
Mesoporous NiCu–CeO2 oxide catalysts for high-temperature water–gas shift reaction†
Ajay Jhaa,
Dae-Woon Jeonga,
Won-Jun Janga,
Chandrashekhar V. Rodeb and
Hyun-Seog Roh*a
aDepartment of Environmental Engineering, Yonsei University, 1 Yonseidae-gil, Wonju, Gangwon 220-710, South Korea. E-mail: hsroh@yonsei.ac.kr; Fax: +82-33-760-2571
bChemical Engineering & Process Development Division, CSIR-National Chemical Laboratory, Pune-411008, India
First published on 20th November 2014
Abstract
Mesoporous NiCu–CeO2 oxide catalysts were synthesized by using the evaporation-induced self-assembly method applied to the high-temperature, water–gas shift reaction (HT-WGS) between 350 to 550 °C. Nickel and copper loadings on mesoporous ceria were tailored to achieve high activity and selectivity by suppressing methane formation in HT-WGS. Among the prepared catalysts, NiCu(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4)–CeO2 exhibited the highest selectivity to CO2 and H2 with 85% CO conversion at a very high GHSV of 83665 h−1. The higher activity of the catalysts was due to the mesoporous architecture, which provides more accessible active sites for the WGS reaction. Powder X-ray diffraction (XRD), small angle X-ray scattering (SAXS), N2-adsorption/desorption isotherm, high-resolution transmission electron microscopy (HR-TEM), and H2-temperature-programmed reduction (TPR) techniques were used to understand the role of mesoporosity and bimetallic composition of various NiCu–CeO2 oxides in enhancing catalytic activity for HT-WGS.
:4)–CeO2 exhibited the highest selectivity to CO2 and H2 with 85% CO conversion at a very high GHSV of 83665 h−1. The higher activity of the catalysts was due to the mesoporous architecture, which provides more accessible active sites for the WGS reaction. Powder X-ray diffraction (XRD), small angle X-ray scattering (SAXS), N2-adsorption/desorption isotherm, high-resolution transmission electron microscopy (HR-TEM), and H2-temperature-programmed reduction (TPR) techniques were used to understand the role of mesoporosity and bimetallic composition of various NiCu–CeO2 oxides in enhancing catalytic activity for HT-WGS.
Introduction
Production of hydrogen has been a major focus of academic as well as industrial research because hydrogen is a promising sustainable and clean energy carrier. Hydrogen is produced from steam reforming of natural gas, partial oxidation of petroleum oil and steam gasification of coal.1 However, the CO level in the output gas of these processes is very high (>10%) and sufficient to poison the Pt electrode in the fuel cell. This triggered immense interest in the water–gas shift reaction (WGS; CO + H2O → CO2 + H2). The WGS reaction is an industrially important reaction to produce hydrogen for chemical processing and to remove CO contamination in feed streams for fuel cell applications.2,3 The reaction involves CO oxidation and H2O reduction to give CO2 and H2. As the WGS reaction is limited by equilibrium, at an industrial scale the reaction is implemented in two stages: the first, at high temperatures (HT-WGS) using Fe and Cr based catalysts; and the second, at low temperatures (LT-WGS), employing Cu–Zn–Al catalysts.4,5Recently, waste to energy has received much attention due to its potential to become a major hydrogen source.6,7 In general, gasification can convert waste into valuable synthesis gas (H2 + CO) including CO2, CH4 and N2, etc. However, CO concentration in the waste-derived synthesis gas is higher than the typical synthesis gas produced from steam reforming of hydrocarbons. In addition, the presence of the CO2 and H2 in the waste-derived synthesis gas, which can drive the reverse WGS reaction as well as higher concentration of CO, makes the system more severe. Therefore, it is necessary to develop a novel HTS catalyst to force the reaction into a forward direction and withstand severe conditions. In our previous study, the CuFe2O4 catalyst supported on mesoporous alumina showed the stable CO conversion (80%) with high selectivity to CO2 and H2 at 500 °C at a GHSV of 41821 h−1 in a waste-derived synthesis gas condition.8 In this work we observed that the mesoporous nature of the catalyst played a major role in controlling catalytic activity via uniform dispersion of the metal oxide. The mesoporous structure of the catalyst facilitates the uninterrupted diffusion of molecules to and from active sites of the catalysts. Moreover, because of the ascendant textural properties of mesoporous materials, well-dispersed nanoparticles over mesoporous support can provide more accessible active sites. In the continuation of this work reported here, mesoporous NiCu–CeO2 oxide catalysts for waste-derived synthesis gas were employed for the HT-WGS reaction.
Ceria (CeO2) has attracted much interest in recent decades in many areas of chemistry, materials science, and physics.9–12 Widespread applications mainly originate from the its outstanding oxygen storage capacity, that is, its ability to repeatedly and rapidly pass through redox (Ce4+/Ce3+) cycles.13,14 This is usually related to the ease of formation of labile oxygen vacancies, and particularly, those that promote relatively high mobility of bulk oxygen species at the surface of solid ceria.15–17 Relative to other oxide supports, ceria also enhances the performance of transition metal catalysts in a variety of reactions, including water–gas shift, steam reforming of oxygenates, and preferential oxidation of CO, all of which hold promise for enabling a hydrogen economy.18,19 In particular, the CuO/CeO2 catalyst has attracted great attention to the WGS due to its ability to firmly anchor Cu, reducing its tendency to sinter. Although the Cu/CeO2 catalyst performed well in the LT-WGS reaction, it is not suitable for the HT-WGS reaction and suffers from rapid deactivation at high temperatures (>300 °C).20–22 On the other hand, nickel-based catalysts are currently providing greater scope in the WGS reaction than the copper catalyst due to its high CO adsorption efficiency.23 However, hydrogenation of CO and CO2 is an undesired side reaction (in methane formation), reducing the efficiency of nickel-based catalysts for the WGS reaction.24
To overcome the drawbacks of lower thermal stability of Cu and side reactions of Ni-based catalysts in the WGS reaction, bimetallic Cu–Ni catalysts were developed for HT-WGS reactions. Lin et al. suggested that the presence of copper in the bimetallic Cu–Ni catalysts suppresses undesirable side effects, whereas the nickel component enhanced WGS activity.25 Saw et al. observed the formation of Ni–Cu alloy in the bimetallic NiCu–CeO2 catalyst that enhanced the CO adsorption at high reaction temperatures and increased its thermal stability.23 Doping of nickel to the copper oxide led to significant changes in the electronic properties of the catalyst via formation of active surface structures, new chemical bonding and surface coordination environment, which subsequently considerably influenced catalytic performance.26,27
The objective of the present work is to study effects of the catalyst's mesoporous architecture catalyst on performance of the NiCu–CeO2 oxide for the HT-WGS reaction with minimization of methane formation. The mesoporous bimetallic NiCu–CeO2 catalysts were prepared by the evaporation-induced self-assembly (EISA) method. A series of NiCu–CeO2 oxide catalysts were prepared by varying the weight percentage ratio of Ni/Cu from (1:1) to (1:4). The amount of CeO2 was fixed to 70 wt% in all cases investigated in this study. Performances of the mesoporous NiCu–CeO2 oxide were compared to CuO–CeO2 and NiO–CeO2 oxide catalysts in HT-WGS with the aim of understanding the effect of Ni doping with respect to catalytic activity and selectivity.
Results and discussion
The catalysts were studied by XRD to analyze their composition and phase purity. Fig. 1A shows the XRD patterns of calcined samples of CuO–CeO2, NiO–CeO2 and NiCu(1:4)–CeO2. The fluorite-type oxide diffraction pattern of CeO2 was present in all catalysts. X-ray powder diffraction patterns of CuO–CeO2 exhibited peaks at 2θ = 35.4° and 38.5°, which correspond to CuO (PDF00-001-1117), while NiO–CeO2 exhibited peaks at 2θ = 37.2°, 43.1°, and 62.6°, which correspond to NiO (PDF00-004-0835).25 The XRD pattern of the NiCu(1:4)–CeO2 catalyst also exhibited the corresponding peaks of both CuO and NiO at the same 2θ values.
 | ||
| Fig. 1 XRD patterns of (A) CuO–CeO2, NiO–CeO2, and NiCu(1:4)–CeO2 oxides, (B) NiCu(1:1)–CeO2, NiCu(1:2)–CeO2, and NiCu(1:4)–CeO2 oxides, and (C) reduced Cu–CeO2, Ni–CeO2, and NiCu(1:4)–CeO2 catalysts. | ||
Fig. 1B shows the XRD patterns of NiCu–CeO2 series catalysts prepared by varying the weight percentage ratio of Ni/Cu. Fig. 1B exhibits that intensity of the diffraction peaks increased with higher Cu loading, indicating greater crystalline nature of the catalysts. The Scherrer equation was applied to estimate the crystallite size of the catalysts by using the peak at 2θ = 28.4° (111) corresponding to the cubic phase of CeO2, and the crystallite size values are given in Table 1. From this table it is evident that the crystallite size of the NiCu–CeO2 catalysts increased along with increased copper loadings. This may be due to agglomeration of excess CuO on the surface of ceria. Fig. 1C represents XRD patterns of the reduced catalysts. Metallic phases of Cu and Ni were observed in the XRD patterns of Cu–CeO2 and Ni–CeO2, respectively, after being tested in the WGS reaction (up to 550 °C). These results indicate that oxide phases of the Cu and Ni were reduced to their corresponding metallic phases under WGS conditions. Crystallite sizes of all catalysts increased after the water–gas shift reaction, which suggests the agglomeration of catalysts that led to a decrease in surface area (Table 1). In the case of NiCu(1:4)–CeO2 oxide after the WGS reaction, three new reflections appeared that belong to the (111), (200), and (220) crystal planes, at 2θ = 43.3°, 50.45°, and 74.1°, respectively, which are related to the metallic phase of copper;27 we did not observe the metallic phase of nickel in the reduced NiCu(1:4)–CeO2 catalyst. This may be because of segregation of copper on the surface layer during the WGS reaction, and thus lower surface energy.11
H2-TPR patterns of NiO–CeO2, CuO–CeO2 and bimetallic NiCu–CeO2 oxide catalysts presented in Fig. 2 show very interesting patterns. In the case of CuO–CeO2, the shoulder peak appears at 125 °C, corresponding to the reduction of CuO nanoparticles highly dispersed on CeO2. The major peak at 204 °C, could be ascribed to the reduction of bulk CuO.28 In contrast, the TPR of NiO–CeO2 exhibited a distinct first peak at the higher temperature of 208 °C followed by another major peak at 300 °C and a small hump at 369 °C. The first peak at 208 °C corresponded to the reduction of NiO nanoparticles highly dispersed on the CeO2, while the second peak at 300 °C could be ascribed to the reduction of bulk NiO.29 The third shoulder peak at 369 °C could be due to the partial reduction of ceria.30 In the case of NiCu–CeO2 oxide catalysts, reduction of CuO and NiO shifted towards lower temperature as compared to CuO–CeO2 and NiO–CeO2 catalysts. This indicates that Ni and Cu together on mesoporous CeO2 have a combined influence on the reducibility of NiCu–CeO2 oxide. This is clearly evident from the fact that the reduction temperature of highly dispersed CuO in the NiCu–CeO2 catalysts decreased significantly with the increase in copper loadings (Fig. 2). Interestingly, the reduction peak of bulk CuO and NiO remained at the same position in the bimetallic series.31 The lowest reduction temperature in the range of 82–168 °C observed for the NiCu(1:4)–CeO2 oxide indicates the easy reducibility of the catalyst. Several authors attribute the strong interaction between highly dispersed metal oxides (CuO and NiO) and CeO2 for shifting reduction peaks toward lower temperatures.32,33 Doping of Ni- and Cu- together in the ratio of 1:4 into CeO2 produced a complex TPR profile (Fig. 2), giving four maxima at 82, 107, 168 and 259 °C. The first two reduction peaks at 82 and 107 °C were due to the reduction of interfacial and highly dispersed CuO, respectively.34 The two remnant peaks at 168 and 259 °C were possibly due to reduction of bulk CuO and NiO on the reducible mesoporous CeO2.
 | ||
| Fig. 2 H2-TPR patterns of the catalysts. | ||
A typical ordered mesoporous material is characterized by a SAXS (Fig. 3), indicating the existence of a meso-structure and a type-IV adsorption isotherm obtained through N2 adsorption/desorption measurements, indicating the textural property of catalysts. SAXS of CuO–CeO2 exhibited a broad peak while NiO–CeO2 showed a sharp peak and shifted to the higher 2θ value. Interestingly, the peak of NiCu(1:4)–CeO2 oxide was similar to that of NiO–CeO2 in spite of higher copper loading. This observation suggested that the incorporation of Cu- and Ni- together into the CeO2 did not influence the mesoporous framework of the catalyst.
 | ||
| Fig. 3 SAXS patterns of CuO–CeO2, NiO–CeO2, and NiCu(1:4)–CeO2 catalysts. | ||
Fig. 4 displays the N2-adsorption/desorption isotherm plots of the CuO–CeO2, NiO–CeO2 and NiCu(1:4)–CeO2 oxide catalysts. Isotherms of CuO–CeO2 reveal that with increasing relative pressure (p/po), an uptake of N2-adsorption volume increased slowly, which is the characteristic feature of type-I isotherm, typical of microporous materials.35 CuO–CeO2 sample displayed H4-shaped hysteresis loop, implying the existence of the micropores in the sample.36 However, N2 adsorption–desorption isotherms of NiO–CeO2 and NiCu(1:4)–CeO2 oxide demonstrated typical type IV isotherms and an uptake of N2-adsorbed volume reflected in the adsorption branches at the relative pressure range p/po = 0.5–1.0, due to capillary condensation of nitrogen into mesopores, which is a characteristic feature of ordered mesoporous material.37 This result is consistent with the SAXS shown in Fig. 3. NiO–CeO2 and NiCu(1:4)–CeO2 samples displayed an H3-shaped hysteresis loop, implying the presence of sheet-like aggregates of metal oxides.36 Interestingly, a significant change in the surface area and textural properties of the CeO2 was observed upon doping with Ni- or Cu-. Doping of Cu- into CeO2 increased the surface area; however, doping with Ni- led to a decrease in surface area. In contrast, NiCu–CeO2 oxides exhibited higher surface area than pure Ni–CeO2, and the surface area was found to further increase with greater Cu- loadings (Table 1). Enhanced surface areas could be correlated to formation of ordered mesoporous structure of the sample with increased Cu- loadings in the case of bimetallic catalysts.
 | ||
| Fig. 4 N2-adsorption/desorption isotherms of (a) CuO–CeO2, (b) NiO–CeO2, and (c) NiCu(1:4)–CeO2 catalysts. | ||
Fig. 5 shows SEM and HR-TEM images of the fresh mesoporous NiCu(1:4)–CeO2 oxide. The SEM image displayed agglomerated particles having uniform spherical morphology with the diameter in the range of 8–12 nm, which is well matched with the XRD result. Fig. 5B shows the lattice fringe patterns of the fresh catalyst, which exhibits the interplaner spacing of 0.28 nm and 0.31 nm corresponding to the (1 0 0) and (1 1 1) planes of the ceria cubic phase,37 while 0.19 nm corresponds to the monoclinic phase of the CuO. The inset in Fig. 5B represents the hexagonal lattice fringe pattern with a d-spacing value of 0.29 nm corresponding to the ceria oxide phase.
 | ||
| Fig. 5 (A) SEM and (B) HR-TEM image of fresh NiCu(1:4)–CeO2 catalyst. | ||
Catalytic performance for WGS reaction
The catalytic activities of mesoporous NiCu–CeO2 oxide catalysts were compared with the monometallic catalysts for WGS reaction in the temperature range of 350–550 °C, the results of which are shown in Fig. 6A. Nickel-based catalysts were found to be more highly active than Cu–CeO2. The higher activity of the catalysts was related to higher CO adsorption efficiency of Ni- than the Cu-.23 The catalytic activity at 350 °C occurs in the following order: CuO–CeO2 < NiCu(1:4)–CeO2 < NiCu(1:2)–CeO2 < NiCu(1:1)–CeO2 < NiO–CeO2. This observation indicates that CO conversion was directly proportional to the nickel loadings. In general, Ni supported on metal oxide is a well-known catalyst for methane formation.38 Since formation of CH4 consumes H2, the higher the CH4 yield, the lower the H2 yield in the WGS reaction. Fig. 6B shows methane selectivity during WGS reaction for all prepared catalysts as a function of reaction temperature. Fig. 6A and B display that Ni–CeO2 catalyst was more active but gave the highest selectivity to CH4 (28%) at 350 °C. In contrast, selectivity to CH4 was negligible for the Cu–CeO2 and NiCu(1:4)–CeO2 catalysts at all reaction temperatures employed. For NiCu–CeO2 oxide catalysts, selectivity to CH4 was found to progressively decrease with increases in Cu- loadings (Fig. 6B). Among the NiCu–CeO2 oxide catalysts, NiCu(1:4)–CeO2 was found to be a better catalyst in the WGS reaction, where we observed that the addition of only 6 wt% of Ni– to Cu–CeO2 increased the CO conversion from 30% to 65%, with complete selectivity to CO2 and H2. NiCu–CeO2 catalysts showed reduction in CH4 formation compared to Ni–CeO2 that may be related to formation of a copper-rich surface on the top of the catalyst after activation that suppressed the methanation reaction. This fact is also supported by XPS analysis of the fresh NiCu(1:4)–CeO2 catalyst (Fig. 7). Based on the area calculation we observed that the surface of the NiCu(1:4)–CeO2 oxide was composed of 21% Ni and 79% Cu, which is almost equal to the 1:4 ratio of Ni/Cu into the catalyst (Table S1†). The HR-TEM result of the NiCu(1:4)–CeO2 catalyst also showed the Cu-rich surface with the (2 0 2) plane of the CuO.
 | ||
| Fig. 6 (A) CO conversion and (B) selectivity to CH4 as a function of reaction temperature (H2O/(CH4 + CO + CO2) = 2.0; GHSV = 83665 h−1). | ||
 | ||
| Fig. 7 XPS spectrum of fresh (A) Cu 2p3/2 and (B) Ni 2p3/2 of NiCu(1:4)–CeO2 catalyst. | ||
The importance of the catalyst's mesoporous architecture was proven by comparing reaction results with the same catalytic composition, that is, Ni/Cu(1:4), but prepared through the impregnation method (I–NiCu/CeO2) and also without ceria (NiCu oxide). Fig. 8 depicts the CO conversion profile as a function of reaction temperature over mesoporous NiCu(1:4)–CeO2, I–NiCu/CeO2 and NiCu-oxide catalysts. It was found that the CO conversion increased with increasing temperature, but the shape of conversion versus temperature curve was affected by the catalyst nature reflecting the influence of catalyst composition. The CO conversion over mesoporous NiCu(1:4)–CeO2 catalyst was approximately four times higher at 350 °C as compared to the catalyst prepared by the impregnation method (I–NiCu(1:4)/CeO2, surface area = 14 m2 g−1). The higher WGS activity of the NiCu(1:4)–CeO2 catalyst could be explained by the presence of high surface area and mesoporous nature of the catalysts, which can allow uniform distribution of the active sites on the catalyst surface. TPR results showed that the NiCu(1:4)–CeO2 could be reduced at lower temperature than the I–NiCu(1:4)/CeO2 catalyst (Fig. S1†). This indicates that the mesoporous architecture of the catalysts helps to enhance the redox capability of the Ce4+/Ce3+ via formation of nanocrystallite CeO2, facilitating oxygen mobility. As a consequence, the EISA method helps to increase reducibility of NiCu–CeO2 oxide catalyst, resulting in enhanced activity of the WGS at high temperatures. In the case of the NiCu-oxide without CeO2, conversion of CO increased with rising temperature up to 400 °C; after that the CO conversion began to decrease notably, reaching 0% at 550 °C (Fig. 8). The decrease in activity at higher temperatures was due to sintering of the catalyst, which may lead to substantial reduction in surface area (from 11 to 0.6 m2 g−1). The poor performance of the bare NiCu-oxide in the WGS reaction could be due to its lower capability for water dissociation, which is one of the important steps in the WGS reaction assisted by ceria.39 Supporting the NiCu oxide on CeO2 facilitates the water dissociation, and easily continues the catalytic cycle in the production of hydrogen as we observed in the case of NiCu(1:4)–CeO2 catalyst. Furthermore, ceria possesses oxygen vacancies, which stabilize the transition metal nanoparticles supported on oxide surfaces against sintering. Our results clearly demonstrate that presence of ceria in the catalyst formulation is absolutely necessary for activity and stability in the WGS reaction.
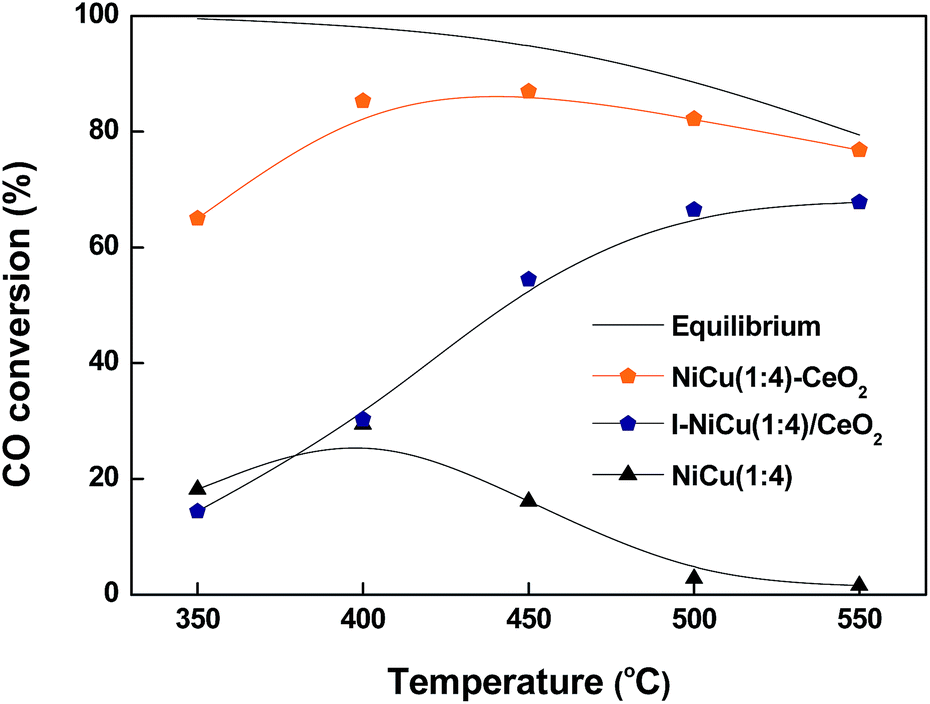 | ||
| Fig. 8 CO conversion as a function of reaction temperature over NiCu(1:4), I–NiCu–CeO2, and NiCu–CeO2 catalysts (H2O/(CH4 + CO + CO2) = 2.0; GHSV = 83665 h−1). | ||
For possible industrial applications, both activity and stability of the catalyst are necessary. To compare stability of NiCu(1:4)–CeO2, I–NiCu(1:4)/CeO2 and Cu–CeO2 catalysts in WGS, CO conversion data were collected at 450 °C and at a very high GHSV of 83665 h−1 for 45 h; corresponding results are displayed in Fig. 9. Under these conditions, the NiCu(1:4)–CeO2 catalyst had higher stability than I–NiCu(1:4)/CeO2 and Cu–CeO2 catalysts, showing only a slight decrease in the CO conversion (85–80% for CO conversion after 45 h). This result proved the potential application of the catalyst's mesoporous architecture, which makes it highly active, and the doping of Ni stabilizes the NiCu(1:4)–CeO2 oxide during the HT-WGS reaction.
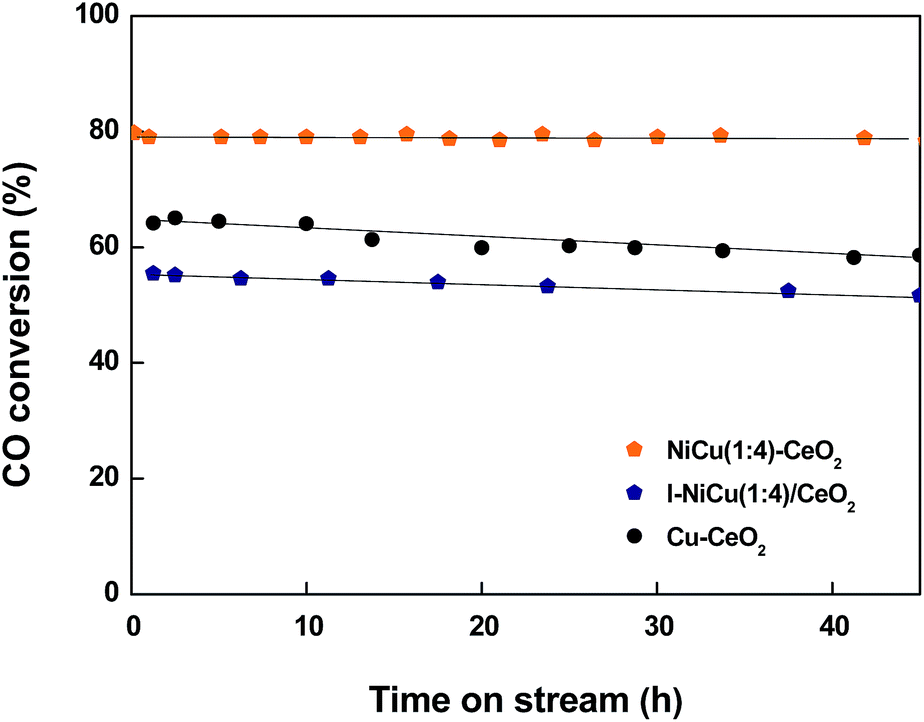 | ||
| Fig. 9 CO conversion with time on stream over NiCu(1:4)–CeO2, I–NiCu/CeO2 and Cu–CeO2 catalysts (H2O/(CH4 + CO + CO2) = 2.0; T = 450 °C; GHSV = 83665 h−1). | ||
Conclusions
A series of mesoporous NiCu–CeO2 oxides and CuO–CeO2 and NiO–CeO2 catalysts were prepared by the evaporation-induced self-assembly method using a non-ionic P123 template to understand the effect of mesoporosity on catalytic activity and selectivity towards the HT-WGS reaction. Among these catalysts, NiCu(1:4)–CeO2 was found to be more highly active than Cu–CeO2 and more selective than Ni–CeO2, NiCu(1:1)–CeO2 and NiCu(1:2)–CeO2 catalysts towards the WGS reaction. The higher selectivity of the NiCu(1:4)–CeO2 catalyst was due to the formation of Cu- rich surface that suppressed the methanation, which was also supported by the XPS and HR-TEM results. Activity comparison results between the NiCu–CeO2, I–NiCu–CeO2 and NiCu-oxide catalysts exhibited that the mesoporous NiCu–CeO2 was highly active compared to I–NiCu–CeO2 and NiCu-oxide catalysts. The higher activity of NiCu–CeO2 was due to the lowest reduction temperature and mesoporous architecture of the catalyst. The catalyst without ceria (NiCu-oxide) was least active and showed deactivation at higher temperature. N2-adsorption/desorption and SAXS measurements showed that CuO–CeO2 has a microporous nature, and doping of Ni increased the surface area as well as mesoporosity of the samples. H2-TPR of the NiCu(1:4)–CeO2 exhibited the lowest reduction temperature peak for the highly dispersed CuO compared to pure CuO–CeO2 catalysts, which could be the result of strong interaction between the metal oxide and CeO2. Furthermore, the NiCu(1:4)–CeO2 catalyst exhibited stable activity with 80% CO conversion after 45 h on stream at a high GHSV of 83665 h−1. The NiCu(1:4)–CeO2 catalyst should be considered a promising catalyst for the high-temperature WGS reaction.
Experimental section
Catalyst preparation
The mesoporous NiCu–CeO2 oxide catalysts were prepared by the evaporation-induced self-assembly method using triblock copolymers (EO)20(PO)70(EO)20, P123. A series of NiCu–CeO2 oxide catalysts were prepared by varying the weight percentage ratio of Ni/Cu from (1:1) to (1:4). The amount of CeO2 was fixed to 70 wt% in all cases. In a typical synthesis, solution “A” was prepared by adding 1.0 g of P123 in 10 ml of iso-butanol. Similarly, solution “B” was made with the combination of 10 ml of iso-butanol, 1.0 g P123 and corresponding nitrates precursors of copper and nickel. Both solutions A and B were allowed to stir separately for 1 h. Meanwhile, 4.32 g of Ce(NO3)3·6H2O was dissolved into 10 ml of iso-butanol and 1.6 ml of 67 wt% HNO3. Once dissolved it was added to solution A, and then solution B was also mixed with solution A. To get a homogeneous solution, the entire mixture solution was kept 5 h for stirring. After that the gel formed was kept at 120 °C for 48 h in an oven for solvent evaporation. The final catalyst NiCu–CeO2 was obtained after calcination at 500 °C for 5 h. The CuO–CeO2 and NiO–CeO2 were also prepared by the same method. The NiCu-oxide (without ceria) was prepared by using only solution “B,” keeping remaining conditions the same. NiCu(1:4)-oxide supported on ceria (I–NiCu/CeO2) was also prepared by the impregnation method. CeO2 used for this purpose was prepared by the precipitation method having a surface area of 117 m2 g−1.4
Catalyst characterization
X-ray diffractograms of the catalysts were recorded in the 2θ range of 20–80° using a Rigaku D/MAX-IIIC diffractometer (Ni-filtered Cu-Kα radiation, 40 kV, 100 mA). The crystallite size was estimated using the Debye–Scherrer equation.40,41 Small angle X-ray scattering (SAXS) was collected on the same instrument over a 2θ range of 0.3–5°. The BET surface area and the type of isotherm were determined by the N2 adsorption/desorption method at 77 K using an ASAP 2010 Micromeritics. Hydrogen-temperature programmed reduction (H2-TPR) experiments were conducted on an Autochem 2910 (Micromeritics). Typically, 0.1 g of sample was loaded into a quartz reactor. The H2-TPR was performed using 10% H2 in Ar with a heating rate of 10 °C min−1, from room temperature to 800 °C. The sensitivity of the detector was calibrated by reducing a known weight of NiO.42,43 Scanning electron microscopy (SEM) was carried out using JSM-7001F. Transmission electron microscopy (TEM) images were obtained via a JEOL JEM-2100F microscope.Catalytic activity measurement
Catalyst activity tests were performed from 350 to 550 °C at atmospheric pressure in a fixed-bed micro-tubular quartz reactor with an inner diameter of 4 mm. The catalyst charge was 0.035 g. A T-union was employed at the exit of the quartz reactor to install a thermocouple. A thermocouple was inserted into the catalyst bed to measure the reaction temperature. Prior to each catalytic measurement, the catalyst was reduced in 5% H2/N2 from room temperature to 400 °C at a heating rate of 4.6 °C min−1 and then the temperature was maintained for 1 h. Afterward, the temperature was decreased to 350 °C. The simulated reformed gas consisted of 17.02 vol.% CO, 9.55 vol.% CO2, 1.03 vol.% CH4, 13.14 vol.% H2, 55.20 vol.% H2O, and 4.06 vol.% N2, which represents a typical syngas from a waste gasifier that might enter the WGS reactor in a waste gasification system.44,45 The feed H2O/(CH4 + CO + CO2) ratio was intentionally fixed at 2.0 to avoid coke formation. The GHSV of 83665 h−1 was used to screen all catalysts. Water was fed using a syringe pump and vaporized at 180 °C upstream of the reactor. The product gas was chilled, passed through a trap to condense residual water, and then analyzed online using an Agilent micro-gas chromatograph.46
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2013R1A1A1A05007370). This work is financially supported by Korea Ministry of Environment (MOE) as “Knowledge-based environmental service (waste to energy recycling) human resource development project”.Notes and references
- L. Alibardi and A. Muntoni, Waste Manage. DOI:10.1016/j.wasman.2014.09.001.
- R. B. Mane, D.-W. Jeong, A. V. Malawadkar, H.-S. Roh and C. V. Rode, ChemCatChem, 2014, 6, 1698 CrossRef CAS.
- S. C. Ammal and A. Heyden, J. Catal., 2013, 306, 78 CrossRef CAS PubMed.
- D.-W. Jeong, H. S. Potdar, J.-O. Shim, W.-J. Jang and H.-S. Roh, Int. J. Hydrogen Energy, 2013, 38, 4502 CrossRef CAS PubMed.
- S.-C. Huang, C.-H. Lin and J.-H. Wang, J. Phys. Chem. C, 2010, 114, 9826 CAS.
- D. L. Klass, Biomass for renewable energy, fuels, and chemicals, Academic Press, San Diego, 1998 Search PubMed.
- C. Ludwig, S. Hellweg, S. Stucki, Municipal solid waste management, Springer-Verlag, Berlin, 2002 Search PubMed.
- V. Subramanian, E. S. Gnanakumar, D.-W. Jeong, W.-B. Han, C. S. Gopinath and H.-S. Roh, Chem. Commun., 2013, 49, 11257 RSC.
- P. Sudarsanam, B. Mallesham, D. N. Durgasri and B. M. Reddy, RSC Adv., 2014, 4, 11322 RSC.
- L. Katta, B. M. Reddy, M. Muhler and W. Grünert, Catal. Sci. Technol., 2012, 2, 745 CAS.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949 CrossRef CAS PubMed.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475 CAS.
- R. Farra, M. G. Melchor, M. Eichelbaum, M. Hashagen, W. Frandsen, J. Allan, F. Girgsdies, L. Szentmiklósi, N. López and D. Teschner, ACS Catal., 2013, 3, 2256 CrossRef CAS.
- N. Pal, E.-B. Cho and D. Kim, RSC Adv., 2014, 4, 9213 RSC.
- W. Song and E. J. M. Hensen, ACS Catal., 2014, 4, 1885 CrossRef CAS.
- M. A. Henderson, C. L. Perkins, M. H. Engelhard, S. Thevuthasan and C. H. F. Peden, Surf. Sci., 2003, 526, 1 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulo, Science, 2003, 301, 935 CrossRef CAS PubMed.
- M. Manzoli, G. Avgouropoulos, T. Tabakova, J. Papavasiliou, T. Ioannides and F. Boccuzzi, Catal. Today, 2008, 138, 239 CrossRef CAS PubMed.
- D.-W. Jeong, W.-J. Jang, J.-O. Shim, W.-B. Han, H.-S. Roh, U. H. Jung and W. L. Yoon, Renewable Energy, 2014, 65, 102 CrossRef CAS PubMed.
- P. Djinović, J. Batista and A. Pintar, Appl. Catal., A, 2008, 347, 23 CrossRef PubMed.
- G. C. de Araújo and M. do Carmo Rangel, Catal. Today, 2000, 62, 201 CrossRef.
- E. T. Saw, U. Oemar, X. R. Tan, Y. Du, A. Borgna, K. Hidajat and S. Kawi, J. Catal., 2014, 314, 32 CrossRef CAS PubMed.
- S. D. Senanayake, J. Evans, S. Agnoli, L. Barrio, T.-L. Chen, J. Hrbek and J. A. Rodriguez, Top. Catal., 2011, 54, 34 CrossRef CAS.
- J.-H. Lina, P. Biswas, V. V. Guliants and S. Misture, Appl. Catal., A, 2010, 387, 87 CrossRef PubMed.
- Y. H. Choi and W. Y. Lee, J. Mol. Catal. A: Chem., 2001, 174, 193 CrossRef CAS.
- Q. Wu, L. D. L. Duchstein, G. L. Chiarello, J. M. Christensen, C. D. Damsgaard, C. F. Elkjær, J. B. Wagner, B. Temel, J.-D. Grunwaldt and A. D. Jensen, ChemCatChem, 2014, 6, 301 CrossRef CAS.
- J. Kugai, J. T. Miller, N. Guo and C. Song, J. Catal., 2011, 277, 46 CrossRef CAS PubMed.
- P. Maitarad, J. Han, D. Zhang, L. Shi, S. Namuangruk and T. Rungrotmongkol, J. Phys. Chem. C, 2014, 118, 9612 CAS.
- V. M. Shinde and G. Madras, Appl. Catal., B, 2013, 132–133, 28 CrossRef CAS PubMed.
- P. Li, J. Liu, N. Nag and P. A. Crozier, J. Catal., 2009, 262, 73 CrossRef CAS PubMed.
- E. P. Fraccari, F. Mariňo, M. Laborde and G. Baronetti, Appl. Catal., A, 2013, 460–461, 15 CrossRef PubMed.
- P. Ratnasamy, D. Srinivas, C. V. V. Satyanarayana, P. Manikandan, R. S. Senthil Kumaran, M. Sachin and V. N. Shetti, J. Catal., 2004, 221, 455 CrossRef CAS PubMed.
- W.-P. Dow, Y.-P. Wang and Ta-J. Huang, J. Catal., 1996, 160, 155 CrossRef CAS.
- S. Storck, H. Bretinger and W. F. Maier, Appl. Catal., A, 1998, 174, 137 CrossRef CAS.
- K. Kaneko, J. Membr. Sci., 1994, 96, 59 CrossRef CAS.
- T. X. T. Sayle and D. C. Sayle, J. Am. Chem. Soc., 2014, 136, 4056 CrossRef CAS PubMed.
- P. I. Ravikovitch, D. Wei, W. T. Chueh, G. L. Haller and A. V. Neimark, J. Phys. Chem. C, 1997, 101, 3671 CrossRef CAS.
- F. Zhao, Z. Liu, W. Xu, S. Yao, A. Kubacka, A. C. Johnston-Peck, S. D. Senanayake, A.-Q. Zhang, E. A. Stach, M. F. García and J. A. Rodriguez, J. Phys. Chem. C, 2014, 118, 2528 CAS.
- D.-W. Jeong, H.-S. Na, J.-O. Shim, W.-J. Jang, H.-S. Roh, U. H. Jung and W. L. Yoon, Int. J. Hydrogen Energy, 2014, 39, 9135 CrossRef CAS PubMed.
- D.-W. Jeong, H. S. Potdar and H.-S. Roh, Catal. Lett., 2012, 142, 439 CrossRef CAS.
- W.-J. Jang, D.-W. Jeong, J.-O. Shim, H.-S. Roh, I. H. Son and S. J. Lee, Int. J. Hydrogen Energy, 2013, 38, 4508 CrossRef CAS PubMed.
- D.-W. Jeong, W.-J. Jang, J.-O. Shim, H.-S. Roh, I. H. Son and S. J. Lee, Int. J. Hydrogen Energy, 2013, 38, 13649 CrossRef CAS PubMed.
- D.-W. Jeong, W.-J. Jang, J.-O. Shim, W.-B. Han, K.-W. Jeon, Y.-C. Seo, H.-S. Roh, J. H. Gu and Y. T. Lim, J. Mater. Cycles Waste Manage., 2014, 16, 650 CrossRef CAS PubMed.
- D.-W. Jeong, V. Subramanian, J.-O. Shim, W.-J. Jang, Y.-C. Seo, H.-S. Roh, J. H. Gu and Y. T. Lim, Catal. Lett., 2013, 143, 438 CrossRef CAS.
- D.-W. Jeong, W.-J. Jang, J.-O. Shim, W.-B. Han, H.-M. Kim, Y.-L. Lee, J. W. Bae and H.-S. Roh, Renewable Energy DOI:10.1016/j.renene.2014.07.041.
Footnote |
| † Electronic supplementary information (ESI) available: H2-TPR patterns, surface composition, and WGS reaction results. See DOI: 10.1039/c4ra13142h |
| This journal is © The Royal Society of Chemistry 2015 |