
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution versus solid-state dual emission of the Au(I)-alkynyl diphosphine complexes via modification of polyaromatic spacers†
Andrey
Belyaev
 *a,
Ilya
Kolesnikov
b,
Alexei S.
Melnikov
c,
Vladislav V.
Gurzhiy
d,
Sergey P.
Tunik
e and
Igor O.
Koshevoy
*a
*a,
Ilya
Kolesnikov
b,
Alexei S.
Melnikov
c,
Vladislav V.
Gurzhiy
d,
Sergey P.
Tunik
e and
Igor O.
Koshevoy
*a
aDepartment of Chemistry, University of Eastern Finland, Joensuu, 80101, Finland. E-mail: andreib@uef.fi; igor.koshevoy@uef.fi
bCenter for Optical and Laser Materials Research, St. Petersburg State University, St. Petersburg, 198504, Russia
cSt. Petersburg State Polytechnical University, St. Petersburg, 195251, Russia
dCrystallography Department, St. Petersburg State University, St. Petersburg, 198504, Russia
eInstitute of Chemistry, St. Petersburg State University, St. Petersburg, 198504, Russia
First published on 8th August 2019
Abstract
Single molecule luminophores capable of multiple emissions are essential for the development of new materials with unconventional photophysical behavior. In this work, a family of diphosphine ligands PPh2–PAH–PPh2 with variable polyaromatic hydrocarbon (PAH) spacers (PAH = 9,10-anthracene L1, 1,4-naphthalene L2, 2,6-naphthalene L3, and their diethynyl congeners L4–L6) were employed to prepare gold(I) complexes (RC2Au)PPh2–PAH–PPh2(AuC2R) (1–22), containing a selection of alkynyl groups. Investigation of their optical properties indicates that compounds with anthracene-based diphosphines (1–4 and 13–16) display only 1IL (ππ*) fluorescence with Φem up to 93%. The naphthalene and diethynyl-naphthalene diphosphine complexes (5–12 and 17–22), however, demonstrate panchromatic emission in the solid state and in solution featuring well-separated low and high energy signals, which originate from 1IL (ππ*) and 3IL (ππ*) transitions along with certain contribution from metal to ligand and ligand to ligand charge transfers.
Introduction
The nature of the ground and excited states of luminescent molecules primarily determines their photophysical characteristics, such as absorption and emission, the rates of radiative and non-radiative processes and, consequently, the excited state lifetimes. Rational design of the emitting molecular skeleton is expected to provide control over the energies of electronic transitions by manipulating the Frontier molecular orbitals. Such target modulation of luminescence properties results in solvatochromism, thermally activated delayed fluorescence, stimuli-responsive behavior, long-lived phosphorescence, and panchromatic emission, which are suitable for a number of practical applications including electroluminescent devices, sensors and bioimaging.1,2One approach to novel light emitting materials involves the incorporation of late transition metal atoms (OsII, ReI, IrIII, PtII and AuI/III) into an organic chromophore.3 In particular, the formation of the σ-bond between the carbon skeleton and the gold(I) ion by means of a heavy atom effect enhances spin–orbit coupling (SOC) and accelerates the rate of intersystem crossing (ISC, i.e. singlet–triplet transition S1 → T1). This leads to a rapid population of the triplet excited state and further can induce “forbidden” phosphorescence (T1 → S0). However, the reports of pure room temperature fluorescence (S1 → S0) from the molecules containing gold atoms indicate that the presence of this heavy element does not necessarily ensure a fast ISC rate, and the nature of the ligands also plays an important role in the excited-state dynamics and deactivation mechanisms.3a,4 Conversely, in the case of moderate ISC rate constant, compatible with that of fluorescence relaxation, room temperature fluorescence/phosphorescence dual emission (S1 + T1 → S0) can be observed for this sort of gold compound. Construction of such emitters remains a challenge due to the need to keep a delicate balance of populating the S1 and T1 excited states, but potential benefits of this photophysical phenomenon comprise ratiometric oxygen and pH monitoring,5 white-light generation,5b,6 and fluorescence/phosphorescence lifetime imaging.7
Dually emissive gold(I) organometallic species predominantly have low nuclearity and belong to the well-studied LAuX type with a linear two-coordinate geometry of the metal center. These complexes are conventionally composed of isolobal to the proton LAu+ (L = phosphine or N-heterocyclic carbene) cationic fragments,8 which are complemented by σ-bonded aromatic,4c,9 alkynyl,10 or halide11 X ligands. Among accessible variations of the constituents, phosphine-alkynyl complexes (R3PAu–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)n–R′ prevail in the family of dual gold(I) luminophores due to facile synthesis and functionalization (Fig. 1A). Their photophysical properties are mainly regulated by intraligand transitions localized on the alkynyl ligands, which may contain a wide range of conjugated organic groups. As a representative illustration, simultaneous ligand-centered dual fluorescence (prompt and delayed) and phosphorescence of the conjugated (poly)phenylethylene–gold(I) phosphine complexes was described by the group of Che.10a Another molecular design, which is less common, implies the incorporation of a π-chromophore spacer into the diphosphine moiety. Utilizing this strategy, we reported a series of dinuclear gold(I) alkynyl complexes based on oligophenylene diphosphine ligands. The rate of ISC for these compounds systematically decreases upon an increase of the effective distance between the heavy gold atom and the center of the chromophore fragment, which allows for fine-tuning the fluorescence/phosphorescence ratio by varying the length of the oligophenylene spacer (Fig. 1B).6a,12 Moreover, the electronic properties of the ancillary alkynyl substituents (Fig. 1C) were shown to affect significantly the rate of the ISC process by means of altering the contribution of charge transfer transitions, and therefore influence the probability of singlet vs. triplet emission.13
C)n–R′ prevail in the family of dual gold(I) luminophores due to facile synthesis and functionalization (Fig. 1A). Their photophysical properties are mainly regulated by intraligand transitions localized on the alkynyl ligands, which may contain a wide range of conjugated organic groups. As a representative illustration, simultaneous ligand-centered dual fluorescence (prompt and delayed) and phosphorescence of the conjugated (poly)phenylethylene–gold(I) phosphine complexes was described by the group of Che.10a Another molecular design, which is less common, implies the incorporation of a π-chromophore spacer into the diphosphine moiety. Utilizing this strategy, we reported a series of dinuclear gold(I) alkynyl complexes based on oligophenylene diphosphine ligands. The rate of ISC for these compounds systematically decreases upon an increase of the effective distance between the heavy gold atom and the center of the chromophore fragment, which allows for fine-tuning the fluorescence/phosphorescence ratio by varying the length of the oligophenylene spacer (Fig. 1B).6a,12 Moreover, the electronic properties of the ancillary alkynyl substituents (Fig. 1C) were shown to affect significantly the rate of the ISC process by means of altering the contribution of charge transfer transitions, and therefore influence the probability of singlet vs. triplet emission.13
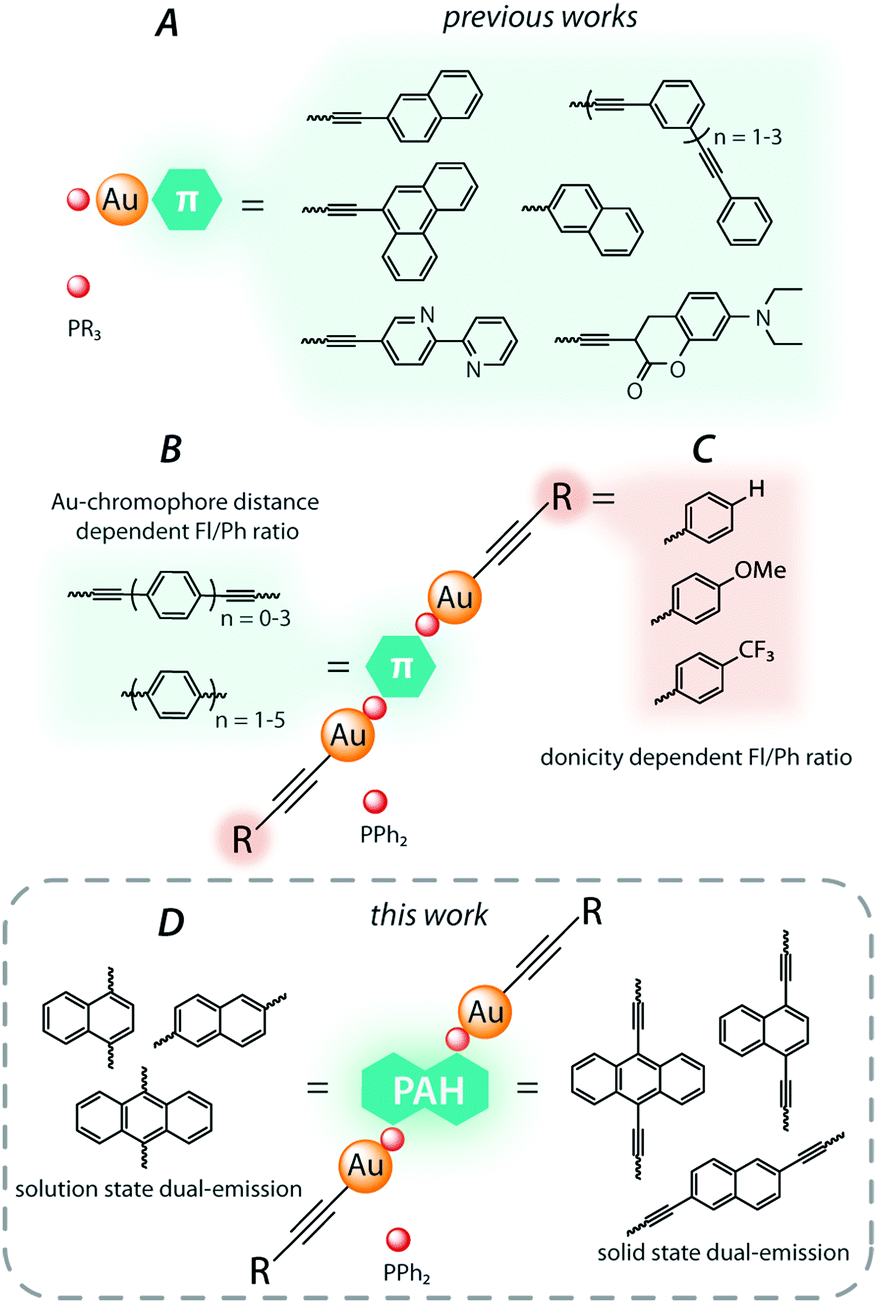 | ||
| Fig. 1 The representative families of previously studied dually emissive phosphine–gold(I) alkynyl complexes (A–C), and the scope of the current work (D). | ||
In the continuation of our studies, herein, we employ a family of diphosphine ligands based on the polyaromatic spacers (anthracene, naphthalene and their diethynyl derivatives, Fig. 1D). These P-donor modified chromophores were utilized for the preparation of novel gold(I) alkynyl complexes, the luminescence behavior of which was analyzed in solution and in the solid state to correlate with their molecular structures.
Experimental section
General comments
9,10-Bis-(diphenylphosphino)anthracene (L1),4a (AuC2CR)n (R = Ph; 4-C6H4–X, X = CF3, OMe, NH2, NMe2, Ph; C(CH3)2OH; C6H10OH; C(C6H5)2OH),14 and diethynylarenes (arene = 9,10-anthracene; 1,4- and 2,6-naphthalene)15 were synthesized according to the published procedures. Tetrahydrofuran (THF) and diethyl ether were distilled over Na-benzophenone ketyl under a nitrogen atmosphere prior to use. Other reagents and solvents were used as received. The solution 1H, 31P{1H} and 1H–1H COSY NMR spectra were recorded on a Bruker 400 Avance spectrometer. Mass spectra were recorded with a Bruker maXis HD ESI-QTOF instrument in the ESI+ mode. Microanalyses were carried out in the analytical laboratories of the University of Eastern Finland and of Saint-Petersburg State University.Photophysical measurements
Absorption spectra were recorded using Lambda 1050 and Shimadzu UV 1800 spectrophotometers; excitation and emission spectra in solution and the solid-state were measured on Fluoromax 4 and Fluorolog 3 spectrofluorimeters. LEDs (maximum of emission at 265 nm, 340 nm and 390 nm) were used in the pulse mode to pump luminescence in fluorescence lifetime measurements (pulse width ∼1 ns and repetition rate 100 MHz to 10 kHz). DTL-399QT Laser-export Co. Ltd (351 nm, 50 mW, pulse width 6 ns and repetition rate 0.01–1 kHz), MUM monochromator (LOMO, 1 nm bandwidth), counting head for photons H10682 (Hamamatsu) and digital time transducer P7887 (FAST ComTec GmbH) were used to measure the phosphorescence lifetime. The emission quantum yield in solution was determined by Vavilov's method16 using LED (385 nm, continuous mode) and xenon lamp pumping, with coumarine 102 (Φem = 0.76) and tryptophan (Φem = 0.13) as standards. The solutions were carefully degassed by purging Ar for 25 min. The integration sphere Quanta-φ (6-inches) was used to measure the solid state emission quantum yields for the complexes by an absolute method.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v) and further recrystallized by slow evaporation of a hexanes/CH2Cl2 solution of L2 at +5 °C to afford a colorless crystalline material (0.62 g, 55%). 31P{1H} NMR (CDCl3, 298 K; δ): −13.4 (s). 1H NMR (CDCl3, 298 K; δ): 8.45–8.48 (m, 3,6-H C10H6, 2H), 7.44–7.48 (m, 4,5-H C10H6, 2 H), 7.30–7.36 (m, Ph, 20 H), 6.86–6.87 (m, 1,2-H C10H6, 2H). Anal. calc. for C34H26P2: C, 82.25; H, 5.28. Found: C 81.98; H 5.34.
:1 v/v). The pure sample was obtained by additional treatment of L4 with activated charcoal in dichloromethane and recrystallization by slow evaporation of a toluene/CH2Cl2 solution of L4 at +5 °C to afford bright yellow needles (1.24 g, 63%). 31P{1H} NMR (CDCl3, 298 K; δ): −32.7 (s). 1H NMR (CDCl3, 298 K; δ): 8.55–8.57 (m, 1,4,5,8-H C14H8, 4H), 7.81–7.85 (m, ortho-H Ph, 8H), 7.57–7.59 (m, 2,3,6,7-H C14H8, 4H), 7.41–7.47 (m, meta + para-H Ph, 12H). Anal. calc. for C42H28P2: C, 84.84; H, 4.75. Found: C, 84.64; H, 4.81.
:4 v/v) and further recrystallized by slow evaporation of a hexanes/CH2Cl2 solution of L2 at +5 °C to afford a colorless crystalline material (0.62 g, 55%). 31P{1H} NMR (CDCl3, 298 K; δ): −13.4 (s). 1H NMR (CDCl3, 298 K; δ): 8.45–8.48 (m, 3,6-H C10H6, 2H), 7.44–7.48 (m, 4,5-H C10H6, 2 H), 7.30–7.36 (m, Ph, 20 H), 6.86–6.87 (m, 1,2-H C10H6, 2H). Anal. calc. for C34H26P2: C, 82.25; H, 5.28. Found: C 81.98; H 5.34.
:1 v/v). The pure sample was obtained by additional treatment of L4 with activated charcoal in dichloromethane and recrystallization by slow evaporation of a toluene/CH2Cl2 solution of L4 at +5 °C to afford bright yellow needles (1.24 g, 63%). 31P{1H} NMR (CDCl3, 298 K; δ): −32.7 (s). 1H NMR (CDCl3, 298 K; δ): 8.55–8.57 (m, 1,4,5,8-H C14H8, 4H), 7.81–7.85 (m, ortho-H Ph, 8H), 7.57–7.59 (m, 2,3,6,7-H C14H8, 4H), 7.41–7.47 (m, meta + para-H Ph, 12H). Anal. calc. for C42H28P2: C, 84.84; H, 4.75. Found: C, 84.64; H, 4.81.
General procedure for the preparation of complexes 1–22
(AuC2R)n (0.3 mmol) was mixed with the corresponding diphosphine (0.16 mmol) in dichloromethane (10 ml). The reaction mixture was stirred for 45 min in the absence of light. The resulting solution was filtered through a Celite pad and vacuum dried and the solid residue was purified by recrystallization. The crystallization and spectroscopic details are given in the ESI.†Results and discussion
Synthesis and characterization
Two families of diphosphine ligands containing polyaromatic hydrocarbon (PAH) backbones (9,10-anthracene (L1), 1,4-naphthalene (L2), 2,6-naphthalene (L3), and their PAH-diethynyl congeners (L4–L6), Scheme 1) were conventionally prepared in good yields via lithiation of dihalo- or diethynyl PAH precursors and coupling the metalated derivatives with a stoichiometric amount of PPh2Cl.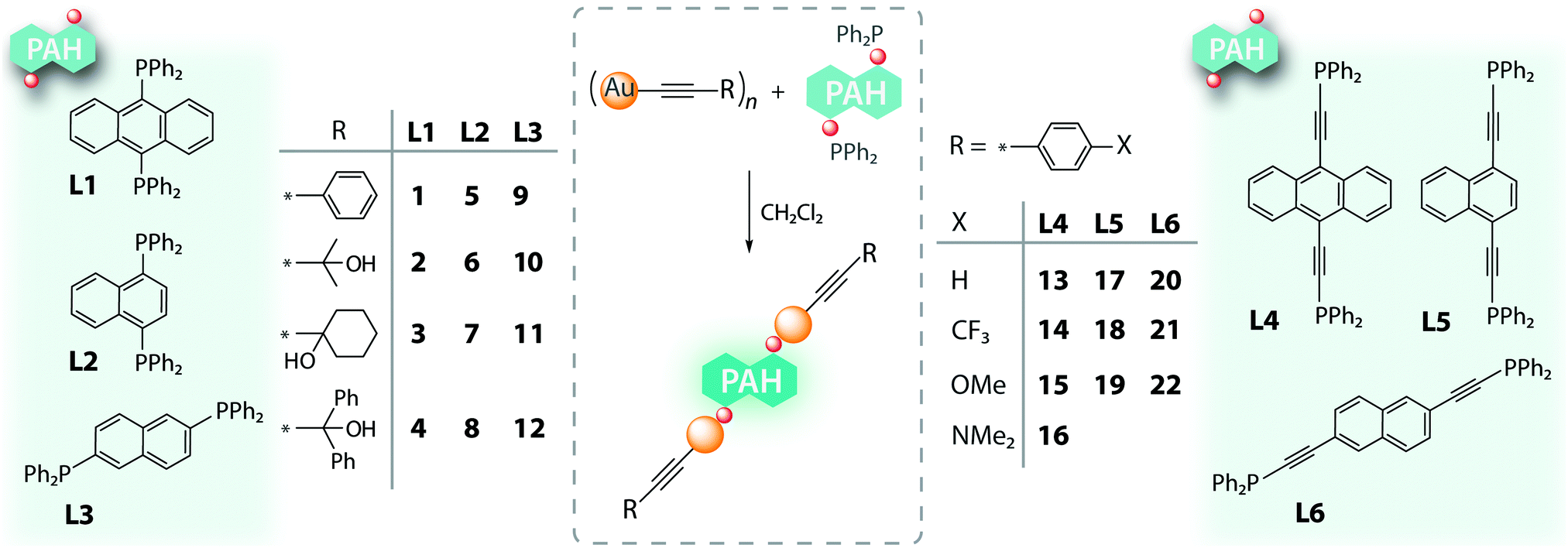 | ||
| Scheme 1 Polyaromatic phosphine ligands and their gold(I) alkynyl complexes 1–22. | ||
The dinuclear complexes (RC2Au)PPh2–spacer–PPh2(AuC2R) were readily obtained by reacting (AuC2R)n species with the corresponding bidentate ligands under ambient conditions (Scheme 1), analogously to a number of earlier reported congener species.13,14,17 The resulting compounds form two series (1–12) and (13–22), which are distinguished by the type of stereochemically and electronically different phosphine ligands (tertiary aromatic L1–L3 and ethynyl-aromatic L4–L6, respectively).
In solution, all the title complexes 1–22 were characterized using ESI+ mass spectrometry and 1H and 31P NMR spectroscopy. The ESI MS spectrum of 1–12 (Fig. S1, ESI†) shows the dominating signals of positively charged ion signals generated either by dissociation of alkynyl ligands or by association of the corresponding molecules with Na+ ions. The 31P{1H} NMR spectra of 1–22 display singlet resonances with chemical shifts in the ranges of 36.3–43.0 ppm (1–12) and 16.4–17.1 ppm (13–22), which are typical for the gold(I) compounds containing the related phosphines13 and are virtually insensitive to the nature of alkynyl ligands. These data indicate that complexes 1–22 exist in solution in their molecular forms of idealized symmetry that is additionally supported by the 1H NMR spectroscopic patterns, completely compatible with molecular arrangements shown in Scheme 1.
The solid-state structures of 1–3, 7, 9, 12, 13 and 19 have been elucidated by the XRD analysis (Fig. 1–4 and Fig. S2, ESI†); selected structural parameters are summarized in Table S2 (ESI†). Complexes 1 and 9 containing –C2Ph ligands display intermolecular Au–Au interactions to form a dimeric structure and an infinite polymeric structure.
 | ||
| Fig. 2 Molecular views of dimer 1 and polymer 9 (thermal ellipsoids are shown at 50% probability). | ||
The metal–metal distances in 1 (3.092 Å) and 9 (3.212 Å) are typical for aurophilic bonding frequently encountered in the crystals of gold(I) phosphine compounds.18
The Au(I) species with hydroxyaliphatic alkynes (2, 3, 7 and 12) do not feature metallophilic contacts (Fig. 3 and Fig. S2, ESI†). Alternatively, complexes 2 and 3 with anthracene diphosphine L1 demonstrate extensive intermolecular O–H⋯O hydrogen bonding (O⋯O separations are 2.76–2.88 Å) that evidently affects molecular packing for these species (Fig. S2 and S3, ESI†). The visible bending of anthracene motifs in 1–3 is similar to that observed for other gold compounds based on L1, and has been tentatively attributed to intramolecular steric hindrance.4a,19
 | ||
| Fig. 3 Molecular views of complexes 3, 7 and 12 (thermal ellipsoids are shown at 50% probability). | ||
 | ||
| Fig. 4 Molecular views of dimers 13 and 19 (thermal ellipsoids are shown at 50% probability). | ||
Photophysical properties
 | ||
| Fig. 5 (A) UV-vis absorption (dotted lines) and normalized emission (solid lines) spectra of 1–4 in CH2Cl2 (298 K, λex = 415 nm); (B) normalized solid-state excitation (dotted lines) and emission (solid lines) spectra of 1–4 (298 K, λex = 415 nm). | ||
| Solution (CH2Cl2, 298 K) | Solid | ||||||
|---|---|---|---|---|---|---|---|
| λ abs, nm (ε × 10−3, cm−1 M−1) | λ em,a nm | Φ em,b % | τ obs, ns | λ em,a nm 298 K | λ em,a nm 77 K | Φ em,c % | |
| a λ ex = 415 nm. b Measured in degassed solution. c In the KPF6 tablet. | |||||||
| L1 | 277, 355sh, 374, 396, 419 | ||||||
| 1 | 270 (86), 281sh (57), 368sh (4), 389 (8), 415 (12), 440 (13) | 485 | 3 | 2.3 | 522 | ∼554 | <1 |
| 2 | 271 (63), 367sh (5), 389 (10), 415 (15), 440 (15) | 475 | 4 | 3 | 489 | 467, 490, 506sh | <1 |
| 3 | 271 (80), 366 (6), 388 (13), 415 (19), 439 (20) | 480 | 5 | 3 | 492 | 464, 488, 522sh | <1 |
| 4 | 270 (83), 366 (6), 389 (13), 415 (20), 439 (20) | 480 | 4 | 3.2 | 489 | 477, 508, 545sh | <1 |
Complexes 1–4 are luminescent in solution at room temperature (Fig. 5A). Their emission profiles, small Stokes shifts and lifetimes of ca. 3 ns fit well with the fluorescence behavior of cationic complexes [Au3(L1)3]3+, [Au4(L1)2(μ-bipy)2]4+ and [Au4(L1)(diethyldithiocarbamate)3]+,4a,19b,21 and of the oxide derivative O = L1.22 Thus, the observed emission of 1–4 has mainly an intraphosphine character (1IL) and is virtually independent of the nature of ancillary ligands. The unstructured fluorescence signals for these complexes, which are red shifted in comparison to that of the parent anthracene, point to some charge transfer contribution to the emissive excited state due to the presence of coordinated PPh2 groups. The luminescence quantum yields of a few percent for 1–4 can only be attributed to the gold-induced heavy atom effect that facilitates fast ISC and leads to the population of the dark triplet state. Furthermore, larger rates of S1 → T1 transition are known to increase the radiationless internal conversion S1 → S0.10e
The emission and excitation characteristics of 2–4 at room temperature in the solid state resemble those measured in solution (Fig. 5B). The emission band maxima are slightly red shifted with respect to the fluid medium, whereas the quantum yield decreases considerably pointing to the effective aggregation-caused quenching effect, which often operates for organic luminophores.
In contrast to 2–4, complex 1 displays an ca. 40 nm bathochromic shift of luminescence in the solid state, that is presumably determined by the dimeric structure and π-stacking of the anthracene chromophores, see Fig. 1. These intermolecular interactions apparently increase the ground state energy and decrease the energy gap between the S0 and S1 states. For all complexes of this group, the excited state lifetimes fall in the nanosecond domain that confirms the singlet origin of emission.
At 77 K the emission bands of 2–4 exhibit vibronic progressions of ca. 1000–1200 cm−1 without a substantial shift of the band center that clearly points to intraligand L1 origin of fluorescence (Fig. S4A, ESI†). The broad emission of 1 at 77 K suggests that more than one excited state operate upon cooling. This features evidently the crystal packing effect, i.e. the influence of π-stacking and metal–metal interactions, as the spectrum of 1 in frozen solution (CH2Cl2) resembles those of 2–4 (Fig. S4B, ESI†).
| Solution (CH2Cl2, 298 K) | Solid | |||||
|---|---|---|---|---|---|---|
| λ abs, nm (ε × 10−3, cm−1 M−1) | λ em,a nm | λ em,b nm 298 K | λ em,b nm 77 K | τ av,c μs | Φ em,d % | |
| a λ ex = 310 nm for 5–8 and 300 nm for 9–12. b λ ex = 330 nm. c Average emission lifetimes for the two-exponential decay determined using the equation τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2), where Ai is the weight of the i-exponent. d In KPF6 tablet. | ||||||
| L2 | 274 (6), 332 (6) | |||||
| 5 | 268 (32.5), 280 (32), 296sh (19), 310 (15), 326 (10) | 350, 503, 538, 580 | 506, 536, 582 | 511, 524, 540, 590sh | 13.2 | 2 |
| 6 | 275 (7), 297 (10), 310 (11), 326 (8.5) | 351, 501, 536, 583 | 497, 509sh, 536, 580, 632 | 497, 510, 535, 549, 576 | 2.4 | <1 |
| 7 | 274 (6.5), 297 (9.5), 310 (11), 326 (7) | 351, 501, 536, 586 | 510, 536, 580sh | 516, 533, 550 | 2.1 | <1 |
| 8 | 273 (7), 297 (10), 310 (11), 326 (8.5) | 352, 502, 540, 582 | 498, 510, 536, 578, 630 | 514, 523sh, 553, 600 | 86.3 | <1 |
| L3 | 270 (17), 310 (9) | |||||
| 9 | 258 (72), 283 (46), 293 (33), 310 (15), 320 (2.3), 336 (2) | 341, 356, 516, 555, 600 | 478, 520sh, 557, 605sh | 488, 524, 555, 605sh | 225.9 | <1 |
| 10 | 258 (72), 275 (19), 288 (16), 298 (11), 320 (2.3), 336 (2) | 340, 356, 373, 514, 555, 604 | 475, 521, 554, 605, 664sh | 486sh, 519, 552, 600sh | 205.9 | 2 |
| 11 | 258 (72), 275 (19), 288 (16), 298 (11), 320 (2.3), 336 (2) | 341, 356, 514, 555, 603 | 513, 551, 597, 654 | 507, 546, 592, 647 | 1694.4 | 5 |
| 12 | 258 (72), 275 (19), 288 (16), 298 (11), 320 (2.3), 336 (2) | 342, 357, 517, 557, 603 | 509, 548, 593, 648 | 504, 515, 543, 589, 645 | 1407.4 | 7 |
 | ||
| Fig. 6 Room temperature UV-vis absorption (dotted lines) and normalized emission (solid lines) spectra of 5–8 (A, λex = 310 nm) and 9–12 (B, λex = 300 nm) in degassed CH2Cl2; color-filled profiles correspond to the spectra of 5 (peach, A) and 9 (blue, B) in aerated solutions. | ||
In solution, complexes 5–8 and 9–12 display dual emission, which comprises the HE band centered at ca. 350 nm and a structured LE band with wavelength above 500 nm. The vibronic progression of the LE band at ca. 1300–1500 cm−1 is normal for the gold-bound aromatic chromophores9b,23 and therefore points to a ligand centered (L2 naphthalene backbone) nature of this emission. A large Stokes shift together with oxygen quenching of the LE signal as depicted in Fig. 5, is indicative of the triplet origin of this band (phosphorescence), while the HE one, which also shows vibronic structures for 9–12 series (Δν ∼ 1200 cm−1) and is not sensitive to the presence of molecular oxygen, is associated with the intraligand fluorescence of L2 (5–8) and L3 (9–12) diphosphines, visibly perturbed for 5–8 by the –PPh2AuR motifs. Unfortunately, the low intensity of luminescence (Φem < 0.1%) did not allow the accurate determination of the lifetimes of singlet and triplet excited states to confirm their multiplicities.
The positions of the emission maxima are very similar within each group of complexes and the spectra are only different in relative intensities of HE fluorescence and LE phosphorescence bands (Fig. 6). The invariance of the emission energies and thus of the lowest lying excited states (S1 and T1) is in line with the poor involvement of the alkynyl ligands in their composition. However, as we have shown earlier, the ancillary –C2R groups are capable of affecting the rate of intersystem crossing S1 → Tn (n ≥ 1), altering the contribution of MLCT/L′LCT transitions.13 The non-innocent role of alkynyl ligands in populating the T1 state is also seen for 5–8 and 9–12 (see the excitation spectra in Fig. S5, ESI†), in which phenylalkynyl complexes 5 and 9 show the largest phosphorescence vs. fluorescence ratio within each series.
It is worth comparing the photophysical performance of 5–12 with that of the structurally related complexes of isomeric 1,8-bis(diphenylphosphino)naphthalene studied by Yam.20 In the latter case, the close disposition of the phosphorus substituents on the naphthalene backbone results in a virtually complete suppression of the singlet emission in solution to give phosphorescence in a red region (λem > 700 nm) and microsecond lifetimes. These emission characteristics drastically differ from those of 5–12 and illustrate the effect of substitutional isomerism of the diphosphines (i.e. 1,4-naphthalene (L2), 2,6-naphthalene (L3) and 1,8-naphthalene20) in the modulation of the lowest lying excited state.
In the solid state, the luminescence efficiency of 5–8 and 9–12 series is enhanced compared to that in solution, and the quantum yields fall in the range of 1–7% (Table 2). All complexes of these groups both at room temperature and at 77 K display structured LE bands with line shapes, which resemble the profiles of the phosphorescence profiles revealed in solution (Fig. S6, ESI†). The long lifetimes (2.1–86.3 μs for 5–8 and 206–1694 μs for 9–12) imply that only triplet emission retains in solids.
Complexes (AuC2C6H4X)2L4 (X = H, 13; CF3, 14; OMe, 15; and NMe2, 16) with diethynyl-anthracene phosphine show absorption spectra, which are mainly derived from that of the free ligand L4 (Table 3 and Fig. 7). In addition to the absorption bands of the phosphines, complexes 13–16 display moderately intense absorption shoulders in the range of 286–311 nm, which can be attributed to CCR intraligand transitions. The emission profiles for 13–16 are nearly the same irrespective of the CCR ligand and also match the spectra of (PR3Au)2(9,10-diethynylanthracene) complexes meaning that the PAH core of L4 determines the luminescence properties.20,25 The structured signals, high intensity (Φem = 87% and 93% for 13 and 14) and nanosecond lifetimes prove the 1IL (anthracene) nature of the excited state. The excitation spectra of 13–16 (Fig. 7B) are essentially similar to the absorption spectrum of L4, meaning that LL′ (π C2R → π* L4)/ML (Au dπ → π* L4) charge transfers play a minor role in the lowest lying excited state S1. Despite the fact that the modulation of the electron donating ability of –C2C6H4X alkynyl ligands does not change the shape 14 (X = CF3) is the most intense fluorophore among the studied compounds (Φem = 93%), gradually increasing the basicity of X substituents leads to a drastic drop of the quantum yield for 16 (X = NMe2, Φem = 1%). A similar trend was observed for Au(I) complexes with oligophenylene π-chromophores, for which, however, changing X = CF3 for OMe primarily enhances phosphorescence vs. fluorescence emission.13 To gain additional experimental proof that alkynyl ligands can govern the radiationless decay pathways, the photophysics of complex 16 has been studied in the presence of trifluoroacetic acid. Indeed, protonation of the NMe2 group results in an ca. 35-fold increase of luminescence intensity (Table 3 and Fig. 7C) and disappearance of the ∼300 nm absorption band assigned to C2C6H4NMe2 localized transitions. The latter evidently shifts to higher energies due to stabilization of the alkynyl-aniline π orbitals caused by switching the electron-rich NMe2 function to the electron deficient NMe2H+ ammonium derivative.26
| Solution (CH2Cl2, 298 K) | Solid | |||||||
|---|---|---|---|---|---|---|---|---|
| λ abs, nm (ε × 10−3, cm−1 M−1) | λ em,a nm | Φ em, % | τ obs, ns | λ em,b nm 298 K | λ em,b nm 77 K | Φ em, % | τ obs, ns | |
| a λ ex = 430 nm. b λ ex = 350 nm. c 16 in the presence of 0.1 M CF3COOH in CH2Cl2. | ||||||||
| L4 | 273, 381, 406, 430, 458 | — | — | — | — | — | — | |
| 13 | 270 (103), 363 (3), 382 (7), 407 (12), 431 (28), 460 (39) | 465, 495, 530 | 87 | 4.2 | 541 | 530, 575sh | 4 | 3.8 |
| 14 | 271 (106), 286 (53), 363 (2), 382 (7), 407 (11), 431 (27), 460 (39) | 465, 495, 530 | 93 | 4.4 | 535 | 516, 555 | 5 | 5.5 |
| 15 | 270 (106), 286sh (56), 363 (5), 382 (9), 407 (14), 431 (31), 460 (44) | 465, 495, 530 | 2 | 4.2 | 541 | 525, 562 | 1 | 11.8 |
| 16 | 271 (104), 289sh (53), 311 (46), 363 (7), 382 (10), 407 (15), 431 (33), 460 (46) | 465, 495, 530 | 1 | 4.0 | — | — | — | |
| 16H+ | 271 (105), 384 (9), 408 (12), 433 (26), 461 (36) | 465, 495, 530 | 39 | 4.7 | — | — | — | |
 | ||
| Fig. 7 (A) UV-vis absorption spectra of 13–16, color-filled profile corresponds to L4 (CH2Cl2, 298 K); (B) normalized excitation (dotted lines) and emission (solid lines) spectra of 13–16 (CH2Cl2, 298 K); and (C) changes in absorption and emission spectra of 16 upon protonation by CF3COOH. | ||
The solid-state emissions of complexes 13–15 are substantially red shifted (ca. 75 nm) with respect to their solution spectra (Fig. S7, ESI†), and 16 is not luminescent. Additionally, the loss of the vibronic structure, dramatically lower intensity (Φem up to 5% for 14) and short emission lifetimes of several nanoseconds evidence fluorescence, probably arising from intermolecular charge transfer between the PAH chromophores, which tend to aggregate in the solid state.
The photophysical data for the series 17–19 and 20–22 with 1,4- and 2,6-diethynylnaphthalene emitting centers, respectively, are listed in Table 4. In solution both groups of complexes reveal singlet emission HE signals in the deep blue to violet region with maximum quantum yields reached by CF3-substituted alkynyl species 18 (Φem = 24%) and 21 (Φem = 11%). Analogously to the anthracene-based family 13–16 described above, the naphthalene compounds 17–22 manifest a steep dependence of fluorescence intensity on the electron richness of the alkynyl ligands, which do not affect the energies of the bands (Fig. 8).
| Solution (CH2Cl2, 298 K) | Solid | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| λ abs, nm (ε × 10−3, cm−1 M−1) | λ em,a nm | Φ em, % | τ obs, ns | λ em,a nm 298 K | λ em,a nm 77 K | Φ em,b % | τ f,c ns | τ ph(av),d μs | |
| a λ ex = 330 nm. b Total quantum yield measured in air at 298 K. c For the HE fluorescence band at 298 K. d For the LE phosphorescence band at 298 K, average emission lifetimes for the two-exponential decay determined using the equation τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2), where Ai is the weight of the i-exponent. | |||||||||
| L5 | 250 (39), 327 (14), 348 (25), 368 (28) | — | — | — | — | — | — | ||
| 17 | 253 (65), 268 (60), 281 (57), 332 (28), 348 (47), 367 (55) | 377, 397 | 8 | 0.5 | 432, 622, 680 | 454, 472sh, 628, 689, 758 | 3 | 5.3 | 249.4 |
| 18 | 254 (47), 273 (52), 286 (51), 333 (21), 348 (34), 367 (41) | 377, 397 | 24 | 0.6 | 414, 601, 658, 723 | 414, 438, 614, 666, 733 | 2 | 3.7 | 54.8 |
| 19 | 253 (48), 272sh (41), 284 (44), 332 (23), 348 (36), 367 (41) | 380, 396 | 1 | 1.6 | 414, 603, 660, 727 | 414, 438, 602, 620, 664, 730 | 3 | 4.0 | 85.1 |
| L6 | 267 (39), 328 (23), 342 (25) | ||||||||
| 20 | 261 (90), 271 (123), 284sh (55), 296 (39), 310 (48), 316 (51), 324 (67), 331 (69), 338 (37), 357 (8) | 361, 380, 400 | 4 | 1.1 | 400, 551 | 550, 562, 600, 621, 650 | <1 | 3.4 | 5.9 |
| 21 | 262 (87), 272 (121), 286 (69), 310 (44), 316 (49), 324 (64), 331 (66), 338 (37), 357 (8) | 361, 380, 400 | 11 | 1.2 | 390, 542, 590, 647 | 385, 544, 556, 593, 644 | 1 | 2.9 | 91.6 |
| 22 | 262 (74), 272 (101), 286 (50), 298 (50), 305 (52), 310 (54), 316 (52), 324 (61), 331 (60), 338 (37), 357 (8) | 361, 380, 400 | <1 | 2.7 | 410, 540, 590 | 536, 550, 587, 631 | <1 | 3.7 | 10.6 |
 | ||
| Fig. 8 Room temperature UV-vis absorption (dotted lines) and normalized emission (solid lines) spectra of 17–19 (A) and 20–22 (B) in degassed CH2Cl2; and (C) normalized solid-state emission spectra of 17–19 under an Ar atmosphere; the color-filled profile corresponds to the spectra of 18 in air. | ||
In the solid state, complexes 17–22 behave differently than in solution and display two emission bands, which are particularly pronounced for CF3-containing compounds 18 and 21 (Fig. 8C, Fig. S8 and S9, ESI†). The broadened unresolved HE signals, which have the lifetimes of few nanoseconds, are red shifted for ca. 30 nm with respect to the corresponding fluorescence bands in solution. The intensity of long-lived LE emissions (λem > 600 nm for 17–19 and >540 for 20–22) is readily reduced in air, the extent of quenching is supposedly dependent on the morphology of the solid sample and is facilitated by its intrinsic porosity.13 Notably, the Commission Internationale de l’Eclairage (CIE) coordinates of compound 21 in the solid state (Table S3, ESI†) correspond to nearly pure white color (0.32, 0.32). The fluorescence vs. phosphorescence ratio for 17–19 at 298 K can be correlated with the donicity of alkynyl ligands, the increase of which favors transition to the T1 state, and therefore explained in terms of variable MLCT/L′LCT contributions.13 However, triad 20–22 does not obey the given trend (Fig. S9, ESI†) that testifies to the presence of subtle effects, which operate in the solid phase and have fine influence on the optical characteristics.
It is pertinent to remark the discrepancy in the emission behavior for 17 and 20 and of their aryl relatives 5 and 9 bearing the same phenyl alkynyl ligands. The latter complexes are dually emissive in solution, but in the solid state demonstrate pure phosphorescence. In a simplified approach, the difference in inducing triplet luminescence in 17, 20vs.5, 9 stems from the nature of the coordinating group, which links the chromophore PAH center to the gold ion and therefore determines the electronic communication and the distance between them. The smaller separation PAH⋯Au in 5 and 9 presumably facilitates spin–orbit coupling primarily via the heavy atom effect and increases the rate of intersystem crossing S1 → Tn so that phosphorescence starts to compete with fluorescence in fluid medium and prevails in solid samples. In line with this rationalization, the direct bonding of naphthalene to {AuPR3} fragments almost completely suppresses singlet emission already in solution.4c In the case of 17–22 the additional ethynyl spacers of the diphosphine backbones increase both the PAH⋯Au gap and fluorescence quantum efficiency, but prevent triplet emission in solution due to poor ISC. On the other hand, for these compounds dual luminescence is attained in the solid state that provides a route to molecular materials for panchromatic light generation.
Conclusions
In this work, we have synthesized a series of luminescent dinuclear gold(I) alkynyl complexes composed of phosphine-functionalized polyaromatic (PAH) chromophores. Systematic alteration of PAH moieties, the position and electronic features of P-donor connectivities reveal an essential influence on the optical behavior of the metal compounds. The anthracene-based gold(I) complexes 1–4 and 13–16 apparently demonstrate intraligand fluorescence localized on the diphosphine backbone with negligible contribution of the alkynyl and metal d orbitals to the emissive excited state. An extension of the PAH spacer (anthracene → diethynylanthracene) appears to be an efficient tool for increasing the quantum yield in solution; compounds 1–4 show Φem up to 5%, whereas for 13–16Φem reaches 93%. Increasing the basicity of substituents X in alkynyl ligands –C2C6H4X leads to a drastic drop of the emission intensity (X = CF3, Φem = 93% → X = NMe2, Φem = 1%) complexes 13–16. The replacement of the anthracene motif for the naphthalene core produces distinct dual emission in solution for L2 and L3-based complexes 5–12. Their luminescence profiles comprise the high-energy fluorescence band centered at ca. 350 nm and a vibronically structured phosphorescence signal with the wavelength maxima above 500 nm. In the solid state, the fluorescence signals for these species are completely suppressed and only triplet emission is observed. In contrast to 5–12, their ethynyl-phosphine congeners 17–22 (L4 and L5-based compounds) in solution exhibit only structured HE fluorescence, as manifested by nanosecond lifetimes of the excited state, with maximum quantum yields attained for 18 (Φem = 24%) and 21 (Φem = 11%) with CF3-substituted alkynyl groups. However, as solid powders complexes 17–22 are dually luminescent at room temperature. This effect is tentatively ascribed to the influence of ethynyl fragments between the phosphorus atoms and the π-spacers, which increase the PAH⋯Au distance and consequently diminish spin–orbit coupling, resulting in radiative relaxation of both S1 and T1 states only in the solid state.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Academy of Finland (grant 317903, I. O. K.) and Russian Science Foundation (spectral characterization and photophysical studies, grant 19-13-00132, S. P. T.) is gratefully acknowledged. This work was performed using the equipment of the Analytical Centre for Nano- and Biotechnologies (Peter the Great St. Petersburg Polytechnic University with financial support from the Ministry of Education and Science of Russian Federation) and Centers for Magnetic Resonance, for Optical and Laser Materials Research, for Chemical Analysis and Materials Research, Research Park of St. Petersburg State University.References
- (a) C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS; (b) N. Siraj, B. El-Zahab, S. Hamdan, T. E. Karam, L. H. Haber, M. Li, S. O. Fakayode, S. Das, B. Valle, R. M. Strongin, G. Patonay, H. O. Sintim, G. A. Baker, A. Powe, M. Lowry, J. O. Karolin, C. D. Geddes and I. M. Warner, Anal. Chem., 2016, 88, 170–202 CrossRef CAS PubMed.
- (a) X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed; (b) S.-C. Lee, J. Heo, H. C. Woo, J.-A. Lee, Y. H. Seo, C.-L. Lee, S. Kim and O. P. Kwon, Chem. – Eur. J., 2018, 24, 13706–13718 CrossRef CAS PubMed; (c) L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS PubMed.
- (a) Y. Y. Chia and M. G. Tay, Dalton Trans., 2014, 43, 13159–13168 RSC; (b) H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC; (c) F. N. Castellano, Acc. Chem. Res., 2015, 48, 828–839 CrossRef CAS PubMed; (d) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- (a) J. H. K. Yip and J. Prabhavathy, Angew. Chem., Int. Ed., 2001, 40, 2159–2162 CrossRef CAS; (b) V. W.-W. Yam, K.-L. Cheung, S.-K. Yip and N. Zhu, Photochem. Photobiol. Sci., 2005, 4, 149–153 RSC; (c) L. Gao, M. A. Peay, D. V. Partyka, J. B. Updegraff, T. S. Teets, A. J. Esswein, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2009, 28, 5669–5681 CrossRef CAS; (d) M.-H. Nguyen and J. H. K. Yip, Organometallics, 2010, 29, 2422–2429 CrossRef CAS; (e) L. Gao, D. S. Niedzwiecki, N. Deligonul, M. Zeller, A. D. Hunter and T. G. Gray, Chem. – Eur. J., 2012, 18, 6316–6327 CrossRef CAS PubMed; (f) S. Goswami, R. W. Winkel and K. S. Schanze, Inorg. Chem., 2015, 54, 10007–10014 CrossRef CAS PubMed; (g) A. M. Christianson and F. P. Gabbaï, Inorg. Chem., 2016, 55, 5828–5835 CrossRef CAS PubMed; (h) J. R. Shakirova, O. A. Tomashenko, E. V. Grachova, G. L. Starova, V. V. Sizov, A. F. Khlebnikov and S. P. Tunik, Eur. J. Inorg. Chem., 2017, 4180–4186 CrossRef CAS; (i) A. Gutiérrez-Blanco, V. Fernández-Moreira, M. C. Gimeno, E. Peris and M. Poyatos, Organometallics, 2018, 37, 1795–1800 CrossRef.
- (a) J. C. Lima and L. Rodriguez, Chem. Soc. Rev., 2011, 40, 5442–5456 RSC; (b) Y. Liu, H. Guo and J. Zhao, Chem. Commun., 2011, 47, 11471–11473 RSC; (c) T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS PubMed; (d) T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS PubMed; (e) P. W. Zach, S. A. Freunberger, I. Klimant and S. M. Borisov, ACS Appl. Mater. Interfaces, 2017, 9, 38008–38023 CrossRef CAS PubMed.
- (a) Y.-C. Chang, K.-C. Tang, H.-A. Pan, S.-H. Liu, I. O. Koshevoy, A. J. Karttunen, W.-Y. Hung, M.-H. Cheng and P.-T. Chou, J. Phys. Chem. C, 2013, 117, 9623–9632 CrossRef CAS; (b) M. Bachmann, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2017, 23, 9451–9456 CrossRef CAS PubMed; (c) M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Chem. Rev., 2018, 118, 8889–8935 CrossRef CAS PubMed.
- (a) H. Shi, H. Sun, H. Yang, S. Liu, G. Jenkins, W. Feng, F. Li, Q. Zhao, B. Liu and W. Huang, Adv. Funct. Mater., 2013, 23, 3268–3276 CrossRef CAS; (b) Q. Zhao, X. Zhou, T. Cao, K. Y. Zhang, L. Yang, S. Liu, H. Liang, H. Yang, F. Li and W. Huang, Chem. Sci., 2015, 6, 1825–1831 RSC; (c) F. Liu, J. Wen, S.-S. Chen and S. Sun, Chem. Commun., 2018, 54, 1371–1374 RSC.
- H. G. Raubenheimer and H. Schmidbaur, Organometallics, 2012, 31, 2507–2522 CrossRef CAS.
- (a) W. Y. Heng, J. Hu and J. H. K. Yip, Organometallics, 2007, 26, 6760–6768 CrossRef CAS; (b) D. V. Partyka, T. S. Teets, M. Zeller, J. B. Updegraff, A. D. Hunter and T. G. Gray, Chem. – Eur. J., 2012, 18, 2100–2112 CrossRef CAS PubMed.
- (a) W. Lu, W.-M. Kwok, C. Ma, C. T.-L. Chan, M.-X. Zhu and C.-M. Che, J. Am. Chem. Soc., 2011, 133, 14120–14135 CrossRef CAS PubMed; (b) J. Gil-Rubio, V. Cámara, D. Bautista and J. Vicente, Organometallics, 2012, 31, 5414–5426 CrossRef CAS; (c) K. T. Chan, G. S. M. Tong, W.-P. To, C. Yang, L. Du, D. L. Phillips and C.-M. Che, Chem. Sci., 2017, 8, 2352–2364 RSC; (d) M. Ferrer, L. Giménez, A. Gutiérrez, J. C. Lima, M. Martínez, L. Rodríguez, A. Martín, R. Puttreddy and K. Rissanen, Dalton Trans., 2017, 46, 13920–13934 RSC; (e) E. Aguiló, A. J. Moro, M. Outis, J. Pina, D. Sarmento, J. S. Seixas de Melo, L. Rodríguez and J. C. Lima, Inorg. Chem., 2018, 57, 13423–13430 CrossRef PubMed; (f) S. Wan and W. Lu, Angew. Chem., Int. Ed., 2017, 56, 1784–1788 CrossRef CAS PubMed; (g) E. C. Constable, C. E. Housecroft, M. K. Kocik, M. Neuburger, S. Schaffner and J. A. Zampese, Eur. J. Inorg. Chem., 2009, 4710–4717 CrossRef CAS; (h) X.-L. Li, K.-J. Zhang, J.-J. Li, X.-X. Cheng and Z.-N. Chen, Eur. J. Inorg. Chem., 2010, 3449–3457 CrossRef CAS.
- (a) T. L. Stott, M. O. Wolf and B. O. Patrick, Inorg. Chem., 2005, 44, 620–627 CrossRef CAS; (b) Z. Chen, G. Liu, R. Wang and S. Pu, RSC Adv., 2017, 7, 15112–15115 RSC.
- I. O. Koshevoy, C.-L. Lin, C.-C. Hsieh, A. J. Karttunen, M. Haukka, T. A. Pakkanen and P.-T. Chou, Dalton Trans., 2012, 41, 937–945 RSC.
- I. Kondrasenko, K.-y. Chung, Y.-T. Chen, J. Koivistoinen, E. V. Grachova, A. J. Karttunen, P.-T. Chou and Igor O. Koshevoy, J. Phys. Chem. C, 2016, 120, 12196–12206 CrossRef CAS.
- (a) G. E. Coates and C. Parkin, J. Chem. Soc., 1962, 3220–3226 RSC; (b) I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, S. I. Selivanov, J. Jänis, M. Haukka, T. A. Pakkanen, S. P. Tunik and P.-T. Chou, Inorg. Chem., 2012, 51, 7392–7403 CrossRef CAS PubMed.
- Y.-P. Ou, C. Jiang, D. Wu, J. Xia, J. Yin, S. Jin, G.-A. Yu and S. H. Liu, Organometallics, 2010, 30, 5763–5770 CrossRef.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- (a) V. W.-W. Yam, S. W.-K. Choi and K.-K. Cheung, Organometallics, 1996, 15, 1734–1739 CrossRef CAS; (b) M. Ferrer, A. Gutiérrez, L. Rodríguez, O. Rossell, J. C. Lima, M. Font-Bardia and X. Solans, Eur. J. Inorg. Chem., 2008, 2899–2909 CrossRef CAS.
- (a) E. R. T. Tiekink and J.-G. Kang, Coord. Chem. Rev., 2009, 253, 1627–1648 CrossRef CAS; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- (a) K. Zhang, J. Prabhavathy, J. H. K. Yip, L. L. Koh, G. K. Tan and J. J. Vittal, J. Am. Chem. Soc., 2003, 125, 8452–8453 CrossRef CAS PubMed; (b) R. Lin, J. H. K. Yip, K. Zhang, L. L. Koh, K.-Y. Wong and H. K. Piu, J. Am. Chem. Soc., 2004, 126, 15852–15869 CrossRef CAS PubMed.
- V. W.-W. Yam and S. W.-K. Choi, J. Chem. Soc., Dalton Trans., 1996, 4227–4232 RSC.
- J. Li, X.-F. Zhu, L.-Y. Zhang and Z.-N. Chen, RSC Adv., 2015, 5, 34992–34998 RSC.
- (a) Z. Fei, N. Kocher, C. J. Mohrschladt, H. Ihmels and D. Stalke, Angew. Chem., Int. Ed., 2003, 42, 783–787 CrossRef CAS PubMed; (b) Y. Zhao, L. Duan, X. Zhang, D. Zhang, J. Qiao, G. Dong, L. Wang and Y. Qiu, RSC Adv., 2013, 3, 21453–21460 RSC.
- L. Gao, D. V. Partyka, J. B. I. Updegraff, N. Deligonul and T. G. Gray, Eur. J. Inorg. Chem., 2009, 2711–2719 CrossRef CAS.
- V. F. Moreira, J. Cámara, E. S. Smirnova, I. O. Koshevoy, A. Laguna, S. P. Tunik, M. C. Blanco and M. C. Gimeno, Organometallics, 2016, 35, 1141–1150 CrossRef.
- V. Mishra, A. Raghuvanshi, A. K. Saini and S. M. Mobin, J. Organomet. Chem., 2016, 813, 103–109 CrossRef CAS.
- I. O. Koshevoy, A. J. Karttunen, S. P. Tunik, J. Jänis, M. Haukka, A. S. Melnikov, P. Y. Serdobintsev and T. A. Pakkanen, Dalton Trans., 2010, 39, 2676–2683 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 993387–993389, 1054718–1054720, 1937321 and 1937322. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj03426a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |