interestingly, 100:0 and 90:10 also showed full self-healing capacity, which we did not observe in the visual macroscopic self-healing test. This shows that the shear rheology and visual self-healing test can result in contradicting results; macroscopic visual self-healing tests should be conducted for investigating the macroscopic self-healing capacity. The self-healing behaviour within these BTA based hydrogels can be associated with reversible and dynamic supramolecular interactions.
2.5. Cell studies
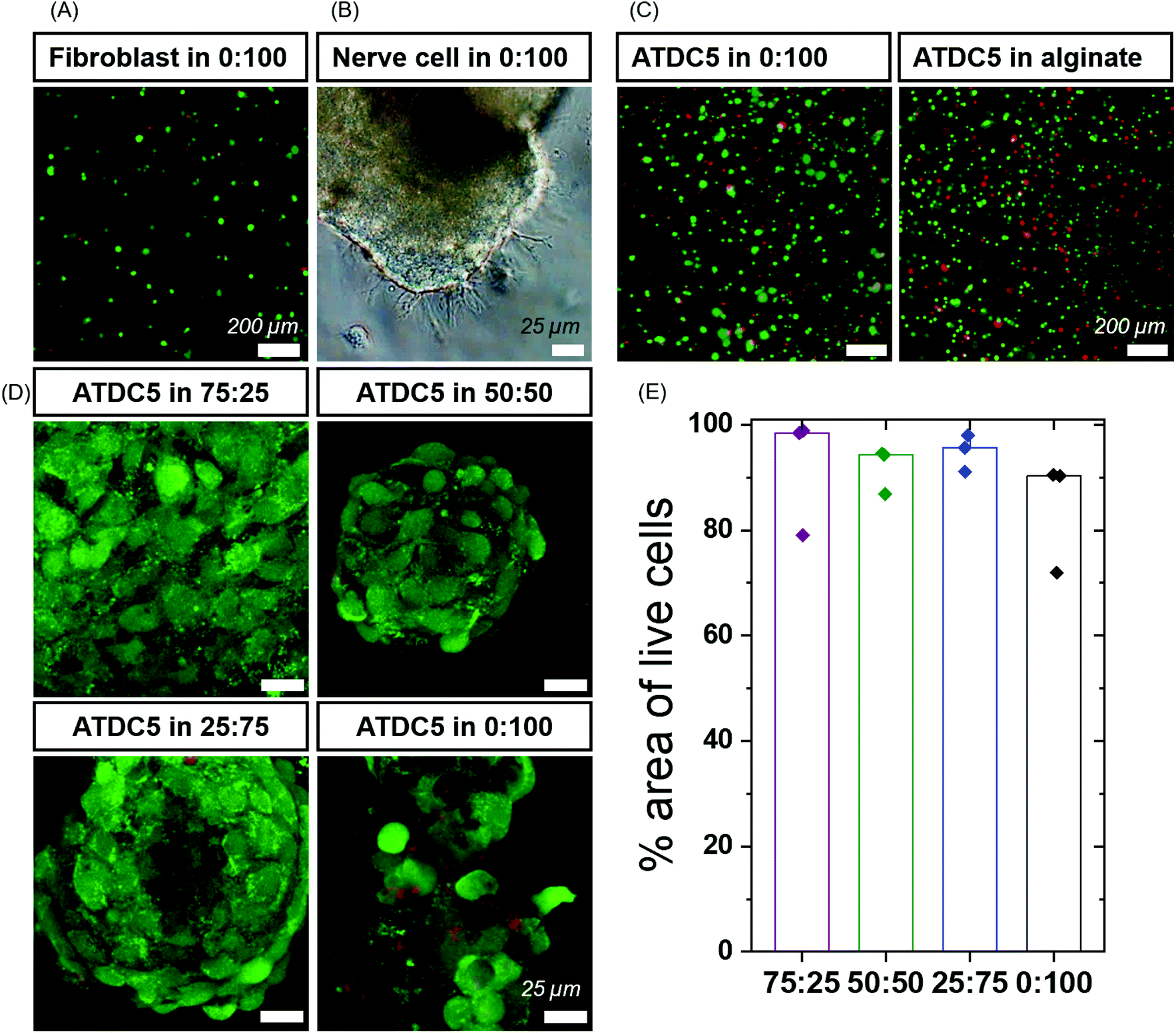 | ||
| Fig. 3 High cell viability in BTA formulations: (A) fibroblasts showed high cell viability using a live/dead assay after 24 hours. Green represents live cells and red represents dead cells. (B) Dorsal root ganglion (DRG) cells showed outgrowth in BTA-PEG-BTA (0:100) gels after 48 hours. (C) ATDC5 chondrocytes were around 80% viable (green) using a live/dead assay and the cell viability was comparable to alginate hydrogels cross-linked using calcium ions. (D) ATDC5 chondrocytes viability in BTA formulations after 7 days. Green represents live cells and red represents dead cells. (E) % area of live cells calculated using the analyze particle function in Image J software for BTA formulations; the cell area for three images (top, middle and bottom of an aggregate) was calculated. Minimum two biological replicates were used for each experiment. | ||
In the initial trials, we observed aggregation of fibroblasts (Fig. S6C and Video 1†) within the BTA-PEG-BTA hydrogels likely due to fast exchange dynamics. The aggregation of cells is required for many cell types, and multicellular aggregates are beneficial en route to tissue formation; however, the cell aggregation is usually prevented by covalently cross-linked hydrogels due to their static mesh network. For example, the aggregation of chondrocytes is desired to produce cartilage-like ECM for the creation of cartilage tissue. Correspondingly, we were interested to see if these different hydrogels could control the cell aggregation of chondrocytes in a 3D culture. First, we compared the viability of chondrocytes (ATDC5) in alginate, a standard tissue engineering hydrogel,18,54,55 to our BTA-PEG-BTA hydrogels (Fig. S7†). We determined the live/dead cell area since accurate single-cell counting was misleading owing to the cell aggregation. Chondrocytes cultured in both the BTA-PEG-BTA and the alginate showed similar live/dead results; the total area for live cells was ∼3× times greater than dead cells, suggesting low (15–20%) cytotoxicity (Fig. S7B†). Cytotoxicity was also monitored using lactate dehydrogenase (LDH) release assay and both the BTA-PEG-BTA and the alginate showed lower (8×) LDH release compared to a cell pellet control, suggesting again greater than 80% viability (Fig. S7C†).
Next, in order to investigate cell viability for the complete BTA series, we attempted cell encapsulation using a chemically dissolved freeze-drying method. The self-healing method for cell encapsulation was not the best choice since 100:0 and 90:10 showed poor cell encapsulation due to the macroscopic non-self-healing (Fig. S5A†). We chemically dissolved the solid polymer in dichloromethane and methanol, dried in a vacuum oven, and then DPBS was added to make a hydrogel. Then the hydrogels were frozen and freeze-dried, where we noticed that the BTA hydrogels have a porous structure after freeze-drying. The cell suspension was added on the top of the freeze-dried BTA hydrogels and incubated for ∼1 hour at 37 °C for hydrogel formation. Though we were able to encapsulate cells within the freeze-dried hydrogels, not all formulations made stable hydrogels, especially those which have a larger percentage of BTA. This might be due to the fact that BTA requires heating for dissolving in water/media for self-assembly. Also, 100:0 formed a foamy lightweight structure, which was soft and most of the cells sedimented at the bottom, resulting in poor cell encapsulation. This procedure of cell encapsulation was not robust; it needed 4–5 days of gel preparation before cell encapsulation and involved toxic chemicals such as dichloromethane and methanol.
Using the chemically dissolved freeze-drying method, we observed greater than 80% viability (% of live cell area) in all the formulations besides 100:0 and 90:10, which showed around 40% dead cells (by cell area) relative to other formulations (Fig. S8A and S8B†). We hypothesized that 100:0 could retain methanol because of the BTA hydrogen bonding interactions with methanol, which likely compromised the cell membrane. In a control experiment, we prepared the 100:0 hydrogel in cell culture media and encapsulated ATDC5s using the self-healing method. While this formulation did not self-heal rapidly enough for efficient cell encapsulations, the live/dead staining showed almost no dead cells for both cells in the hydrogel and on the tissue culture plastic (Fig. S8C and D†). This indicated that the hydrogel, itself, was not toxic, but likely toxicity came from the residual solvent or one of the processing steps during the freeze-drying method.
Lastly, we also explored the quicker heating method for cell encapsulation for 90:10 and 0:100 BTA formulations, where we heated BTA hydrogels until they turned into liquid so that the hydrogel was pipetteable to mix with the cell suspension; however, high temperatures (minimum 60 °C) were required (Fig. S9†). Transferring the hydrogel from a glass vial to a cell culture well plate, gelation in the pipette tip, and rapid gelation (<30 s) upon mixing cell suspension prevented effective cell encapsulation. Compared to the self-healing method, relatively more dead cells were seen (Fig. S10†) and no advantages were found.
We found the self-healing method to be more reliable, applicable and reproducible compared to the other cell encapsulation methods we tested, since the hydrogel could be broken into small pieces and mixed with the cell suspension. Formulations that could not effectively self-heal and encapsulate cells were left out and not studied further. We then investigated the cytotoxicity of the different formulations within our small library of BTA-PEG-BTA and BTA hydrogels. Chondrocytes were encapsulated within the different hydrogels for 7 days and a live/dead assay was performed. All hydrogels showed greater than 80% of live cell area, indicating high cell viability within the modularly mixed BTA hydrogels (Fig. 3D, E and Fig. S11†).
Next, we set out to investigate the capabilty of neural cells to exhibit neurite outgrowth in the BTA based hydrogels using the PC12 cell line. Neuronal cells require a dynamic and cell remodellable microenvironment.56,57 We encapsulated PC12s as dissociated cells in the most dynamic BTA-PEG-BTA hydrogel and observed that the PC12 cells aggregated in the first 48 hours; the neurites showed neurite outgrowth in the dynamic BTA-PEG-BTA hydrogel over 4 days (Fig. S12†). Then we mixed a small amount of either laminin or fibrinogen into the BTA-PEG-BTA hydrogel and observed that the PC12 cells exhibited more neurite projections; from a few in BTA-PEG-BTA to tens of neurites per cell aggregate in the BTA-PEG-BTA hydrogel with either laminin or fibrinogen (Fig. S12†). Next, dorsal root ganglia (DRGs) were embedded within the hydrogel, and they exhibited branching neurite outgrowth when cultured in pure BTA-PEG-BTA hydrogel in just two days (Fig. 3B and Fig. S12†). This showed that both PC12s and DRGs can stay healthy and grow neurite projections in the dynamic BTA-PEG-BTA hydrogels. To further understand the role of materials dynamics, exploration of the complete BTA series to study neurite outgrowth or neuronal sphere formation is of particular interest.
:25 to 0:100 and moved forward to characterize the aggregate behaviour in these hydrogels in more detail (Fig. 4).
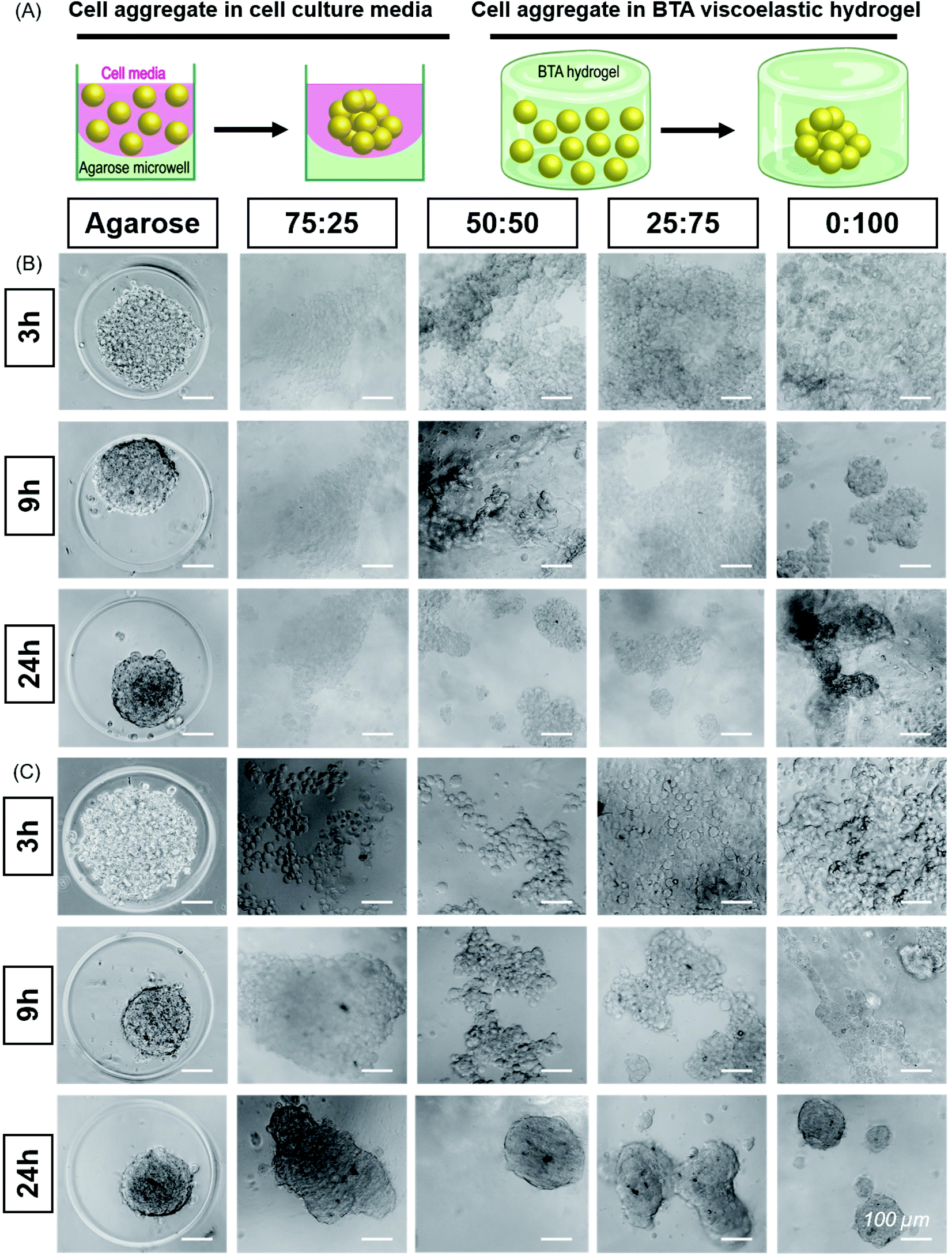 | ||
| Fig. 4 Self-assembly and aggregation of ATDC5s and hMSCs within BTA hydrogels: (A) Cell aggregate formation in cell media in agarose microwell versus cell aggregate formation in BTA viscoelastic hydrogels. (B) ATDC5s formed round and compact aggregates in agarose mould (positive control). ATDC5 chondrocytes tended to aggregate in all BTA formulations; however, cell aggregate were not as compact as compared to agarose mold. Among BTA formulations, 0:100 showed more compact aggregates, scale bar 100 μm, n = 2 biological replicates. (C) Self-assembly and aggregation of hMSCs within BTA hydrogels. Cell aggregates size decrease in agarose mold (positive control) showing that aggregate is getting more compact over time. All BTA formulations showed cells aggregation and spheroids formation. However, it can be seen that cells formed smaller aggregates and that there was more spheroid fusion with increasing % of BTA-PG-BTA, scale bar 100 μm. n = 3 biological replicates. | ||
While several previous studies have shown spheroid culture,24,58 the formation of spheroids in supramolecular nanofibrillar hydrogels,46,47 and pre-made spheroid fusion in supramolecular hydrogels with a vicosus component,59,60 the influence of hydrogel viscoelasticity on cell aggregation/spheroid formation in 3D remains unknown. We further hypothesized that changes in the viscoelasticity/stress relaxation of the matrix could lead to control over cell aggretation – more simply, is this phenomena a viscoelastic timescale dependent phenomena? We chose 75:25, 50:50, 25:75 and 0:100 to study the cell aggregation and compactness of aggregates formed when ATDC5s were encapsulated within the hydrogels (Fig. 4B). An agarose microwell mould was used as a positive control for aggregation, which is a 2.5D cell aggregation model on a well plate. In this microwell mould, the cells are confined by non-adherent agarose but the 3D environment is simply cell media. These agarose microwells allow free self-assembly of cells and are the current standard for the formation of cell aggregates, but is limited to 2D culture platforms.61
We immediately observed that the cells started to aggregate in the BTA hydrogels after 6 hours and formed larger, more compact aggregates in 0:100 compared to the other BTA formulations. The ATDC5s did not form uniform aggregates in the BTA hydrogels over 24 hours, yet aggregates in the 0:100 formulation were more compact and dense when compared to other BTA formulations. In the other BTA formulations, single-cell boundaries could still be identified in the cell aggregates, indicative of less compactness.62 When compared to the BTA formulations, the ATDC5 aggregates were more round and compact in the agarose microwells after 24 hours, likely due to free assembly and the presence of only cell-growth media around the cells.
Having established the ability of these dynamic formulations to allow for chondrocyte aggregation, we wanted to explore this phenomenon with hMSCs (Fig. 4C), which have clinical potential in multiple applications. Studying the self-assembly of primary hMSCs for aggregation and spheroid formation has been linked to cadherin expression and the osteogenic, adipogenic, and chondrogenic phenotypes.63,64 We were interested to investigate if hMSCs aggregate within these hydrogels and the effect of BTA formulations on the cell aggregation and compactness. hMSCs started to aggregate after 6 hours; however, aggregation was slower in the BTA hydrogels compared to the free self-assembly of hMSCs in the agarose mould control. With time, the cell aggregates in the BTA hydrogels became more compact and changed their shape from elliptical to spherical after 24 hours (Fig. 4C). hMSCs formed more compact and spherical aggregates in the 0:100 hydrogel compared to the other BTA formulations, likely due to faster supramolecular dynamics, which supported rapid hMSC aggregation. In addition, a clear trend could be seen where aggregates were more compact and roundly shaped as the percentage of BTA-PEG-BTA increased (after 24 hours). Overall, the images revealed that the dynamicity and viscoelastic/stress relaxation properties of the BTA hydrogels supported cell aggregation without the presence of proteolytically degradable cross-linkers.
We hypothesized that the observed cell aggregation behavior was controlled by the dynamic properties of the hydrogel (viscoelasticity/stress relaxation), with the main driving force as the exchange dynamics of the BTA molecules. However, the differences in nutrient availability within the series could also influence the cell aggregation. To determine if nutrient diffusion is similar across all BTA formulations, FRAP measurements were performed using fluorescently labelled dextran of 3–5 and 70 kDa. Dextran of 3–5 kDa showed a two-fold higher diffusion coefficient compared to 70 kDa, likely due to differences in the hydrodynamic radii. Interestingly, no significant difference in the diffusion coefficient was observed for each molecular weight dextran (20 μm2 s−1 for 3–5 kDa and around 10 μm2 s−1 for 70 kDa) when comparing across all the BTA formulations (Fig. S14†). The FRAP data indicated that, even though the diffusion coefficient was different for the different sizes of dextran, the diffusion of macromolecules stayed constant across the BTA series. This FRAP data supported the idea that the differences in cellular self-assembly are not likely due to differences in nutrient diffusion.
We next explored if pre-formed spheroids would fuse within the hydrogels. We continued hMSC culture and observed the fusion of aggregates at 48 hours in all the BTA formulations except 0:100 (Fig. 5A). The fusion behaviour in the BTA hydrogels could be attributed to reversible supramolecular interactions and viscoelasticity of the hydrogels. The observed stable aggregate formation and hMSC spheroid fusion after 48 hours gave a strong indication of healthy and viable cells.
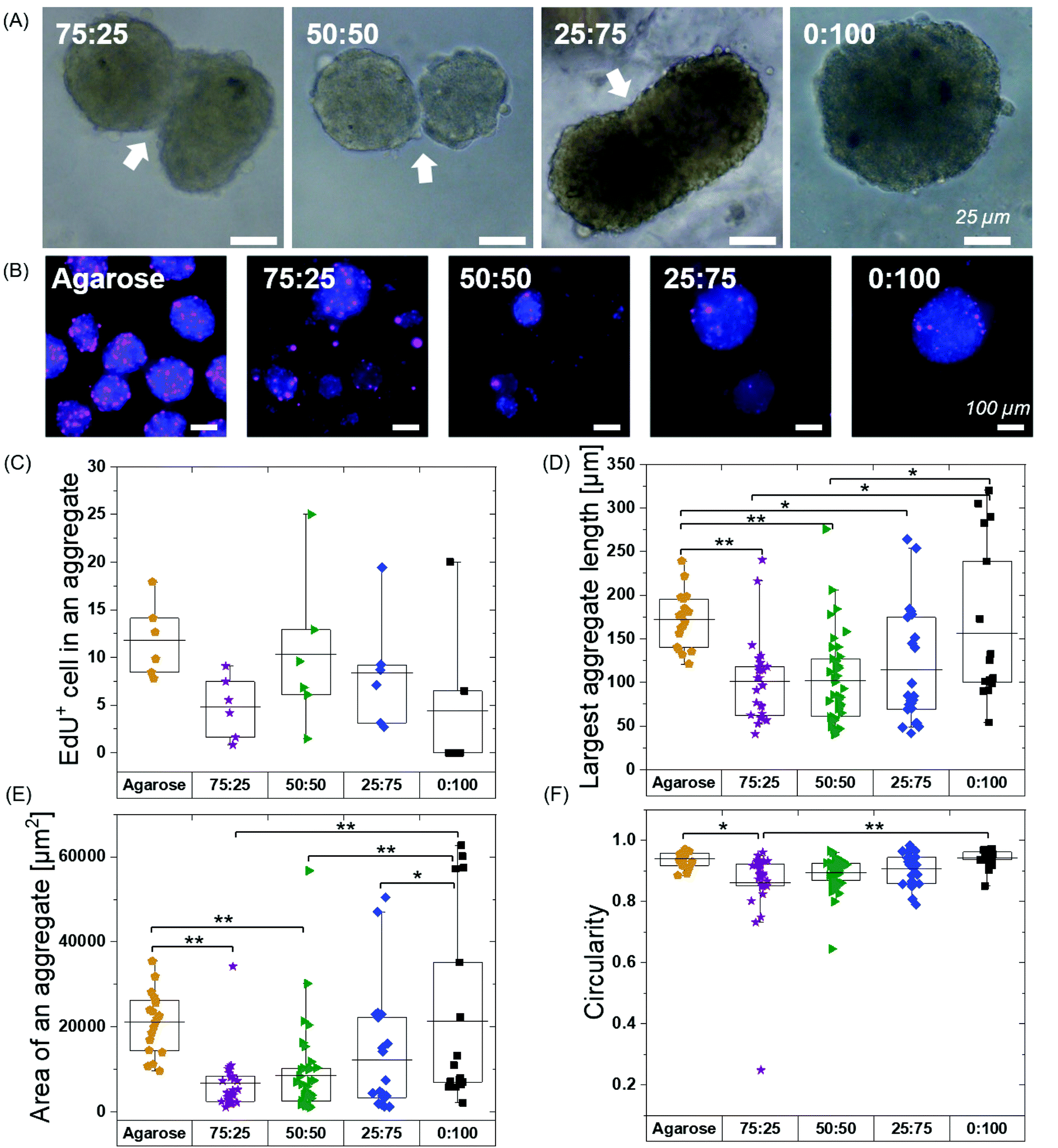 | ||
| Fig. 5 HMSCs spheroid fusion, proliferation and morphology after 48 hours. (A) The fusion of spheroids at 48 hours in BTA hydrogels. (B) Proliferating hMSCs cultured in BTA formulations stained for EdU. All formulations showed EdU positive cells in cell aggregates; however, the fewest EdU positive cells per spheroids were seen in the 0:100 formulation. Agarose control (allow free directed self-assembly of hMSCs) showed highest number of proliferating cells (EdU positive) compared to BTA formulations. (C) Quantitative analysis of EdU positive cells per aggregate in BTA based hydrogels versus agarose. EdU+ cells were counted in six aggregates. (D) Largest length of an aggregate in BTA based hydrogel versus agarose; aggregates mean length value was smaller in BTA based hydrogels compared to agarose after 48 hours. (E) Mean value of an aggregate area is smaller in BTA based hydrogels compared to agarose. (F) hMSCs aggregates are more circular in agarose compared to BTA based hydrogels. n = 3 biological replicates were used for D–F and between 20–40 aggregates were randomly selected and analysed. *p < 0.05, and **p < 0.005. Statistical significant test was performed using one-way ANOVA with Tukey's post-hoc test. | ||
Overall, we have observed that the hydrogel dynamicity supported ATDC5 and hMSC cell aggregation without the presence of degradable cross-linkers. The hMSC aggregates formed well-defined spheroids compared to the less defined aggregates for ATDC5s in 24 hours, which indicated cell-dependent aggregation behaviour facilitated by supramolecular dynamics in the stress relaxing BTA hydrogels.
Considering the increasing need for platforms to develop 3D building blocks for tissue engineering and animal alternatives for scaleable drug testing models, these dynamic and viscoelastic BTA hydrogels can be promising candidates to overcome the current limitations of 2D culture. For example, by removing dimensional constraints (e.g. 2D or 2.5D), such dynamic hydrogels offer new flexibility in the scaling and size of aggregated cells and tissues that would be more physiologically relevant when compared to current 2D models. In addition, the tunable mechanical properties and controlled fibrous structures make these BTA hydrogels ideal as a biomimetic environment. In short, our results suggest that these BTA hydrogels, and potentially other dynamic hydrogels, can provide a scalable platforms where cells in physiologically relevant cell densities, mechanical properties, and architectures can reorganize, assemble into desired morphologies, and developed into complex tissues.
Free self-assembled spheroids in the agarose mould showed more proliferating cells when compared to the BTA formulations, suggesting that physical confinement in the BTA hydrogels reduced proliferation (Fig. 5B and C). The agarose microwells showed a mean value for EdU positive hMSCs at around 12%. All the tested BTA formulations showed proliferating cells (EdU positive), and the mean value for EdU positive hMSCs varied from 0.5 to 10%. The lowest number of proliferating cells in the BTA fomulations was found in the 0:100 formulation and the highest in the 50:50. A decrease in proliferating cells in 0:100 could also be attributed to the clumping of hMSCs. These results showed that the 3D confinement still allowed for significant cell proliferation, yet there was reduced proliferation when compared to the free self-assembly of cells in suspension (in the agarose mould).
We further determined shape descriptors, such as spheroid size (largest length), area, and morphology, of the hMSC spheroids in the BTA hydrogels after 48 hours. Quantification revealed that the longest length of an aggregate was significantly reduced in the BTA hydrogels compared to agarose, except in 0:100 which was similar (Fig. 5D). Next, we evaluated the aggregate area and observed similar trends: BTA hydrogels showed a smaller aggregate area compared to agarose (Fig. 5E). BTA-PEG-BTA (0:100) showed a significantly higher area compared to other BTA formulations. The smaller length, reduced spheroid area, and lower circularity of other BTA formulations compared to 0:100 likely could be due to slow supramolecular dynamics; for example, fewer MSCs would be able to find each other and form aggregates, resulting in smaller aggregates. In contrast, for 0:100, the faster supramolecular exchange dynamics would support the formation of larger aggregates.
Next, we determined circularity (Fig. 5F) in order to uncover any differences in spheroids morphology. The free self-assembled spheroids in the agarose mould showed the highest circularity mean value of 0.94 and the lowest circularity value was 0.86 for 75:25. The circularity mean value of the spheroids increased with increasing % of BTA-PEG-BTA and reached 0.94 (similar to free self-assembly in an agarose mould) for 0:100. The significant differences in the circularity found between 75:25 (0.86) and 0:100 (0.94) could be due to slower supramolecular dynamics. We observed that 75:25 supported slower cell aggregation and the shape was relatively elliptical when compared to 0:100 that showed near-spherical aggregates after 24 hours. This observation suggested that faster supramolecular dynamics supported faster aggregation into more spherical shapes.
3. Conclusions
The findings from this study showed that the modular mixing of supramolecular architectures with slow and fast exchange dynamics provides quick access to tunable biomaterials. These copolymerised hydrogels resulted in materials with tunable stiffness and viscoelasticity in the physiological range of soft tissues and ECM. We were able to encapsulate cells in these hydrogels and showed that the dynamic properties of the hydrogels allowed cell aggregation, spheroid formation, and spheroid fusion—features that are not readily possible in traditional covalent hydrogels. Furthermore, the different hydrogel formulations were able to steer the cell aggregation speed and aggregate fidelity, as attributed to changes in the dynamic character and viscoelasticity of the hydrogel. FRAP experiments showed that differences in diffusion were not seen in the different formulations, which does not support an alternative hypothesis attributing this behavior to differences in nutrient diffusion. In the effort to find more complex and tunable ECM replacements, supramolecular systems like this offer significant advantages to rapidly increase the materials space investigated by leveraging modular mixing. For the creation of multi-cellular aggregates, supramolecular systems give significant advantages over 2D cell aggregate models since they recapitulate fundamental properties of the ECM, including fibrous morphology and tunable viscoelasticity. This work introduces these BTA hydrogels as promising supramolecular biomaterials for spheroid formation. Furthermore, this concept may be generalizable to other dynamic and supramolecular hydrogels. Control over viscoelastic timescales may aid in the quest to create scaleable environments for directing cell aggregation/spheroid formation and towards developing larger tissues for organ development and drug screening.4. Materials and methods
4.1. Synthesis of BTA architectures
The synthesis of BTA44 and BTA-PEG-BTA36 architectures was performed according to previous reports and were provided by SyMOChem B.V. Eindhoven, The Netherlands.4.2. Preparation of hydrogels for rheology and cell culture
A heating and cooling procedure was adopted to make BTA hydrogels. The solid polymer was weighed in a glass vial and Milli Q water/DPBS/media was added. Water was used for rheology and DPBS and Dulbecco's Modified Eagle's Medium (DMEM) media with 1% penicillin/streptomycin (P/S) was used to make hydrogels for cell culture. All hydrogels were made at a final concentration of 10 wt%. This was followed by a 2× heating to go above lower critical solution temperature (LCST, ∼80 °C) of polyethylene glycol (PEG), vortexing, and cooling. The last (3rd step) included more controlled cooling where all hydrogels were placed on a heating plate at 80 °C and slowly cooled to room temperature.For nerve cells (PC12 and DRGs) growth, hydrogels were made by incubating pure solid BTA-PEG-BTA (0:100) overnight in DPBS with either laminin or fibrinogen at 37 °C. BTA-PEG-BTA formed a transparent hydrogel after overnight incubation in DPBS. Fibrinogen final concentration in the hydrogel is 3.5 mg mL−1 and laminin final concentration in the hydrogel is 20 μg mL−1.
4.3. Preparation of hydrogels by chemically dissolving and freeze-drying
BTA hydrogels were also made by chemically dissolving BTA formulations in DCM using cosolvent methanol between 10–50%. Chemically dissolved BTA was added into a well plate and solvents were removed overnight using a vacuum oven at room temperature. Upon solvent drying, the solids form a layer at the bottom of the well plate to which the required amount of DPBS was added to adjust hydrogel concentration to 10% (w/v). The well plate was left at 37 °C overnight for hydrogelators to form hydrogels. Subsequently, hydrogels were frozen and freeze-dried. After freeze-drying, a porous foam structure was obtained to which required cell suspension in cell culture media (high glucose DMEM with 1% P/S) was added and the final concentration was adjusted to 10% (w/v).4.4. Rheological measurements
Rheological measurements were performed on a DHR-2 rheometer (TA instruments). The rheometer is equipped with an evaporation control chamber. A 20 mm 2.002° cone plate geometry was used. Samples were loaded at room temperature (RT) ∼20 °C and trimmed. The final gap was set to 53 μm.An amplitude sweep (ω = 10 rad s−1, γ = 0.01–400%) was performed on each sample to act as a pre-shear for all samples to erase any mechanical history and confirm the material's linear viscoelastic region. Afterwards, a time sweep at 0.1% strain, and 10 rad s−1, was run until the material's G′ was stabilized which allowed the material to set before a subsequent measurement. A frequency sweep from 100 to 0.01 rad s−1 with 0.1% strain was performed next followed by another time sweep to ensure the material was stable before stress relaxation measurement. The relaxation modulus was recorded via a step relaxation experiment for 1000–10000 s where we used 1% strain and a rise time of 0.01 s.
4.5. Vial inversion and macroscopic self-healing experiment
Hydrogels were made in Milli Q water using the standard procedure described above and vials were inverted and imaged at time zero and after 24 hours. For the self-healing test, the same formulation was coloured either red or green using food colour dyes for visibility. Two pieces of the hydrogels were placed side by side adjacent to each other in a petri-dish closed environment. Water was added around the hydrogel making sure that no water touched the hydrogel in order to control humidity. Self-healing of the hydrogel was tested after 24 hours by looking at how uniformly the interface had healed and then the hydrogel was lifted using a spatula and observed if the hydrogel's two pieces can fall apart under gravity.4.6. Agarose microwell fabrication for aggregate formation
Agarose microwell arrays were prepared as previously described.61 Briefly, 3% ultra-pure agarose solution (Invitrogen) was cast onto a poly(dimethylsiloxane) stamp with microstructures to imprint microwells, de-moulded upon solidification, cut to size, and inserted into 12-well plates. Each well of the microwell array contained 450 microwells with a diameter of 400 μm.4.7 Fluorescent recovery after photobleaching (FRAP) experiment
Hydrogels were made using the standard heating–cooling procedure described above. A volume of 200 μL of all formulations were transferred in 35 mm diameter 4 compartment cell culture disk (VWR 391-0225, Greiner bio-one), 1 mL of FITC-labelled dextran solution (3–5 or 70 kDa at 0.1 mg mL−1, Sigma-Aldrich, FD4-250MG and 46945-100MG-F) was added and incubated overnight at 4 °C in the dark. Fluorescent recovery after photobleaching (FRAP) imaging was performed on a Leica TCS SP8 STED using the FRAP modules of Leica Application Suite X software (LAS X FRAP). FRAP bleaching was performed on a z-height of 20 μm in each hydrogel. Parameters were set as followed: bleaching point of 60 μm diameter, bleaching laser at 100%, pre/post bleaching laser at 5%/488 nm/800 gain, and a time per frame of 0.223 s. After 5 frames pre-bleach (1.2 s), samples were bleached for 90 frames (21.2 s) and fluorescent recovery was gathered for 400 frames post-bleaching (110.5 s). 5 different areas within the hydrogels were bleached (N = 5). ROI data were extracted in Fiji ImageJ-win64. Area and mean gray values were obtained for the bleach, total and background ROIs. The obtained values were imported (.cvs files) in the open-source application FrapBot68 to obtain the τ1/2 (half-time) of the fluorescent recovery curve. Obtained half times (τ1/2) were used to calculate the diffusion coefficients by the Soumpasis equation, with D = diffusion coefficient, r = radius of the bleaching area and τ1/2 = the halftime of recovery (eqn (4)). Statistical analysis were performed in GraphPad Prism 8.2.0, one-way ANOVA.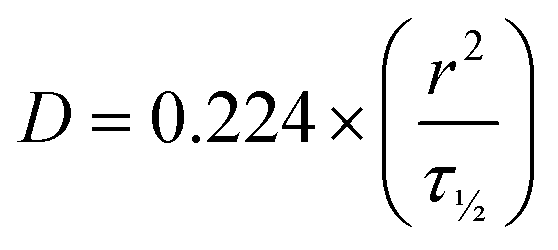 | (4) |
4.8 Cell Culture and cell encapsulation in hydrogels
:0 and 90:10 formed very soft foamy structure after freeze-drying and upon adding cell suspension most of the cells sediment to the bottom of the hydrogel.
:10 and 0:100) was maintained at 60 °C in a clear glass vial (4 mL storage capacity, VWR, Netherlands) and then using a gel pipette (Gilson, Netherlands) transferred to 24 tissue culture well plate. Cell suspension in 50 μL was added and mixed quickly using a gel pipette. We noticed that 100:0 was not in solution state at 60 °C and quickly gelled as soon as was transferred to the well plate while 90:10 was in solution state. Both formulations started to gel as soon as the gel pipette touch them in the glass vial and then in 24-well tissue culture plate rendering ineffective cell encapsulation.
4.9. Live/dead cell viability assay
BTA hydrogelators were tested for cytotoxicity with fibroblasts, and chondrocytes. Cytotoxicity was evaluated by staining cultured cells with calcein-AM and ethidium homodimer-1, following the manufacturer's kit instructions. Chondrocytes spreading/growth within hydrogels were evaluated using Alexa Fluor 488-Phalloidin and DAPI staining. Images were acquired using inverted epifluorescence microscope (Nikon Eclipse Ti-e) or confocal laser scanning microscope (Leica TCS SP8).4.10. EdU cell proliferation detection
To assess cell proliferation, 5-ethynyl-2′-deoxyuridine (EdU) staining was conducted using the Click-iT EdU Alexa Fluor 647 Imaging Kit (Thermo Fisher Scientific), according to the manufacturer's protocol. HMSCs were incubated with 50 μM EdU for 48 h in the incubator before fixation for 30 min in 4% (v/v) formaldehyde (Sigma-Aldrich) in PBS at ambient temperature. Fixed samples were permeabilized with 0.5% (v/v) Triton X-100 (VWR) in PBS for 1 h and the incorporated EdU was labelled using a click reaction with Alexa Fluor 647 azide for 1 h according to the manufacturer's protocol. The nuclear DNA was counterstained by DAPI (0.1 μg ml−1) for 1 h. The fluorescence images were acquired on a Nikon E600 inverted microscope.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
S. H. and C. v. B. would like to thank the European Research Council (ERC) for funding under the European Union's Horizons 2020 research and innovation programme (grant agreement no. 694801). S. H., P. W., and M. B. B. would like to thank the Dutch Research Council (NWO) for funding under the “Open Mind” granting scheme (grant agreement no. 18263) A. J. F. and M. B. B. would like to thank InSciTe for funding under the “EyeSciTe” consortium. F. A. A. R., V. L. S. L., and M. B. B. would like to thank the partners of Regenerative Medicine Crossing Borders (RegMed XB), financed by the Dutch Ministry of Economic Affairs by means of the PPP Allowance made available by the Top Sector Life Sciences & Health to stimulate public-private partnerships. R. P. M. L. and N. M. M. would like to thank the Dutch Ministry of Education, Culture, and Science (Gravity program 024.001.035) and the NWO/DPI program NEWPOL (project 731.015.503) for funding. We would like to thank Prof. Egbert Willem Meijer from Eindhoven University of Technology for supporting R. P. M. L. and N. M. M. and providing materials support during this study. The 3D hydrogel schematic was partially created using Biorender.com.References
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 1–15 CrossRef PubMed.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
- D. E. Discher, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- C. F. Guimarães, L. Gasperini, A. P. Marques and R. L. Reis, Nat. Rev. Mater., 2020, 5, 351–370 CrossRef.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- J. R. Tse and A. J. Engler, PLoS One, 2011, 6, e15978 CrossRef CAS PubMed.
- A. D. Theocharis, S. S. Skandalis, C. Gialeli and N. K. Karamanos, Adv. Drug Delivery Rev., 2016, 97, 4–27 CrossRef CAS PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- W. L. Murphy, T. C. McDevitt and A. J. Engler, Nat. Mater., 2014, 13, 547–557 CrossRef CAS PubMed.
- W. P. Daley, S. B. Peters and M. Larsen, J. Cell Sci., 2008, 121, 255–264 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, M. Darnell, D. Klumpers, S. A. Bencherif, J. C. Weaver, N. Huebsch and D. J. Mooney, Nat. Commun., 2015, 6, 6364 CrossRef PubMed.
- X. Zhao, N. Huebsch, D. J. Mooney and Z. Suo, J. Appl. Phys., 2010, 107, 063509 CrossRef PubMed.
- S. D. Sommerfeld and J. H. Elisseeff, Cell Stem Cell, 2016, 18, 166–167 CrossRef CAS PubMed.
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef CAS PubMed.
- B. M. Richardson, D. G. Wilcox, M. A. Randolph and K. S. Anseth, Acta Biomater., 2019, 83, 71–82 CrossRef CAS PubMed.
- J. Lou, R. Stowers, S. Nam, Y. Xia and O. Chaudhuri, Biomaterials, 2018, 154, 213–222 CrossRef CAS PubMed.
- Z. Gong, S. E. Szczesny, S. R. Caliari, E. E. Charrier, O. Chaudhuri, X. Cao, Y. Lin, R. L. Mauck, P. A. Janmey, J. A. Burdick and V. B. Shenoy, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2686–E2695 CAS.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- C. Loebel, R. L. Mauck and J. A. Burdick, Nat. Mater., 2019, 18, 883–891 CrossRef CAS PubMed.
- H. P. Lee, L. Gu, D. J. Mooney, M. E. Levenston and O. Chaudhuri, Nat. Mater., 2017, 16, 1243–1251 CrossRef CAS PubMed.
- E. E. Charrier, K. Pogoda, R. G. Wells and P. A. Janmey, Nat. Commun., 2018, 9, 449 CrossRef PubMed.
- Y. C. Yeh, E. A. Corbin, S. R. Caliari, L. Ouyang, S. L. Vega, R. Truitt, L. Han, K. B. Margulies and J. A. Burdick, Biomaterials, 2017, 145, 23–32 CrossRef CAS PubMed.
- A. R. Cameron, J. E. Frith, G. A. Gomez, A. S. Yap and J. J. Cooper-White, Biomaterials, 2014, 35, 1857–1868 CrossRef CAS PubMed.
- M. Diba, S. Spaans, S. I. S. Hendrikse, M. M. C. Bastings, M. J. G. Schotman, J. F. van Sprang, D. J. Wu, F. J. M. Hoeben, H. M. Janssen and P. Y. W. Dankers, Adv. Mater., 2021, 33, 2008111 CrossRef CAS PubMed.
- B. Marco-Dufort, R. Iten and M. W. Tibbitt, J. Am. Chem. Soc., 2020, 142, 15371–15385 CrossRef CAS PubMed.
- H. W. Ooi, S. Hafeez, C. A. Van Blitterswijk, L. Moroni and M. B. Baker, Mater. Horiz., 2017, 4, 1020–1040 RSC.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Mater. Today Adv., 2019, 3, 100021 CrossRef.
- M. Rizwan, A. E. G. Baker and M. S. Shoichet, Adv. Healthcare Mater., 2021, 2100234, 1–23 Search PubMed.
- A. J. Feliciano, C. van Blitterswijk, L. Moroni and M. B. Baker, Acta Biomater., 2021, 124, 1–14 CrossRef CAS PubMed.
- I. A. Marozas, K. S. Anseth and J. J. Cooper-White, Biomaterials, 2019, 223, 119430 CrossRef CAS PubMed.
- V. Yesilyurt, A. M. Ayoob, E. A. Appel, J. T. Borenstein, R. Langer and D. G. Anderson, Adv. Mater., 2017, 29, 1605947–1605947 CrossRef PubMed.
- S. Hafeez, H. Ooi, F. Morgan, C. Mota, M. Dettin, C. van Blitterswijk, L. Moroni and M. Baker, Gels, 2018, 4, 85 CrossRef CAS PubMed.
- F. L. C. Morgan, J. Fernández-Pérez, L. Moroni and M. B. Baker, Adv. Healthcare Mater., 2022, 11, 2101576 CrossRef CAS PubMed.
- L. Zou, A. S. Braegelman and M. J. Webber, ACS Appl. Mater. Interfaces, 2019, 8–13 Search PubMed.
- B. Yang, K. Wei, C. Loebel, K. Zhang, Q. Feng, R. Li, S. H. D. Wong, X. Xu, C. Lau, X. Chen, P. Zhao, C. Yin, J. A. Burdick, Y. Wang and L. Bian, Nat. Commun., 2021, 12, 3514 CrossRef CAS PubMed.
- E. Vereroudakis, M. Bantawa, R. P. M. Lafleur, D. Parisi, N. M. Matsumoto, J. W. Peeters, E. Del Gado, E. W. Meijer and D. Vlassopoulos, ACS Cent. Sci., 2020, 6, 1401–1411 CrossRef CAS PubMed.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef PubMed.
- J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- H. H. Susapto, D. Alhattab, S. Abdelrahman, Z. Khan, S. Alshehri, K. Kahin, R. Ge, M. Moretti, A. H. Emwas and C. A. E. Hauser, Nano Lett., 2021, 21, 2719–2729 CrossRef CAS PubMed.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. Van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- C. M. A. Leenders, T. Mes, M. B. Baker, M. M. E. Koenigs, P. Besenius, A. R. A. Palmans and E. W. Meijer, Mater. Horiz., 2014, 1, 116–120 RSC.
- S. I. S. Hendrikse, L. Su, T. P. Hogervorst, R. P. M. Lafleur, X. Lou, G. A. Van Der Marel, J. D. C. Codee and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 13877–13886 CrossRef CAS PubMed.
- C. Tong, T. Liu, V. Saez Talens, W. E. M. Noteborn, T. H. Sharp, M. M. R. M. Hendrix, I. K. Voets, C. L. Mummery, V. V. Orlova and R. E. Kieltyka, Biomacromolecules, 2018, 19, 1091–1099 CrossRef CAS PubMed.
- J. Y. Chia, T. Miki, H. Mihara and H. Tsutsumi, Bioorg. Med. Chem., 2021, 46, 116345 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- R. C. van Gaal, A. B. C. Buskermolen, B. D. Ippel, P. P. K. H. Fransen, S. Zaccaria, C. V. C. Bouten and P. Y. W. Dankers, Biomaterials, 2019, 224, 119466 CrossRef CAS PubMed.
- B. B. Mollet, M. Comellas-Aragonès, A. J. H. Spiering, S. H. M. Söntjens, E. W. Meijer and P. Y. W. Dankers, J. Mater. Chem. B, 2014, 2, 2483–2493 RSC.
- K. L. Niece, J. D. Hartgerink, J. J. J. M. Donners and S. I. Stupp, J. Am. Chem. Soc., 2003, 12524, 7146–7147 Search PubMed.
- S. I. S. Hendrikse, S. P. W. Wijnands, R. P. M. Lafleur, M. J. Pouderoijen, H. M. Janssen, P. Y. W. Dankers and E. W. Meijer, ChemComm, 2017, 53, 2279–2282 RSC.
- S. Varela-aramburu, G. Morgese, L. Su, S. M. C. Schoenmakers, M. Perrone, L. Leanza, C. Perego, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Biomacromolecules, 2020, 21, 4105–4115 CrossRef CAS PubMed.
- J. A. Rowley, G. Madlambayan and D. J. Mooney, Biomaterials, 1999, 20, 45–53 CrossRef CAS PubMed.
- N. Huebsch, E. Lippens, K. Lee, M. Mehta, S. T. Koshy, M. C. Darnell, R. M. Desai, C. M. Madl, M. Xu, X. Zhao, O. Chaudhuri, C. Verbeke, W. S. Kim, K. Alim, A. Mammoto, D. E. Ingber, G. N. Duda and D. J. Mooney, Nat. Mater., 2015, 14, 1269–1277 CrossRef CAS PubMed.
- C. M. Madl, B. L. Lesavage, R. E. Dewi, C. B. Dinh, S. Ryan, M. Khariton, K. J. Lampe, D. Nguyen, O. Chaudhuri, A. Enejder and S. C. Heilshorn, Nat. Mater., 2018, 16, 1233–1242 CrossRef PubMed.
- C. M. Madl, B. L. LeSavage, R. E. Dewi, K. J. Lampe and S. C. Heilshorn, Adv. Sci., 2019, 6, 1801716 CrossRef PubMed.
- J. P. Wojciechowski, A. D. Martin, A. F. Mason, C. M. Fife, S. M. Sagnella, M. Kavallaris and P. Thordarson, ChemPlusChem, 2017, 82, 383–389 CrossRef CAS PubMed.
- A. C. Daly, M. D. Davidson and J. A. Burdick, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed.
- C. Loebel, R. L. Mauck and J. A. Burdick, Nat. Mater., 2019, 18, 883–891 CrossRef CAS PubMed.
- E. Vrij, J. Rouwkema, V. Lapointe, C. Van Blitterswijk, R. Truckenmüller and N. Rivron, Adv. Mater., 2016, 28, 4032–4039 CrossRef CAS PubMed.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS PubMed.
- F. R. Passanha, T. Geuens, S. Konig, C. A. van Blitterswijk and V. L. LaPointe, Biomaterials, 2020, 254, 120127 CrossRef CAS PubMed.
- B. D. Cosgrove, K. L. Mui, T. P. Driscoll, S. R. Caliari, K. D. Mehta, R. K. Assoian, J. A. Burdick and R. L. Mauck, Nat. Mater., 2016, 15, 1297–1306 CrossRef CAS PubMed.
- K. Bott, Z. Upton, K. Schrobback, M. Ehrbar, J. A. Hubbell, M. P. Lutolf and S. C. Rizzi, Biomaterials, 2010, 31, 8454–8464 CrossRef CAS PubMed.
- V. Hongisto, S. Jernström, V. Fey, J. P. Mpindi, K. Kleivi Sahlberg, O. Kallioniemi and M. Perälä, PLoS One, 2013, 8, 1–16 CrossRef PubMed.
- F. R. Passanha, D. B. Gomes, J. Piotrowska, L. Moroni, M. B. Baker and V. L. S. LaPointe, J. Tissue Eng. Regener. Med., 2022, 16, 14–25 CrossRef CAS PubMed.
- R. Kohze, C. E. J. Dieteren, W. J. H. Koopman, R. Brock and S. Schmidt, Cytometry, Part A, 2017, 91, 810–814 CrossRef CAS PubMed.
- A. Malheiro, F. Morgan, M. Baker, L. Moroni and P. Wieringa, Biomaterials, 2020, 257, 120230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00312k |
| This journal is © The Royal Society of Chemistry 2022 |