
Synthesis and evaluation of analogs of tetracyclic Ganoderma alkaloids as inhibitors of the enzyme α-glucosidase
L. Javier
Cala Gomez
ab,
Natalia L.
Calvo
ab,
Mario O.
Salazar
b,
Sebastian O.
Simonetti
 ab,
Ricardo L. E.
Furlan
b,
Andrea B. J.
Bracca
ab and
Teodoro S.
Kaufman
*ab
ab,
Ricardo L. E.
Furlan
b,
Andrea B. J.
Bracca
ab and
Teodoro S.
Kaufman
*ab
aInstituto de Química Rosario (IQUIR, CONICET-UNR), Universidad Nacional de Rosario, Suipacha 531, 2000, Rosario, Argentina
bFacultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, 2000, Rosario, Argentina. E-mail: kaufman@iquir-conicet.gov.ar
First published on 17th December 2025
Abstract
A series of synthetic analogs of tetracyclic Ganoderma alkaloids were prepared via short routes in moderate overall yields by the reaction of 3-formylchromene derivatives with enamines prepared from cyclic 1,3-dicarbonyl compounds. Single-crystal X-ray analysis of one of the tetracyclic products confirmed that they embody original tetrahydro-6H-chromeno[4,3-b]quinoline (and tetrahydrochromeno[4,3-b] cyclopenta[e]pyridine) skeletons. When tested as α-glucosidase inhibitors, these analogs proved to be up to 40 times more potent than acarbose, used as the reference inhibitor.
Introduction
Type 2 diabetes mellitus is a chronic metabolic disease characterized by persistent hyperglycemia, resulting from insulin resistance and pancreatic β-cell dysfunction.1a It is a major global health issue that affects over 500 million individuals and is the fourth leading cause of death in most developed countries.1b Therefore, great efforts have been made to achieve a better understanding of the disease and improve its clinical management.One of the pharmacological approaches devised to treat this condition entails the inhibition of key carbohydrate digestive enzymes, such as α-amylase1c and α-glucosidase.1d However, despite the high quality of the modern pharmacological arsenal,1e the prevalence of the disease continues to grow and some of its complications remain challenging to control. Hence, the search for new inhibitors of these enzymes with novel structures is an active field of research.
Ganoderma is a genus of basidiomycete fungi that have been used for centuries in many Asian cultures for the treatment of several diseases.2 Many species are known; among them, G. lucidum, G. sinense, G. capense, G. cochlear, and G. tsugae play important roles in folk medicine in various Southeast Asian countries.
For example, G. lucidum has long been used in traditional Chinese medicine to treat diabetes mellitus and recently it has been shown to enhance the blood glucose-lowering effects of modern drugs.3 In addition, some fungal polysaccharides4a and proteoglucans have been demonstrated to possess activity in type 1 diabetic mice,4b,c increasing blood insulin levels, stimulating the activity of liver enzymes related to carbohydrate metabolism, and thus accelerating the metabolism of glucose.4d
The chemical constituents and biological activities of 25 species of Ganoderma have been recently reviewed.5 Different meroterpenoids (such as ganomycins)6a,b have been shown to display aldose reductase7 or α-glucosidase3c,8 inhibition, among other effects.7b,9
These activities have also been found in some lanostane triterpenoids,6c such as ganoderic acids (Fig. 1), ganodeweberiols, lucidenic acid esters, ganoleucins, and ganoleuconins, among others.
 | ||
| Fig. 1 Some anti-diabetic triterpenoids and meroterpenoids along with selected tetracyclic alkaloids isolated from different species of Ganoderma. | ||
Despite their abundance in nature and wide range of biological and pharmacological activities, which often attract considerable attention, alkaloids remain among the least evaluated classes of compounds in the context of diabetes mellitus.10 Studies referring to Ganoderma alkaloids are relatively few; hence, this class of natural products and their bioactivities remain unknown or largely unexplored.
Ganoines I and II, isolated from G. capense in 1990, were the first alkaloids found in this genus,11 whereas the tricyclic ganocochlearines A and B were the first alkaloids isolated from G. cochlear.12 In 2017, additional ganocochlearines (C–I) were isolated from the latter species, with ganocochlearines H and I having skeletons that differ from those of their tetracyclic congeners.13 Most of these heterocycles were shown to lack toxicity toward normal cells and did not inhibit the proliferation of NRK-49F fibroblasts under TGF-β1-induction.
However, Chen and Lan recently synthesized the related lucidimines B and C (isolated from G. lucidum) and examined their bioactivity.14 They found that lucidimine B exhibited antiproliferative properties toward MCF-7 cells; in addition, it proved to be a better antioxidant than lucidimine C. On the other hand, ganocalicine A, from G. calidophilum, was shown to be an anti-allergic agent.15
Therefore, with a view to exploring the chemical space of structures inspired by this class of understudied compounds, and in continuation of our research work on the synthesis and biological evaluation of heterocyclic natural products and their analogs,16 herein we disclose our results related to the synthesis of analogs of tetracyclic Ganoderma alkaloids and their evaluation as α-glucosidase inhibitors.
Results and discussion
Retrosynthetic analysis
The heterocycles were synthesized according to the guidelines obtained from the retrosynthetic analysis displayed in Scheme 1. It was envisaged that the proposed analogs could be accessed through the late-stage construction of the central pyridine ring, followed by modifications of the functionalities attached to rings A and D. Accordingly, it was conjectured that the key heterocyclic ring C could be formed through a 6π-1-azaelectrocyclization.17 | ||
| Scheme 1 Retrosynthetic analysis of the projected analogs of Ganoderma alkaloids. | ||
On this basis, one of the C–N bonds of the prototypic target 1 was strategically disconnected, revealing 1-azatriene 2 as a potential advanced intermediate. A more in-depth analysis suggested disconnection of the triene at its central bond, assuming that the latter could be built by direct condensation between an enamine (3) and a 3-formylchromene (4).
In turn, enamine 3 could be derived from the corresponding cyclic 1,3-dicarbonyl precursor (5), where the additional carbonyl group could add a point of variation and mimic the oxygen functionalities found in the natural products. On the other hand, the required 3-formylchromene partner could be formed by the reaction of a conveniently functionalized salicylaldehyde derivative (6) and acrolein (7), through a tandem oxa-Michael addition/aldol condensation process.
Chemical synthesis
According to the retrosynthetic plan, which followed an A → AB → ABD → ABCD constructive approach, the synthesis commenced with the preparation of the starting aldehyde 9 (Scheme 2). | ||
Scheme 2 Reagents and conditions: (a) Me2SO4, NaHCO3, PhMe, 80 °C, 4 h (44%); (b) NaOH, H2O, CHCl3 (8 equiv.), 70 °C, 4 h (62%); (c) 7 (1.5 equiv.), dioxane, 100 °C, 6 h (9 → 12, 56%; 8 → 11, 80%); (d) MeI, K2CO3, DMF, rt, overnight (96%); (e) NH4OAc, Si(OEt)4, MeCN, reflux (100%); (f) FeCl3, MeCN, MW (150 °C), 60 min (13, 17%, 16, 20%; 17, 19%; 18, 10%); (g) NaBH4, MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 (1:10, v/v), rt, 18 h (75%); (h) I2/Al–DMSO, MeCN rt, or NaStBu, DMF, 90 °C, 1.5 h (decomposition). :CH2Cl2 (1:10, v/v), rt, 18 h (75%); (h) I2/Al–DMSO, MeCN rt, or NaStBu, DMF, 90 °C, 1.5 h (decomposition). | ||
Initially, it was observed that the O-methylation of aldehyde 8 (Me2SO4, NaHCO3, PhMe, reflux) proved to lack selectivity, whereas the Casnati–Skattebøl and Duff formylations of 4-methoxyphenol (10) gave unsatisfactory results (low yields, diformylation). In contrast, the Reimer–Tiemann reaction cleanly provided the expected product 9 in 62% yield when 8 equiv. of CHCl3 were employed, and the reaction was carried out at 70 °C; not surprisingly, however, the yield dropped to 15% with the use of only 2 equiv. of CHCl3.
Next, the transformation of 9 into formylchromene 12 was studied18 and the product was obtained in 56% yield when K2CO3 was used as a base in the presence of freshly prepared acrolein (7)19 in refluxing dioxane.
The reaction is likely to involve an initial conjugate addition of phenol to 7, a strong Michael acceptor, to furnish a 1,7-dicarbonyl system intermediate (i). In turn, the resulting acidic α-carbonyl methylene unit would cyclize by attacking the carbonyl of the benzaldehyde motif (ii), and this should ultimately dehydrate to furnish the formylchromene,20 completing the tandem oxa-Michael addition–aldol condensation sequence.
Interestingly, the analogous heterocycle 11 was obtained in 80% yield when the reaction was carried out with the commercial aldehyde 8 at 100 °C (0.18 M level). It was observed that the performance of this transformation is sensitive to the concentration of the reagents, with its yields diminishing to 53% when conducted at the 0.45 M level. Methylation of 11 with MeI in DMF with K2CO3 as the base21 furnished 12 in 96% yield, turning the latter into an improved and more versatile route toward the 3-formylchromene fragment.
On the other hand, the formation of enamine 3 was first attempted mechanochemically by the reaction of 1,3-dione 5 with NH4OAc under KHSO4/SiO2 promotion.22 This resulted in a 2:1 mixture of the product with the starting ketone, as per 1H NMR integration of the diagnostic H-2 signals (δ = 5.29, s, 1H). For this reason, the crude enamine preparation was used in the reaction with 3-formylchromene 12. When it was carried out in MeCN using FeCl3 as the promoter and oxidant,23 it afforded a single isolated product in 17% yield, after heating under microwave irradiation at 150 °C for 1 h (Table 1, entry 1).
a
|
|
|||
|---|---|---|---|
| Entry no. | Conditions | Yield (%) | |
| Enamine formation | Cyclization | ||
| a When the reactions were run in two steps, enamine formation was carried out first by the reaction between 5 and NH4OAc under the stated conditions, before running the cyclization stage with the crude enamine preparation. b The volatiles were removed in vacuo before running the cyclization stage. | |||
| 1 | KHSO4/SiO2, solventless, 0.25 h | FeCl3, MeCN, MW, 150 °C, 1 h | 17 |
| 2 | Si(OEt)4, MeCN, reflux, 1 h | FeCl3, MW, 80 °C, 1 h | 18 |
| 3 | Si(OEt)4, MeCN, MW, 150 °C, 1 h | FeCl3, O2, MW, 150 °C, 0.5 h | 12 |
| 4 | Si(OEt)4, MeCN, MW, 150 °C, 1 h | CuCl2, MW, 120 °C, 2 h | 11 |
| 5 | 1. InCl3 (10 mol%), EtOH, rt, 2 h | 16 | |
| 2. 80 °C, 36 h (dihydropyridine 13a, 84%) | |||
| 3. DDQ, THF, −30 °C → −5 °C | |||
| 6 | Si(OEt)4, PhMe, reflux, 48 h | 26 | |
| 7 | Si(OEt)4, PhMe, reflux, 48 h (purif. on Al2O3) | 40 | |
| 8 | Si(OEt)4, EtOH, reflux, 1 hb | Zn(OTf)2, DMF, 75–100 °C, 48 h | 45 |
| 9 | DABCO, MeCN, rt, 1 h | 70 °C, 3 h | 20 |
| 10 | Dioxane, rt, 0.5 h | DABCO, K2CO3, O2, reflux, 5 d | 24 |
| 11 | DCE, 80 °C, 2 d | K2CO3, I2, O2, reflux, 5 d | 17 |

Suspecting that the observed result could be due to incomplete enamine formation, Si(OEt)4 was employed as a H2O scavenger, and the reaction was performed in refluxing MeCN,24 which enabled a facile and efficient preparation of the required intermediate. However, when the latter was reacted with 12 under FeCl3 promotion at 80 °C, the product was obtained in an analogous 18% yield (entry 2).
The product was subjected to a careful structural analysis. The conventional mechanism of a typical Stork's enamine alkylation of α,β-unsaturated carbonyls involves a Michael addition of the enamine to the β-carbon atom of the latter favored by the electrons of the nitrogen of the enamine,25 resulting in the formation of a new C–C bond, which in the case of 12 and 3 would ultimately lead to the skeleton of 19, reminiscent of ganocochlearine I (Scheme 3).
 | ||
| Scheme 3 Proposed cyclization mode toward 19 based on the typical reactivity of the Stork's enamine alkylation of 12 with 3. | ||
In light of previous results,26 an analogous outcome would be expected from the conjugate addition of the 1,3-dicarbonyl precursor (5) to the enal moiety. Therefore, it was deemed necessary to carefully examine the reaction product by NMR to ensure unequivocal differentiation between tetracycle 19 and its isomer 13. In this case, the signals of the Csp2–H motif of the pyridine ring (C7/H7 in 19) were considered the best diagnostic elements.
Examination of the 1H NMR spectrum of the product revealed the presence of four hydrogens attached to sp2 carbon atoms, from which it was possible to unequivocally assign three of them to the isocyclic aromatic ring, based on comparison with its precursor 12 and analysis of the 2D spectra (COSY, HSQC and HMBC). The remaining Csp2–H signal [δH = 8.00 ppm (1H, s) and δC = 130.9 ppm] clearly indicated that 13 was the cyclization product (Scheme 2) because a more deprotected signal, such as δC7 ∼ 150 ppm, would be expected for 19, which has this Csp2–H motif attached to the pyridine N-atom.
Considering that suitable crystals of the product were obtained from EtOAc, in order to get definitive structural evidence, single crystal X-ray diffraction analysis of the heterocycle was undertaken (Fig. 2). It was observed that the tetracycle crystallized in the monoclinic system, in the P21/n space group with four molecules per unit cell (Z = 4). In addition, at room temperature, the solid exhibited some disorder in the isocyclic ring (Fig. 2A), specifically at C2, C3, and C4 and in the heavy atoms of the OMe motif; however, the refinement data clearly pointed out to 13 as the structure of the tetracyclic reaction product.
 | ||
| Fig. 2 ORTEP diagrams of tetracycle 13 with the thermal ellipsoids drawn at their 50% level. (A) At room temperature, displaying distortion at C2–C4 and the attached OMe group (the C2–O20–C21 bonds are colored for the sake of clarity) and (B) at 80 K. | ||
This conjecture was confirmed by performing the diffraction at 80 K (Fig. 2B), which improved accuracy, giving a definitive picture of the product structure and confirming that its room temperature disorder is thermal in nature (dynamic disorder). Under these conditions, it was observed that compound 13 displays an essentially planar structure, where both aromatic rings are slightly twisted, as evidenced by their small interplanar angle (10.31°). It was also noticed that the methyl moiety of the OMe group is oriented toward the C3 ortho side and is slightly out of plane [dihedral angle C3–C2–O20–C21 = −26.83(16)°]. In addition, C7, C13 and C21 are observed on the same side of the pyridine ring plane, whereas the carbonyl oxygen faces the opposite direction [C9–C10–C11–O19 = −6.87(16)°].
Having established the identity of the tetracyclic product 13, an optimization was undertaken (Table 1). It was observed that the use of aerobic conditions led to a diminished yield (12%; entry 3), while the performance did not improve when CuCl2 was used as the promoter and oxidant27 (11%, entry 4). On the other hand, when the reaction was performed as a one-pot process with InCl3 as the promoter, the dihydropyridine intermediate 13a was formed in a good yield (84%);28 however, further oxidation of the latter with DDQ in THF resulted in only 16% yield of the tetracyclic product 13 (entry 5), accompanied by a series of unidentified compounds.
In view of the above results, one-pot procedures were attempted (entries 6 and 7) in refluxing toluene. Unexpectedly, chromatographic product purification on an alumina column improved the yields up to 40% (entry 10). In addition, slightly better results (45% yield) were obtained when the enamine was prepared in refluxing EtOH and the cyclization was performed in DMF under Zn(OTf)2 promotion,29 after changing the solvent (entry 8).
Similarly, the use of bases (K2CO3, DABCO) to promote enamination (entry 9) and further cyclization (entries 9–11) failed to significantly improve the yield of the product, even when run under aerobic conditions30 (entry 11).
Next, the reaction was tested with enamines 3a and 3b, derived from the related 5,5-dimethyl cyclohexane-1,3-dione (dimedone, 5a) and 4,4-dimethyl cyclohexane-1,3-dione (5b). It was observed that their performances were similar, since they afforded analogs 16 and 17 in 20% and 19% yields, respectively (Scheme 2). However, a diminished product yield was recorded (18, 10%) when 3c, the enamine derivative of cyclopentane-1,3-dione (5c), was employed instead.
The case of 5b is interesting, because only one of the two possible regioisomeric tetracycles (17) was isolated. This is a result of the selective formation of a single enamine (3b) through the reaction of the added NH4AcO with the less hindered carbonyl moiety,31 as evidenced by the diagnostic signal of H-2 observed as a singlet in its 1H NMR spectrum at δ = 5.14 ppm.
On the other hand, when the regioisomeric tetracycles 16 and 17 were compared, it was observed that the carbon atoms of the gem-dimethyl motif of 16 were less protected in their 13C NMR spectra than those of tetracycle 17 (δ = 28.5 vs. 24.3 ppm). However, the hydrogen nuclei of the methyl groups of the latter compound were more deshielded than those in 16 (δ = 1.24 vs. 1.14 ppm), as a result of their proximity to the carbonyl moiety.
The tetracyclic ketone 13 was uneventfully reduced with NaBH4 in MeOH:CH2Cl2 (10:1, v/v) to afford alcohol 14. However, attempts to access phenol 15 by demethylation of 14 with the I2/Al–DMSO system in MeCN at room temperature32 or with sodium mercaptides (NaSEt or NaStBu in DMF, 90 °C, 1.5 h) met with failure, leading to decomposition of the tetracycle into unidentifiable products. This was attributed to the presence of a competing ether moiety in the oxygen heterocyclic motif.
In addition, attempts at performing the cyclization with 3-formylchromene 11 bearing a free phenol resulted in the decomposition of the starting material. Therefore, an alternative protecting group was sought that could withstand the cyclization conditions and be more easily removable (Table 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry no. | Protection | Cyclization | |||||
| Enal no. | R | Yield (%) | 1,3-Diketone | n | Product no. | Yield (%) | |
| a The reaction was carried out in DMF, in the presence of Zn(OTf)2, at 75–100 °C for 48 h. | |||||||
| 1 | 20 | Ac | 93 | 5 | 1 | Decomposition | |
| 2 | 21 | TBS | 99 | 5 | 1 | 24 | 27 |
| 3 | 22 | Bn | 85 | 5 | 1 | Decomposition | |
| 4 | 23 | MOM | 91 | 5 | 1 | 25 | 30 |
| 5 | 5 | 1 | 25 | 30a | |||
| 6 | 5c | 0 | 26 | 15 | |||
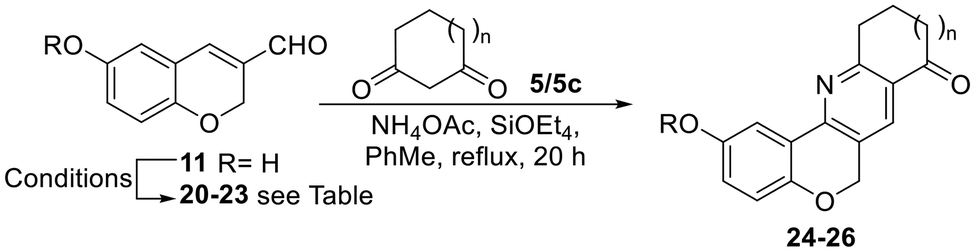
TBS, Bn and MOM ethers along with the acetate derivative of 11 were prepared in 85–99% yields.33 However, both the acetate (entry 1) and the benzyl ether (entry 3) underwent decomposition when exposed to the cyclization conditions and the remaining protecting groups gave meagre yields of the products (entries 2, 4 and 6). Unfortunately, the yields did not improve under other conditions; for example, cyclization of the MOM derivative 23 in the presence of Zn(OTf)2 (as in Table 1, entry 8) gave 25 in only 30% yield (Table 2, entry 5).
Next, the ketone moiety of tetracycle 25 was reduced with NaBH4 in a MeOH/CH2Cl2 (10:1, v/v) medium, affording 27 in 95% yield (Scheme 4), while deprotection of the phenol by transacetalization with refluxing iPrOH under Amberlyst-15 promotion for 18 h (ref. 34) provided 28 in 54% yield (62% overall yield when the sequence was performed as a one-pot process).
 | ||
| Scheme 4 Reagents and conditions: (a) MeOH, CH2Cl2 (10:1, v/v), 8 h (95%); (b) Amberlyst-15, iPrOH, reflux, 18 h (54%); (c) Amberlyst-15, iPrOH, reflux, 48 h (25 → 29, 88%; 26 → 30, 15%). | ||
Similarly, when the MOM protecting group was removed from 25 and 26, 29 and 30 were afforded in 88 and 15% yields, respectively. The yield of the deprotection of 29 was unexpectedly low, compared with similar transformations (25 → 29, 88%; 27 → 28, 54%). Although the ultimate causes for this behavior remain unknown, it resembles the differences observed during the cyclization stage, which was more efficient with the six-membered 1,3-dicarbonyl compounds than with their five-membered congeners.
Mechanistic considerations
The low product yields and selectivity observed during the transformations, even at temperatures as low as 70 °C, prompted an investigation into their possible causes. Although no identifiable products besides the expected tetracycles were detected under the different conditions, the use of copper(II) catalysis and milder conditions enabled the isolation of a small amount of chromene 31, suggesting that deformylation may be one of the operating degradation routes for the starting material (Scheme 5). This event does not seem to be uncommon, in light of the fact that the deformylation of 3-formylchromenes under noble metal catalysis (Pd, Rh) has been repeatedly reported.35 | ||
| Scheme 5 Possible decomposition pathways of the starting 3-formyl chromene 12 under the cyclocondensation conditions. | ||
On the other hand, it has been proposed that under thermal conditions, chromenes of this type can undergo a dearomatizative retro-6π-oxaelectrocyclization;36 this would result in a highly reactive conjugated tetraene-dicarbonyl species (32), which would likely be prone to undergo further decomposition.
An additional explanation was derived from careful inspection of the possible reaction mechanism (Scheme 6, Fig. 3), related to the strategic disconnections made during the retrosynthetic analysis. Under the assistance of the pair of electrons of the nitrogen atom of the enamine (3), the direct attack of the latter on the carbonyl carbon atom of 12 and the corresponding [1,5]H-shift would result in intermediate 34 through TS33. Next, dehydration of the thus formed allylic alcohol moiety would afford the 1-azatriene intermediate.
 | ||
| Scheme 6 A possible reaction mechanism for the synthesis of tetracycle 13 from 12 and 3. Energies are in kcal mol−1. | ||
 | ||
| Fig. 3 Energy diagram for the formation of 34en route to the cyclized product 13a, and its competitive alternative 38. Energies are in kcal mol−1. Bond forming/breaking distances shown in TS33, TS36 and TS37 are in angstroms (Å) and Wiberg indexes are shown in parentheses. | ||
Interestingly, although dehydration could result in a pair of geometric isomers, only those with the E-configuration (35E) could then be able to undergo a 6π-1-azaelectrocyclization through TS36 to form a 1,2-dihydropyridine (13a); in contrast, the Z-configured congener is proposed to undergo decomposition, consistent with the fact that despite many efforts, intermediates of this type could not be isolated. Upon oxidation in the reaction medium, 13a would afford 13. Dihydropyridines have been oxidized with an ample variety of reagents.37a In our case, however, the use of added oxidants such as Cu(II) and Fe(III) salts, as well as DDQ and O2 did not result in improved yields (Table 1).
Although efforts have been made to identify the required “internal” oxidant, they have not been successful; however, it is likely that the starting materials or the product itself may be involved in this process, adding a new explanation for the observed diminished yields. This is not unlikely because 1,4-dihydropyridines formed from 1,5-diketones and primary amines are known to disproportionate in AcOH to the corresponding tetrahydropyridine and pyridine derivatives,37b hydrogen transfer reactions among pyridine derivatives have been documented,37c and it has been observed that certain α,β-unsaturated carbonyl derivatives can oxidize Hantzsch dihydropyridines into the corresponding pyridines in a reaction driven by aromatization.37d
To better understand the reaction selectivity, a theoretical study was performed using DFT calculations at the m062x/6-311 g(d,p) theoretical level and employing toluene as the solvent. It was speculated that the carbon atom of 3-aminocyclohexanone (3) can attack the 3-formylchromene (12) either at the carbonyl moiety (1,2-addition) or at C4 of the chromene moiety (1,4-addition). Therefore, two attacks were modelled (Fig. 3, Scheme 6), observing the involvement of proton transfers from the amine to the oxygen of the carbonyl moiety in both cases, and an activation energy difference of 3.21 kcal mol−1 between TS33 (favored) and TS37 (disfavored).37e
The alternative transfer of H-4 to the carbonyl oxygen was also modelled and considered unlikely because the process has a free energy difference of 10.81 kcal mol−1. Additional calculations further demonstrated that the 1,2-adduct (34, ΔG = 11.82 kcal mol−1) is thermodynamically favored over the 1,4-adduct (38, ΔG = 15.61 kcal mol−1).
The molecular orbitals (MO) of the starting materials, along with natural bond orbitals and the thermodynamic parameters of the reaction were also examined. In the MO analysis, it was observed that both carbons can act as acceptors in 12, with coefficients of 0.1134 and 0.2072 for the carbonyl and β-carbon atom, respectively.
These partial results seemed to be somehow in disagreement with the experimental and calculated activation energies. However, further analysis of the natural population indicated that the charge difference on the carbonyl carbon in the starting material and TS is −0.1232 for the 1,2-addition pathway, while an analogous charge difference on the β-carbon is −0.004316 for the 1,4-addition pathway. This charge transfer difference is significant and sufficient to explain the observed preference for the 1,2-addition route.
In the 1,2-addition transition state, the NBO analysis of TS33 identified the C–C bond as partially formed, appearing as a BD(C–C) orbital interacting with its corresponding BD*(C2–C19) orbital. This indicates that electron donation from the nucleophile to the antibonding orbital of the nascent C–C bond stabilizes the transition state. The associated second-order perturbation energy is 4.28 kcal mol−1, significantly higher than the interaction observed in TS37 (1.73 kcal mol−1). Thus, although the frontier orbital coefficients initially suggested reactivity at the β-carbon atom, NBO analysis of the transition states revealed a stronger orbital interaction in the 1,2-addition pathway, justifying its lower activation barrier and the kinetic preference.
After ruling out the 1,4-addition pathway, the cyclization process toward the pyridine moiety was examined, beginning with the dehydration of 34. It was observed that the formation of the E-configured triene 35E, suited for subsequent electrocyclization, is endothermic (ΔG = 2.76 kcal mol−1) and only slightly energetically preferred (ΔΔG = 0.50 kcal mol−1) over the dehydration toward its Z-configured congener 35Z. This result provides an additional explanation for the observed moderate reaction yields, as the latter undergoes decomposition instead of the 6π-electrocyclization.
Regarding the electrocyclization step (Scheme 6), the pathway presents an activation energy of ΔG* = 16.82 kcal mol−1 (TS36) and a thermodynamic value of ΔG = −18.13 kcal mol−1 for the cyclized intermediate (13a). This result suggests that the formation of the tetracyclic motif takes place with a lower activation energy than that calculated for the formation of isoquinolines in aromatic systems.38 The HOMO analysis showed orbitals located in the azatriene system, corresponding to a disrotatory electrocyclization.
Biological activity
The inhibitory activity of a small library of tetracycles displaying variability at the C2 functionality and the D-ring (including size, substitution pattern, and degree of oxidation) was studied against α-glucosidase.39a In the assay, the enzymatic activity was quantified by measuring the release of p-nitrophenol from the enzymatic hydrolysis of p-nitrophenyl-α-O-D-glucopyranoside, used as a synthetic substrate.39a α-Cyclodextrin was added to the reaction medium to enhance the assay sensitivity39b and acarbose was employed as the reference inhibitor.After normalization of the results obtained at different concentrations of the test compounds (initially evaluated with eight concentration points), it was observed (Table 3) that except for compounds 13 and 14 (entries 1 and 3), the tetracycles showed higher inhibitory activity than acarbose. Therefore, for a more accurate assessment, the experiment was repeated using 11 concentration points.
|
|
|||
|---|---|---|---|
| Entry no. | Comp. no. | Enzyme inhibition (IC50, μM) | Potency vs. acarbose (×)b |
| a Assay conditions: α-glucosidase and p-nitrophenyl-α-O-D-glucopyranoside (substrate) were employed in a medium containing 0.1 M phosphate buffer (pH 7) and α-cyclodextrin. Absorbance readings were performed at 405 nm. b Referred to the IC50 of acarbose in the 11-points test (entry 9). | |||
| 1 | 13 | 761.0 ± 0.16 | 0.7 |
| 2 | 13a | 266.1 ± 0.13 | 1.9 |
| 3 | 14 | 500.7 ± 0.06 | 1.0 |
| 4 | 16 | 105.5 ± 0.11 | 4.8 |
| 5 | 17 | 152.3 ± 0.17 | 3.3 |
| 6 | 18 | 31.0 ± 0.09 | 16.3 |
| 7 | 24 | 32.5 ± 0.05 | 15.6 |
| 8 | 28 | 93.3 ± 0.11 | 5.4 |
| 9 | 29 | 25.1 ± 0.12 | 20.2 |
| 10 | 30 | 12.3 ± 0.05 | 41.2 |
| 11 | Acarbose | 505.2 ± 0.05 | 1.0 |

The results of the second assay confirmed that compound 30 (entry 10) displayed the lowest IC50 value, being over 40 times more potent than acarbose and approximately 2–2.5 times more potent than compounds 18, 24 and 29 (entries 6, 7 and 9), which showed similar potencies and also displayed promising activity. A more in-depth analysis revealed that the heterocycles derived from cyclopentane-1,3-dione (18 and 30; entries 6 and 10) and those containing a free phenol in their 2-position, such as 29 and 30 (entries 9 and 10), exhibited the highest potency. It was also found that the substituent on ring A modulates the activity of the tetracycles, with the methyl ether 13 (entry 1) being less active that the related TBS ether (24, entry 7) and the free phenol (29, entry 9).
In addition, it was observed that ketones 16 and 17 (entries 4 and 5), characterized by the presence of the gem-dimethyl moieties on the D-ring, were 4–6 times more potent than their related ketone 13, which lacks these substituents (entry 1). On the other hand, the dihydropyridine derivative 13a (entry 2) was 2.7 times more active than its unsaturated congener 13 (entry 1).
To evaluate promiscuity and selectivity, the inhibitory properties of the series were also assessed against two alternative hydrolases, namely acetylcholinesterase and β-glucosidase. However, no significant inhibition was observed for either enzyme within the experimental concentration range.
Taken together, these results provided a better understanding of the biological profile of the tested compounds and their enzyme selectivity. These findings offer valuable clues for the design of new α-glucosidase inhibitors as potential anti-diabetic drugs based on these previously unknown skeletons.
Conclusions
In conclusion, we have developed short routes toward tetracyclic analogs of Ganoderma alkaloids, which were based on the reaction between cyclic enamines derived from 1,3-dicarbonyl compounds and 3-formylchromene derivatives. Exhaustive optimization of the reaction conditions leading to the tetracyclic compounds was performed and DFT calculations provided sound explanation for the regioselectivity of the enamine attack on the α,β-unsaturated system. In this way, ten compounds were prepared in moderate overall yields, with original skeletons and several aspects of structural diversity. The tetracyclic analogs proved to be inactive as β-glucosidase and acetylcholinesterase inhibitors; however, they displayed selective inhibition of α-glucosidase, being up to 41 times more potent than acarbose.Experimental
General information
The reactions were performed under a dry argon atmosphere using oven-dried glassware. Anhydrous dioxane was obtained by refluxing the analytical grade reagent over sodium (benzophenone as the indicator) and distilling from the deep blue solution of the sodium benzophenone ketyl. Anhydrous MeOH, EtOH and iPrOH were obtained by treatment of the corresponding analytical grade solvent with clean magnesium turnings and a crystal of iodine, followed by a 4 h reflux and distillation from the resulting magnesium alkoxide suspension. Anhydrous MeCN and CH2Cl2 were prepared by refluxing the corresponding analytical solvent over P2O5 for 4 h, followed by distillation. Anhydrous DMF was obtained by heating the analytical grade product over BaO for 4 h, followed by distillation under reduced pressure. The anhydrous solvents were transferred to dry Young ampoules containing molecular sieves where they were kept for use. All other reagents were used as received.The reactions were monitored by TLC (Merck's silica gel 60 GF254) run in hexanes/EtOAc mixtures of different polarities. The chromatographic spots were revealed by exposure to UV light (254 and 365 nm) and spraying with phosphomolybdic acid solution, followed by careful heating to improve selectivity. For the workup procedure, in general, the reaction mixture was diluted with brine, and the products were extracted three times with EtOAc. The combined organic extracts were then washed once with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography (Merck's silica gel 60 H, particle size <55 μm or alumina), employing hexane/EtOAc polarity gradient techniques, under positive pressure of air.
Atom numbering and naming of the compounds were performed in agreement with the IUPAC guidelines.
Equipment
The melting points (uncorrected) were measured on an Ernst Leitz Wetzlar model 350 hot-stage microscope. The FT-IR spectra were recorded on a Shimadzu Prestige 21 spectrophotometer, with the samples prepared as solid dispersions in KBr disks. The NMR spectra were recorded with a Bruker Avance 300 FT-NMR spectrometer (300.13 MHz for 1H NMR and 75.48 MHz for 13C NMR) in CDCl3, MeOH-d4 or DMSO-d6, as required. The chemical shifts are reported in ppm in the δ scale, and TMS was used as the internal standard; the residual solvent peaks of CDCl3 (δH = 7.26 ppm, δC = 77.16 ppm), MeOH-d4 (δH = 3.31 ppm, δC = 49.00 ppm) and DMSO-d6 (δH = 2.54 ppm, δC = 40.45 ppm) were used as internal references. The assignments of signals marked with an asterisk (*) may be interchanged. In special cases, NOE and 2D NMR experiments (COSY, HSQC, TOCSY, and HMBC) were also employed. The HRMS data were obtained with a Bruker MicroTOF-Q II instrument (Bruker Daltonics, Billerica, MA) from UMyMFOR (Buenos Aires, Argentina). Detection of the ions was performed by electrospray ionization in the positive ion mode.The single crystal X-ray diffraction data were collected using a Bruker D8 QUEST ECO diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) equipped with a PHOTON II CPAD area detector with MoK radiation (λ = 0.71073 Å) at room temperature. For the measurement at 80 K, an open-flow Oxford Cryosystems nitrogen gas chiller (Oxford Cryosystems Ltd, Long Hanborough, UK) was employed. The data collection strategy and data reduction followed standard procedures implemented in Bruker's APEX4 software. Structures were solved using SHELXS-9740a and refined using the full-matrix least squares procedure with SHELXL2018/3.40b All H atoms were initially found in a difference map; those that bonded to C were further idealized [C–H range = 0.93–0.99 Å] and refined using a conduction model. Anisotropic displacement parameters were employed for non-hydrogen atoms, and H atoms were treated isotropically with Uiso(H) = 1.5Ueq. (X) (methyl H) or 1.2Ueq. (X) (aromatic and other H) times the equivalent displacement parameter (Ueq.) of the parent atoms. Multi-scan absorption correction was performed employing SADABS 2016/2.40c WinGX40d and PLATON40e were used to prepare the material for publication. The selection of suitable single crystals for measurement was carried out with a Leica stereoscopic magnifier model S9D (Leica, Wetzlar, Germany) with 1–5× objectives. The crystal structure data of 13 (Tables S1–S9) were deposited as cif files at the Cambridge Crystallographic Data Centre (CCDC).41
Computational methods
Conformational searches for the reactants, TS and products were run using the conformational search module of Hyperchem v.7.52 with the MM+ method. Selected structures were then successively optimized at the M062X/6-311G** level with toluene as the solvent.Frequency calculations were made to confirm the nature of the stationary points and to evaluate their thermochemical properties. Transition states were confirmed by the presence of unique imaginary frequencies and by intrinsic reaction coordinate (IRC) calculations. The molecular orbitals of the reactants and natural bonding orbitals of the transition state were calculated to analyze the frontier orbital interactions at the M062X/6-311G** level of theory.
Biological activity
The assays were carried out in 96-well, flat-bottomed, clear acrylic microplates. Once initiated by addition of the substrate, the microplates were shaken for 2 seconds and, while being maintained at 37 °C, the increase in absorbance at 405 nm was recorded every 30 seconds for 10–15 minutes using a VERSAMax microplate reader (Molecular Devices, Sunnyvale, USA). Wells containing DMSO, without an inhibitor, were used as a negative assay control and served as a reference for maximum enzyme activity.Each reaction was performed in triplicate, and the mean values with their corresponding standard deviations were used for data analysis. For IC50 determination, 8 or 11 serial dilutions of each test compound were prepared as equally spaced points on a natural logarithmic (Neperian) scale. The IC50 values were obtained by non-linear regression fitting of the log [inhibitor] versus normalized response data.
Synthesis and characterization
General procedure for the synthesis of enaminones
A stirred mixture of the given cyclic 1,3-dione (0.21 mmol) and NH4OAc (20 mg, 0.26 mmol) in MeCN (1 mL) was treated with Si(OEt)4 (57.3 µL, 0.26 mmol), and the mixture was heated under reflux for 20 h affording a yellow oil. Product formation was confirmed by TLC, and the products were identified by spectroscopy of the unstable crude products, which were used without further purification.:10, v/v; 1.1 mL) was treated with NaBH4 (15.4 mg, 0.28 mmol) and stirred again for 18 h at rt. The reaction was diluted with brine (5 mL) and the products were extracted with EtOAc (3 × 15 mL); the combined organic extracts were dried (MgSO4) and then concentrated under reduced pressure. Chromatography of the residue provided 14 (15 mg, 75%) as an orange solid; mp: 78–80 °C. IR (film, ν): 3417, 2938, 2851, 1647, 1564, 1493, 1273, 1204, 1040, 939, 870, 800 and 737 cm−1. 1H NMR (300 MHz, CDCl3): δ = 7.75–7.72 (1H, m, H-1), 7.50 (1H, s, H-7), 6.90–6.86 (2H, m, H-3 and H-4), 5.12 (2H, s, H-6), 4.81 (1H, t, J = 5.5, H-8), 3.87 (3H, s, OMe), 3.08–2.87 (2H, m, H-11), 2.15–2.02 (2H, m, H-9, H-10) and 1.94–1.80 (2H, m, H-9, H-10). 13C NMR (75 MHz, CDCl3): δ = 157.0 (C-11a), 155.1 (C-2), 150.7 (C-4a), 147.6 (C-12a), 133.3 (C-7a), 132.3 (C-7), 124.6 (C-6a), 123.8 (C-12b), 118.4 (C-3*), 118.0 (C-4*), 108.0 (C-1), 68.2 (C-8), 68.0 (C-6), 56.1 (OMe), 32.6 (C-11), 32.5 (C-9) and 19.0 (C-10). HRMS (ESI-TOF): m/z calcd for C17H17NO3 [(M + H)+]: 284.1281; m/z found: 284.1287.
:EtOH; 1% gradient), affording 28 (11 mg, 63%) as a brown solid; mp: 114–116 °C. IR (film, ν): 3418, 2928, 1645, 1537, 1470, 1452, 1337, 1200, 1065, 1014, 943 and 665 cm−1. 1H NMR (300 MHz, MeOH-d4): δ = 7.89 (1H, s, OH-2), 7.60 (1H, s, H-7), 7.53 (1H, d, J = 2.3, H-1), 6.82–6.72 (2H, m, H-3, H-4), 5.08 (2H, s, H-6), 4.74 (1H, s, H-8), 4.58 (1H, s, OH-8), 2.98–2.86 (2H, m, H-11), 2.05 (2H, dd, J = 13.8 and 8.4, H-9 and H-10) and 1.84 (2H, dd, J = 8.4 and 4.2, H-9 and H-10). 13C NMR (75 MHz, MeOH-d4): δ = 157.1 (C-11a), 152.6 (C-4a), 150.5 (C-2), 147.7 (C-12a), 134.4 (C-7a), 133.2 (C-7), 125.7 (C-6a), 124.0 (C-12b), 118.4 (C-3), 117.9 (C-4), 110.4 (C-1), 67.9 (C-6), 67.5 (C-8), 32.2 (C-11*), 32.1 (C-9*) and 19.0 (C-10). HRMS (ESI-TOF): m/z calcd for C16H15NO3 [(M + H)+]: 270.1125; m/z found: 270.1138.
Author contributions
All authors have agreed to their individual contributions. L. Javier Cala Gomez: formal analysis, validation, visualization, writing (original draft; review & editing); Natalia L. Calvo: data curation, formal analysis, investigation, validation, visualization, writing (original draft; review & editing); Mario O. Salazar: data curation, formal analysis, investigation, validation, visualization, writing (original draft; review & editing); Sebastián O. Simonetti: data curation, formal analysis, investigation, validation, visualization, writing (original draft; review & editing); R. L. E. Furlan: data curation, formal analysis, investigation, validation, visualization, writing (original draft; review & editing); A. B. J. Bracca: conceptualization, data curation, formal analysis, funding acquisition, investigation, project administration, supervision, validation, visualization, writing (original draft; review & editing); and T. S. Kaufman: conceptualization, data curation, formal analysis, funding acquisition, investigation, project administration, supervision, validation, visualization, writing (original draft; review & editing).Conflicts of interest
There are no conflicts to declare.Data availability
All data supporting this article have been included in the supplementary information (SI). Supplementary information: spectral compound characterization data [1H NMR, 13C NMR, 2D spectra (COSY, HSQC, HMBC)], details of DFT calculations and single-crystal XRD data. See DOI: https://doi.org/10.1039/d5ob01787d.CCDC 2501681 and 2501682 contain the supplementary crystallographic data for this paper.43a,b
Acknowledgements
The authors gratefully acknowledge Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, PIP 0765) and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT 2021-0162 and PICT 2019-3969) for financial support. L. J. C. G. also thanks CONICET for his doctoral fellowship. The authors are grateful to the FAIRE programme provided by the Cambridge Crystallographic Data Centre (CCDC) for the opportunity to use the Cambridge Structural Database (CSD) and associated software. The results of the mechanistic calculations have been obtained by using the facilities of the CCT-Rosario Computational Center, member of the High Performance Computing National System (SNCAD, MincyT-Argentina).References
- (a) J. Flannick, S. Johansson and P. R. Njølstad, Nat. Rev. Endocrinol., 2016, 12, 394–406 Search PubMed; (b) International Diabetes Federation, IDF Diabetes Atlas, ed. IDF, 10th edn, 2021. https://www.diabetesatlas.org/ (accessed November 1, 2025) Search PubMed; (c) M. Telagari and K. Hullatti, Indian J. Pharmacol., 2015, 47, 425–429 CrossRef CAS PubMed; (d) S. Kumar, S. Narwal, V. Kumar and O. Prakash, Pharmacogn. Rev., 2011, 5, 19–29 CrossRef CAS PubMed; (e) A. Chaudhury, C. Duvoor, V. S. R. Dendi, S. Kraleti, A. Chada, R. Ravilla, A. Marco, N. S. Shekhawat, M. T. Montales and K. Kuriakose, Front. Endocrinol., 2017, 8, 1–12 CrossRef.
- (a) B. Boh, M. Berovic, J. Zhang and L. Zhi-Bin, Biotechnol. Annu. Rev., 2007, 13, 265–301 CrossRef CAS; (b) S. Wachtel-Galor, J. Yuen, J. A. Buswell and I. F. F. Benzie, Ganoderma lucidum (Lingzhi or Reishi). A Medicinal Mushroom, in Herbal Medicine: Biomolecular and Clinical Aspects, 2nd edn, ed. I. F. F. Benzie and S. Wachtel-Galor, CRC Press, Boca Raton USA, 2011, ch. 9 Search PubMed; (c) R. Yangzom and P. Wangchuk, Ganoderma lucidum (Lingzhi Mushroom): Its Medicinal Uses, Biomolecules and Therapeutic Applications, in Phytochemistry and Nutritional Composition of Significant Wild Medicinal and Edible Mushrooms. Traditional Uses and Pharmacology, ed. A. Sharma, G. Bhardwaj and G. A. Nayik, RSC, London, UK, 2023 Search PubMed; (d) Therapeutic Mushrooms for Diabetes Mellitus. Current Evidences and Future Scope, ed. U. Azeem and K. R. Hakeem, Apple Academic Press, Inc., Palm Bay, USA, 2023 Search PubMed.
- (a) W. L. Li, H. C. Zheng, J. Bukuru and N. De Kimpe, J. Ethnopharmacol., 2004, 92, 1–21 CrossRef CAS; (b) B. S. Teng, C. D. Wang, H. J. Yang, J. S. Wu, D. Zhang, M. Zheng, Z. H. Fan, D. Pan and P. Zhou, J. Agric. Food Chem., 2011, 59, 6492–6500 CrossRef CAS; (c) K. Winska, W. Maczka, K. Gabryelska and M. Grabarczyk, Molecules, 2019, 24, 4075 CrossRef CAS PubMed.
- (a) C. Xiao, Q. Wu, J. Zhang, Y. Xie, W. Cai and J. Tan, J. Ethnopharmacol., 2017, 196, 47–57 CrossRef CAS PubMed; (b) H. Hikino, C. Konno, Y. Mirin and T. Hayashi, Planta Med., 1985, 339–340 CrossRef CAS PubMed; (c) M. Tomoda, R. Gonda, Y. Kasahara and H. Hikino, Phytochemistry, 1986, 25, 2817–2820 CrossRef CAS; (d) H. Hikino, M. Ishiyama, Y. Suzuki and C. Konno, Planta Med., 1989, 55, 423–428 CrossRef CAS PubMed.
- (a) M. C. A. Galappaththi, N. M. Patabendige, B. M. Premarathne, K. K. Hapuarachchi, S. Tibpromma, D.-Q. Dai, N. Suwannarach, S. Rapior and S. C. Karunarathna, Biomolecules, 2023, 13, 24 CrossRef CAS; (b) Q. Xia, H. Zhang, X. Sun, H. Zhao, L. Wu, D. Zhu, G. Yang, Y. Shao, X. Zhang, X. Mao, L. Zhang and G. She, Molecules, 2014, 19, 17478–17535 CrossRef PubMed.
- (a) J. Zhang, K. Ma, H. Chen, K. Wang, W. Xiong, L. Bao and H. Liu, J. Antibiot., 2017, 70, 915–917 CrossRef CAS; (b) K. Wang, L. Bao, K. Ma, J. Zhang, B. Chen, J. Han, J. Ren, H. Luo and H. Liu, Eur. J. Med. Chem., 2017, 127, 1035–1046 Search PubMed; (c) H. T. Ma, J. F. Hsieh and S. T. Chen, Phytochemistry, 2015, 114, 109–113 CrossRef CAS.
- (a) T. Kubota, Y. Asaka, I. Miura and H. Mori, Helv. Chim. Acta, 1982, 65, 611–619 CrossRef CAS; (b) S. Fatmawati, R. Kondo and K. Shimizu, Bioorg. Med. Chem. Lett., 2013, 23, 5900–5903 CrossRef CAS; (c) S. Fatmawati, K. Shimizu and R. Kondo, Planta Med., 2010, 76, 1691–1693 Search PubMed; (d) S. Fatmawati, K. Shimizu and R. Kondo, Fitoterapia, 2010, 81, 1033–1036 Search PubMed.
- (a) N. P. D. Nguyen, P. T. Quy, D. C. To, T. Q. Bui, N. V. Phu, T. T. A. My, P. H. Nguyen, N. H. Kien, N. T. T. Hai and N. T. A. Nhung, Trop. J. Nat. Prod. Res., 2023, 7, 3421–3432 Search PubMed; (b) L. Yang, D.-X. Kong, N. Xiao, Q.-Y. Ma, Q.-Y. Xie, J.-C. Guo, C. Y. Deng, H.-X. Ma, Y. Hua, H.-F. Dai and Y.-X. Zhao, Bioorg. Chem., 2022, 127, 106025 CrossRef CAS PubMed; (c) K. Wang, L. Bao, W. Xiong, K. Ma, J. Han, W. Wang, W. Yin and H. Liu, J. Nat. Prod., 2015, 78, 1977–1989 CrossRef CAS.
- (a) C. W. Phan, V. Sabaratnam and U. R. Kuppusamy, Antidiabetic Effects of Ganoderma Prospects and Challenges, in Ganoderma: Cultivation, Chemistry and Medicinal Applications, ed. K. Acharya and S. Khatua, CRC Press, Boca Raton, USA, 2024, vol, 1 Search PubMed; (b) X.-R. Peng, S. B. Unsicker, J. Gershenzon and M.-H. Qiu, Nat. Prod. Rep., 2023, 40, 1354–1392 RSC; (c) S. Fatmawati, K. Shimizu and R. Kondo, Phytomedicine, 2011, 18, 1053–1055 CrossRef CAS PubMed.
- (a) H. Rasouli, R. Yarani, F. Pociot and J. Popović-Djordjević, Pharmacol. Res., 2020, 155, 104723 CrossRef CAS; (b) S. Sukhikh, O. Babich, A. Prosekov, O. Kalashnikova, S. Noskova, A. Bakhtiyarova, O. Krol, E. Tsvetkova and S. Ivanova, Metabolites, 2023, 13, 513 CrossRef CAS PubMed; (c) S. B. Gaikwad, G. K. Mohan and M. S. Rani, Pharm. Crops, 2014, 5(Supp. 1: M2), 11–28 CrossRef.
- J. J. Yang and D. Q. Yu, Acta Pharm. Sin., 1990, 25, 555–559 CAS.
- L. Tian, X.-L. Wang, Y.-Z. Wang and Y.-X. Cheng, Nat. Prod. Res. Dev., 2015, 27, 1325–1328 CAS.
- X.-L. Wang, M. Dou, Q. Luo, L.-Z. Cheng, Y.-M. Yan, R.-T. Li and Y.-X. Cheng, Fitoterapia, 2017, 116, 93–98 Search PubMed.
- Y. Chen and P. Lan, ACS Omega, 2018, 3, 3471–3481 CrossRef CAS.
- S. Z. Huang, B. H. Cheng, Q. Y. Ma, Q. Wang, F. D. Kong, H. F. Dai, S. Q. Qiu, P. Y. Zheng, Z. Q. Liu and Y.-X. Zhao, RSC Adv., 2016, 6, 21139–21147 RSC.
- (a) I. Cortés, E. Cordisco, T. S. Kaufman, M. A. Sortino, L. A. Svetaz and A. B. J. Bracca, RSC Adv., 2021, 11, 19587–19597 RSC; (b) L. Fernández, A. L. Reviglio, D. A. Heredia, G. M. Morales, M. Santo, L. Otero, F. Alustiza, A. C. Liaudat, P. Bosch, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, Heliyon, 2021, 7, e06436 Search PubMed; (c) I. Cortés, C. M. Borini Etichetti, J. E. Girardini, A. B. J. Bracca and T. S. Kaufman, Synthesis, 2019, 433–440 Search PubMed; (d) J. L. Pergomet, M. G. Di Liberto, M. G. Derita, A. B. J. Bracca and T. S. Kaufman, Fitoterapia, 2018, 125, 98–105 Search PubMed; (e) E. L. Larghi, M. A. Operto, R. Torres and T. S. Kaufman, Eur. J. Med. Chem., 2012, 55, 74–84 CrossRef CAS.
- (a) D. F. Vargas, E. L. Larghi and T. S. Kaufman, Nat. Prod. Rep., 2019, 39, 354–401 RSC; (b) D. A. Heredia, E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2016, 1397–1404 CrossRef CAS; (c) C. C. Silveira, E. L. Larghi, S. R. Mendes, A. B. J. Bracca, F. Rinaldi and T. S. Kaufman, Eur. J. Org. Chem., 2009, 4637–4645 CrossRef CAS.
- (a) Y. Satoh, J. L. Stanton, A. J. Hutchison, A. H. Libby, T. J. Kowalski, W. H. Lee, D. H. White and E. F. Kimble, J. Med. Chem., 1993, 36, 3580–3594 Search PubMed; (b) Y. Zhou, Y. Zhu, S. Yan and Y. Gong, Angew. Chem., Int. Ed., 2013, 52, 10265–10269 CrossRef CAS; (c) S. Y. Shin, H. Jung, S. Ahn, D. Hwang, H. Yoon, J. Hyun, Y. Yong, H. J. Cho, D. Koh, Y. H. Lee and Y. Lim, Bioorg. Med. Chem., 2014, 22, 1809–1820 CrossRef CAS.
- (a) H. Adkins and W. H. Hartung, Org. Synth. Coll., 1941, 1, 15 Search PubMed; (b) W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, 4th edn, Butterworth-Heinemann, Oxford, UK, 2017 Search PubMed.
- T. G. F. Marsh, A. Santambrogio, B. J. Murray, L. T. Boulton, J. A. Aguilar, D. S. Yufit, G. Sandford and W. D. G. Brittain, Eur. J. Org. Chem., 2023, e202300058 Search PubMed.
- Y. Satoh, J. L. Stanton, A. J. Hutchison, A. H. Libby, T. J. Kowalski, W. H. Lee, D. H. White and E. F. Kimble, J. Med. Chem., 1993, 36, 3580–3594 CrossRef CAS.
- S. L. Xu, C. P. Li and J. H. Li, Synlett, 2009, 0818–0822 CAS.
- (a) S. Hirai, Y. Horikawa, H. Asahara and N. Nishiwaki, Chem. Commun., 2017, 53, 2390–2393 RSC; (b) M. Arita, S. Yokoyama, H. Asahara and N. Nishiwaki, Synthesis, 2019, 2007–2013 Search PubMed.
- Y. Zhao, J. Zhao, Y. Zhou, Z. Lei, L. Li and H. Zhang, New J. Chem., 2005, 29, 769–772 RSC.
- S. Hirai, Y. Horikawa, H. Asahara and N. Nishiwaki, Chem. Commun., 2017, 53, 2390–2393 RSC.
- (a) S. Rana, H. S. Hong, L. Barrigan, L. W. Jin and D. H. Hua, Bioorg. Med. Chem. Lett., 2009, 19, 670–674 CrossRef CAS; (b) F. Liéby-Muller, C. Allais, T. Constantieux and J. Rodriguez, Chem. Commun., 2008, 4207–4209 RSC.
- G. Bartoli, K. Babiuch, M. Bosco, A. Carlone, P. Galzerano, P. Melchiorre and L. Sambri, Synlett, 2007, 2897–2901 CAS.
- (a) V. P. A. Raja, G. Tenti, S. Perumal and J. C. A. Menéndez, Chem. Commun., 2014, 50, 12270–12272 RSC; (b) M. Venkati and G. L. D. Krupadanam, Synth. Commun., 2001, 31, 2589–2598 CrossRef CAS.
- (a) J. Lombard, S. Romain, S. Dumas, J. Chauvin, M. N. Collomb, D. Daveloose, A. Deronzier and J. C. Leprêtre, Eur. J. Inorg. Chem., 2005, 3320–3330 CrossRef CAS; (b) G. Serdaroğlu, N. Uludag and E. Üstün, J. Mol. Liq., 2023, 375, 121364 CrossRef.
- (a) F. Zou, F. Pei, L. Wang, Z. Ren, X. Cheng, Y. Sun, J. Wu and P. He, Synlett, 2019, 717–720 CAS; (b) A. Kundu, B. Bhattacharyya, K. Dhara, S. Paul, I. Majumdere and R. Kundue, New J. Chem., 2020, 44, 4898–4906 RSC; (c) X. Chang, X. Yang, Z. Chen and W. Zhong, Synlett, 2019, 1431–1436 CAS.
- (a) S. Kantevari, D. Addla and B. Sridhar, Synthesis, 2010, 3745–3754 CrossRef CAS; (b) P. N. Ngubane, T. Papo, S. Zamisa and B. Omondi, J. Mol. Struct., 2025, 1322, 140567 Search PubMed.
- (a) D. Sang, C. Yi, C. He, J. Wang, J. Tian, M. Yao and H. Shi, Tetrahedron Lett., 2018, 59, 1469–1472 Search PubMed; (b) J. Tian, H. Yue, P. Yang and D. Sang, ChemistrySelect, 2019, 4, 38–41 CrossRef CAS; (c) T. S. Kaufman, J. Chem. Soc., Perkin Trans. 1, 1996, 2497–2505 RSC.
- (a) Y. Ishihara, S. Azuma, T. Choshi, K. Kohno, K. Ono, H. Tsutsumi, T. Ishizu and S. Hibino, Tetrahedron, 2011, 67, 1320–1333 CrossRef CAS; (b) R. S. Porto, M. L. A. A. Vasconcellos, E. Ventura and F. Coelho, Synthesis, 2005, 2297–2306 CAS; (c) M. Süsse, S. Johne and S. Hesse, Helv. Chim. Acta, 1992, 75, 457–470 CrossRef; (d) Y. Satoh, J. L. Stanton, A. J. Hutchison, A. H. Libby, T. J. Kowalski, W. H. Lee, D. H. White and E. F. Kimble, J. Med. Chem., 1993, 36, 3580–3594 CrossRef CAS.
- (a) T. S. Kaufman, R. P. Srivastava, R. D. Sindelar, S. M. Scesney and H. C. Marsh Jr., J. Med. Chem., 1996, 38, 1437–1445 CrossRef; (b) T. S. Kaufman, R. P. Srivastava, R. D. Sindelar, S. M. Scesney and H. C. Marsh Jr., Bioorg. Med. Chem. Lett., 1995, 5, 501–506 CrossRef CAS.
- (a) M. C. Bröhmer, N. Volz and S. Bräse, Synlett, 2009, 1383–1386 Search PubMed; (b) A. Modak, A. Deb, T. Patra, S. Rana, S. Maity and D. Maiti, Chem. Commun., 2012, 48, 4253–4255 RSC; (c) Z. Selakovic, A. M. Nikolic, V. Ajdacic and I. M. Opsenica, Eur. J. Org. Chem., 2022, e202101265 CrossRef CAS; (d) B. Gutmann, P. Elsner, T. Glasnov, D. M. Roberge and C. O. Kappe, Angew. Chem., Int. Ed., 2014, 53, 11557–11561 CrossRef CAS PubMed.
- (a) M. C. Bröhmer, N. Volz and S. Bräse, Synlett, 2009, 1383–1386 Search PubMed; (b) G. Hall, A. Samat, R. Guglielmetti, L. Van Parys, W. Saeyens, D. De Keukeleir, K. Lorenz and A. Mannscbreck, Helv. Chim. Acta, 1997, 80, 1122–1132 CrossRef.
- (a) J. Xia and G.-W. Wang, Synthesis, 2005, 2379–2383 Search PubMed; (b) K. V. Maslov, T. I. Akimova and V. A. Kaminski, Chem. Heterocycl. Compd., 2002, 38, 417–421 CrossRef CAS; (c) W. Shang, S. Li, Y. Ding, Z. Pan, X. Tong and C. Xia, Org. Chem. Front., 2017, 4, 1789–1793 RSC; (d) S. J. Garden, C. R. Werneck Guimaraes, M. B. Corréa, C. A. Fernandes de Oliveira, A. da Cunha Pinto and R. Bicca de Alencastro, J. Org. Chem., 2003, 68, 8815–8822 Search PubMed; (e) S. Bahmanyar and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 11273–11283 CrossRef CAS PubMed.
- Y. Cao, J. S. M. Perry, E. Zhang, A. Trinh, A. Kacker, S. Cruz, H. Ceballos, A. Pan, W. Huang and K. G. M. Kou, JACS Au, 2025, 5, 1429–1438 CrossRef CAS.
- (a) M. I. Osella, M. O. Salazar, M. D. Gamarra, D. M. Moreno, F. Lambertucci, D. E. Frances and R. L. E. Furlan, RSC Med. Chem., 2020, 11, 518–527 RSC; (b) M. R. Eftink and J. C. Harrison, Bioorg. Chem., 1981, 10, 388–398 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (c) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalk, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed; (d) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS; (e) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- M. O. Salazar, M. I. Osella, I. A. Ramallo and R. L. E. Furlan, RSC Adv., 2018, 8, 36209–36218 RSC.
- (a) CCDC 2501681: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pz6d9; (b) CCDC 2501682: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pz6fb.
| This journal is © The Royal Society of Chemistry 2026 |