
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis, conformation and hydrolytic stability of an N,S-bridging thiophosphoramidate analogue of thymidylyl-3′,5′-thymidine†
Louis P.
Conway
a,
Satu
Mikkola
b,
AnnMarie C.
O'Donoghue
a and
David R. W.
Hodgson
 *a
*a
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: d.r.w.hodgson@durham.ac.uk
bDepartment of Chemistry, University of Turku, Vatselankatu 2, 20014 Turku, Finland
First published on 11th July 2016
Abstract
A 3′-N,5′-S-bridging thiophosphoramidate analogue of thymidylyl-3′,5′-thymidine was synthesised under aqueous conditions. 1H NMR conformational measurements show that the 3′-N-substituted deoxyribose ring is biased towards the ‘north’, RNA-like conformation. Rate constants for hydrolysis of the analogue were measured at 90 °C in the pH range 1.3–10.9. The pH-log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kobs profile displays a pH-independent region between approximately pH 7 and 10 (t1/2 ∼13 days). Under acidic conditions, kobs displays a first order dependence on [H3O+].
kobs profile displays a pH-independent region between approximately pH 7 and 10 (t1/2 ∼13 days). Under acidic conditions, kobs displays a first order dependence on [H3O+].
A Introduction
Phosphate analogues, where oxygen atoms have been replaced by nitrogen or sulfur, are important mechanistic enzymology probes.1 These heteroatom substitutions serve to alter the electrophilicity at P, change leaving group properties, and the interactions between these atoms and metal ions at the active sites of enzymes and ribozymes. Sulfur substitutions, in particular, offer the ability to study cation binding through metal ion rescue experiments, and the use of phosphorothiolates has proven the existence of general acid–base catalysed cleavage of phosphodiester bonds in ribozymes.1–5 Oligonucleotides containing modified phosphate groups show increased resistance towards (ribo)nucleases and enhanced therapeutic effects.6–8 Modified phosphates also offer the potential for modulating the structural properties of nucleic acid assemblies such as the i-motif.9Substitution of the O-heteroatoms of phosphoryl groups has also been used to facilitate phosphorylation and nucleic acid ligation technologies. To detect single nucleotide polymorphisms and facilitate signal amplification, Kool employed the nucleophilicity of sulfur anions to allow templated ligation through S-alkylation.8,10–17 We have exploited the nucleophilicity of amines to facilitate the efficient aqueous N-phosphorylation of amines,18–20 and N-thiophosphorylation plus S-alkylation of thiophosphoramidates.21–23 Amine nucleophilicity has also been harnessed to promote enzyme-free templated-ligation between 3′-amino-substituted oligonucleotides and 5′-activated phosphodiester nucleosides.24,25
We now demonstrate the application of aqueous N-thiophosphorylation plus S-alkylation towards the ligation of two nucleosides to afford thiophosphoramidate analogue TnpsT 1 of the dinucleotide, TpT 2 (Fig. 1). We study its conformational preference and hydrolytic stability across a broad pH range, with a view towards applying our aqueous strategies in nucleic acid ligations and bioconjugations.
 | ||
| Fig. 1 3′-Amino-3′-deoxythymidylyl-(3′→5′)-5′-deoxy-5′-thiothymidine (TnpsT 1) and its natural counterpart, thymidylyl-(3′→5′)-thymidine (TpT 2). | ||
B Synthesis
3′-Amino-3′-deoxythymidylyl-(3′→5′)-5′-deoxy-5′-thiothymidine (TnpsT, 1) was chosen as a target because starting materials for our proposed synthetic route (Scheme 1) were readily accessible. The amine, 3′-amino-3′-deoxythymidine 3 was accessed as a hydrochloride salt via the reduction of 3′-azido-3′-deoxythymidine (AZT). 5′-Deoxy-5′-iodothymidine 5 was prepared by 5′-tosylation of thymidine,26 followed by conversion to the iodide using sodium iodide in acetone.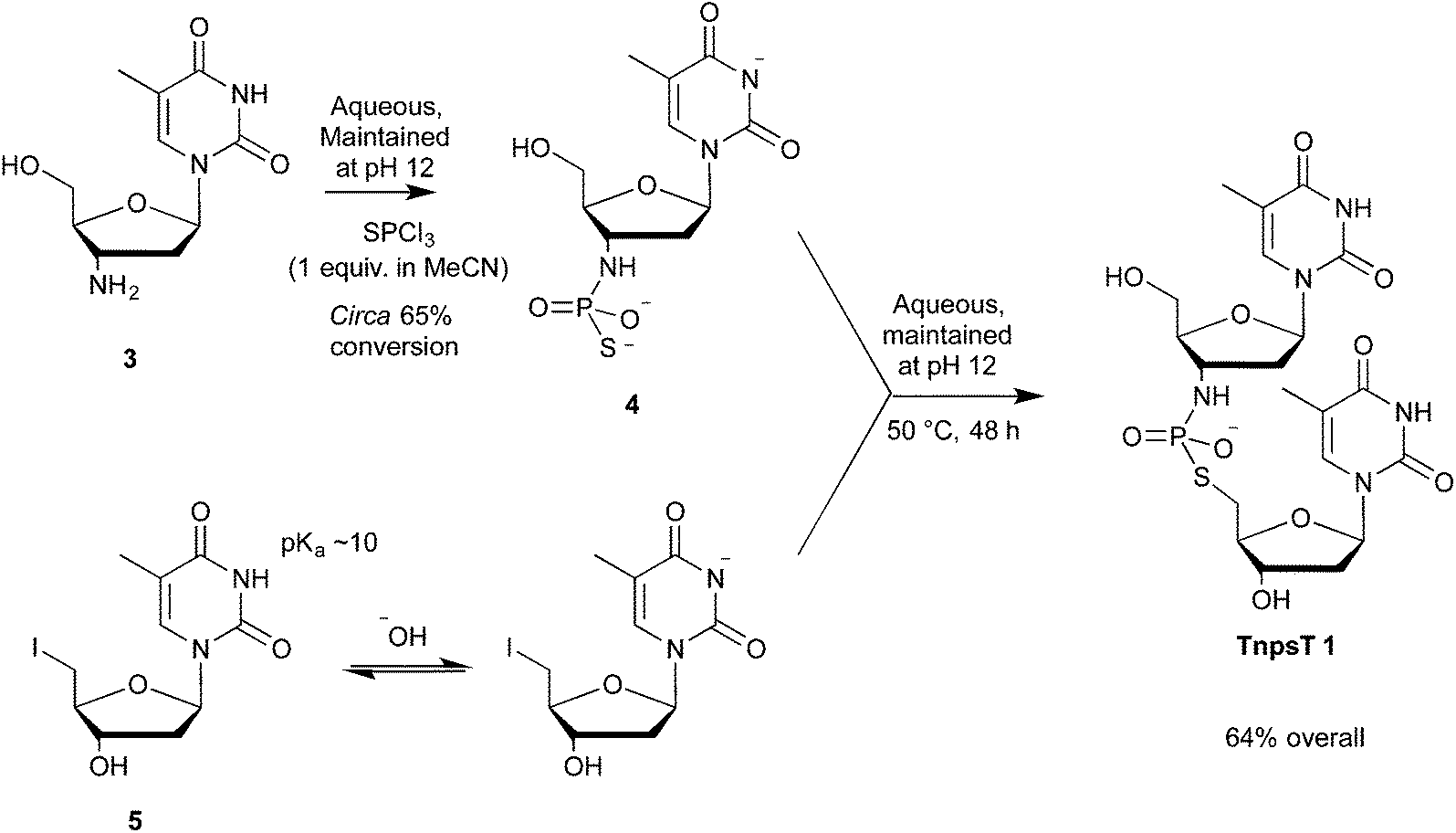 | ||
| Scheme 1 Thiophosphorylation of 3′-amino-3′-deoxythymidine 3 and subsequent reaction with 5′-deoxy-5′-iodothymidine 5. | ||
3′-Amino-3′-deoxythymidine 3 was N-thiophosphorylated using our pH-optimised protocol,22 where an aqueous solution of hydrochloride salt 3·HCl was maintained at pH 12 using KOH(aq) from an autotitrator during the addition of 1.0 equiv. of PSCl3 dissolved in MeCN. This afforded crude thiophosphoramidate 4 (approximately 65% conversion by 31P NMR spectroscopy).
The alkylating agent, 5′-deoxy-5′-iodothymidine 5, showed poor solubility at neutral pH, however, deprotonation of the thymine base (pKa ∼10) increased solubility markedly. Thus pH 12 was maintained throughout the addition of 5′-deoxy-5′-iodothymidine 5 to thiophosphoramidate 4. Under these conditions, 98% of the crude thiophosphoramidate 4 was converted to TnpsT 1 as determined by 31P NMR spectroscopy. TnpsT 1 was isolated by ion exchange chromatography in 64% yield (based on starting amine 3) as a triethylammonium salt, which was converted to the K+ salt for subsequent conformational and kinetic studies.
C Conformational analysis of TnpsT 1
Modified (deoxy)riboses display perturbed north/south conformational preferences (Fig. 2).27 Given our long-term intention to employ our synthetic approach in nucleic acid templated ligations, that would generate ligated products containing a thiophosphoramidate motif, we sought to gauge the conformational preference of TnpsT 1 using NMR methods. | ||
| Fig. 2 The north (N) and south (S) conformations of a deoxyribose ring. | ||
The elucidation of solution-state ribose conformations by means of 1H NMR J-coupling values was pioneered by Altona and Sundaralingam,28,29 and the effect on the conformation of thiophosphate-containing dinucleosides was investigated by Beevers et al.30 using similar methods. Rinkel and Altona found the sum of the J1′–2′ and J1′–2′′ coupling constants, ΣH1′, to be linearly correlated with the proportion of the south conformer through eqn (1), where PS is the proportion of the ‘south’ conformer:
 | (1) |
Many of the 1H NMR signals of TnpsT 1 are coincident, and second-order couplings complicate the extraction of J values of the deoxyribose protons. The signals for both C1′ protons, however, are well separated, and the J1′–2′ and J1′–2′′ coupling values were 7.5 and 4.0 Hz, respectively for the 3′-amino-3′-deoxythymidine (Tnp) residue of 1. The C1′–H signal at 6.28 ppm for the 5′-deoxy-5′-thiothymidine fragment (psT) presents as an apparent triplet with a coupling constant of 6.7 Hz (Fig. 3).
 | ||
| Fig. 3 The signals corresponding to the C1′ protons in TnpsT 1. The signal at 6.27 ppm corresponds to the 5′-linked nucleoside (psT), while the signal at 6.19 ppm corresponds to the 3′-linked nucleoside (Tnp). | ||
Application of eqn (1) yields values of 29% south for the Tnp ribose ring, and 61% south for the psT ribose ring. Comparison with TpT 2 (Tp: 74.2% south; pT: 62.7% south)30 indicates that the substitution of nitrogen for oxygen brings about a greater population of the ‘north’, C3′-endo, or ‘RNA-like’ conformer in the Tnp fragment, while the psT ribose ring retains its ‘DNA-like’ conformation. This is not surprising, as the conformation of the furanose ring is largely dictated by the anomeric and gauche effects, where the lower electronegativity of C3′–nitrogen compared to oxygen reduces the gauche effect of donation from the C2′–H bonding orbital into the C3′–N/O antibonding orbital and thus disfavours the south conformation. The thiophosphoramidate group is not linked directly to the ribose ring of the psT fragment and so has no apparent influence on its conformation. The Tnp conformational change is similar to that observed by Beevers et al. in the dideoxynucleoside 3′-phosphorothiolate analogue (TspT),30 however, a more detailed analysis,31 particularly in the context of extended and double-stranded nucleic acid structures will be required to confirm this result.
D Kinetics and mechanism of TnpsT hydrolysis
Hydrolysis experiments on TnpsT 1 were carried out at 90 °C in buffered aqueous solutions with pHs ranging from 1.32 to 10.91, at intervals of approximately one pH unit (pH values calculated at 90 °C based on values measured at 25 °C, see ESI†). Aliquots of substrate 1 in each buffered solution were sealed into vials, and heated at 90 °C. Samples were removed from the heating bath or block at suitable intervals, the remaining starting material and products were resolved by HPLC, and the ratios of the integrals of the substrate and hydrolysis products relative to an internal standard were assessed.At each pH, separate experiments were performed, using 10 and 100 mM buffer concentrations in order to check for buffer-promoted hydrolysis pathways. Acetate and formate buffered-experiments afforded rate constants that appeared to be dependent on buffer concentration. Thus, additional experiments were performed using 40 and 70 mM buffers, and kobs-buffer concentration plots were extrapolated to estimate the buffer-dependent and buffer-independent rate constants (see ESI†).
The logkobs-pH profile for the hydrolysis of TnpsT 1 displayed a pH-independent ‘plateau’ from ∼pH 7 to 10 with t1/2 ∼13 days, while at pH 1.32 the half-life was nine seconds (red trace in Fig. 4). The rapidity of reaction at lower pH values put practical limits on our ability to further explore this region. At the high pH extreme, the appearance of insoluble materials in the reaction mixture suggested etching of the glass vials, which precluded the straightforward exploration of the higher pH extreme within the format of our experimental design.
 | (2) |
 | ||
| Fig. 4 The pH-log(kobs) profiles for the hydrolyses at 90 °C of: TnpsT 1; and related systems Tnp(s)T 6 and TnpT 7 studied by Ora et al.32 Kinetic data are fitted to eqn (2). | ||
Data for the disappearance of TnpsT 1 were found to fit eqn (2), where rate coefficients k0 and kH represent the contributions to kobs of the neutral/zwitterionic forms 1(neutral) and monocationic form 1H+ respectively (Scheme 2). The acid dissociation constant between 1H+ and the kinetically indistinguishable neutral forms 1(neutral) is captured in Ka1. A single data point at pH 10.91 suggests a potential downward trend in reactivity towards higher pHs. This may be associated with nucleobase ionisation (Ka2) of 1(neutral), however, there are insufficient data to substantiate this hypothesis.
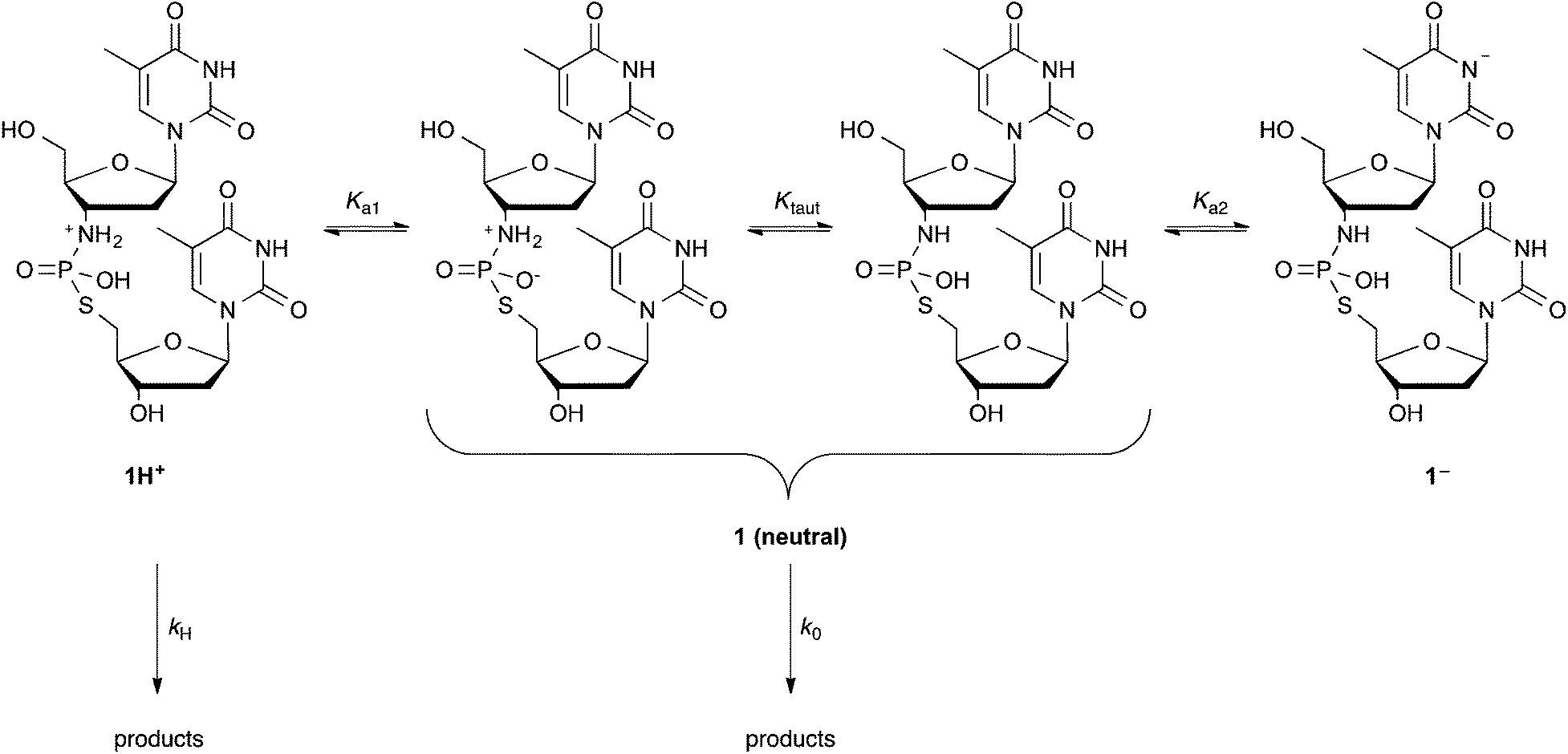 | ||
| Scheme 2 pH-Dependent speciation of TnpsT 1. | ||
The reactivity on the pH plateau, with k0 = 6.3 × 10−7 s−1, is similar to the related analogues Tnp(s)T 6 and TnpT 7 that were studied by Ora et al. (blue and green traces in Fig. 4).32 The lack of an observed plateau in reactivity at lower pH values limits our ability to unequivocally assign values to the interdependent variables kH and Ka1. Based on the fitting of the available data, however, values of kH < 0.15 M−1 s−1 and pKa1 < 1 were estimated, which align with those observed for TnpT 7.
The similarity in reactivity profiles of TnpsT 1 and TnpT 7 suggests that mechanisms are likely to be similar. This is borne out in product analysis studies, and illustrative examples are discussed below (Scheme 3).
 | ||
| Scheme 3 Mechanistic pathways for the hydrolysis of TnpsT 1. | ||
At pH 7.0 and 7.7 the largest detected peak by HPLC is thymine 8, derived from initial depyrimidinylation (route A) of either thymidine site within TnpsT 1 and subsequent fragmentation of the resulting species, as seen by Ora et al. for Tnp(s)T 6 and TnpT 7.32 Given the remoteness of the phosphoryl-sites from the C1′ sites where de-pyrimidininylations occur, it is unsurprising that the reactivities of TnpsT 1, Tnp(s)T 6 and TnpT 7 in the pH independent regions are similar. At pH 6, some depyrimidinylation is observed, however, P–N cleavage is now evident, with amine 3 being observed at a similar retention time to thymine 8 (routes A and B). Another product appears at a much longer retention time, with a lag in its formation. We believe this to be disulfide 11 formed from thiol 10 through oxidation, which is expected to be relatively facile at this pH, and has been reported in a similar system.33 We were, however, unable to confirm this by HPLC in a MS-compatible buffer system. Thiol 10 is formed by rapid dephosphorylation of phosphorothiolate 9, which arises from acid promoted P–N scission. At pH 3.2, the product chromatograms are simpler, displaying only two major peaks. Amine 3 represents one of these peaks, derived from P–N scission, whereas the second peak is consistent with thiol 10, which is formed rapidly from phosphorothiolate 9 (route B). Thiol 10 is expected to be relatively stable towards oxidation under these conditions. The overlap of the pH-logkobs profiles of TnpsT 1 and TnpT 7 in the acidic region suggests that either the values of kH and Ka1 are identical for these species, or that ionisation and reactivity compensate each other to arrive at identical kobs values.
E Conclusions
TnpsT 1, which is an analogue of thymidyl-3′,5′-thymidine 2, was successfully synthesised under aqueous conditions, without protecting groups. NMR-based analyses revealed a predominantly ‘north’, ‘RNA-like’ C3′-endo conformational preference for the 3′-aza-substituted deoxyribose (Tnp) fragment of TnpsT 1. Hydrolytic studies on TnpsT 1 yielded a near-identical profile to non-thio-analogue TnpT 7, where for pH > 7, de-pyrimidinylation dominates, and P–N scission is dominant for lower pHs. The combination of simple aqueous synthesis, knowledge of conformational preference and stability of the linkage will allow us to exploit N,S-bridging nucleotide systems in future applications.F Experimental
Synthesis
HPLC standard – potassium p-nitrobenzenesulfonate
p-Nitrobenzenesulfonyl chloride (222 mg, 1.00 mmol) was placed in a flask with potassium hydroxide solution (2 ml, 1.000 M) and water (20 ml). The solution was heated at reflux for 3 h before being lyophilised to yield a mixture of the desired product and potassium chloride in a 1:1 molar ratio. (231 mg, 98%) δH (400 MHz, (D2O)) 8.00 (2H, d, J 8.9, 1H m- to sulfonate), 8.35 (2H, d, J 8.9, 1H o- to sulfonate); δC (101 MHz, (D2O)) 124.3, 126.9, 148.1, 149.1.
Hydrolysis studies
See ESI† for details of the hydrolysis studies.Acknowledgements
This work was supported by EPSRC DTA funding to LPC (EP/P505488/1) and the Royal Society (chromatography system).Notes and references
- H. J. Korhonen, L. P. Conway and D. R. W. Hodgson, Curr. Opin. Chem. Biol., 2014, 21, 63 CrossRef CAS PubMed.
- N. S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257 CrossRef CAS PubMed.
- T. J. Wilson, N. S. Li, J. Lu, J. K. Frederiksen, J. A. Piccirilli and D. M. J. Lilley, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11751 CrossRef CAS PubMed.
- J. E. Deweese, A. B. Burgin and N. Osheroff, Nucleic Acids Res., 2008, 36, 4883 CrossRef CAS PubMed.
- K. H. Jung and A. Marx, Cell. Mol. Life Sci., 2005, 62, 2080 CrossRef CAS PubMed.
- R. O. Bak, A. Hendel, J. T. Clark, A. B. Kennedy, D. E. Ryan, S. Roy, I. Steinfeld, B. D. Lunstad, R. J. Kaiser, A. B. Wilkens, R. Bacchetta, A. Tsalenko, D. Dellinger, L. Bruhn and M. H. Porteus, Hum. Gene Ther., 2015, 26, A11 Search PubMed.
- A. Hendel, R. O. Bak, J. T. Clark, A. B. Kennedy, D. E. Ryan, S. Roy, I. Steinfeld, B. D. Lunstad, R. J. Kaiser, A. B. Wilkens, R. Bacchetta, A. Tsalenko, D. Dellinger, L. Bruhn and M. H. Porteus, Nat. Biotechnol., 2015, 33, 985 CrossRef CAS PubMed.
- G. Zon, New J. Chem., 2010, 34, 795 RSC.
- J. A. Brazier, J. Fisher and R. Cosstick, Angew. Chem., Int. Ed., 2006, 45, 114 CrossRef CAS PubMed.
- M. K. Herrlein and R. L. Letsinger, Nucleic Acids Res., 1994, 22, 5076 CrossRef CAS PubMed.
- Y. Xu and E. T. Kool, Nucleic Acids Res., 1999, 27, 875 CrossRef CAS PubMed.
- S. Sando and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 2096 CrossRef CAS PubMed.
- H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 13980 CrossRef CAS PubMed.
- S. Sando and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 9686 CrossRef CAS PubMed.
- S. Sando, H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 1081 CrossRef CAS PubMed.
- E. M. Harcourt and E. T. Kool, Nucleic Acids Res., 2012, 40, e65 CrossRef CAS PubMed.
- J. Michaelis, A. Roloff and O. Seitz, Org. Biomol. Chem., 2014, 12, 2821 CAS.
- R. J. Delley, A. C. O'Donoghue and D. R. W. Hodgson, J. Org. Chem., 2012, 77, 5829 CrossRef CAS PubMed.
- D. Williamson and D. R. W. Hodgson, Org. Biomol. Chem., 2008, 6, 1056 CAS.
- D. Williamson, M. J. Cann and D. R. W. Hodgson, Chem. Commun., 2007, 5096, 10.1039/b717896d.
- M. Trmcic and D. R. W. Hodgson, Chem. Commun., 2011, 47, 6156 RSC.
- L. P. Conway, R. J. Delley, J. Neville, G. R. Freeman, H. J. Maple, V. Chan, A. J. Hall and D. R. W. Hodgson, RSC Adv., 2014, 4, 38663 RSC.
- M. Trmcic, F. L. Chadbourne, P. M. Brear, P. W. Denny, S. L. Cobb and D. R. W. Hodgson, Org. Biomol. Chem., 2013, 11, 2660 CAS.
- M. Rothlingshofer, E. Kervio, T. Lommel, U. Plutowski, A. Hochgesand and C. Richert, Angew. Chem., Int. Ed., 2008, 47, 6065 CrossRef PubMed.
- A. Kaiser and C. Richert, J. Org. Chem., 2013, 78, 793 CrossRef CAS PubMed.
- D. M. Williams and V. H. Harris, Practical Approach in Chemistry: Organophosphorus Reagents, OUP, 1st edn, 2004 Search PubMed.
- W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1984 Search PubMed.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 94, 8205 CrossRef CAS PubMed.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1973, 95, 2333 CrossRef CAS PubMed.
- A. P. G. Beevers, E. M. Witch, B. C. N. M. Jones, R. Cosstick, J. R. P. Arnold and J. Fisher, Magn. Reson. Chem., 1999, 37, 814 CrossRef CAS.
- J. Jakhlal, S. Coantic-Castex, C. Denhez, C. Petermann, A. Martinez, D. Harakat, D. Guillaume and P. Clivio, Chem. Commun., 2015, 51, 12381 RSC.
- M. Ora, M. Murtola, S. Aho and M. Oivanen, Org. Biomol. Chem., 2004, 2, 593 CAS.
- J. B. Thomson, B. K. Patel, V. Jimenez, K. Eckart and F. Eckstein, J. Org. Chem., 1996, 61, 6273 CrossRef CAS PubMed.
- P. Cano-Soldado, M. Molina-Arcas, B. Alguero, I. Larrayoz, M. P. Lostao, A. Grandas, F. J. Casado and M. Pastor-Anglada, Biochem. Pharmacol., 2008, 75, 639–648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C and, where applicable, 31P NMR spectra of synthetic intermediates and analogue 1, HPLC calibration data, pH-temperature corrections, further details of individual kinetics experiments, example chromatograms, and tabulated HPLC data. See DOI: 10.1039/c6ob01270a |
| This journal is © The Royal Society of Chemistry 2016 |