 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A quinoxaline-fused tetrathiafulvalene-based sensitizer for efficient dye-sensitized solar cells†
Anneliese
Amacher
a,
Chenyi
Yi
*b,
Jiabao
Yang
bc,
Martin Peter
Bircher
a,
Yongchun
Fu
a,
Michele
Cascella
d,
Michael
Grätzel
b,
Silvio
Decurtins
a and
Shi-Xia
Liu
*a
aDepartement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: liu@dcb.unibe.ch; Fax: +41 31 6314399; Tel: +41 31 6314296
bLaboratory of Photonics and Interfaces, Institute of Chemical Science and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), Station 6, CH-1050 Lausanne, Switzerland. E-mail: chenyi.yi@epfl.ch; Fax: +41 21 693 61 00; Tel: +41 21 693 31 12
cKey Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, Shanghai 200237, People's Republic of China
dDepartment of Chemistry and Centre for Theoretical and Computational Chemistry (CTCC), University of Oslo, PO Box 1033 Blindern, N-0315 Oslo, Norway
First published on 2nd May 2014
Abstract
A new quinoxaline-fused tetrathiafulvalene-based sensitizer has been prepared and characterized. The resulting power conversion efficiency of 6.47% represents the best performance to date for tetrathiafulvalene-sensitized solar cells.
Dye-sensitized solar cells (DSSCs) have attracted interest for over two decades as low cost and low environmental impact alternatives to traditional silicon photovoltaics.1,2 Current research is focusing on the development of efficient organic dyes overtaking Ru(II)-based dyes which show a power conversion efficiency (PCE) of up to 12%.2 Such organic dyes offer many advantages, such as the ease of synthesis, a vast flexibility in design strategy, and excellent light-harvesting ability. Most of them have a donor–π–acceptor (D–π–A) structure as the electron displacement from D to A units upon photoexcitation facilitates the electron injection into the TiO2 electrode.1–3 To tailor the photo-accessible excited states of organic sensitizers, a variety of molecular scaffolds such as triarylamine,4 phenothiazine,5 benzo[1,2-b:4,5-b′]difuran6 and porphyrin7 have been used as donor components. Tetrathiafulvalene (TTF), as a strong π-donor, has been intensively investigated within the context of materials chemistry towards molecular (opto)electronics.8 However, only one publication on π-extended TTF-sensitized solar cells has appeared in the literature, showing a moderate efficiency of 3.8%.9 The little application so far of TTF-based dyes in DSSCs is mainly due to the fact that they absorb only in the UV region and have an energetically high-lying HOMO, and therefore the dye-regeneration after electron-injection is thermodynamically unfavorable. In response, we have developed a new strategy for achieving rigid and planar D–A ensembles by annulation of TTF to acceptor moieties via a Schiff-base reaction.10 Such compactly fused D–π–A systems exhibit intense optical intramolecular charge transfer (ICT) absorbances over a wide spectral range and a substantially stabilized HOMO. In the present work, we have reported the synthesis, characterization and electronic properties of a quinoxaline-fused tetrathiafulvalene-based sensitizer (1, Scheme 1) and explored its application in DSSCs.
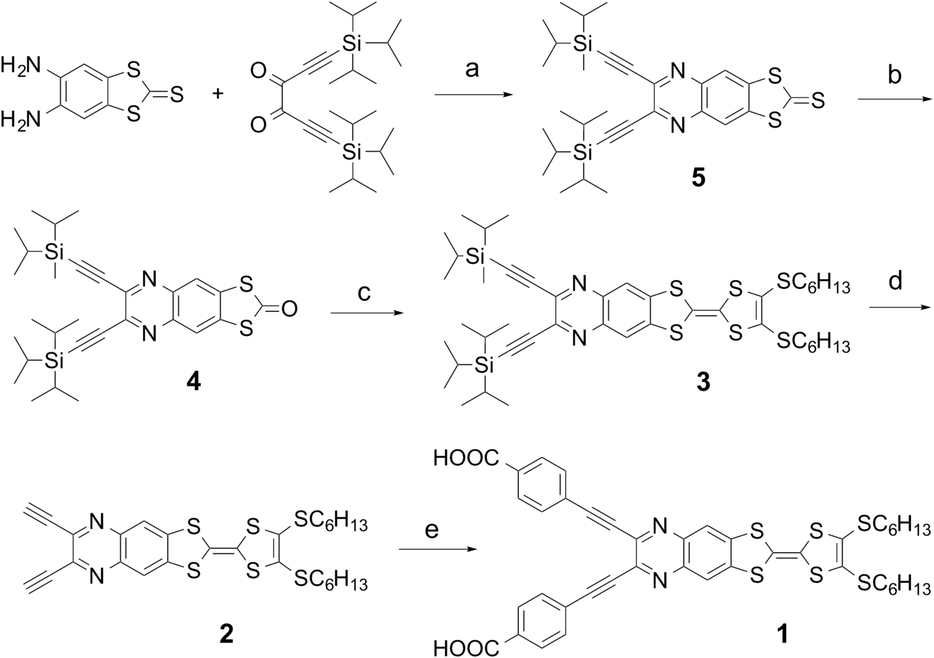 | ||
| Scheme 1 A synthetic route to the target dye 1: (a) ethanol, at reflux, 68%; (b) mercuric acetate, CH2Cl2, 98%; (c) 4,5-bis(hexylsulfanyl)-1,3-dithiol-2-thione (2 equiv.), triethylphosphite/toluene, 130 °C, 71%; (d) tetrabutylammonium fluoride, THF, −88 °C, 93%; (e) 4-iodobenzoic acid (5 equiv.), Pd(PPh3)2Cl2, diisopropylamine, 80 °C, 12%. | ||
The synthetic pathway to the target dye 1 is illustrated in Scheme 1, and involved the Sonogashira coupling of the TTF–quinoxaline diyne precursor 2 with 4-iodobenzoic acid. The former was prepared via a TIPS-deprotection reaction of 3 in the presence of TBAF. The synthesis of 3 is based on a phosphite-mediated cross-coupling reaction of 4 with 4,5-bis(hexylsulfanyl)-1,3-dithiol-2-thione. The key precursor 4 was achieved by the direct condensation of 5,6-diamino-1,3-benzodithiole-2-thione with 1,6-bis(triisopropylsilyl)-hexa-l,5-diyne-3,4-dione followed by the oxidation with Hg(OAc)2. All precursors are quite soluble in common organic solvents, which allowed the easy purification using standard chromatographic techniques and full characterization.
To estimate the HOMO and LUMO energy levels and thus to evaluate the feasibilities of two processes including electron injection from the photoexcited dyes to the TiO2 conduction band and the dye regeneration, the electrochemical and optical properties of organic dyes are of prime importance. The electrochemical properties of 1 in THF were investigated by cyclic voltammetry. Two reversible one-electron oxidations at E11/2 = 0.47 V and E21/2 = 0.64 V (vs. Fc/Fc+) for the successive formation of the TTF radical cation and dication were observed (Fig. 1). From the onset of the first oxidation potential according to the equation EHOMO = [−e(Eonset + 4.8)] eV, where 4.8 eV is the energy level of ferrocene below the vacuum level, the HOMO level is calculated to be −5.2 eV, which is lower than the energy level of the iodine/iodide redox shuttle. Such a relatively low-lying HOMO energy level ensures efficient regeneration of the oxidized dye and also good air stability in the DSSC device.
 | ||
| Fig. 1 Cyclic voltammogram of 1 in THF (0.1 M Bu4NPF6; Pt working electrode; scan rate 50 mV s−1). | ||
The optical absorption spectrum of 1 in THF solution is shown in Fig. 2. Strikingly, strong electronic transitions cover the whole spectral region from the UV out to the red range around 610 nm. In particular, we observe an intense and broad absorption band in the middle of the visible region peaking at 526 nm (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1) with a molar extinction coefficient close to 2 × 104 M−1 cm−1. Such an optical absorption pattern is typical for fused donor–acceptor molecules and it is a manifestation of the occurrence of a variety of excited electronic charge-transfer (CT) states. Furthermore, the spectrum reveals an optical HOMO–LUMO gap, based on the absorption red-edge, of about 2.3 eV; thus the LUMO energy level of 1 can be estimated to be −2.9 eV according to the equation ELUMO = [Eg,opt + EHOMO] eV, providing a sufficient driving force for the electron injection from the dye to TiO2.
000 cm−1) with a molar extinction coefficient close to 2 × 104 M−1 cm−1. Such an optical absorption pattern is typical for fused donor–acceptor molecules and it is a manifestation of the occurrence of a variety of excited electronic charge-transfer (CT) states. Furthermore, the spectrum reveals an optical HOMO–LUMO gap, based on the absorption red-edge, of about 2.3 eV; thus the LUMO energy level of 1 can be estimated to be −2.9 eV according to the equation ELUMO = [Eg,opt + EHOMO] eV, providing a sufficient driving force for the electron injection from the dye to TiO2.
 | ||
| Fig. 2 Electronic absorption spectrum of 1 in THF solution, together with the calculated S0 → Sn transitions using the PBE0 xc-functional. | ||
In order to assign the orbital nature of the electronic transitions of 1, quantum mechanical calculations were performed at the DFT/PBE0 level for geometry optimization, and employing TDDFT/PBE0 for the corresponding optical excitations (see ESI†). Geometry optimization reveals a virtually planar π-conjugated skeleton with only a slight bending, which is, however, typical for neutral TTF units. This result compares well with a X-ray structure of a fused TTF–quinoxaline type of molecule that has recently been published.11 Moreover, both phenyl groups exhibit only small dihedral angles of ≈18° with respect to the main molecular skeleton. The planarity renders electronic coupling through the dye molecule quite efficient. Upon photo-excitation, the electron is transferred from the TTF to the carboxylic acid moieties. This can ensure good coupling to the conduction band of TiO2, resulting in efficient electron injection. The principal frontier Kohn–Sham orbitals involved in the calculated electronic transitions and their respective energies are shown in Fig. 3. The π-type HOMO exhibits its main electron density localization on the TTF unit, whereas the coefficients for the π-type LUMO are essentially spread over the quinoxaline moiety while also reaching all the way out to both carboxylic acid groups. A similar picture holds for the LUMO + 1 but with even higher coefficient values on the peripheral anchoring groups. Here it already seems clear that the light-induced CT excitations will push the electron density in the “right” direction, namely towards both anchoring groups. Indeed, the TDDFT calculations show that the S0 → S1 excitation corresponds to an ICT transition with a one-electron HOMO to LUMO promotion (Table S4, ESI†). With its high oscillator strength of 0.47 at the wavelength of 566 nm, this excitation describes perfectly the absorption band centred at 526 nm. As is to be expected, this excited S1 CT state reveals a large dipole moment of 12.7 D (calc.). The stick plot of Fig. 2 together with the information given in Table S4 (ESI†) demonstrates the good overall agreement between the experimental spectral data and all the calculated electronic excitations up to S10.
 | ||
| Fig. 3 Molecular orbitals of 1 that are involved in the main optical transitions. | ||
DSSC experiments were performed using the synthesized dye 1 in the presence and absence of a prototype coabsorbent, chenodeoxycholic acid (CDCA). It is suspected that dye 1 with a rigid, planar and large aromatic system has a strong tendency to aggregate on the TiO2 film through π–π stacking. As depicted in Fig. 4, the DSSC device performances are to some extent affected by dye aggregation. With CDCA, the integrated IPCE values between 500 nm and 600 nm exceed 70%, indicating that 1 shows enhanced photon to current conversion efficiency. The efficiency increases from 5.19% with a short-circuit current (Jsc) of 12.56 mA cm−2, open circuit voltage (Voc) of 580 mV and fill factor (FF) of 0.70 to 6.47% (Jsc = 13.76 mA cm−2, Voc = 617 mV, fill factor (FF) = 0.75). These results can be accounted for by the fact that the presence of CDCA prevents both the dye aggregation and the side-on attachment of the dyes, hence effectively increasing charge injection efficiency and retarding the back electron transfer.3a,6,12
 | ||
| Fig. 4 Photovoltaic performance of 1 with (red) and without (black) a CDCA coabsorbent. (a) Photocurrent action spectra showing the incident photon-to-current conversion efficiency as a function of excitation wavelength. (b) Photocurrent density (J) as a function of voltage (V) measured under standard air mass 1.5 and simulated sunlight at 1000 W m−2 intensity. | ||
In conclusion, we have developed a new strategy for the attainment of a quinoxaline-fused TTF-based sensitizer 1 for DSSC application. Such a compactly fused D–π–A system exhibits an intense optical ICT absorbance over a wide spectral range while the HOMO is substantially stabilized. As a consequence, DSSC experiments with 1 exhibit a broad IPCE exceeding 70% between 500 and 600 nm and high photocurrent densities reaching 13.76 mA cm−2, leading to a power conversion efficiency close to 6.5%, which represents the best performance to date for TTF-sensitized solar cells. We believe that this work will open up new opportunities in the design of high-performance organic sensitizers.
This work was supported by the Swiss National Science Foundation (No. 200021-147143) as well as by the Norwegian Research Council through the CoE Centre for Theoretical and Computational Chemistry (CTCC) Grant Nos. 179568/V30 and 171185/V30.
Notes and references
- (a) Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC; Z. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170 Search PubMed; (b) J. N. Clifford, E. Martinez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635 RSC; (c) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- A. Mishra, M. K. Fischer and P. Baeuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed.
- (a) L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC; (b) Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC.
- (a) G. Zhang, H. Bala, Y. Cheng, D. Shi, X. Lv, Q. Yu and P. Wang, Chem. Commun., 2009, 2198 RSC; (b) W. D. Zeng, Y. Cao, Y. Bai, Y. H. Wang, Y. S. Shi, M. Zhang, F. F. Wang, C. Y. Pan and P. Wang, Chem. Mater., 2010, 22, 1915 CrossRef CAS; (c) N. Satoh, T. Nakashima and K. Yamamoto, J. Am. Chem. Soc., 2005, 127, 13030 CrossRef CAS PubMed.
- J. Lee, J. Kwak, K. C. Ko, J. H. Park, J. H. Ko, N. Park, E. Kim, D. H. Ryu, T. K. Ahn, J. Y. Lee and S. U. Son, Chem. Commun., 2012, 48, 11431 RSC.
- H. Li, C. Yi, S. Moussi, S.-X. Liu, C. Daul, M. Grätzel and S. Decurtins, RSC Adv., 2013, 3, 19798 RSC.
- (a) A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed; (b) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- (a) M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153 RSC; (b) J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS; (c) J. Liao, J. S. Agustsson, S. Wu, C. Schönenberger, M. Calame, Y. Leroux, M. Mayor, O. Jeannin, Y.-F. Ran, S.-X. Liu and S. Decurtins, Nano Lett., 2010, 10, 759 CrossRef CAS PubMed; (d) D. Canevet, M. Salle, G. X. Zhang, D. Q. Zhang and D. B. Zhu, Chem. Commun., 2009, 2245 RSC; (e) J. Yamada and T. Sugimoto, TTF Chemistry. Fundamentals and applications of Tetrathiafulvalene, Springer Verlag, Berlin, 2004 Search PubMed; (f) P. Batail, (ed.), Chem. Rev., 2004, 104, special issue No. 11 “Molecular Conductors”; (g) R. Pfattner, E. Pavlica, M. Jaggi, S.-X. Liu, S. Decurtins, G. Bratina, J. Veciana, M. Mas-Torrent and C. Rovira, J. Mater. Chem. C, 2013, 1, 3985 RSC.
- S. Wenger, P.-A. Bouit, Q. Chen, J. Teuscher, D. Di Censo, R. Humphry-Baker, J.-E. Moser, J. L. Delgado, N. Martin, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 5164 CrossRef CAS PubMed.
- (a) X. Guégano, A. L. Kanibolotsky, C. Blum, S. F. Mertens, S.-X. Liu, A. Neels, H. Hagemann, P. J. Skabara, S. Leutwyler, T. Wandlowski, A. Hauser and S. Decurtins, Chem. – Eur. J., 2009, 15, 63 CrossRef PubMed; (b) C.-Y. Jia, S.-X. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804 CrossRef CAS PubMed; (c) C.-Y. Jia, S.-X. Liu, C. Tanner, C. Leiggener, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. Commun., 2006, 1878 RSC; (d) M. Jaggi, C. Blum, N. Dupont, J. Grilj, S.-X. Liu, J. Hauser, A. Hauser and S. Decurtins, Org. Lett., 2009, 11, 3096 CrossRef CAS PubMed; (e) S. Delahaye, C. Loosli, S.-X. Liu, S. Decurtins, G. Labat, A. Neels, A. Loosli, T. R. Ward and A. Hauser, Adv. Funct. Mater., 2006, 16, 286 CrossRef CAS.
- Y. Geng, C. Fiolka, K. Krämer, J. Hauser, V. Laukhin, S. Decurtins and S.-X. Liu, New J. Chem., 2014, 38, 2052 RSC.
- (a) J. Yang, F. Guo, J. Hua, X. Li, W. Wu, Y. Qu and H. Tian, J. Mater. Chem., 2012, 22, 24356 RSC; (b) D. Daphnomili, G. Landrou, S. Prakash Singh, A. Thomas, K. Yesudas, K. Bhanuprakash, G. D. Sharma and A. G. Coutsolelos, RSC Adv., 2012, 2, 12899 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization data for all new compounds and the detailed DFT calculations on 1. See DOI: 10.1039/c4cc02696a |
| This journal is © The Royal Society of Chemistry 2014 |