
Modified strontium titanates: from defect chemistry to SOFC anodes
M. C. Verbraeken*a,
T. Ramosb,
K. Agerstedb,
Q. Mac,
C. D. Savaniua,
B. R. Sudireddyb,
J. T. S. Irvinea,
P. Holtappelsb and
F. Tietzc
aSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: mcv3@st-andrews.ac.uk
bDTU Energy Conversion and Storage, Technical University of Denmark, Frederiksborgvej 399, DK-4000 Roskilde, Denmark
cForschungszentrum Jülich GmbH, Institut für Energieforschung (IEK-1), DE-52425 Jülich, Germany
First published on 26th November 2014
Abstract
Modified strontium titanates have received much attention recently for their potential as anode material in solid oxide fuel cells (SOFC). Their inherent redox stability and superior tolerance to sulphur poisoning and coking as compared to Ni based cermet anodes could improve durability of SOFC systems dramatically. Various substitution strategies can be deployed to optimise materials properties in these strontium titanates, such as electronic conductivity, electrocatalytic activity, chemical stability and sinterability, and thus mechanical strength. Substitution strategies not only cover choice and amount of substituent, but also perovskite defect chemistry, distinguishing between A-site deficiency (A1−xBO3) and cation-stoichiometry (ABO3+δ). Literature suggests distinct differences in the materials properties between the latter two compositional approaches. After discussing the defect chemistry of modified strontium titanates, this paper reviews three different A-site deficient donor (La, Y, Nb) substituted strontium titanates for their electrical behaviour and fuel cell performance. Promising performances in both electrolyte as well as anode supported cell designs have been obtained, when using hydrogen as fuel. Performances are retained after numerous redox cycles. Long term stability in sulphur and carbon containing fuels still needs to be explored in greater detail.
1. Introduction
Due to their ability to directly convert chemical energy into electricity solid oxide fuel cells (SOFC) offer an efficient alternative to combustion technology.1 Their high operating temperatures offers the option of utilising exhaust heat in gas turbines or for combined heat/electricity generation.2 Additionally, the high operating temperatures make SOFC fuel flexible, with fuels ranging from hydrogen to various gaseous as well as liquid hydrocarbons (e.g. methane, butane, ethanol, LPG, etc.).3 The good scalability, from stacks producing a few kW up to several MW, makes this technology very flexible in terms of its applications, ranging from decentralised domestic electricity and heat generation to power plant scale energy production.2,4,5Due to their excellent performance in hydrogen fuel, Ni/YSZ cermets are the state-of-the-art anodes for both electrolyte as well as anode supported cell designs. However, they still suffer from some drawbacks, such as Ni agglomeration at high temperatures,6 coking when using hydrocarbon fuels under low steam to carbon ratios,7 sulphur poisoning,8,9 and especially instability upon redox cycling (cyclic reduction and oxidation).10 These provide the motivation for developing an alternative, more robust anode. In this light, ceramic anodes have received increased interest, as they are expected to be less prone to the typical issues associated with Ni based anodes. Perovskite materials containing transition metals (e.g. Ti, Nb, V, Co, Mn, Mo, etc.) are of particular interest due to the availability of multiple oxidation states, higher tolerance to coking and poisoning, and good dimensional stability upon redox cycling.11 These types of materials have inherently limited electronic conductivity however, which is a major drawback. Currently it is believed that the minimum requirement for electronic conductivity in SOFC anode materials should be at least 10–100 S cm−1; this may have to be even higher when using an anode supported cell design.12,13
Within the vast family of perovskite materials, SrTiO3-based compounds have attracted particular attention over the past years, due to their decent n-type conductivity and good stability in various atmospheres. This work aims to review the latest developments in material design of such SrTiO3 based anodes, with special emphasis on substitution with lanthanum (LST), yttrium (YST) and niobium (STN), covering important topics such as defect chemistry, electrical properties, as well as their performance as potential SOFC anode materials.
2. Defect chemistry of modified SrTiO3
The effectiveness of ceramic electrodes in SOFC depends on an intricate interplay between the material's crystal structure and defect chemistry. Especially materials containing transition metals are of interest, since the availability of multiple oxidation states can facilitate bulk transport properties, and provide mechanisms for electrocatalytic processes. For example, under reducing atmospheres the transition metal ions change to lower oxidation states, effectively creating electronic carriers to pass current. Also, the formation of oxygen vacancies on reduction is likely to yield an increase in the oxide ion conductivity according to eqn (1), in Kröger–Vink notation:
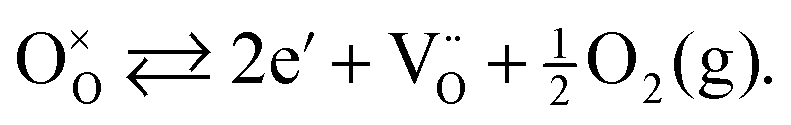 | (1) |
Perovskite materials have received much attention as potential anodes for SOFC, due to their adaptable defect chemistry through compositional tailoring. In particular aliovalent substitution, i.e. exchange of ABO3 cations with similar sized cations of different valence, is an effective way to introduce defects, control defect concentrations and hence tailor anode functionality. For instance, A-site vacancies can be introduced by substitution of A-site ions with cations of higher valence (donors) giving compounds of stoichiometry of A(1−x−y)A′xBO3, while the substitution of A-site ions with lower valence cations (acceptors) could be used to introduce oxygen vacancies, giving compounds of stoichiometry A(1−x)A′xBO(3−δ) if the B-site valence remains constant.14 Alternatively, acceptor or donor-substitution can also be performed on the B-site, with the most important criterion for a possible substitution of A- or B-site ions by dopants being the ionic radius of the considered species.
Strontium titanate (SrTiO3) is an excellent example of a perovskite exhibiting a wide range of defect chemistry that can illustrate the influence of different factors on conductivity. Due to the reducibility of Ti4+ to Ti3+ it exhibits n-type semiconducting behaviour in reducing atmospheres. Introducing donor and/or acceptor defects has shown to be an effective way of modifying the oxygen and cation content in the perovskite structure, thereby altering the material's conductivity behaviour under various atmospheres.15,16
To fully appreciate the various defect chemistries in modified SrTiO3 and their effect on the material's electrical properties, it is important to first discuss the effect of donor and acceptor substitution under oxidising conditions.
Cation and oxygen stoichiometric SrTiO3
Due to different defect chemistries achievable in strontium titanates, ambiguities arise in literature as to the nature of the non-stoichiometry, since this is not always evident from the composition alone. For this reason, Savaniu and Irvine have proposed a new oxide nomenclature to describe these perovskite phase compositions.17 The oxides are represented using normal case capital letters corresponding to the present cations, the oxide composition being normalized to one cation per formula unit, e.g. LaO1.5 → L, SrO → S, TiO2 → T, NbO2.5 → N, etc. The deviation from the ABO3 stoichiometry is marked as a final subscript, for example A- for A-site deficiency, O+ for oxygen excess and so on.Many reports and reviews on modified SrTiO3 focus on cation-stoichiometric perovskites. This is an important distinction (as compared to A-site deficient perovskites), which is sometimes overlooked in literature. Acceptor (A) or donor (D) substitution may affect the oxygen stoichiometry according to eqn (2) and (3) (substitution arbitrarily chosen on B- and A-site, respectively). It has been argued that interstitial oxygen is highly unlikely in the densely packed perovskite lattice.15 Therefore the excess oxygen is rather expected to be accommodated in secondary SrO phases or oxygen rich extended defects.18–20 In turn it has generally been accepted that eqn (4) is more correct in these materials, whereby the positive charge of the donor is compensated by strontium vacancies.
 | (2) |
 | (3) |
 | (4) |
McColm and Irvine noticed that acceptor substitution on the B-site in fact results in a decrease in conductivity under reducing atmosphere.16 However, a marked improvement of the p-type conductivity under oxidising conditions was noticed, suggesting that holes are the predominant compensation under these conditions. This can be explained by eqn (5), where intrinsic oxygen vacancies (created due to eqn (2)) are filled with ambient oxygen generating a pair of holes. It seems therefore that donor-substitution of SrTiO3 is the preferred strategy for making superior SOFC anode materials with good electrical properties under reducing conditions.
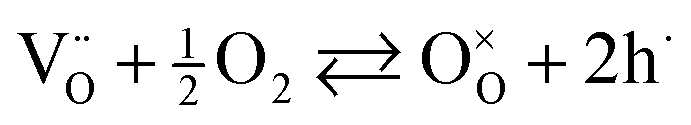 | (5) |
Cation-stoichiometric titanates with nominal oxygen excess (e.g. LSTO+) are of remarkable interest due to their very high electronic conductivity, the presence of extended defects rich in oxygen and high stability in reducing conditions.18,21 The presence of extra oxygen beyond the normal stoichiometry increases the ease and amount of reduction of Ti4+ to Ti3+, in turn increasing the conductivity. The oxygen excess plays a critical role in both the structural and electrochemical properties, although this is often ignored in the literature. On consideration of strontium titanate solid state chemistry, it is likely that for compositions such as LaxSr1−xTiO3+δ, δ is generally positive and will only be nil or negative under very reducing conditions, probably more reducing than the actual fuel cell operating conditions. Flandermeyer et al.22 calculated, using thermogravimetric measurements, that up to an extra x/2 moles of oxygen can be accommodated by LaxSr1−xTiO3+δ formula. It has been shown that the presence of disordered oxygen-rich defects affects to a great extent the redox characteristics of an oxide as indicated by very significant changes in conductivity under certain conditions.18,23 The conductivity data indicate that oxygen excess compositions are more conductive than the ones presenting oxygen deficiency, at the same oxygen partial pressure.24,25
Alternative to being cation-stoichiometric, strontium titanates which are stoichiometric in oxygen have been developed.26 Here A-site cation vacancies are purposely introduced into the material's composition, which will now adopt the general formula XySr1−3/2yTiO3, with X being a trivalent cation. Once again, strontium vacancies are the main compensating mechanism for the introduced donor, according to eqn (4). However, due to the large concentration of  , the formation of intrinsic Schottky defects is now being reduced, pushing eqn (6) to the left and reducing
, the formation of intrinsic Schottky defects is now being reduced, pushing eqn (6) to the left and reducing 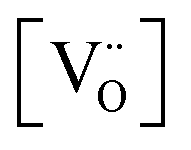 . At any given oxygen partial pressure, eqn (1) now shifts to the right, facilitating oxygen removal from the perovskite lattice associated with the generation of free electrons.
. At any given oxygen partial pressure, eqn (1) now shifts to the right, facilitating oxygen removal from the perovskite lattice associated with the generation of free electrons.
 | (6) |
Titanium reduction
To produce highly conductive anode materials based on modified SrTiO3, elevated temperatures and reducing atmospheres are required, either during the material preparation or prior to testing. A comprehensive review of the defect chemistry of cation-stoichiometric donor-substituted strontium titanates has been given by Moos and Härdtl.27 Fig. 1 shows the conductivity behaviour for this type of donor-substituted SrTiO3 versus pO2 for two different temperatures, T1 and T2. As mentioned earlier, in cation-stoichiometric donor-modified SrTiO3, the extra positive charge of the donor is either compensated by oxygen excess, or by A-site cation vacancies (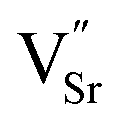 ). This donor compensation mechanism means that under mildly reducing conditions, Ti reduction is compensated by lowering of the oxygen excess or concentration of strontium vacancies, probably according to eqn (7). The SrO in this equation is likely to originate from a Ruddlesden-Popper phase or grain terminations.
). This donor compensation mechanism means that under mildly reducing conditions, Ti reduction is compensated by lowering of the oxygen excess or concentration of strontium vacancies, probably according to eqn (7). The SrO in this equation is likely to originate from a Ruddlesden-Popper phase or grain terminations.
 | (7) |
 | ||
| Fig. 1 Log–log plots of the conductivity of modified SrTiO3 versus oxygen partial pressure (pO2) representing donor-modified SrTiO3, with slopes of −1/6, 0, and −1/4 in the low, intermediate, and high pO2 ranges, respectively.27 (Copyright © 2005, John Wiley and Sons.) | ||
It can easily be derived that under these conditions a pO2−1/4 dependence of the conductivity is to be expected. At intermediate pO2 values, extrinsic donor concentration,  dominates and conductivity is now dependent only on the donor concentration and the temperature-dependent mobility, but not on pO2. This region is therefore often called the “plateau region”, or the region of electronic compensation.
dominates and conductivity is now dependent only on the donor concentration and the temperature-dependent mobility, but not on pO2. This region is therefore often called the “plateau region”, or the region of electronic compensation.
It is only at very low pO2 values, where oxygen vacancies become the predominant ionic defect (eqn (1)), giving rise to a pO2−1/6 dependence of the conductivity. In terms of lattice oxygen stoichiometry, at high pO2 there will be excess oxygen, i.e. A′1−xA′′xBO3+δ, with δ > 0; in the plateau region the perovskite will be oxygen stoichiometric, i.e. δ = 0; only at very low pO2 will δ become negative. Depending on composition and substituents, the pO2 at which δ is negative may even be beyond SOFC operating conditions.
In other words, for cation-stoichiometric donor substituted SrTiO3, titanium reduction is either compensated by a change in strontium vacancy concentration in oxidising atmospheres, or by the formation of oxygen vacancies under very reducing conditions. The substitution strategy and donor concentration determines the initial strontium vacancy concentration, effectively enhancing the reducibility of titanium, thereby increasing the charge carrier concentration.
The situation is expected to be different for A-site deficient donor substituted strontium titanates, owing to a different oxygen stoichiometry. Since there is no oxygen excess, titanium reduction should primarily be compensated through eqn (1), i.e. the formation of oxygen vacancies and the pO2−1/4 dependence of conductivity should therefore be absent. Slater et al.26 indeed describe a pO2−1/6 conductivity behaviour for A-site deficient La substituted SrTiO3. Plateau regions have been reported for A-site deficient Nb modified SrTiO3, but this could be due to slow reduction kinetics in dense samples.28
One of the drawbacks of using A-site deficient strontium titanates is the large driving force for phase segregation of TiO2 under reducing conditions.29 Although this behaviour seems mainly driven by non-stoichiometry, several authors observed segregation during sintering and cooling of cation-stoichiometric compounds as well.30–32 This may be ascribed to formation of space-charge potentials in individual grains as function of differences in individual defect formation energies.33 Whereas extrinsic acceptor dopants tend to segregate at grain boundaries already under oxidising conditions, donor dopants have recently also been found to show this behaviour when sintered under reducing atmospheres.33,34 Results particularly suggest formation of a double space-charge layer, which results in an increased B/A-cation ratio ±10 nm around the grain boundaries.
It is furthermore found by several people, that A-site deficient donor substituted titanates have superior sintering characteristics and thus mechanical strength over their cation-stoichiometric counterparts.35,36 This is most likely due to the formation of SrO rich phases in cation-stoichiometric titanates, hindering grain growth. Instabilities on reduction–oxidation (redox) cycles have also been found for cation-stoichiometric titanates, which can also be related to the formation/dissolution of these secondary phases.
As the cation mobility is very low in strontium titanates27,37 structural and compositional changes in the bulk phases during short-term repeated oxidation–reduction cycles are expected to be negligible. In contrast, grain boundary phases seem much more sensitive to variations of the pO2, as their conductivity seems closely related to the more mobile oxygen vacancies, and accumulated B-site acceptors. Hence, to some extent, it may only be possible to form the desired bulk structure at high temperature during sintering, and then regard it as “frozen”. From a processing point of view, the Sr-deficiency is desirable as it improves mechanical properties of the material by reducing the formation of “SrO” phases.35 This should also help avoiding formation of insulating SrZrO3 phases. However, A-site deficiency does lead to increased possibility of TiO2 segregation into grain boundaries during sintering, especially under reducing atmospheres as stated earlier. Moreover, Burnat et al. recently published a report clearly showing Ti diffusion into common zirconia based electrolyte, when A-site deficient La substituted strontium titanates are used.38 Here diffusion seems much more pronounced in samples fired in air however, whereas firing under reducing conditions seems an effective way to limit this diffusion phenomenon. Similar behaviour was found for heavily A-site deficient Y-substituted SrTiO3.39
The focus in the following sections is on three materials which are all donor substituted A-site deficient perovskites, i.e. La, Y and Nb modified SrTiO3. Despite the fact that La and Y substitute on the perovskite A-site, whilst Nb modifies the B-site, their defect chemistry should be equivalent. Some comparisons will however be drawn with compositions that are cation stoichiometric to illustrate the effect of different defect chemistry on especially electrical properties and redox stability.
3. Electrical properties of modified strontium titanates
As reported by several authors,19,40,41 the conductivity of strontium titanate based materials strongly depends on the thermal history of the sample. Studies on the defect chemistry of substituted SrTiO3 (with La, Nb or Y as typical substituent) demonstrate that the charge compensation mode strongly depends on the oxygen partial pressure. The effects of changing oxygen partial pressure upon unmodified and differently modified strontium titanates are shown in Fig. 2, in which we can observe the p-type behaviour of the unmodified sample and the extended p-type behaviour of the acceptor modified sample at higher pO2, as well as the very high n-type conductivity of the A-site deficient sample at lower pO2 values.16,26 | ||
| Fig. 2 Conductivity variation for SrTiO3 in different substitution scenarios. Solid line: A-site deficient donor substituted; squares: unmodified; diamonds: acceptor substituted.16,17 With kind permission from Springer Science and Business Media (original caption: SrTiO3 and SrTi0.95Mg0.05O2.95: conductivity variation with oxygen partial pressure at 930 °C). | ||
Since the thermal and atmospheric history of (modified) SrTiO3 samples has such a large influence on the measured materials properties and in particular the electrical properties, attention to sample preparation is critical. The importance of firing conditions is often overlooked in literature. For A-site deficient LST, YST and STN sintered in air, changes in electrical conductivity as function of the oxygen partial pressure have been observed to occur only very slowly at temperatures below approximately 1000 °C;17,28,42,43 this has been ascribed to the afore mentioned slow cation diffusion in the crystal lattice. In spite of somewhat contrasting reports in the literature on this matter, it may therefore be expected that once an electrical conductivity has been established through pre-reduction of dense air sintered samples, this conductivity will stay virtually unchanged with redox cycling at temperatures below 1000 °C.44 The behaviour of porous samples may however differ from this, as diffusion lengths in the bulk will be smaller and changes in the bulk conductivity might be observed with changing pO2. In situ reduction is certainly desirable from a processing point of view, as it eliminates a potentially expensive preparation step. In this review, we therefore present results on both pre-reduced samples as well as samples reduced in situ at SOFC temperatures, along with their redox behaviour. Also both dense and porous samples will be discussed, the latter being more relevant for SOFC anode applications.
Electrical properties of pre-reduced donor-modified SrTiO3
Firstly, to show the effect of reductive sintering, the electrical properties of Y substituted SrTiO3 (YST) will be discussed. The samples discussed have been sintered under reducing conditions (4% H2/96% argon, at 1400 °C) in order to try and ‘freeze’ in the metallic bulk properties.39,45,46 Subsequently, the electrical properties of pre-reduced Nb and La substituted SrTiO3 will be presented, including the effect of redox cycling. | ||
| Fig. 3 Temperature dependence of the electrical conductivity in Ar/4% H2/3% H2O for samples with the general formula Sr1−xY0.07TiO3−δ ranging from x = 0.13 to x = 0.01 (Sr87 to Sr99) sintered in Ar/4% H2 at 1400 °C for 5 h. Reprinted from Q. L. Ma, F. Tietz and D. Stover, Nonstoichiometric Y-substituted SrTiO3 materials as anodes for solid oxide fuel cells, Solid State Ionics, 2011, 192, 535–539, with permission from Elsevier.46 | ||
According to Mott's theory,47 if the concentration of electrons in the conduction band – which means the concentration of Ti3+ in the present materials – exceeds a critical value, a transition from semiconducting to metal-like behaviour may take place. A-site deficient samples are expected to have a higher number of charge carriers (Ti3+) through eqn (1) and (6). On the atomic scale, A-site deficiency will weaken the bond strength of the adjacent TiO6 octahedra, which facilitates the reduction of Ti.40 This may well be the main reason for the low conductivity and semiconducting behaviours of B-site deficient samples. And quite clearly, A-site deficient samples can reach the conductivity requirement for anode supports in SOFC.
In order to compensate for the low ionic conductivity of YST materials, YSZ can be added to form a composite, similarly to what is routinely done on Ni/YSZ cermets. However, Ti has been found in the YSZ electrolyte after co-sintering with an anode substrate made of Sr0.89Y0.07TixO3−δ (YST).39,45 It is known that Ti additions to YSZ increase unwanted electronic conduction in the electrolyte,48,49 which will be discussed in next paragraphs. And moreover, the electrical properties of YST in the YST–YSZ anode functional layer are also different from those of pure YST. Because of the Ti diffusion into the electrolyte, the stoichiometry of YST in YST–YSZ may change from A-site deficiency to B-site deficiency, which will result in a decrease of conductivity, as previously mentioned. Based on this assumption, YST samples with additional TiO2 were also prepared. Fig. 4 shows the conductivity of Sr0.89Y0.07TixO3−δ/YSZ mixtures (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume ratio, YSTx–YSZ, x = 1.00–1.20) in humidified (3% H2O) 4% H2/96% argon atmospheres. As can be seen, the conductivity increases with increasing x value in YSTx–YSZ up to x = 1.10, and this composition was found suitable for an anode functional layer for YST-based SOFC.
:1 in volume ratio, YSTx–YSZ, x = 1.00–1.20) in humidified (3% H2O) 4% H2/96% argon atmospheres. As can be seen, the conductivity increases with increasing x value in YSTx–YSZ up to x = 1.10, and this composition was found suitable for an anode functional layer for YST-based SOFC.
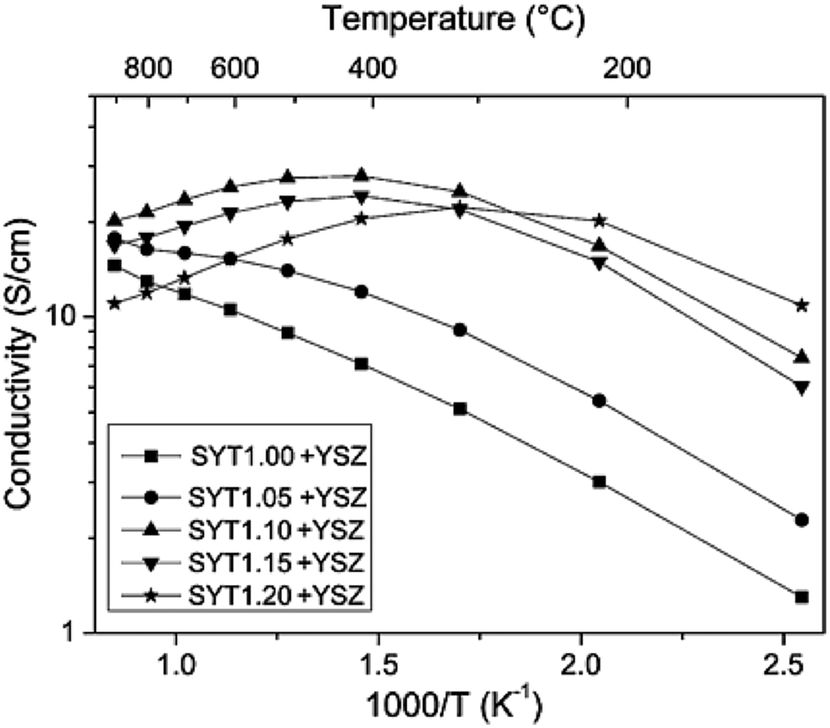 | ||
| Fig. 4 Electrical conductivity in Ar/4% H2/3% H2O of samples with the general formula Sr0.89Y0.07TixO3−δ with 1.0 < x < 1.2, and mixed with YSZ in volume ratio 2:1. Reprinted from Q. Ma, F. Tietz, D. Sebold and D. Stover, Y-substituted SrTiO3–YSZ composites as anode materials for solid oxide fuel cells: Interaction between YST and YSZ, Journal of Power Sources, 2010, 195, 1920–1925, with permission from Elsevier.39 | ||
 | ||
| Fig. 5 Measured points of electrical conductivity for bulk STN94, overlaid with schematic graphs from modeling results.28 | ||
First measurements on pre-reduced porous electrode backbones were mainly conducted at 1000 °C, and at oxygen partial pressures in the range of 0.21–10−18 atm at this temperature. The backbones were applied onto pre-sintered YSZ, sintered at 1250 °C in air and afterwards re-reduced at 1250 °C for 3 hours in 9% H2/91% argon. During testing, samples were kept at each pO2-step for >30 h in order to allow the sample to equilibrate. The measurements started in the most reducing environment (wet H2, pO2 ≈ 10−18 atm). The pO2 was then increased stepwise up to air, and then back to reducing environment. The results are shown in Fig. 6.
 | ||
| Fig. 6 Conductivity of printed STN94 electrodes with different layer thicknesses at 1000 °C and different pO2. Each data point was recorded after at least 30 h equilibration time at 1000 °C. | ||
At low pO2, the conductivity values of the porous samples correlate fairly well with the conductivity measured on dense samples, though not being corrected for porosity. However, where conductivity of the dense samples seemed independent of pO2, conductivity of porous samples show a slope close to −1/6 as expected for A-site deficiency, as shown by the lines in Fig. 6. Although only indicative, conductivity does not seem to fully recover on re-reduction of the samples, as also observed previously by Kolodiazhnyi and Petric.19 This hypothesis is further supported by the conductivity measurements performed on porous STN94 samples that received different reduction profiles. One set of samples was pre-reduced at 1250 °C for 4 h in 9% H2/91% argon, whereas another set was reduced in situ, during testing at 850 °C. Conductivity started at ∼60 S cm−1 and ∼10 S cm−1 for the pre-reduced and the in situ reduced samples, respectively, and seems to respond with changing oxygen partial pressure fairly uniformly among the two tested sets. None of the samples regain the initial conductivity on re-reduction, with the pre-reduced material suffering the largest degree of degradation, as both sets ended up at 6–8 S cm−1 when brought again to low pO2 from atmospheric conditions. Inspection by SEM did not reveal any post-test changes of the STN94 microstructures.
The effect of defect chemistry on redox properties is shown by a direct comparison between STN94 (Sr0.94Ti0.9Nb0.1O3) and STN99 (Sr0.99Ti0.9Nb0.1O3), the latter of which has a much decreased A-site deficiency. STN99 samples, sintered under reducing atmosphere, showed an even stronger dependence on the environmental and thermal history, as may be deduced from conductivity results in Fig. 7, where STN99 powders were calcined in air (1300 °C/4 h) and sintered in 9% H2/91% argon (1450 °C/10 h) under identical conditions, but where one of the powders was reduced at 1300 °C (10 h) prior to sintering. The STN94 powder was also sintered similarly, under reducing conditions. All samples were sintered to near full density. The fact that STN94 after sintering in reducing atmosphere reaches conductivity levels close to STN99 samples is interesting from a point of how fast equilibrium establishes in STN-materials on sintering, as different conductivity values might have been expected considering the powder stoichiometry.
 | ||
| Fig. 7 DC conductivity behaviour of STN99 and STN94, all sintered to nearly full density at 1450 °C for 10 h in reducing atmosphere (9% H2/91% Ar). “STN99 Pre-reduced” was furthermore calcined under reducing conditions. | ||
Measuring DC-conductivity on sintered STN99 based half-cells shows a surprisingly low redox-stability, as conductivity drops almost two decades on redox-cycling from the initial value of ∼100 S cm−1 at 850 °C. In line with all the pre-reduced STN compounds, the STN99 half-cell showed metallic conductivity, however with a lower transition temperature. The maximum conductivity at 850 °C in highly reducing conditions was about 100 S cm−1. This conductivity decreased drastically with increasing pO2 reaching less than 0.01 S cm−1 at pO2 = 10−5 atm. Upon a new decrease in the pO2, regaining of conductivity was rather slow, and the maximum obtained at 10−24 atm was ∼2.5 S cm−1. After the test, the cell was partly cracked, but conductivity readings did not show discontinuity, and the cracking may not completely destroy the mechanical integrity of the sample. Due to the near cation-stoichiometry in STN99, cracking may have resulted from the formation of secondary SrO phases on oxidation, such as Ruddlesden Popper phases incorporated into the perovskite structure, and subsequent segregation of Nb and Ti-oxide on on reduction.19
The conductivity as a function of pO2 was also measured for a STN99 porous single layer, sintered at 1300 °C for 4 h in 9% H2/91% argon at 850 °C, and a value of 140 S cm−1 was obtained at 10−20 atm. The pO2 was then cycled between 10−19 atm and 10−12 atm, where the conductivity reverted almost to its initial level. However, after subjecting the sample to a pO2 value of ∼10−5 atm, the STN99 layer was unable to regain the original conductivity initially exhibited at 10−20 atm. Internal cracking was also seen for this sample.
 | ||
| Fig. 8 Electrical conductivity for pre-reduced dense samples of La0.2Sr1−xCaxTiO3 at 900 °C and pO2 = 10−19 atm (reproduced from ref. 50). | ||
During SOFC operation it should be expected that redox cycles to near atmospheric conditions occur on several occasions during a system's lifespan. The above results show that pre-reduction of donor-modified SrTiO3 samples only guarantees superior conductivity values when the samples are kept within a narrow pO2 range, i.e. ∼10−20 to 10−10 atm. These conductivity values cannot be recovered when the samples have been exposed to air (pO2 = 0.2 atm).
Electrical properties of in situ reduced donor-modified SrTiO3
In this section, a brief overview will be given for the properties of materials which have been reduced in situ. Because of the limited reduction kinetics of modified strontium titanates at SOFC operating conditions (800–900 °C), few papers actually report conductivity values obtained in this way. Some examples of A-site deficient La substituted SrTiO3 will be discussed and a brief comparison with pre-reduced conductivity values will also be given to show the effect of thermal history, which may help understand the large range of reported conductivities and apparent discrepancies found in literature.The conductivity of dense in situ reduced LaxSr1−3x/2TiO3 samples, where x = 0.2–0.6, was studied in detail by Slater et al.26 Highest conductivities of 7 S cm−1 were reported for x = 0.6 at 930 °C and pO2 = 10−20 atm. after equilibrating for 24 hours. Similar values are reported by Neagu and Irvine36 on porous samples (62% TD), with x = 0.4. Despite the porosity, equilibration is still slow and takes more than 20 hours. Savaniu and Irvine report conductivity values of ∼3 S cm−1 for both dense and porous La0.2Sr0.7TiO3 samples, in identical atmosphere and after similar equilibration times.17 Pre-reduction of these samples at 1050–1100 °C (in 5% H2/95% argon), increases their conductivity by nearly an order of magnitude, i.e. 20–25 S cm−1.
Both Slater and Savaniu report an approximate pO2−1/6 dependency of the conductivity at pO2 < 10−15 atm, suggesting an oxygen vacancy compensated mechanism, according to eqn (1). This is to be expected in A-site deficient strontium titanates as explained in the defect chemistry section. The formation of oxygen vacancies might furthermore be beneficial for its application as SOFC anode material, as it may create some oxygen ion mobility, thus increasing the thickness of the active anode for fuel oxidation.
Neagu and Irvine also showed that substitution on the B-site with gallium (La0.4Sr0.4Ti1−xGaxO3−x/2, x < 0.15), improves both the conductivity in oxidising as well as reducing conditions. It is suggested that Ga enhances the reduction of Ti, due to weakening of the BO bonds. This also leads to greatly improved reduction kinetics at any given temperature. However, some Ga loss was observed with time, due to the volatility in the form of Ga2O and GaOH species. Fig. 9 shows the variation of conductivity of La0.4Sr0.4Ti1−xGaxO3−x/2 with time at 880 °C and pO2 = 10−18 atm.
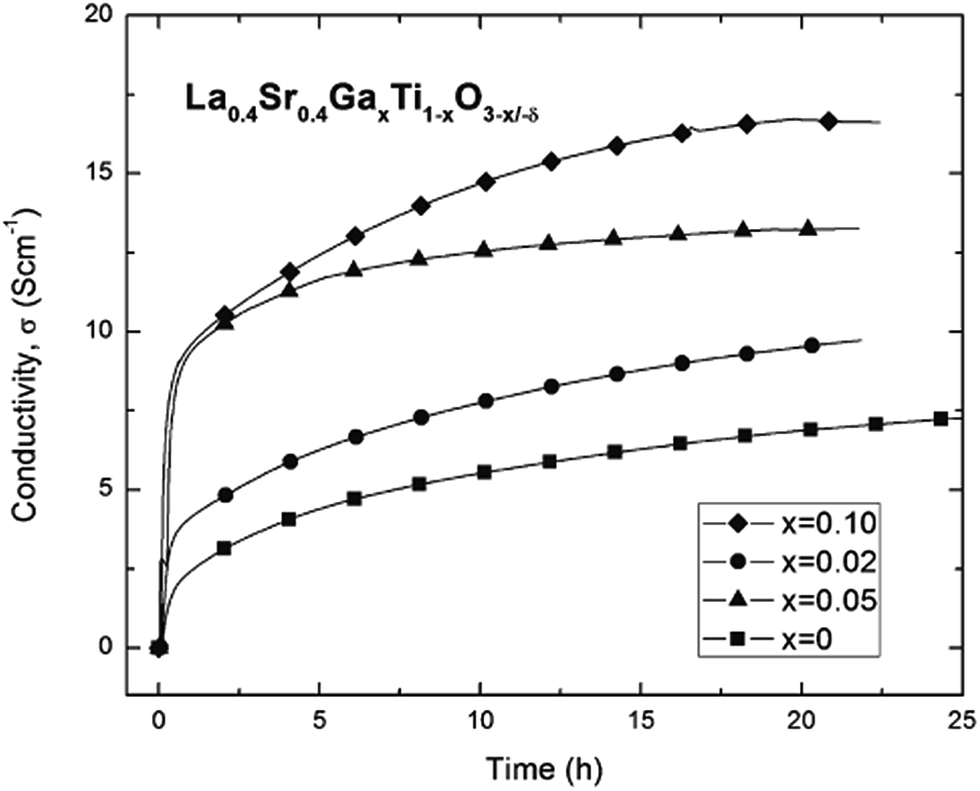 | ||
| Fig. 9 Conductivity of in situ reduced La0.4Sr0.4Ti1−xGaxO3−x/2 at 880 °C and pO2 = 10−18 atm.51 Reprinted with permission from “D. Neagu and J. T. S. Irvine, Chemistry of Materials, 2011, 23, 1607–1617”. Copyright 2011 American Chemical Society. | ||
In situ reduction at 850 °C in 5% H2/95% argon of porous La0.2Sr1−xCaxTiO3 electrodes with x = 0.45 has resulted in conductivity values in excess of 5 S cm−1.52 As mentioned earlier, pre-reduction of dense samples at 1050 °C results in conductivity values of 28 S cm−1 at 900 °C. Yaqub et al. performed redox cycles on in situ (880 °C, 5% H2/95% argon) reduced porous bars with the same composition (La0.20Sr0.25Ca0.45TiO3).53 The porosity and lack of pre-reduction results in a drop of conductivity of approximately an order of magnitude, but constant conductivity values are obtained on redox cycling between pO2 = 10−17 to 0.21 atm, with no sign of sample degradation or mechanical failure. The authors further noticed a marked increase in the redox kinetics, when impregnating the bars with ceria. Whilst un-impregnated bars required ∼3 hours of reduction to attain reasonable conductivity values, upon ceria impregnation this time reduced by about 50%.
4. Fuel cell performance of A-site deficient La, Y and Nb substituted SrTiO3
This review is concluded with a short overview of the use of said titanate materials in SOFC anodes. Where available, the effect of A-site deficiency on important stability aspects, such as redox cycling and mechanical robustness, will be described.LSTA- based anodes
A comprehensive review was recently written on the fuel cell performance of many La substituted SrTiO3 based anode materials by Zhou et al.54 Here we limit ourselves to the performance of some A-site deficient lanthanum substituted strontium titanates.Savaniu and Irvine showed promising fuel cell results for both electrolyte and anode supported cell (ESC and ASC, respectively) designs comprising La0.2Sr0.7TiO3 (LSTA-) anodes.17 Using LSTA- as the anode backbone impregnated with nanosized metal (oxide) catalysts (CeO2 and Cu), they achieved good performance using humidified hydrogen at temperatures as low as 600 °C. Power densities in excess of 0.4 W cm−2 were obtained at 750 °C in an anode supported cell configuration, as shown in Fig. 10. They noticed that for both cell designs, performance was limited by ohmic losses, originating from both the electrolyte as well as electrodes. Fig. 11 shows the impedance spectra for the anode supported cell at different temperatures. To achieve these performances at relatively low temperatures for SOFC, the anodes had been pre-reduced prior to the experiments. The anode performance is furthermore comparable to those of infiltrated YSZ scaffolds with an LSTO- (cation-stoichiometric La substituted SrTiO3) current collecting layer55 and to composite anodes in which (LSTO-) has been impregnated into a porous YSZ scaffold with further enhancement from infiltrated CeO2 and Pd.56
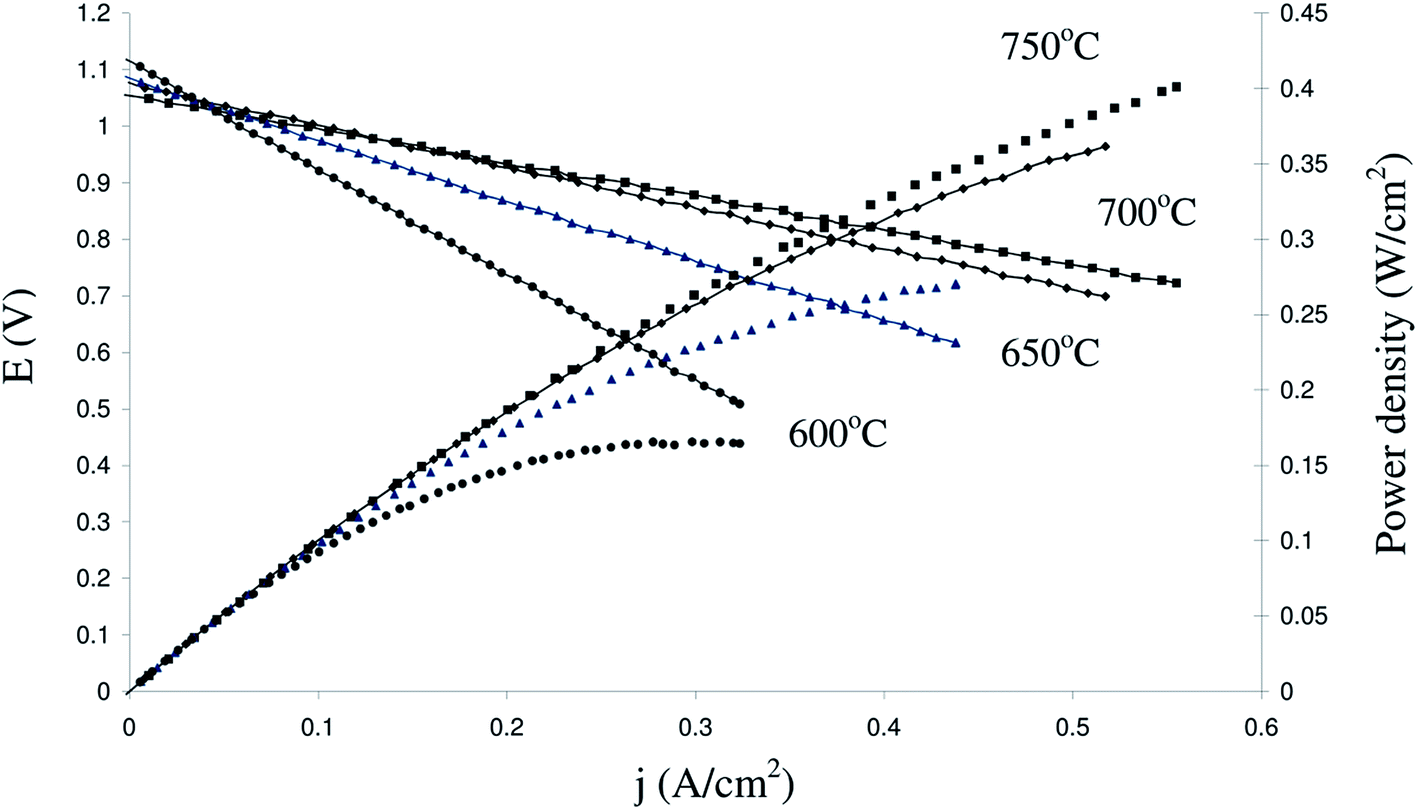 | ||
| Fig. 10 Fuel cell performance of anode supported LSTA- cell at different temperatures in pure humidified H2 (2.3% H2O) and pure O2 (reproduced from ref. 17). | ||
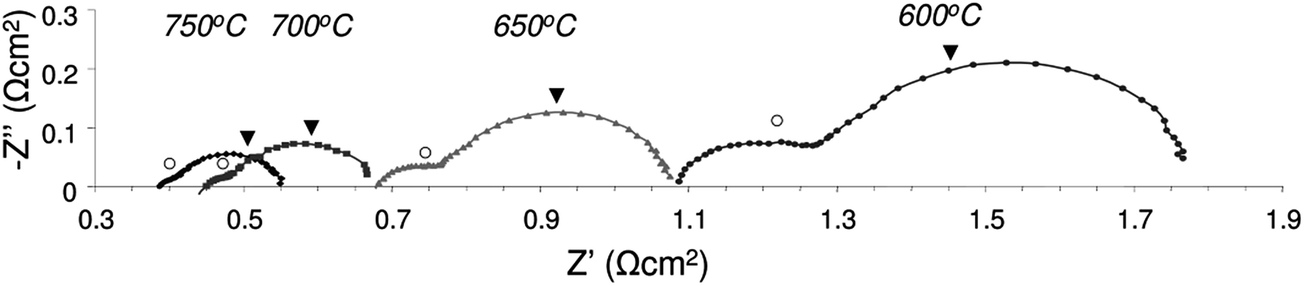 | ||
| Fig. 11 Impedance spectra for fuel cell comprising anode supported LSTA- impregnated with ceria and copper. Tests performed in humidified (2.3% H2O) pure H2 and pure O2 (reproduced from ref. 17). | ||
Similarly promising results have been found using a Ca substituted A-site deficient La0.2Sr0.7TiO3 as anode backbone in an electrolyte supported cell, impregnated with a combination of ceria and nickel catalysts. A stable area specific resistance of 0.37 Ω cm2 was achieved after 20 redox cycles and 250 hours of operation at 900 °C in H2 with 8% H2O, showing excellent redox stability. Power densities in excess of 0.5 W cm−2 could be obtained for these cells.52 This shows the promise for this type of material as a potential conductive backbone, whilst modification by infiltration can provide the necessary electrocatalytic activity.
YSTA- based anodes
Forschungzentrum Juelich has developed an anode supported cell based on YSTA-, demonstrating the mechanical robustness of A-site deficient titanates. Fig. 12 shows the cross section of such a cell.57 The anode supports with electrolyte layer, undergo a final firing step under reducing conditions.58 About 3 wt% NiO is subsequently impregnated into the anode structure to provide catalytic activity for the fuel oxidation reaction. | ||
| Fig. 12 Cross section of YST-ASC composed of YST/YST1.10–YSZ/YSZ/PVD-CGO/LSC. Reprinted from Q. L. Ma, F. Tietz, A. Leonide and E. Ivers-Tiffee, Anode-supported planar SOFC with high performance and redox stability, Electrochemistry Communications, 2010, 12, 1326–1328, with permission from Elsevier.57 | ||
Analysis of the electrolyte by Energy Dispersive Spectroscopy confirms the likelihood of Ti diffusion from the YSTA- into zirconia, although this may be exaggerated due to the addition of 10 wt% TiO2 to the anode support. A gradient of the Ti content from 4.5 to 12.5 atm% in the electrolyte layer was noticed, decreasing from the anode side to cathode side, suggesting possible electronic conduction in the co-sintered electrolyte.
The performance of the YSTA- ASC was characterized electrochemically by means of current voltage (I/U), and impedance measurements. Typical I/U characteristics recorded at different temperatures is shown in Fig. 13. At 800 °C, a typical operation temperature for SOFC systems, an OCV of 1.09 V is obtained, very close to the theoretical value and indicating that the Ti content in the electrolyte layer, especially for the region close to cathode with only 4–5 at% of Ti, does not cause significant electronic conduction. At the same temperature, a current density of 1.22 A cm−2 at 0.7 V is achieved, which corresponds to a power density of 0.85 W cm−2. The actual data for all the tested cells so far varied from 1.0 to 1.5 A cm−2 at 0.7 V and 800 °C. Although compared with the state-of-the-art cells the performance of YST-based cells is still lower (see Table 1), it has already reached the level of practical use and has a valid prospect for commercial application.
 | ||
| Fig. 13 Current–voltage curves of an YST ASC for temperatures ranging from 600 to 850 °C. Reprinted from Q. L. Ma, F. Tietz, A. Leonide and E. Ivers-Tiffee, Anode-supported planar SOFC with high performance and redox stability, Electrochemistry Communications, 2010, 12, 1326–1328, with permission from Elsevier.57 | ||
| Temperature | YST-ASC | SoA-ASC |
|---|---|---|
| 800 °C | 855 | 1314 |
| 750 °C | 551 | 931 |
| 700 °C | 294 | 600 |
| 650 °C | 145 | 356 |
| 600 °C | 62 | 193 |
Fig. 14 shows the changes of OCV and performance at 0.7 V, of the cell as a function of the numbers of redox cycles. Impressively, after 200 redox cycles, the OCV only decreased by 1.3%, indicating the high robustness and stability of the cell. In contrast, state-of-the-art SOFCs based on Ni cermets usually collapsed, or had apparent losses of OCV after just one redox cycle.9 However, the performance of the cell decreased by 35% after 200 redox cycles. The reason is most likely the gradual loss of electrical conductivity after each redox cycle either due to an irreversible decrease of charge carriers, or due to the slow kinetics between the reduced and oxidized state of YST materials. As previously mentioned, the conductivity of YST sintered in reducing atmosphere, as well as other SrTiO3 based materials, depends significantly on the oxygen partial pressure during testing. The values are nearly 100 S cm−1 after sintering at 1400 °C in H2/argon, but only about 0.01 S cm−1 in air, both measured at 800 °C. If the atmosphere is changed from air back to argon/H2, the conductivity can be restored to some extent, but this may take considerably longer than the cycles used in this study. It is very likely that the 10 minutes interval for reduction during the redox cycle is not long enough for recovery of the conductivity after the oxidizing period (also 10 minutes). Hence, this results in a continuous performance decrease with continued redox cycling. This may be further investigated by redox tests with longer periods of reduction. Fig. 14 also shows the change of performance of a different cell, at 0.7 V and 800 °C, as a function of the numbers of redox cycles. For this test the reducing time during the cycles was prolonged to 2 hours, and the oxidizing period remained 10 minutes. As anticipated, there is no apparent decrease in performance after 50 redox cycles. More studies will be required however to establish whether any adverse changes in the electrical properties occur on longer oxidation periods. Nevertheless, performance values for cells based on YST anode are remarkably high, and among the best yet reported for cells with a ceramic anode (see Table 2).
 | ||
| Fig. 14 OCV (open circles) and current density at 0.7 V (closed circles) as a function of the number of redox cycles (10 minutes in H2 and 10 minutes in air) at 750 °C as well as current density at 0.7 V (closed squares) as a function of the number of redox cycles at 800 °C applying 2 h in H2 and 10 minutes in air. Reprinted from Q. L. Ma, F. Tietz, A. Leonide and E. Ivers-Tiffee, Anode-supported planar SOFC with high performance and redox stability, Electrochemistry Communications, 2010, 12, 1326–1328, with permission from Elsevier.57 | ||
| Cell type | Dimension | Anode | Performance (maximum power density) | Redox testing | Ref. |
|---|---|---|---|---|---|
| ESC | 20 mm diameter, 0.6 mm 8-YSZ electrolyte | (La0.75Sr0.25)0.9Cr0.5Mn0.5O3 | 0.30 W cm−2 at 850 °C | No test | 65 |
| ESC | 20 mm diameter, 160 μm 6ScSZ electrolyte | Ceria and Ni infiltrated La0.20Sr0.25Ca0.45TiO3 | >0.5 W cm−2 at 900 °C | 20 cycles | 52 |
| CSC | ∼20 mm diameter | Ceria and Ru-infiltrated Sr0.88Y0.08TiO3/YSZ | 0.51 W cm−2 at 800 °C | No test | 66 |
| CSC | ∼1 cm2, 0.4 mm thickness | Ceria and Pd-infiltrated La0.3Sr0.7TiO3/YSZ | 0.43 W cm−2 at 800 °C | No test | 67 |
| ASC | 20 mm diameter | Ceria and Cu infiltrated La0.2Sr0.7TiO3 | >0.4 W cm−2 at 750 °C | No test | 17 |
| ASC | ∼15 mm diameter, 0.8 mm thickness | La0.2Sr0.8TiO3 support, NiO–Ce0.8Sm0.2O2/NiO-YSZ anode | 0.85 W cm−2 at 800 °C | 7 cycles | 68 |
| ASC | 5 cm × 5 cm, 1.5 mm thickness | Ni infiltrated YST/YST1.10–YSZ | >1 W cm−2 at 800 °C | 200 cycles | 57 |
STNA- based anodes
Fuel cell tests on STNA- based anodes have so far been limited and mainly involve symmetrical cell testing in single (wet) hydrogen atmospheres. A first generation of these symmetrical cells comprised STN94 (single phase or composite with YSZ, w/w 1:1) electrodes spray deposited onto 8-YSZ electrolyte substrates. After sintering in air at 1250 °C, the anodes with thicknesses of 15–30 μm were infiltrated with 20–30 wt% Ce0.8Gd0.2O1.9 (CGO). The resulting crystallite size of the infiltrate was found to vary strongly with calcination temperature, namely 5 nm, 14 nm and 40 nm at 350 °C, 650 °C and 850 °C, respectively.28,60 Promising results were obtained by measuring the cells' electrical impedance. Negligible series resistances were found when using Pt current collection and polarisation resistances were comparable with state-of-the-art Ni/YSZ anodes. The impedance spectra at various temperatures are shown in Fig. 15. In addition, the CGO infiltrated electrode was shown to be redox stable and even slightly activated by redox cycling at 650–850 °C.
 | ||
| Fig. 15 Impedance spectra for STN94 infiltrated with CGO at various temperatures in humidified (3% H2O) hydrogen. Spectra have been corrected for Rs60 (reproduced with permission from ECS Transactions, 2008, 13, 181–194. Copyright 2008, The Electrochemical Society). | ||
A combination of Ni and CGO was also infiltrated into STN94 electrodes. Here the nickel was applied on already deposited CGO. Symmetrical cell measurements at open circuit voltage (OCV) in a one-atmosphere set-up showed an electrochemical activity comparable to the current state-of-the-art Ni/YSZ fuel electrode in SOFC applications, i.e. ∼0.12 Ω cm2 at 850 °C. Due to an apparently low activation energy of the electrode (around 0.5–0.7 eV), high performance was achieved at lower temperatures, where the best electrode, a Ni/CGO infiltrated electrode, showed a polarisation resistance of 0.22 Ω cm2, in H2 with ∼3% steam at 650 °C.61 A micrograph of a CGO infiltrated STN94 electrode is shown in Fig. 16.
 | ||
| Fig. 16 SEM micrograph of CGO-electrode after impregnation from an ethanol-based solution and pre-calcination at 350 °C on STN-YSZ composite backbone.62 | ||
A Ni/CGO infiltrated electrode supported by a STN99 backbone sintered for 4 hours at 1320 °C in 9% H2/91% argon was subjected to a single redox cycle of 91 hours to evaluate the electrode degradation behaviour at 800 °C, under high steam concentration (85%). Its series resistance, Rs, increased by 37% and polarisation resistance, Rp, by 36%, which corresponds to degradation rates at ∼400% per 1000 h for these conditions. The electrodes were re-reduced after the exposure to steam and although the summit frequencies visualised through a Bode plot of impedance data returned to their original values and some recovery took place, neither Rs nor Rp returned to their original values, leaving a permanent degradation of 20% and 19% for Rs and Rp values, respectively. Longer exposure to reducing conditions may lead to more recovery, but it still indicates that the performance of the Ni/CGO electrode is sensitive to the steam concentration, which may be linked to the fact that protons associate with the oxygen sub-lattice in CGO.
 | (8) |
The DC-conductivity measurements mentioned earlier showed that a 200 μm thick STN99-backbone of similar size will change resistance (Rs) between 2.7 mΩ and 16 mΩ, when subjected to p(O2)-variations from 10−20 to ∼10−15 bar, which corresponds to the 3% steam and 85% steam, respectively, at 800 °C. The change is practically reversible in this range of oxygen partial pressures and may account for some of the Rs variations observed.
Very promising results have recently been obtained by infiltrating STN94 backbones with a combination of CGO and ruthentium.63 Negligible degradation was observed in Rs and Rp when testing for 200 hours in high steam atmosphere (50% H2O, 50% H2) at 850 °C. In comparison, the Ni/CGO infiltrate combination gave rise to a threefold increase in Rp in these conditions within the same time.
5. Summary
A-site deficient strontium titanates have the potential to replace nickel based cermet anodes in both electrolyte and anode supported cells. Their electronic conductivity under reducing conditions seems sufficient to provide adequate current collection. Their electrocatalytic activity towards fuel oxidation however is very limited, which necessitates the use of infiltrated nanosized catalysts. This approach has so far produced very promising fuel cell performances, with the addition of redox stability at low steam concentrations due to the absence of a structural nickel phase. Some of the fuel cell performances of A-site deficient strontium titanates have been summarised in Table 2. Literature reports on other ceramic anodes have been listed as well for comparison. It can be seen that strontium titanates show promising power outputs and seem to be redox stable. Literature reports on their long term stability is still scant however, so their durability in various configurations remains to be tested. Strontium titanates have also been reported to be sulphur tolerant, at least up to reasonable concentrations,8,64 but suitable catalysts will still have to be found that show a similar tolerance towards sulphur poisoning.Acknowledgements
The authors gratefully acknowledge funding from the Fuel Cells and Hydrogen Joint Undertaking under grant agreement no. 256730.References
- J. H. Hirschenhofer, D. B. Stauffer, R. R. Engleman and M. G. Klett, Fuel Cell Handbook, US Department of Energy, Morgantown WV, 4th edn, 1998 Search PubMed.
- P. Holtappels and U. Stimming, in Handbook of Fuel Cells, John Wiley & Sons, Ltd, 2010 Search PubMed.
- M. Cimenti and J. M. Hill, Energies, 2009, 2, 377–410 CrossRef CAS PubMed.
- A. Mai, B. Iwanschitz, U. Weissen, R. Denzler, D. Haberstock, V. Nerlich, J. Sfeir and A. Schuler, ECS Trans., 2009, 25, 149–158 CrossRef CAS PubMed.
- B. A. Haberman, C. Martinez Baca and T. R. Ohrn, ECS Trans., 2011, 35, 451–464 CrossRef CAS PubMed.
- H. Y. Tu and U. Stimming, J. Power Sources, 2004, 127, 284–293 CrossRef CAS PubMed.
- S. McIntosh and R. J. Gorte, Chem. Rev., 2004, 104, 4845–4865 CrossRef CAS.
- Z. Cheng, J. H. Wang, Y. M. Choi, L. Yang, M. C. Lin and M. L. Liu, Energy Environ. Sci., 2011, 4, 4380–4409 CAS.
- S. W. Zha, Z. Cheng and M. L. Liu, J. Electrochem. Soc., 2007, 154, B201–B206 CrossRef CAS PubMed.
- J. Malzbender, E. Wessel and R. W. Steinbrech, Solid State Ionics, 2005, 176, 2201–2203 CrossRef CAS PubMed.
- B. A. Boukamp, Nat. Mater., 2003, 2, 294–296 CrossRef CAS PubMed.
- A. Atkinson, S. Barnett, R. J. Gorte, J. T. S. Irvine, A. J. Mcevoy, M. Mogensen, S. C. Singhal and J. Vohs, Nat. Mater., 2004, 3, 17–27 CrossRef CAS PubMed.
- Q. X. Fu and F. Tietz, Fuel Cells, 2008, 8, 283–293 CrossRef CAS.
- T. Ishihara, Perovskite Oxide for Solid Oxide Fuel Cells, Springer, 2009 Search PubMed.
- N. G. Eror and U. Balachandran, J. Solid State Chem., 1981, 40, 85–91 CrossRef CAS.
- T. D. McColm and J. T. S. Irvine, Ionics, 2001, 7, 116–121 CrossRef CAS.
- C. D. Savaniu and J. T. S. Irvine, J. Mater. Chem., 2009, 19, 8119–8128 RSC.
- J. C. Ruiz-Morales, J. Canales-Vazquez, C. Savaniu, D. Marrero-Lopez, W. Z. Zhou and J. T. S. Irvine, Nature, 2006, 439, 568–571 CrossRef CAS PubMed.
- T. Kolodiazhnyi and A. Petric, J. Electroceram., 2005, 15, 5–11 CrossRef CAS PubMed.
- U. Balachandran and N. G. Eror, J. Electrochem. Soc., 1982, 129, 1021–1026 CrossRef CAS PubMed.
- D. N. Miller and J. T. S. Irvine, J. Power Sources, 2011, 196, 7323–7327 CrossRef CAS PubMed.
- B. F. Flandermeyer, A. K. Agarwal, H. U. Anderson and M. M. Nasrallah, J. Mater. Sci., 1984, 19, 2593–2598 CrossRef CAS.
- D. N. Miller and J. T. S. Irvine, ECS Trans., 2007, 7, 1447–1454 CAS.
- J. Canales-Vazquez, W. Z. Zhou and J. T. S. Irvine, Ionics, 2002, 8, 252–255 CrossRef CAS.
- V. Vashook, L. Vasylechko, H. Ullmann and U. Guth, Solid State Ionics, 2003, 158, 317–325 CrossRef CAS.
- P. R. Slater, D. P. Fagg and J. T. S. Irvine, J. Mater. Chem., 1997, 7, 2495–2498 RSC.
- R. Moos and K. H. Härdtl, J. Am. Ceram. Soc., 1997, 80, 2549–2562 CrossRef CAS PubMed.
- P. Blennow, PhD thesis, Lund University, 2007.
- P. Blennow, K. K. Hansen, L. R. Wallenberg and M. Mogensen, Electrochim. Acta, 2006, 52, 1651–1661 CrossRef CAS PubMed.
- L. F. Zagonel, N. Barrett, O. Renault, A. Bailly, M. Baeurer, M. Hoffmann, S. J. Shih and D. Cockayne, Surf. Interface Anal., 2008, 40, 1709–1712 CrossRef CAS.
- M. B. Park and N. H. Cho, Solid State Ionics, 2002, 154, 175–181 CrossRef.
- S. J. Shih, S. Lozano-Perez and D. J. H. Cockayne, J. Mater. Res., 2010, 25, 260–265 CrossRef CAS.
- Y. M. Chiang and T. Takagi, J. Am. Ceram. Soc., 1990, 73, 3278–3285 CrossRef CAS PubMed.
- S. Y. Chung, S. J. L. Kang and V. P. Dravid, J. Am. Ceram. Soc., 2002, 85, 2805–2810 CrossRef CAS PubMed.
- A. Ianculescu, A. Braileanu, M. Zaharescu, S. Guillemet, I. Pasuk, J. Madarasz and G. Pokol, J. Therm. Anal. Calorim., 2003, 72, 173–180 CrossRef CAS.
- D. Neagu and J. T. S. Irvine, Chem. Mater., 2010, 22, 5042–5053 CrossRef CAS.
- P. Pasierb, S. Komornicki and M. Rekas, J. Phys. Chem. Solids, 1999, 60, 1835–1844 CrossRef CAS.
- D. Burnat, A. Heel, L. Holzer, E. Otal, D. Kata and T. Graule, Int. J. Hydrogen Energy, 2012, 37, 18326–18341 CrossRef CAS PubMed.
- Q. Ma, F. Tietz, D. Sebold and D. Stover, J. Power Sources, 2010, 195, 1920–1925 CrossRef CAS PubMed.
- O. A. Marina, N. L. Canfield and J. W. Stevenson, Solid State Ionics, 2002, 149, 21–28 CrossRef CAS.
- S. Q. Hui and A. Petric, J. Electrochem. Soc., 2002, 149, J1–J10 CrossRef CAS PubMed.
- S. Hashimoto, F. W. Poulsen and M. Mogensen, J. Alloys Compd., 2007, 439, 232–236 CrossRef CAS PubMed.
- S. G. Cho and P. F. Johnson, Ferroelectrics, 1992, 132, 115–127 CrossRef CAS.
- P. Blennow, A. Hagen, K. K. Hansen, L. R. Wallenberg and M. Mogensen, Solid State Ionics, 2008, 179, 2047–2058 CrossRef CAS PubMed.
- Q. X. Fu, F. Tietz, D. Sebold, S. W. Tao and J. T. S. Irvine, J. Power Sources, 2007, 171, 663–669 CrossRef CAS PubMed.
- Q. L. Ma, F. Tietz and D. Stover, Solid State Ionics, 2011, 192, 535–539 CrossRef CAS PubMed.
- N. F. Mott, Rev. Mod. Phys., 1968, 40, 677–683 CrossRef CAS.
- M. T. Colomer and J. R. Jurado, J. Solid State Chem., 2002, 165, 79–88 CrossRef CAS.
- D. Sarantaridis, R. A. Rudkin and A. Atkinson, J. Power Sources, 2008, 180, 704–710 CrossRef CAS PubMed.
- A. D. Aljaberi and J. T. S. Irvine, J. Mater. Chem. A, 2013, 1, 5868–5874 CAS.
- D. Neagu and J. T. S. Irvine, Chem. Mater., 2011, 23, 1607–1617 CrossRef CAS.
- M. C. Verbraeken, B. Iwanschitz, A. Mai and J. T. S. Irvine, J. Electrochem. Soc., 2012, 159, F757–F762 CrossRef CAS PubMed.
- A. Yaqub, C. Savaniu, N. K. Janjua and J. T. S. Irvine, J. Mater. Chem. A, 2013, 1, 14189–14197 CAS.
- X. Zhou, N. Yan, K. T. Chuang and J. Luo, RSC Adv., 2014, 4, 118–131 RSC.
- M. D. Gross, J. M. Vohs and R. J. Gorte, Electrochem. Solid-State Lett., 2007, 10, B65–B69 CrossRef CAS PubMed.
- S. Lee, G. Kim, J. M. Vohs and R. J. Gorte, J. Electrochem. Soc., 2008, 155, B1179–B1183 CrossRef CAS PubMed.
- Q. L. Ma, F. Tietz, A. Leonide and E. Ivers-Tiffee, Electrochem. Commun., 2010, 12, 1326–1328 CrossRef CAS PubMed.
- D. Simwonis, H. Thulen, F. J. Dias, A. Naoumidis and D. Stover, J. Mater. Process. Technol., 1999, 93, 107–111 CrossRef.
- A. Leonide, Y. Apel and E. Ivers-Tiffee, ECS Trans., 2009, 19, 81–109 CAS.
- P. Blennow, K. K. Hansen, L. R. Wallenberg and M. Mogensen, ECS Trans., 2008, 13, 181–194 CAS.
- A. M. Hussain, J. V. T. Hogh, T. Jacobsen and N. Bonanos, Int. J. Hydrogen Energy, 2012, 37, 4309–4318 CrossRef PubMed.
- P. Blennow, STN94 - YSZ composite backbone with CGO infiltration, ed. K. Agersted, 2010 Search PubMed.
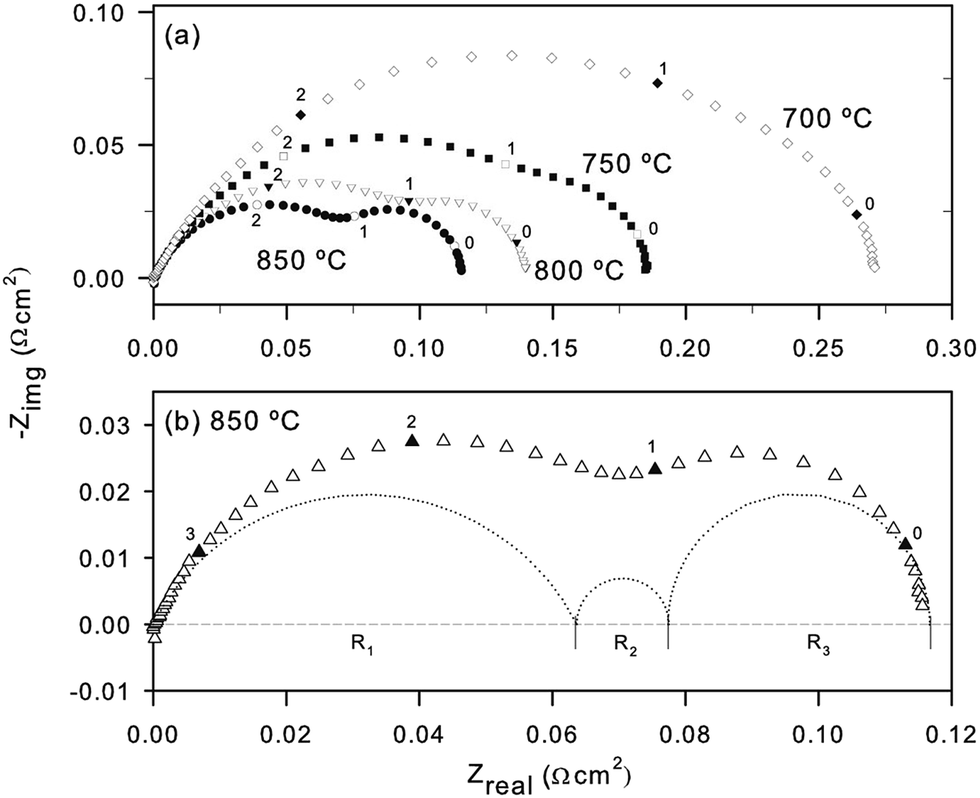
- T. Ramos, S. Veltze, B. R. Sudireddy and P. Holtappels, ECS Electrochem. Lett., 2014, 3, F5–F6 CrossRef CAS PubMed.
- R. Mukundan, E. L. Brosha and F. H. Garzon, Electrochem. Solid-State Lett., 2004, 7, A5–a7 CrossRef CAS PubMed.
- S. Tao, J. T. S. Irvine and J. A. Kilner, Adv. Mater., 2005, 17, 1734–1737 CrossRef CAS.
- H. Kurokawa, L. M. Yang, C. P. Jacobson, L. C. De Jonghe and S. J. Visco, J. Power Sources, 2007, 164, 510–518 CrossRef CAS PubMed.
- G. Kim, M. D. Gross, W. Wang, J. M. Vohs and R. J. Gorte, J. Electrochem. Soc., 2008, 155, B360–B366 CrossRef CAS PubMed.
- M. R. Pillai, I. Kim, D. M. Bierschenk and S. A. Barnett, J. Power Sources, 2008, 185, 1086–1093 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |